Embed Size (px)
Citation preview

Pompe Disease in Infants: Improving the Prognosis byNewborn Screening and Early Treatment
WHAT’S KNOWN ON THIS SUBJECT: Newborn screening programis now technically feasible in ultrarare diseases and cansuccessfully identify patients with Pompe disease before
symptoms have progressed to the point where they would be likely tobe identified through routine pediatric care.
WHAT THIS STUDY ADDS: This is the first report of improvedclinical outcomes in patients with Pompe disease who were identifiedby a national newborn-screening program and were treated before
the onset of clinically recognizable symptoms of Pompe disease.
abstract +
OBJECTIVE: Pompe disease causes progressive, debilitating, and oftenlife-threatening musculoskeletal, respiratory, and cardiac symptoms.Favorable outcomes with early intravenous enzyme-replacement ther-apy and alglucosidase alfa have been reported, but early clinical diag-nosis before the development of severe symptoms has rarely beenpossible in infants.
METHODS: We recently conducted a newborn screening pilot programin Taiwan to improve the early detection of Pompe disease. Six of206 088 newborns screened tested positive and were treated forPompe disease. Five had the rapidly progressive form of Pompe dis-ease, characterized by cardiac and motor involvement, and weretreated soon after diagnosis. The sixth patient was started on treat-ment at 14 months of age because of progressive muscle weakness.Outcomes were compared with treated patients whose disease wasdiagnosed clinically and with untreated historical control subjects.
RESULTS: At the time of this report, patients had been treated for 14 to32 months. The 5 infants who had early cardiac involvement demon-strated normalization of cardiac size and muscle pathology with nor-mal physical growth and age-appropriate gains in motor development.The infant without cardiac involvement also achieved normal motordevelopment with treatment. Survival in patients who had newbornscreening was significantly improved compared with those in the un-treated reference cohort (P � .001). Survival in the treated clinicalcomparators was reduced but not statistically different from that inthe newborn screening group (P� .48).
CONCLUSIONS: Results from this study indicate that early treatmentcan benefit infants with Pompe disease and highlight the advantages ofearly diagnosis, which can be achieved by newborn screening.Pediatrics 2009;124:e1116–e1125
AUTHORS: Yin-Hsiu Chien, MD,a,b Ni-Chung Lee, MD,a,b
Beth L. Thurberg, MD, PhD,c Shu-Chuan Chiang, MSc,b
Xiaokui Kate Zhang, PhD,c Joan Keutzer, PhD,c Ai-ChuHuang, MSc,b Mei-Hwan Wu, MD, PhD,a Pei-Hsin Huang,MD, PhD,d Fuu-Jen Tsai, MD, PhD,e Yuan-Tsong Chen, MD,PhD,f and Wuh-Liang Hwu, MD, PhDa,b,e
Departments of aPediatrics, bMedical Genetics, and dPathology,National Taiwan University Hospital and National TaiwanUniversity School of Medicine, Taipei, Taiwan; cGenzymeCorporation, Cambridge, Massachusetts; eGraduate Institute ofIntegrated Medicine, College of Chinese Medicine, China MedicalUniversity, Taichung, Taiwan; and fInstitute of BiomedicalScience, Academia Sinica, Taipei, Taiwan
KEY WORDSinborn errors of metabolism, neuromuscular disorders,newborn screening, outcome, treatment
ABBREVIATIONSGAA—acid �-glucosidaserhGAA—recombinant human acid �-glucosidaseCRIM—cross-reactive immunologic materialDBS—dried blood spotNTUH—National Taiwan University HospitalNAG—neutral glucosidaseCXR—chest radiographECG—electrocardiogramCK—creatinine kinaseBNP—b-type natriuretic peptideLVMI—left ventricular mass indexPAS—periodic acid-SchiffH&E—hematoxylin and eosinAIMS—Alberta Infant Motor ScalesPDMS-II—Peabody Developmental Motor Scales, Second Edition
www.pediatrics.org/cgi/doi/10.1542/peds.2008-3667
doi:10.1542/peds.2008-3667
Accepted for publication Jun 15, 2009
Address correspondence to Wuh-Liang Hwu, MD, PhD, NationalTaiwan University Hospital, Department of Pediatrics andMedical Genetics, 8 Chung-Shan South Road, Taipei 10041,Taiwan. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2009 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE: Drs Thurberg, Zhang, and Keutzer arefull-time employees of Genzyme Corporation; Dr Chien hasreceived honoraria and research grant funding from Genzyme;Dr Chen has received research grant funding from and servesas a consultant for Genzyme; Dr Hwu has received honoraria,travel funding, and research grant funding from and serves onan advisory board for Genzyme. The other authors haveindicated they have no financial relationships relevant to thisarticle to disclose.
e1116 CHIEN et al by guest on September 12, 2018www.aappublications.org/newsDownloaded from

Pompe disease (glycogen storage dis-ease type II, acid maltase deficiency)was first described by Dr JohannesPompe in 1932 in an infant who hadrapidly progressive cardiomyopathyand was found at autopsy to have mas-sive deposition of glycogen in the heartand skeletal muscle.1 Pompe diseasewas classified as a lysosomal storagedisorder when it was determined thatthe primary pathology resulted from adeficit of the lysosomal enzyme acid�-glucosidase (GAA) (EC 3.2.1.20).2
Worldwide incidence of Pompe diseaseis estimated to be 1 in 40 0002 and maybe higher in certain regions (Israel3
and Taiwan4).
Pompe disease has been classified asinfantile onset or late onset, although acontinuum in disease severity, indica-tive of a single disease with a variableage at onset and rate of progression, isfound. The most severe form is typi-cally diagnosed in infants between 3and 5months of age during the assess-ment of a respiratory infection, cardio-megaly, or hypotonia. In natural his-tory studies in infants, the gapbetween diagnosis and ventilator useor death was 1 to 2 months; nearly allinfants died by the age of 18 months.5,6
A Phase 2/3 trial of enzyme-replacementtherapy with recombinant human GAA(rhGAA) (alglucosidase alfa [GenzymeCorporation, Cambridge, MA]) in 18 pa-tients who were �6 months of age atenrollment (corrected for gestationalage)7 showed that all patients survivedafter 1 year of treatment, and 15 in-fants reached 18 months of age; how-ever, 3 patients required invasive ven-tilatory assistance, 5 were tube-fed,and 5 made no gains in motor develop-ment.7 In a separate clinical trial of in-fants and children who were between3 and 43 months of age at enrollmentand had more advanced disease pro-gression, 15 of 21 patients survived tothe end of the study (median treatmentduration: �2 years), 7 of 16 patients
whowere free of invasive ventilation atbaseline remained so, and 13 of thetotal 21 patients demonstrated im-provements in motor development.8
These results suggest that earliertherapeutic intervention, before exten-sive muscle damage, could result inbetter outcomes.
Cross-reactive immunologic material(CRIM) status may also affect treatmentoutcomes in infantile-onset Pompedisease. CRIM-negative infants haveshown poorer outcomes, even withearly initiation of treatment.7,9,10 CRIM-positive patients have mutations thatresult in the production of endogenousGAA protein with little, if any, enzymeactivity. The presence of endogenousGAA protein may confer some immuno-logic protection or tolerance against thedevelopment of anti-rhGAA antibodies,because the exogenous enzyme is notperceived as foreign protein by the im-mune system. CRIM-negative infantshave no detectable endogenous enzymeand tend to develop high and sustainedlevels of antibodies to exogenously ad-ministered enzyme; therefore, any evalu-ation of the impact of treatment in in-fantswith Pompediseasemust considerCRIM status.
We recently conducted a large-scalenewborn screening pilot program toevaluate the impact of early detection inand treatment of infantswith Pompedis-ease.11,12 Between October 1, 2005, andDecember 31, 2007, �45% of all new-borns in Taiwan had GAA activity mea-sured in dried blood spots (DBSs). Of206 088 newborns screened, 6 cases ofPompediseasewerediagnosed.Herewereport outcomes in those 6 patients.
METHODS
Screening Program
The Pompe disease newborn screen-ing program was approved by the in-stitutional review board of the NationalTaiwan University Hospital (NTUH). Itwas integrated into the preexisting
newborn screening program at theNTUH Screening Center. Taiwan doesnot require informed consent for new-born screening; however, this pilotscreening program was reviewed withparents prenatally, and written in-formed consent for screening from 1parent per infant was required. Theroutine newborn screening DBS speci-men was used for Pompe diseasescreening via enzyme assay, which wasconducted as previously described.11
Briefly, GAA enzyme activity of �55% ofthe populationmean triggered retestingof GAA activity, as well as total neutralglucosidase (NAG) activity in the originalDBS. When the NAG/GAA ratio was�100,the newborn was recalled immediatelyfor confirmatory testing in whole blood;otherwise, when GAA activity was�25%and NAG/GAA ratio was �25, a secondDBS was obtained and tested. When inthe second DBS GAA activity was �8%,NAG/GAA ratio was�60, and percentageof total GAA inhibition was�80%, the in-fant was referred to NTUH for diagnosticconfirmation via GAA activity assay inwhole blood and clinical evaluation.
Clinical Evaluation
For diagnostic confirmation, physical ex-amination, chest radiographs (CXRs),and electrocardiograms (ECGs) wereperformed. When cardiac involvementwas suspected by CXR or ECG, an echo-cardiogram was performed. When hy-pertrophic cardiomyopathy was con-firmed, the infant was admitted to thehospital for additional baseline evalua-tions and initiation of treatment with al-glucosidase alfa. When no cardiac in-volvement was found, the patient wasfollowed closely but no treatment wasinitiated.
Baseline Ancillary Testing
Blood chemistry included creatininekinase (CK) and B-type natriuretic pep-tide (BNP). Ejection fraction, thickness ofthe ventricular walls and interventricu-lar septum, left ventricle end-systolic di-
ARTICLES
PEDIATRICS Volume 124, Number 6, December 2009 e1117 by guest on September 12, 2018www.aappublications.org/newsDownloaded from

mension, left ventricle end-diastolic di-mension, and calculated left ventricularmass index (LVMI) were measuredwith echocardiography. Fibroblastswere obtained by skin biopsy for addi-tional confirmation of GAA deficiencyby enzyme assay. Quadriceps musclebiopsy specimens were obtained afterinformed consent. Muscle specimenswere fixed in glutaraldehyde, pro-cessed for high-resolution light mi-croscopy, and stained with periodicacid-Schiff (PAS) as previously de-scribed.13 Additional tissuewas fixed informalin, paraffin processed, andstained with hematoxylin and eosin(H&E) stain. Informed consent was ob-tained for DNA extraction for mutationanalysis and for data collection.
Treatment Protocol
Patients with confirmed cardiomyop-athy at diagnosis were treated withalglucosidase alfa (manufactured at2000-L bioreactor capacity); asymp-tomatic patients were treated whenPompe-associated symptoms appeared.Treatment costswerereimbursedby theTaiwan Bureau of National Health Insur-ance. After baseline evaluations, intrave-nous alglucosidase alfa (20 mg/kg) wasgiven every other week, infused accord-ing to the product insert. Safety, includ-ing infusion-associated reactions, wasmonitored during the entire treatmentperiod. All patients underwent a physicaltherapy regimen, including extremityand trunk muscle power training; exer-cises to enhance head support fromprone and supine positions; and exer-cises to assist with sitting, standing, andsolid food intake.
GAA Activity, Genotyping, andWestern Blot Analysis
GAA activity in mononuclear cells and/orfibroblasts was measured by using theartificial substrate 4-methylumbelliferyl-�-D-glucopyroside (Fluka Chemical Corp,Ronkonkoma,NY). GenomicDNA frompe-ripheral blood cells was used for muta-
tion analysis of the GAA gene.14 CRIM sta-tus was determined by Western blotanalysis.
Follow-up Clinical and AncillaryTesting
Blood chemistry tests were performedmonthly. Motor and gastrointestinalevaluations were repeated every 3months. Quadriceps muscle biopsy,from the opposite leg used for the base-line biopsy, was repeated 6 months af-ter the first infusion.
The Alberta Infant Motor Scale (AIMS)and the Peabody Developmental MotorScale, Second Edition (PDMS-II), wereused. The AIMS is an observationalmeasure of infant motor performancethat can be administered from birththrough the age of independent walk-ing. It assesses the sequential devel-opment of motor milestones.15 ThePDMS-II is a skill-based measure ofgross and fine motor development forinfants and children from 6 months to6 years of age and consists of 4 grossmotor and 2 fine motor subtests.16
Clinical Comparators
Ten infants who had Pompe disease di-agnosed on the basis of clinical mani-festations rather than NBS and weretreated with rhGAA at NTUH were in-cluded as clinical comparators. Ofthese, 5 received a diagnosis after clin-ical presentation in populations thatwere not covered by the screening pro-gram (CLN1–CLN5), and the otherswere identified and treated betweenDecember 2002 and the start ofthe screening program (CLN6–CLN10).Eleven patients from NTUH who diedbefore the availability of enzyme-replacement therapy and who weredescribed in a previous natural historysurvey5 were used as untreated histor-ical comparators. Survival was com-pared among the 3 cohorts by usingKaplan-Meier analysis.17
RESULTS
Cases Detected by the ScreeningProgram
Six of 206 088 newborns screenedtested positive for Pompe disease(referred to as NBS1–NBS6) by DBSscreen and confirmatory enzyme activ-ity assay in whole blood. The infantswere aged 7 days to 40 days at diagno-sis; baseline characteristics are sum-marized in Table 1. All were born tohealthy, nonconsanguineous parents;none had obvious clinical symptoms atbirth; and all were CRIM-positive (Fig 5,which is published as supporting infor-mation at www.pediatrics.org/content/full/124/6/e1116). The 2 cases identifiedsince the publication of the initial resultsof the screening program,11 NBS5 andNBS6, both were recalled for confirma-tory testing in whole blood immediatelyafter the results of the first DBS testwere known. Five patients (NBS2–NBS6)had cardiomyopathy at the time of diag-nosis; NBS1, who did not, is presentedseparately.
Infants With Cardiac Involvement
Clinical Presentation
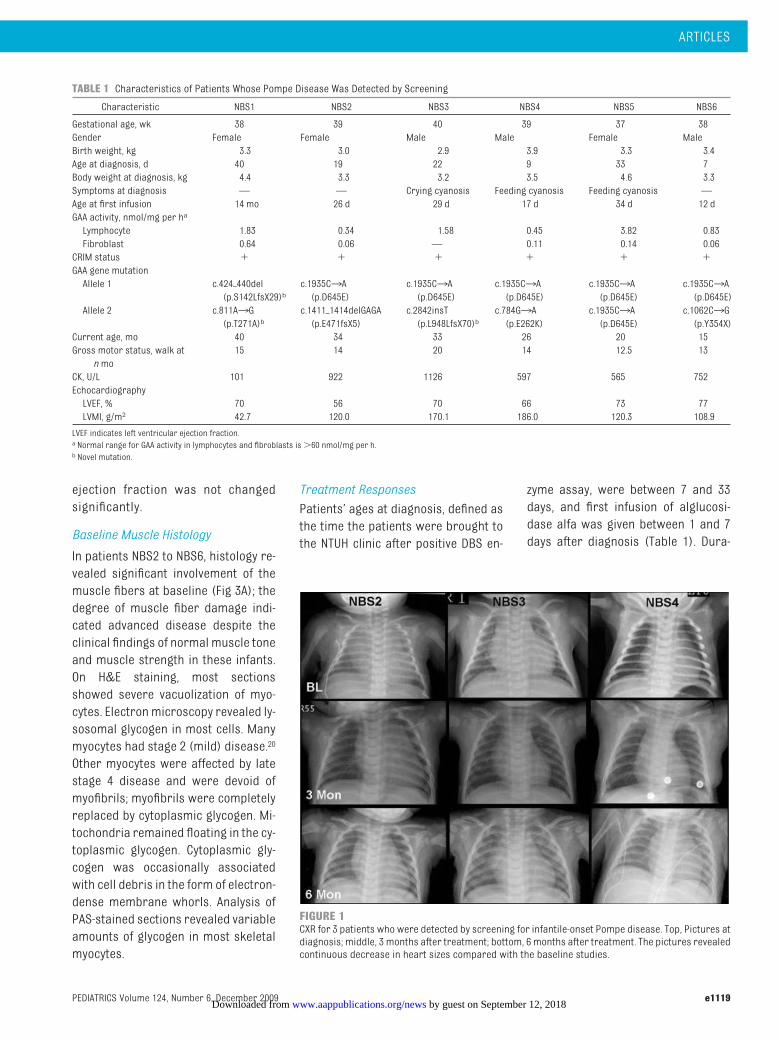
CXR in the 5 patients with hypertrophiccardiomyopathy (NBS2–NBS6) revealedprominent heart shadows (Fig 1). ECGshowed biventricular hypertrophy, al-though the PR intervals were not signifi-cantly shortened. BNPandCK levelsweremarkedly elevated (Fig 2).18 Despite thepresence of significant cardiac disease,these infants showedno signs of cardiacdisease at rest and had normal routinephysical examinations and normalweight gain. Three infants (NBS3–NBS5)demonstrated cyanosis with crying orfeeding. Echocardiography revealedthickened ventricular walls and inter-ventricular septum in all 5 infants. LVMIwassignificantly increased (108.9–186.0g/m2; reference range19: 47.4 � 6.2g/m2), although the left ventricular
e1118 CHIEN et al by guest on September 12, 2018www.aappublications.org/newsDownloaded from
ejection fraction was not changedsignificantly.
Baseline Muscle Histology
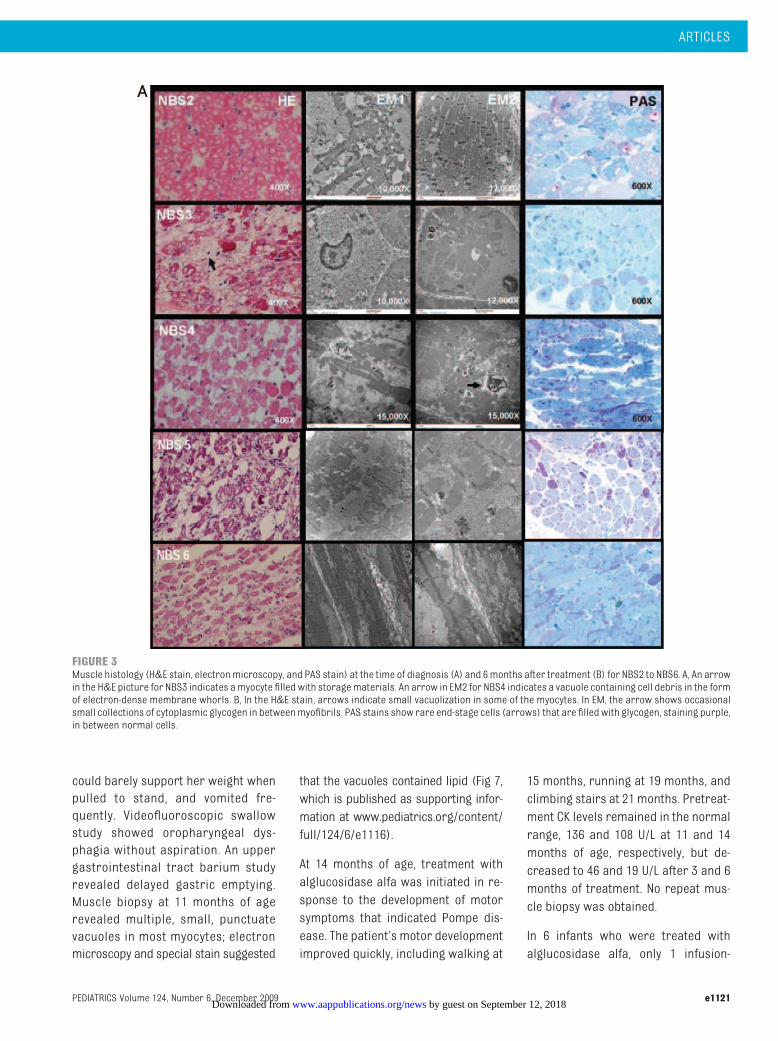
In patients NBS2 to NBS6, histology re-vealed significant involvement of themuscle fibers at baseline (Fig 3A); thedegree of muscle fiber damage indi-cated advanced disease despite theclinical findings of normal muscle toneand muscle strength in these infants.On H&E staining, most sectionsshowed severe vacuolization of myo-cytes. Electronmicroscopy revealed ly-sosomal glycogen in most cells. Manymyocytes had stage 2 (mild) disease.20
Other myocytes were affected by latestage 4 disease and were devoid ofmyofibrils; myofibrils were completelyreplaced by cytoplasmic glycogen. Mi-tochondria remained floating in the cy-toplasmic glycogen. Cytoplasmic gly-cogen was occasionally associatedwith cell debris in the form of electron-dense membrane whorls. Analysis ofPAS-stained sections revealed variableamounts of glycogen in most skeletalmyocytes.
Treatment Responses
Patients’ ages at diagnosis, defined asthe time the patients were brought tothe NTUH clinic after positive DBS en-
zyme assay, were between 7 and 33days, and first infusion of alglucosi-dase alfa was given between 1 and 7days after diagnosis (Table 1). Dura-
FIGURE 1CXR for 3 patients who were detected by screening for infantile-onset Pompe disease. Top, Pictures atdiagnosis; middle, 3 months after treatment; bottom, 6 months after treatment. The pictures revealedcontinuous decrease in heart sizes compared with the baseline studies.
TABLE 1 Characteristics of Patients Whose Pompe Disease Was Detected by Screening
Characteristic NBS1 NBS2 NBS3 NBS4 NBS5 NBS6
Gestational age, wk 38 39 40 39 37 38Gender Female Female Male Male Female MaleBirth weight, kg 3.3 3.0 2.9 3.9 3.3 3.4Age at diagnosis, d 40 19 22 9 33 7Body weight at diagnosis, kg 4.4 3.3 3.2 3.5 4.6 3.3Symptoms at diagnosis — — Crying cyanosis Feeding cyanosis Feeding cyanosis —Age at first infusion 14 mo 26 d 29 d 17 d 34 d 12 dGAA activity, nmol/mg per ha
Lymphocyte 1.83 0.34 1.58 0.45 3.82 0.83Fibroblast 0.64 0.06 — 0.11 0.14 0.06CRIM status � � � � � �GAA gene mutationAllele 1 c.424�440del
(p.S142LfsX29)bc.1935C3A(p.D645E)
c.1935C3A(p.D645E)
c.1935C3A(p.D645E)
c.1935C3A(p.D645E)
c.1935C3A(p.D645E)
Allele 2 c.811A3G(p.T271A)b
c.1411�1414delGAGA(p.E471fsX5)
c.2842insT(p.L948LfsX70)b
c.784G3A(p.E262K)
c.1935C3A(p.D645E)
c.1062C3G(p.Y354X)
Current age, mo 40 34 33 26 20 15Gross motor status, walk atn mo
15 14 20 14 12.5 13
CK, U/L 101 922 1126 597 565 752EchocardiographyLVEF, % 70 56 70 66 73 77LVMI, g/m2 42.7 120.0 170.1 186.0 120.3 108.9
LVEF indicates left ventricular ejection fraction.a Normal range for GAA activity in lymphocytes and fibroblasts is�60 nmol/mg per h.b Novel mutation.
ARTICLES
PEDIATRICS Volume 124, Number 6, December 2009 e1119 by guest on September 12, 2018www.aappublications.org/newsDownloaded from
tion of treatment was between 14months and 33 months for these pa-tients. All patients survived, remainedfree of mechanical ventilation, and hadnormal growth for the duration of thestudy. Heights were within normal Tai-wanese ranges, but NBS3’s weight wasbelow the 10th percentile at all timepoints.
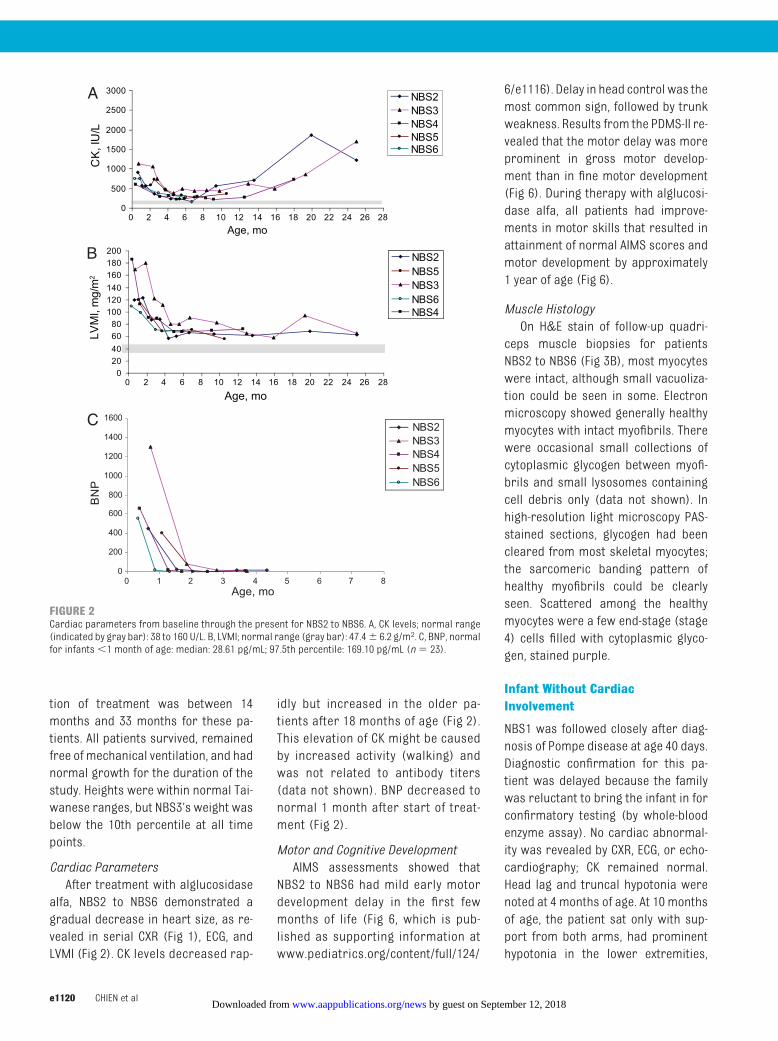
Cardiac ParametersAfter treatment with alglucosidase
alfa, NBS2 to NBS6 demonstrated agradual decrease in heart size, as re-vealed in serial CXR (Fig 1), ECG, andLVMI (Fig 2). CK levels decreased rap-
idly but increased in the older pa-tients after 18 months of age (Fig 2).This elevation of CK might be causedby increased activity (walking) andwas not related to antibody titers(data not shown). BNP decreased tonormal 1 month after start of treat-ment (Fig 2).
Motor and Cognitive DevelopmentAIMS assessments showed that
NBS2 to NBS6 had mild early motordevelopment delay in the first fewmonths of life (Fig 6, which is pub-lished as supporting information atwww.pediatrics.org/content/full/124/
6/e1116). Delay in head control was themost common sign, followed by trunkweakness. Results from the PDMS-II re-vealed that the motor delay was moreprominent in gross motor develop-ment than in fine motor development(Fig 6). During therapy with alglucosi-dase alfa, all patients had improve-ments in motor skills that resulted inattainment of normal AIMS scores andmotor development by approximately1 year of age (Fig 6).
Muscle HistologyOn H&E stain of follow-up quadri-
ceps muscle biopsies for patientsNBS2 to NBS6 (Fig 3B), most myocyteswere intact, although small vacuoliza-tion could be seen in some. Electronmicroscopy showed generally healthymyocytes with intact myofibrils. Therewere occasional small collections ofcytoplasmic glycogen between myofi-brils and small lysosomes containingcell debris only (data not shown). Inhigh-resolution light microscopy PAS-stained sections, glycogen had beencleared from most skeletal myocytes;the sarcomeric banding pattern ofhealthy myofibrils could be clearlyseen. Scattered among the healthymyocytes were a few end-stage (stage4) cells filled with cytoplasmic glyco-gen, stained purple.
Infant Without CardiacInvolvement
NBS1 was followed closely after diag-nosis of Pompe disease at age 40 days.Diagnostic confirmation for this pa-tient was delayed because the familywas reluctant to bring the infant in forconfirmatory testing (by whole-bloodenzyme assay). No cardiac abnormal-ity was revealed by CXR, ECG, or echo-cardiography; CK remained normal.Head lag and truncal hypotonia werenoted at 4 months of age. At 10 monthsof age, the patient sat only with sup-port from both arms, had prominenthypotonia in the lower extremities,
020406080
100120140160180200
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28
Age, mo
LVM
I, m
g/m
2
NBS2
NBS3
NBS4
NBS5
NBS6
0
500
1000
1500
2000
2500
3000
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28Age, mo
CK
, IU
/LNBS2NBS3NBS4NBS5NBS6
0
200
400
600
800
1000
1200
1400
1600
0 1 2 3 4 5 6 7 8Age, mo
BN
P
NBS2NBS3NBS4NBS5NBS6
A
B
C
FIGURE 2Cardiac parameters from baseline through the present for NBS2 to NBS6. A, CK levels; normal range(indicated by gray bar): 38 to 160 U/L. B, LVMI; normal range (gray bar): 47.4� 6.2 g/m2. C, BNP, normalfor infants�1 month of age: median: 28.61 pg/mL; 97.5th percentile: 169.10 pg/mL (n� 23).
e1120 CHIEN et al by guest on September 12, 2018www.aappublications.org/newsDownloaded from
could barely support her weight whenpulled to stand, and vomited fre-quently. Videofluoroscopic swallowstudy showed oropharyngeal dys-phagia without aspiration. An uppergastrointestinal tract barium studyrevealed delayed gastric emptying.Muscle biopsy at 11 months of agerevealed multiple, small, punctuatevacuoles in most myocytes; electronmicroscopy and special stain suggested
that the vacuoles contained lipid (Fig 7,which is published as supporting infor-mation at www.pediatrics.org/content/full/124/6/e1116).
At 14 months of age, treatment withalglucosidase alfa was initiated in re-sponse to the development of motorsymptoms that indicated Pompe dis-ease. The patient’s motor developmentimproved quickly, including walking at
15 months, running at 19 months, andclimbing stairs at 21 months. Pretreat-ment CK levels remained in the normalrange, 136 and 108 U/L at 11 and 14months of age, respectively, but de-creased to 46 and 19 U/L after 3 and 6months of treatment. No repeat mus-cle biopsy was obtained.
In 6 infants who were treated withalglucosidase alfa, only 1 infusion-
FIGURE 3Muscle histology (H&E stain, electronmicroscopy, and PAS stain) at the time of diagnosis (A) and 6months after treatment (B) for NBS2 to NBS6. A, An arrowin the H&E picture for NBS3 indicates amyocyte filled with storagematerials. An arrow in EM2 for NBS4 indicates a vacuole containing cell debris in the formof electron-dense membrane whorls. B, In the H&E stain, arrows indicate small vacuolization in some of the myocytes. In EM, the arrow shows occasionalsmall collections of cytoplasmic glycogen in betweenmyofibrils. PAS stains show rare end-stage cells (arrows) that are filled with glycogen, staining purple,in between normal cells.
ARTICLES
PEDIATRICS Volume 124, Number 6, December 2009 e1121 by guest on September 12, 2018www.aappublications.org/newsDownloaded from

associated reaction occurred, in pa-tient NBS5 6 months after treatmentinitiation. The reaction was mild andmanifested as transient skin rash. Sev-eral episodes of bronchiolitis oc-curred in NBS1, and 1 episode of gas-troenteritis and fever occurred inNBS4, which was assessed to be unre-lated to enzyme administration.
Clinical Comparators
All 10 clinical comparators were symp-tomatic at diagnosis and were CRIM-positive. These patients have been
treated for between 14 months and 76months. Two were treated at 6 monthsof age and died at ages 20 months and48 months with no motor developmentgains (Table 2, which is published assupporting information at www.pedi-atrics.org/content/full/124/6/e1116).Another 2 patients, who were treatedat 6 months of age, were dependent onrespiratory support through tracheos-tomy 24 hours a day by ages 64 and 73months; they achieved no motor devel-opment. One patient’s disease was di-agnosed and treated at the age of 2
months because of an audible heartmurmur. He had near-normal motormilestones; however, another patientwhose disease was diagnosed andtreated at the age of 2months becauseof elevated liver enzymes (alaninetransaminase and aspartate transam-inase) had delayed development. Theother 4 patients, whose disease wasdiagnosed between the ages of 4months and 5months and began treat-ment between the ages of 4 monthsand 6 months, all had delays in motordevelopment.
FIGURE 3Continued.
e1122 CHIEN et al by guest on September 12, 2018www.aappublications.org/newsDownloaded from
Outcomes Comparison AmongNewborn Screening, ClinicalComparators, and UntreatedReference Cohorts
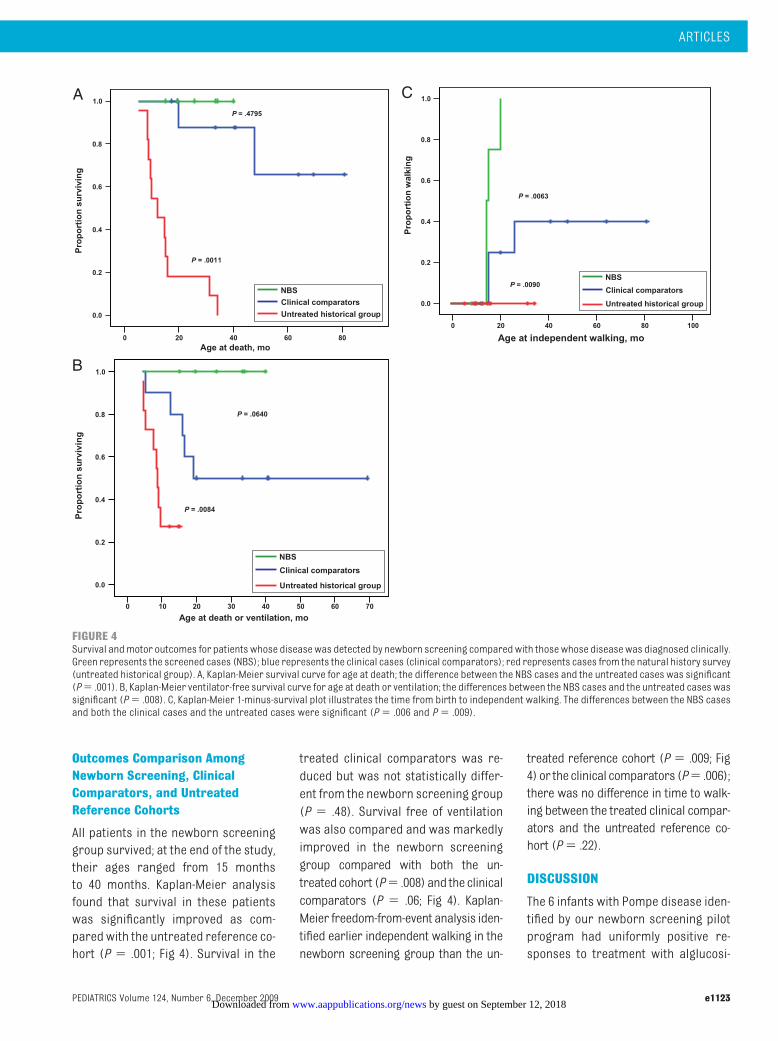
All patients in the newborn screeninggroup survived; at the end of the study,their ages ranged from 15 monthsto 40 months. Kaplan-Meier analysisfound that survival in these patientswas significantly improved as com-pared with the untreated reference co-hort (P � .001; Fig 4). Survival in the
treated clinical comparators was re-duced but was not statistically differ-ent from the newborn screening group(P � .48). Survival free of ventilationwas also compared and was markedlyimproved in the newborn screeninggroup compared with both the un-treatedcohort (P� .008) and the clinicalcomparators (P � .06; Fig 4). Kaplan-Meier freedom-from-event analysis iden-tified earlier independent walking in thenewborn screening group than the un-
treated reference cohort (P � .009; Fig4)or theclinical comparators (P� .006);there was no difference in time to walk-ing between the treated clinical compar-ators and the untreated reference co-hort (P� .22).
DISCUSSION
The 6 infants with Pompe disease iden-tified by our newborn screening pilotprogram had uniformly positive re-sponses to treatment with alglucosi-
0 20 40 60 80
Age at death, mo
0.0
0.2
0.4
0.6
0.8
1.0
NBS
Clinical comparators
Untreated historical group
P = .4795
P = .0011
Pro
po
rtio
n s
urv
ivin
g
0 10 20 30 40 50 60 70
Age at death or ventilation, mo
0.0
0.2
0.4
0.6
0.8
1.0
NBS
Clinical comparators
Untreated historical group
P = .0640
P = .0084
Pro
po
rtio
n s
urv
ivin
g
0 20 40 60 80 100
Age at independent walking, mo
0.0
0.2
0.4
0.6
0.8
1.0
NBS
Clinical comparators
Untreated historical group
P = .0063
P = .0090
Pro
po
rtio
n w
alki
ng
A
B
C
FIGURE 4Survival andmotor outcomes for patients whose diseasewas detected by newborn screening comparedwith thosewhose diseasewas diagnosed clinically.Green represents the screened cases (NBS); blue represents the clinical cases (clinical comparators); red represents cases from the natural history survey(untreated historical group). A, Kaplan-Meier survival curve for age at death; the difference between the NBS cases and the untreated cases was significant(P� .001). B, Kaplan-Meier ventilator-free survival curve for age at death or ventilation; the differences between the NBS cases and the untreated cases wassignificant (P� .008). C, Kaplan-Meier 1-minus-survival plot illustrates the time from birth to independent walking. The differences between the NBS casesand both the clinical cases and the untreated cases were significant (P� .006 and P� .009).
ARTICLES
PEDIATRICS Volume 124, Number 6, December 2009 e1123 by guest on September 12, 2018www.aappublications.org/newsDownloaded from

dase alfa. This response is more con-sistent than has been seen in otherpublished studies of infants who weretreated with rhGAA,8,21 despite that 5 ofthese 6 patients had very early cardiacinvolvement, indicating severe, rapidlyprogressive Pompe disease. All 6 pa-tients had very low GAA activity,comparable to our clinical cases anddata from other studies.6 The infantsseemed to benefit greatly from earlydetection because treatment was initi-ated before the onset of obvious symp-toms and before significant irrevers-ible muscle damage had occurred.The patients demonstrated reductionof LVMI, age-appropriate motor gains,and improvement in muscle mor-phology. The small number of end-stage cells in their follow-up musclebiopsies did not seem to affect ad-versely muscle function, as demon-strated by the achievement of nor-mal motor development.
The infant without cardiac involve-ment, NBS1, also seems to have bene-fited from early treatment andmay nothave been detected until much laterwithout screening. The muscle biopsyresult, although different from thatseen in the other patients, is not incon-sistent with a Pompe disease diagno-sis; in late-onset Pompe disease, somemuscle fibers may show little to no gly-cogen,22 and lipid storage has been re-ported in Pompe disease.23,24 This caseis likely comparable to the patients in-cluded in a trial of older infants andchildren between 3 and 43 months ofage previously described as “nontypi-cal infantile type.”25 Such patients whoshow signs of disease in the first yearsof life, even without cardiac manifesta-tion, tend to have very poor outcomeswhen not treated.26 The patient alsomay represent a juvenile-onset case,because some patients juvenile-onsetPompe disease have been reported tohave had developmental delay in child-hood, motor symptoms in the first
years of life, and CK levels within thenormal range.27,28 Because this patientmay have had a course of Pompe dis-ease that would not necessarily be fa-tal in infancy, we removed NBS1 fromthe survival analysis and recalculatedthe P value, which remained nonsignif-icant when compared with the clinicalcomparators (P� .54) and significantwhen compared with the historicalcontrol subjects (P� .005).
The outcomes of the treated compara-tor infants who were detected afterclinical manifestations in this studyvaried. One infant who began treat-ment at 2 months of age had outcomessimilar to the NBS infants who weretreated early, whereas another patientwho began treatment at 2 months ofage had a similar response to thatseen in cases of Pompe diseasetreated at �6 months of age7,21; how-ever, their outcomes aremarkedly bet-ter than the known natural history ofpatients with untreated infantilePompe disease.5
It is important to note that all of theinfants reported here were CRIM-positive. The Taiwanese population hasa high incidence of themissensemuta-tion p.D645E, which is consistent withCRIM-positive status; 5 (83%) of the 6patients identified in this study hadthis mutation. Other world populationsmay have a higher incidence of CRIM-negativemutations; therefore, the clin-ical results obtained in this study maynot be entirely predictive of expectedresults in other populations.
This study has several potential limita-tions. The sample of treated patients issmall and the length of follow-up is stilllimited; although our results arepromising, it is possible that even withvery early identification and treat-ment, not all infants with Pompe dis-ease will show the type of treatmentresponse seen here. Questions aboutlong-term clinical status in patientswith infantile-onset Pompe disease re-
main; the children in this study con-tinue to develop normally, but there isa shortage of published data on theduration of improvement in muscleand cardiac function in patients whohave infantile-onset Pompe diseaseand are treated with alglucosidasealfa. In addition, althoughwe found sig-nificant differences between the pa-tients who were identified throughnewborn screening and the compara-tor groups of untreated patients andtreated patients whose diagnosis wasmade after clinical symptoms becameevident, the comparison is limited byits retrospective nature.
The cumulative clinical experiencefrom multiple trials performed todate and the availability of a high-throughput testing by using DBS sam-ples29,30 have set the stage for the im-plementation of newborn screeningfor Pompe disease. Results from thisstudy will help to guide the develop-ment of appropriate confirmatory di-agnostic testing to be performed afteran abnormal newborn screening re-sult, as well as clinical protocols thatcan be used to determine the optimaltime to initiate treatment.
CONCLUSIONS
In comparison with findings from pre-vious treated cohorts, the results fromthis study indicate that early treat-ment with alglucosidase alfa is criticalin the treatment of infants with Pompedisease and highlight the need forearly diagnosis, which can be achievedby newborn screening.
ACKNOWLEDGMENTSThe neonatal screening program wassupported by a grant from GenzymeCorporation. This studywas partly sup-ported by National Science Councilgrant NSC 96-2314-B002-044-MY3.
We thank for the Bureau of Health Pro-motion for allowing us to integrate thePompe disease screening programinto the existing screening programs.
e1124 CHIEN et al by guest on September 12, 2018www.aappublications.org/newsDownloaded from

We also thank the many individualsin other hospitals who helped with
screening, referred cases, or tookcare of patients. Katherine Lewis, MA,
of Genzyme provided editorial assis-tance for this manuscript.
REFERENCES
1. Pompe JC. Concerning idiopathic hypertro-phy of the heart [in Dutch]. Ned TijdschrGeneeskd. 1932;76:304–311
2. Hirschhorn R. Glycogen storage diseasetype II: acid alpha-glucosidase (acid mal-tose) deficiency. In: Scriver CR, ed. The Met-abolic and Molecular Bases of InheritedDisease. 8th ed. New York, NY: McGraw-Hill;2001:3389–3420
3. Bashan N, Potashnik R, Barash V, Gutman A,Moses SW. Glycogen storage disease type IIin Israel. Isr J Med Sci. 1988;24(4 –5):224–227
4. Lin CY, Shieh JJ. Molecular study on the in-fantile form of Pompe disease in Chinese inTaiwan. Zhonghua Min Guo Xiao Er Ke Yi XueHui Za Zhi. 1996;37(2):115–121
5. Kishnani PS, Hwu WL, Mandel H, Nicolino M,Yong F, Corzo D. A retrospective, multina-tional, multicenter study on the natural his-tory of infantile-onset Pompe disease. J Pe-diatr. 2006;148(5):671–676
6. van den Hout HM, Hop W, van Diggelen OP, etal. The natural course of infantile Pompe’sdisease: 20 original cases compared with133 cases from the literature. Pediatrics.2003;112(2):332–340
7. Kishnani PS, Corzo D, Nicolino M, et al. Re-combinant human acid alpha-glucosidase:major clinical benefits in infantile-onsetPompe disease. Neurology. 2007;68(2):99–109
8. Nicolino M, Byrne B, Wraith JE, et al. Clinicaloutcomes after long-term treatment withalglucosidase alfa in infants and childrenwith advanced Pompe disease. Genet Med.2009;11(3):210–219
9. Amalfitano A, Bengur AR, Morse RP, et al.Recombinant human ac id a lpha -glucosidase enzyme therapy for infantileglycogen storage disease type II: results ofa phase I/II clinical trial. Genet Med. 2001;3(2):132–138
10. van den Hout JM, Kamphoven JH, WinkelLP, et al. Long-term intravenous treat-ment of Pompe disease with recombinanthuman alpha-glucosidase from milk. Pe-diatrics . 2004;113(5) . Avai lable at :
www.pediatrics.org/cgi/content/full/113/5/e448
11. Chien YH, Chiang SC, Zhang XK, et al. Earlydetection of Pompe disease by newbornscreening is feasible: results from the Tai-wan screening program. Pediatrics. 2008;122(1). Available at: www.pediatrics.org/cgi/content/full/122/1/e39
12. Kemper AR, Hwu WL, Lloyd-Puryear M, Kish-nani PS. Newborn screening for Pompedisease: synthesis of the evidence and de-velopment of screening recommendations.Pediatrics. 2007;120(5). Available at:www.pediatrics.org/cgi/content/full/120/5/e1327
13. Lynch CM, Johnson J, Vaccaro C, ThurbergBL. High-resolution lightmicroscopy (HRLM)and digital analysis of Pompe disease pa-thology. J Histochem Cytochem. 2005;53(1):63–73
14. Ko TM, Hwu WL, Lin YW, et al. Molecular ge-netic study of Pompe disease in Chinese pa-tients in Taiwan. Hum Mutat. 1999;13(5):380–384
15. Piper MC, Pinnell LE, Darrah J, Maguire T,Byrne PJ. Construction and validation of theAlberta Infant Motor Scale (AIMS). Can JPublic Health. 1992;83(suppl 2):S46–S50
16. FolioMR, Fewell RR. Peabody DevelopmentalMotor Scales, 2nd ed. (PDMS-2). Austin, TX:PRO-ED, Inc; 2000
17. Kaplan EL, Meier P. Nonparametric estima-tion from incomplete observations. J AmStat Assoc. 1958;53:457–481
18. Hwu WL, Chien YH, Lee NC, Chen CA, Wu MH.Elevation of BNP in newborns with hypertro-phic cardiomyopathy due to Pompe disease.Heart . 2008 . Ava i lab le at : h t tp : / /heart.bmj.com/cgi/eletters/94/10/1307#8129. Accessed March 30, 2008
19. Vogel M, Staller W, Buhlmeyer K. Left ven-tricular myocardial mass determined bycross-sectional echocardiography in nor-mal newborns, infants, and children. Pedi-atr Cardiol. 1991;12(3):143–149
20. Thurberg BL, Lynch Maloney C, Vaccaro C, etal. Characterization of pre- and post-treatment pathology after enzyme replace-
ment therapy for Pompe disease. Lab In-vest. 2006;86(12):1208–1220
21. Kishnani PS, Nicolino M, Voit T, et al. Chinesehamster ovary cell-derived recombinanthuman acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149(1):89–97
22. Schoser BG, Muller-Hocker J, Horvath R, etal. Adult-onset glycogen storage diseasetype 2: clinico-pathological phenotype revis-ited. Neuropathol Appl Neurobiol. 2007;33(5):544–559
23. Sarnat HB, Roth SI, Carroll JE, Brown BI,DunganWT. Lipid storagemyopathy in infan-tile Pompe’s disease. Arch Neurol. 1982;39(3):180–183
24. Verity MA. Infantile Pompe’s disease, lipidstorage, and partial carnitine deficiency.Muscle Nerve. 1991;14(5):435–440
25. Slonim AE, Bulone L, Ritz S, Goldberg T, ChenA,Martiniuk F. Identification of two subtypesof infantile acid maltase deficiency. J Pedi-atr. 2000;137(2):283–285
26. Hagemans ML, Winkel LP, Hop WC, ReuserAJ, Van Doorn PA, Van der Ploeg AT. Diseaseseverity in children and adults with Pompedisease related to age and disease dura-tion. Neurology. 2005;64(12):2139–2141
27. Winkel LP, Hagemans ML, van Doorn PA, etal. The natural course of non-classicPompe’s disease: a review of 225 publishedcases. J Neurol. 2005;252(8):875–884
28. Van der Beek NA, Hagemans ML, Reuser AJ,et al. Rate of disease progression duringlong-term follow-up of patients with late-onset Pompe disease. Neuromuscul Disord.2009;19(2):113–117
29. Li Y, Scott CR, Chamoles NA, et al. Direct mul-tiplex assay of lysosomal enzymes in driedblood spots for newborn screening. ClinChem. 2004;50(10):1785–1796
30. Zhang XK, Elbin CS, Chuang WL, et al. Multi-plex enzyme assay screening of dried bloodspots for lysosomal storage disorders byusing tandem mass spectrometry. ClinChem. 2008;54(10):1725–1728
ARTICLES
PEDIATRICS Volume 124, Number 6, December 2009 e1125 by guest on September 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2008-36672009;124;e1116Pediatrics
Yuan-Tsong Chen and Wuh-Liang HwuZhang, Joan Keutzer, Ai-Chu Huang, Mei-Hwan Wu, Pei-Hsin Huang, Fuu-Jen Tsai, Yin-Hsiu Chien, Ni-Chung Lee, Beth L. Thurberg, Shu-Chuan Chiang, Xiaokui Kate
Early TreatmentPompe Disease in Infants: Improving the Prognosis by Newborn Screening and
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/124/6/e1116including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/124/6/e1116#BIBLThis article cites 24 articles, 5 of which you can access for free at:
Subspecialty Collections
ubhttp://www.aappublications.org/cgi/collection/metabolic_disorders_sMetabolic Disordershttp://www.aappublications.org/cgi/collection/endocrinology_subEndocrinologyfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on September 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2008-36672009;124;e1116Pediatrics
Yuan-Tsong Chen and Wuh-Liang HwuZhang, Joan Keutzer, Ai-Chu Huang, Mei-Hwan Wu, Pei-Hsin Huang, Fuu-Jen Tsai, Yin-Hsiu Chien, Ni-Chung Lee, Beth L. Thurberg, Shu-Chuan Chiang, Xiaokui Kate
Early TreatmentPompe Disease in Infants: Improving the Prognosis by Newborn Screening and
http://pediatrics.aappublications.org/content/124/6/e1116located on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://pediatrics.aappublications.org/content/suppl/2009/12/03/124.6.e1116.DC1Data Supplement at:
1073-0397. ISSN:60007. Copyright © 2009 by the American Academy of Pediatrics. All rights reserved. Print
the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on September 12, 2018www.aappublications.org/newsDownloaded from





























