Embed Size (px)
Citation preview

intl.elsevierhealth.com/journals/thre
Thrombosis Research (2007) 120, 623–629
REGULAR ARTICLE
Platelet adhesion in atrial fibrillation
Anirban Choudhury, Irene Chung, Andrew Blann, Gregory Y.H. Lip⁎
Haemostasis Thrombosis and Vascular Biology Unit, University Department of Medicine,City Hospital, Birmingham B18 7QH, UK
Received 16 August 2006; received in revised form 6 November 2006; accepted 4 December 2006Available online 23 January 2007
⁎ Corresponding author.E-mail address: [email protected]
0049-3848/$ - see front matter © 200doi:10.1016/j.thromres.2006.12.008
Abstract
Background: Increased platelet activation has been reported in nonvalvular atrial fibril-lation (AF) but it remains unclear whether or not this is due to the underlying cardio-vascular diseases, clinical subtype of AF and the influence of anti-thrombotic therapy.Methods: Platelet adhesion in AF patients was assessed using a ‘platelet adhesionassay’, and compared to both ‘healthy controls’ and ‘disease controls’ (patients withhypertension, coronary artery disease, diabetes or stroke but in sinus rhythm).Results: AF patients on no anti-thrombotic treatment (N=20) had increased plateletadhesion compared to ‘healthy controls’ (N=57) (p=0.044). Initiating treatment withboth aspirin and warfarin resulted in significant reduction in platelet adhesion in AFpatients (p=0.014 and 0.019 respectively). AF patients on optimal anti-thrombotictherapy (N=98) had no significant difference in platelet adhesion compared to‘healthy controls’ and ‘disease controls’ (p=0.312). Platelet adhesion failed todistinguish between ‘high-risk’ (i.e. CHADS score≥2) and ‘low-risk’ (i.e. CHADSscoreb2) AF patients (p=0.352). Amongst the clinical parameters that contribute toincreased stroke risk in AF, platelet adhesion was only correlated with age (r=0.215,p=0.034), and not with other stroke risk factors. There was no significant differencein platelet adhesion between paroxysmal and permanent AF (p=0.618).Conclusion: There is a significant, albeit weak, excess of platelet adhesion in AFpatients on no anti-thrombotic therapy compared to ‘healthy controls’. In optimallytreated AF patients, platelet adhesion is not different to both ‘healthy’ and ‘diseasecontrols’. It is possible that abnormal platelet adhesion does not significantlycontribute to the increased risk of stroke and thromboembolism that persists despiteanti-thrombotic treatment in AF or in identifying ‘high-risk’ AF patients.© 2006 Elsevier Ltd. All rights reserved.
KEYWORDSPlatelet adhesion;Atrial fibrillation
.uk (G.Y.H. Lip).
6 Elsevier Ltd. All rights reserved.

624 A. Choudhury et al.
Introduction
The underlying mechanisms for stroke and throm-boembolism in atrial fibrillation (AF) are complex.The overall risk of stroke depends not only on themere presence of AF but also on the underlying/associated cardiovascular diseases (e.g. hyperten-sion, coronary artery disease or diabetes mellitus)that often themselves lead to the development ofAF [1].
Increasing evidence points to the fulfillment ofVirchow's triad in AF, resulting in a pro-thromboticor hypercoagulable state. AF is characterised byblood flow abnormalities (increased stasis of bloodin left atrium), abnormalities in the vessel wall(macroscopic and microscopic changes in the leftatrium, increased expression of von Willebrandfactor (vWF) and tissue factor in the endocardiumand endothelium) and abnormalities of blood con-stituents (activation of platelets and the coagula-tion cascade) [2,3].
The evidence supporting increased platelet acti-vation in AF has been provided by numerous studies[4–9], but some results have been conflicting, thusreflecting the difference between the assays andthe different aspects of platelet physiology mea-sured by them. Whilst platelets are somewhatactivated in AF, the evidence that it relates dir-ectly to the increased thrombotic risk in AF isuncertain [4,10,11].
We have previously demonstrated a clear linearrelationship between the number of platelets lysedand the mass of P-selectin in the resultant lysate[12]. Based on this technique, we developed anassay of platelet adhesion (using fibrinogen-coatedwells) and applied it to demonstrate that patientswith breast cancer [13] and hypertension [14] hadgreater platelet adhesion, when compared to‘healthy’ controls.
In a cross-sectional study, we tested the hypoth-esis that platelets from patients with AF will exhibitincreased adhesiveness to fibrinogen, compared to‘healthy controls’ and ‘disease controls’ (patients insinus rhythm but with underlying cardiovasculardiseases, e.g. hypertension, coronary artery dis-ease, diabetes and stroke). We also assessed theeffect of initiation of anti-platelet therapy (aspirin)and anticoagulant therapy (warfarin) on the degreeof platelet adhesion in AF patients.
Methods
For our main cross-sectional study, we recruited 98consecutive patients with ECG-defined AF (49paroxysmal AF, 49 permanent AF) from the
outpatient clinic. All were under the care of thecardiology team and had their treatment optimisedprior to recruitment. Exclusion criteria includedany history of malignancy, connective tissuedisease, thyroid disease, renal disease, use ofsteroids, hormone replacement therapy, activesmoking (or those who were smokers in the last3 months) and any major concomitant activecardiovascular disease [e.g. uncontrolled hyper-tension (SBP/DBP≥160/100), left ventricular sys-tolic dysfunction [ejection fractionb40%] or ahistory of unstable angina, myocardial infarctionor stroke in the previous 3 months]. AF patientswere compared to ‘disease controls’ [that is,patients with underlying cardiovascular diseases(e.g. hypertension, coronary artery disease, dia-betes or history of stroke) but in sinus rhythm] and‘healthy controls’. The latter were recruited fromamong hospital staff, patient relatives or thoseattending the hospital preoperative clinic forminor procedures. They were not taking regularmedications and had no clinical evidence ofvascular, metabolic, neoplastic or inflammatorydisease on careful history and examination. Theywere normotensive (b145/85 mmHg) and in sinusrhythm. Clinical and demographic data of thethree groups are presented in Table 1.
We identified and recruited 20 consecutivepatients with ECG-defined AF who were referredto our outpatient AF clinic for initiation of appro-priate anti-thrombotic therapy. Half (n=10) of thesepatients were subsequently started on aspirin whilethe remaining (n=10) were anticoagulated (withwarfarin). We also recruited another 10 AF patientswhose anti-thrombotic treatment was changed fromaspirin to warfarin (as per clinical risk stratifica-tion). Paired data from these 30 patients were usedto determine the effect of anti-thrombotic therapyon platelet adhesion in AF patients.
Laboratory
Venous blood was drawn from an antecubital veinwith minimal stasis into vacutainer tubes (BectonDickinson, UK). Initial aliquots were used to obtainEDTA and serum samples to minimise the chance ofartificial high readings secondary to venepuncture.These samples were analysed in the PathologyLaboratory of City Hospital (which participates inthe United Kingdom External Quality AssuranceScheme and is accredited by the Association ofClinical Pathologists) for total cholesterol, glucose,thyroid function, renal function, haemoglobin,haematocrit, white cell count and total plateletcount. Subsequent aliquots of blood collected into
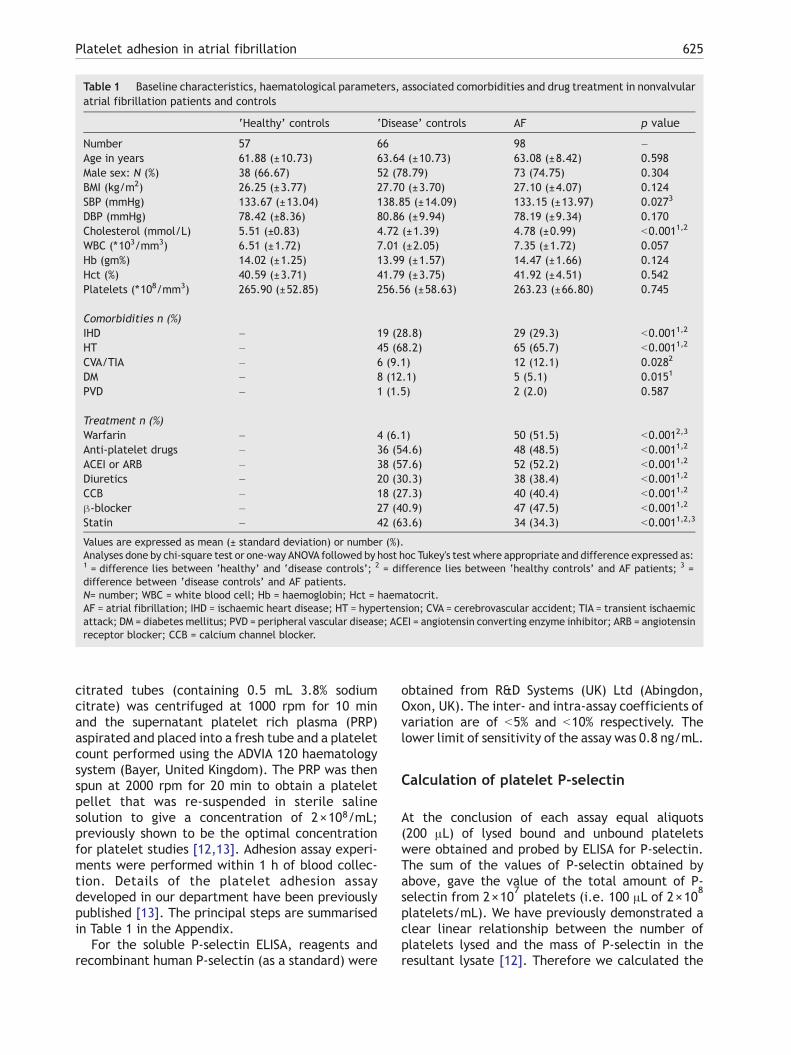
Table 1 Baseline characteristics, haematological parameters, associated comorbidities and drug treatment in nonvalvularatrial fibrillation patients and controls
‘Healthy’ controls ‘Disease’ controls AF p value
Number 57 66 98 –Age in years 61.88 (±10.73) 63.64 (±10.73) 63.08 (±8.42) 0.598Male sex: N (%) 38 (66.67) 52 (78.79) 73 (74.75) 0.304BMI (kg/m2) 26.25 (±3.77) 27.70 (±3.70) 27.10 (±4.07) 0.124SBP (mmHg) 133.67 (±13.04) 138.85 (±14.09) 133.15 (±13.97) 0.0273
DBP (mmHg) 78.42 (±8.36) 80.86 (±9.94) 78.19 (±9.34) 0.170Cholesterol (mmol/L) 5.51 (±0.83) 4.72 (±1.39) 4.78 (±0.99) b0.0011,2
WBC (⁎103/mm3) 6.51 (±1.72) 7.01 (±2.05) 7.35 (±1.72) 0.057Hb (gm%) 14.02 (±1.25) 13.99 (±1.57) 14.47 (±1.66) 0.124Hct (%) 40.59 (±3.71) 41.79 (±3.75) 41.92 (±4.51) 0.542Platelets (⁎108/mm3) 265.90 (±52.85) 256.56 (±58.63) 263.23 (±66.80) 0.745
Comorbidities n (%)IHD – 19 (28.8) 29 (29.3) b0.0011,2
HT – 45 (68.2) 65 (65.7) b0.0011,2
CVA/TIA – 6 (9.1) 12 (12.1) 0.0282
DM – 8 (12.1) 5 (5.1) 0.0151
PVD – 1 (1.5) 2 (2.0) 0.587
Treatment n (%)Warfarin – 4 (6.1) 50 (51.5) b0.0012,3
Anti-platelet drugs – 36 (54.6) 48 (48.5) b0.0011,2
ACEI or ARB – 38 (57.6) 52 (52.2) b0.0011,2
Diuretics – 20 (30.3) 38 (38.4) b0.0011,2
CCB – 18 (27.3) 40 (40.4) b0.0011,2
β-blocker – 27 (40.9) 47 (47.5) b0.0011,2
Statin – 42 (63.6) 34 (34.3) b0.0011,2,3
Values are expressed as mean (± standard deviation) or number (%).Analyses done by chi-square test or one-way ANOVA followed by host hoc Tukey's test where appropriate and difference expressed as:1 = difference lies between ‘healthy’ and ‘disease controls’; 2 = difference lies between ‘healthy controls’ and AF patients; 3 =difference between ‘disease controls’ and AF patients.N= number; WBC = white blood cell; Hb = haemoglobin; Hct = haematocrit.AF = atrial fibrillation; IHD = ischaemic heart disease; HT = hypertension; CVA = cerebrovascular accident; TIA = transient ischaemicattack; DM = diabetes mellitus; PVD = peripheral vascular disease; ACEI = angiotensin converting enzyme inhibitor; ARB = angiotensinreceptor blocker; CCB = calcium channel blocker.
625Platelet adhesion in atrial fibrillation
citrated tubes (containing 0.5 mL 3.8% sodiumcitrate) was centrifuged at 1000 rpm for 10 minand the supernatant platelet rich plasma (PRP)aspirated and placed into a fresh tube and a plateletcount performed using the ADVIA 120 haematologysystem (Bayer, United Kingdom). The PRP was thenspun at 2000 rpm for 20 min to obtain a plateletpellet that was re-suspended in sterile salinesolution to give a concentration of 2×108/mL;previously shown to be the optimal concentrationfor platelet studies [12,13]. Adhesion assay experi-ments were performed within 1 h of blood collec-tion. Details of the platelet adhesion assaydeveloped in our department have been previouslypublished [13]. The principal steps are summarisedin Table 1 in the Appendix.
For the soluble P-selectin ELISA, reagents andrecombinant human P-selectin (as a standard) were
obtained from R&D Systems (UK) Ltd (Abingdon,Oxon, UK). The inter- and intra-assay coefficients ofvariation are of b5% and b10% respectively. Thelower limit of sensitivity of the assay was 0.8 ng/mL.
Calculation of platelet P-selectin
At the conclusion of each assay equal aliquots(200 μL) of lysed bound and unbound plateletswere obtained and probed by ELISA for P-selectin.The sum of the values of P-selectin obtained byabove, gave the value of the total amount of P-selectin from 2×10
7platelets (i.e. 100 μL of 2×10
8
platelets/mL). We have previously demonstrated aclear linear relationship between the number ofplatelets lysed and the mass of P-selectin in theresultant lysate [12]. Therefore we calculated the
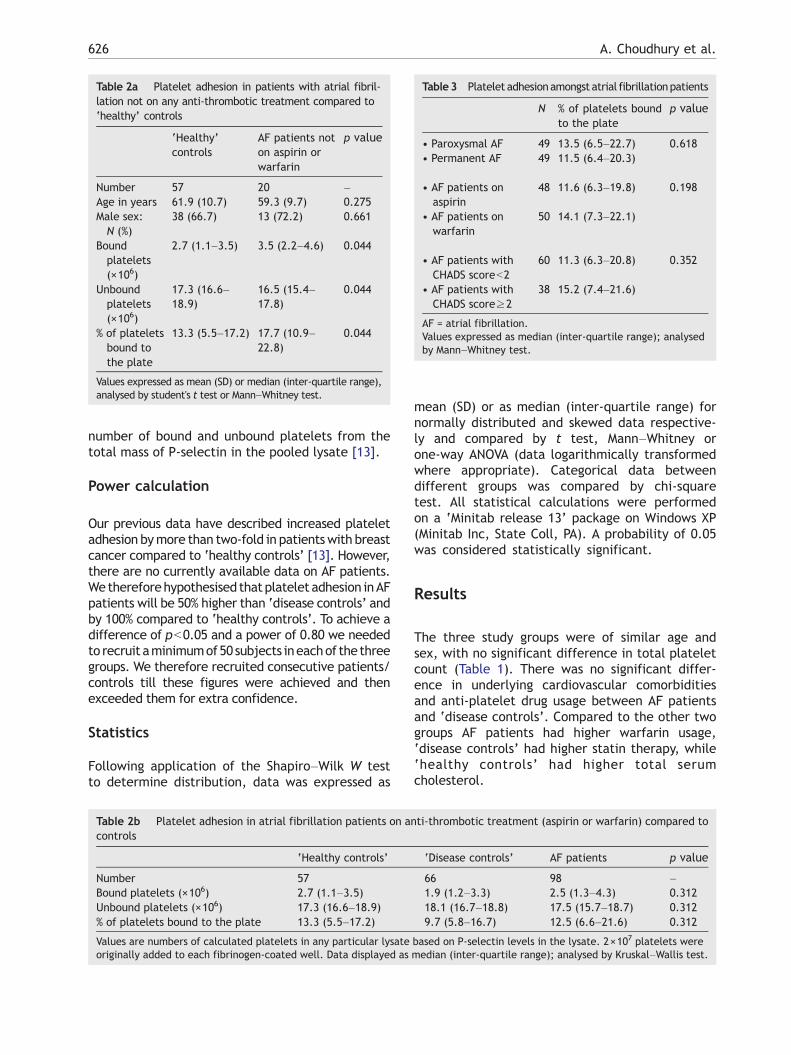
Table3 Platelet adhesionamongst atrial fibrillationpatients
N % of platelets boundto the plate
p value
• Paroxysmal AF 49 13.5 (6.5–22.7) 0.618• Permanent AF 49 11.5 (6.4–20.3)
• AF patients onaspirin
48 11.6 (6.3–19.8) 0.198
• AF patients onwarfarin
50 14.1 (7.3–22.1)
• AF patients withCHADS scoreb2
60 11.3 (6.3–20.8) 0.352
• AF patients withCHADS score≥2
38 15.2 (7.4–21.6)
AF = atrial fibrillation.Values expressed as median (inter-quartile range); analysedby Mann–Whitney test.
Table 2a Platelet adhesion in patients with atrial fibril-lation not on any anti-thrombotic treatment compared to‘healthy’ controls
‘Healthy’controls
AF patients noton aspirin orwarfarin
p value
Number 57 20 –Age in years 61.9 (10.7) 59.3 (9.7) 0.275Male sex:N (%)
38 (66.7) 13 (72.2) 0.661
Boundplatelets(×106)
2.7 (1.1–3.5) 3.5 (2.2–4.6) 0.044
Unboundplatelets(×106)
17.3 (16.6–18.9)
16.5 (15.4–17.8)
0.044
% of plateletsbound tothe plate
13.3 (5.5–17.2) 17.7 (10.9–22.8)
0.044
Values expressed as mean (SD) or median (inter-quartile range),analysed by student's t test or Mann–Whitney test.
626 A. Choudhury et al.
number of bound and unbound platelets from thetotal mass of P-selectin in the pooled lysate [13].
Power calculation
Our previous data have described increased plateletadhesion bymore than two-fold in patientswith breastcancer compared to ‘healthy controls’ [13]. However,there are no currently available data on AF patients.We thereforehypothesised thatplatelet adhesion inAFpatients will be 50% higher than ‘disease controls’ andby 100% compared to ‘healthy controls’. To achieve adifference of pb0.05 and a power of 0.80 we neededto recruit aminimumof50 subjects ineachof the threegroups. We therefore recruited consecutive patients/controls till these figures were achieved and thenexceeded them for extra confidence.
Statistics
Following application of the Shapiro–Wilk W testto determine distribution, data was expressed as
Table 2b Platelet adhesion in atrial fibrillation patients on ancontrols
‘Healthy controls’
Number 57Bound platelets (×106) 2.7 (1.1–3.5)Unbound platelets (×106) 17.3 (16.6–18.9)% of platelets bound to the plate 13.3 (5.5–17.2)
Values are numbers of calculated platelets in any particular lysateoriginally added to each fibrinogen-coated well. Data displayed as
mean (SD) or as median (inter-quartile range) fornormally distributed and skewed data respective-ly and compared by t test, Mann–Whitney orone-way ANOVA (data logarithmically transformedwhere appropriate). Categorical data betweendifferent groups was compared by chi-squaretest. All statistical calculations were performedon a ‘Minitab release 13’ package on Windows XP(Minitab Inc, State Coll, PA). A probability of 0.05was considered statistically significant.
Results
The three study groups were of similar age andsex, with no significant difference in total plateletcount (Table 1). There was no significant differ-ence in underlying cardiovascular comorbiditiesand anti-platelet drug usage between AF patientsand ‘disease controls’. Compared to the other twogroups AF patients had higher warfarin usage,‘disease controls’ had higher statin therapy, while‘healthy controls’ had higher total serumcholesterol.
ti-thrombotic treatment (aspirin or warfarin) compared to
‘Disease controls’ AF patients p value
66 98 –1.9 (1.2–3.3) 2.5 (1.3–4.3) 0.31218.1 (16.7–18.8) 17.5 (15.7–18.7) 0.3129.7 (5.8–16.7) 12.5 (6.6–21.6) 0.312
based on P-selectin levels in the lysate. 2×107 platelets weremedian (inter-quartile range); analysed by Kruskal–Wallis test.

627Platelet adhesion in atrial fibrillation
AF patients not on any anti-thrombotic treatmenthad increased platelet adhesion compared to‘healthy’ controls (p=0.044) (Table 2a). Therewere more unbound platelets in the control wells,and therefore the percentage of platelets adheringwas also significantly different. AF patients onoptimal anti-thrombotic therapy (aspirin or warfarin— as clinically indicated) had no significant differ-ence in platelet adhesion compared to both ‘healthycontrols’ and ‘disease controls’ (p=0.312) (Table2b). There was no significant difference in plateletadhesion between ‘healthy controls’ and ‘diseasecontrols’.
There was no significant differences in plateletadhesion between paroxysmal AF and permanentAF (% of bound platelets, 13.5 (6.5–22.7) vspermanent AF, 11.5 (6.4–20.3); p=0.618) and AFpatients treated with aspirin or warfarin (11.6(6.3–19.8) vs warfarin, 14.1 (7.3–22.1); p=0.198)(Table 3). Paroxysmal AF patients were significant-ly younger compared to their counterparts (inkeeping with day-to-day clinical practice) but thegroups were matched for sex distribution and hadno significant difference in blood pressure [datanot shown]. We also found no significant differencein platelet adhesion between ‘high-risk’ (definedas CHADS score≥ 2) and ‘low-risk’ (CHADSscoreb2) AF patients (11.3 (6.3–20.8)) vs CHADS2score≥2, 15.2 (7.4–21.6); p=0.352) (Table 3)[15,16].
Table 4 Effect of aspirin or warfarin therapy on plateletadhesion in atrial fibrillation
% ofplateletsbound to theplate
Mediandifference[95% CI]
pvalue
• Pretreatment[N=10]
14.6(8.5–28.4)
5.0 0.014[2.5, 7.7]
• 4 week post aspirin 9.1 (6.2–21.5)
• Pretreatment[N=10]
18.9(13.1–22.1)
3.1 0.019[2.9, 6.2]
• 4 week post warfarin 14.9(12.4–22.2)
• On aspirin [N=10] 10.4(6.4–14.6))
2.2 0.076[−4.18,17.62]
• 4 weeks afterstopping aspirin andstarting warfarin
14.1(9.1–19.3)
Values expressed as median (inter-quartile range); paireddata analysed by Wilcoxon test and one-way t test.
Effect of anti-thrombotic treatment onplatelet adhesion
Both aspirin and warfarin significantly reduced thepercentage of platelets binding to the fibrinogen-coated wells (Table 4). Where anti-thrombotictreatment was changed from aspirin to warfarin,there was a trend towards reduced platelet adhe-sion with aspirin compared to warfarin, and this wasnot statistically significant (Table 4).
Correlations
A significant correlation was observed between plate-let adhesion and the age of AF patients (r=0.215,p=0.034). This correlation also observed amongst thecontrols. No other significant correlations were foundbetween platelet adhesion and the clinical character-istics of AF patients that contribute to increased strokerisk (hypertension, coronary artery disease, diabetes orstroke) [data not shown]. Stepwise multiple regressionanalysis on the combined cohort of AF and ‘diseasecontrols’, the presence/absence of AF (Yes/No) wasnot an independent predictor of platelet adhesion(p=0.354).
Discussion
This is the first study of platelet adhesion in AF.Whilstpreliminary, our study raises new potential mechan-isms for further study, to improve our understandingof thrombogenesis in AF. We have shown that AFpatients not on any anti-thrombotic treatment hadincreased platelet adhesion compared to ‘healthycontrols’. Introducing aspirin or warfarin significant-ly reduced platelet adhesiveness so much so that AFpatients on anti-thrombotic treatment (aspirin orwarfarin) had no significant difference in plateletadhesion compared to both ‘healthy controls’ and‘disease controls’. There was also no relationship ofplatelet adhesion to stroke risk, based on the CHADS2risk scoring system [15,16].
The first step of platelet functional response isadhesion to vessel wall, a complex event involving aseries of plasma and subendothelial tissue compo-nents (collagen, fibrinogen, fibronectin, thrombos-pondin and vWF), which bind themselves specificallyto several different platelet membrane glycopro-teins [17–22]. A critical event in platelet adhesion isthe induction of a change in the disposition of thesurface glycoprotein GPIIb/IIIa, which acquires thecapacity to bind fibrinogen. The activation ofplatelets is also associated with expression ofCD62P (membrane bound P-selectin, α-granule

628 A. Choudhury et al.
protein) on the platelet surface. Though P-selectindoes not bind platelets to fibrinogen, it plays a vitalrole in inter-platelet interaction and stabilizing theinitial GPIIb/IIIa–fibrinogen interaction [23].
The adhesion of platelets to vessel wall compo-nents in AF patients has not been investigatedpreviously, although platelet adhesion has beenstudied in other diseases [12,13,24,25]. Moreover,there are considerable data of excess plateletactivation [4–9] and excess fibrin turnover [26–29]in patients with AF compared to ‘healthy controls’.
Other than age, we were unable to identifyany correlation between platelet adhesion andclinical features that contribute to the overall riskof stroke in AF (i.e. hypertension, coronary arterydisease, diabetes and history of stroke) [1,15], norany significant difference in platelet adhesion be-tween ‘high-risk’ and ‘low-risk’ AF patients. There-fore, the possibility arises that platelet adhesion isnot a major contributor to the excess risk of strokein AF, especially so in those on anti-thrombotictreatment.
Interpretation of this data is limited to theadhesive properties of platelets to fibrinogen. It islikely that platelets will adhere to other moleculessuch as thrombospondin, fibronectin and vWF [19–21]. However, the aim of our study was not toinvestigate the mechanistic basis of excess plateletadhesion, but only to gain a better understanding ofwhether this abnormality is present or not. Like anyother ‘indirect’ platelet assay, the possibility of adegree of artifactual platelet activation during‘platelet handling’ cannot be excluded [30]. How-ever, given the overall simplicity of this validatedassay and its reproducibility [13,14], this issue(platelet activation during platelet isolation) isunlikely to significantly affect the overall results.Correlation studies with fibrinogen-binding, asassessed by flow cytometry, could be interestingand would perhaps be the basis for future studies.Platelet reactivity to agonists could also have beenassessed, but this would have replicated previouswork [31,32]. Finally, our simple cross-sectionalapproach and modest numbers of study subjectslimits clinical interpretation, and further work in‘lone AF’ subjects would be necessary to ascertainthe impact of the arrhythmia per se on platelets.
In conclusion, this study provides further evidencethat there is a significant, albeit weak, excess ofplatelet activation (as measured by platelet adhesion)in AF patients compared to ‘healthy controls’. Webelieve that this is probably due to the underlyingcardiovascular diseases [33] and associated activationof the coagulation cascade [34] rather than the rhythmdisturbance itself. Though this abnormality may con-tribute to some extent in the generation of the pro-
thrombotic state in AF, it is possible that abnormalplatelet adhesion does not significantly contribute tothe increased risk of stroke and thromboembolism thatpersists despite anti-thrombotic therapy or in identify-ing ‘high-risk’ AF patients [35,36].
Appendix A
Supplementary data associated with this article canbe found, in the online version, at doi:10.1016/j.thromres.2006.12.008.
References
[1] Lip GYH, Boos CJ. Antithrombotic treatment in atrialfibrillation. Heart 2006;92:155–61.
[2] Fukuchi M, Watanabe J, Kumagai K, Katori Y, Baba S, FukudaK, et al. Increased von Willebrand factor in the endocardiumas a local predisposing factor for thrombogenesis in over-loaded human atrial appendage. J Am Coll Cardiol 2001;37:1436–42.
[3] Nakamura Y, Nakamura K, Fukushima-Kusano K, Ohta K,Matsubara H, Hamuro T, et al. Tissue factor expression inatrial endothelia associated with non-valvular atrial fibril-lation: possible involvement in intracardiac thrombogen-esis. Thromb Res 2003;111:137–42.
[4] Feinberg WM, Pearce LA, Hart RG, Cushman M, Cornell ES,Lip GY, et al. Markers of thrombin and platelet activity inpatients with atrial fibrillation: correlation with strokeamong 1531 participants in the stroke prevention in atrialfibrillation III study. Stroke 1999;30:2547–53.
[5] Gustafsson C, Blomback M, Britton M, Hamsten A, SvenssonJ. Coagulation factors and the increased risk of stroke innonvalvular atrial fibrillation. Stroke 1990;21:47–51.
[6] Lip GY, Lip PL, Zarifis J, Watson RD, Bareford D, Lowe GD,et al. Fibrin D-dimer and betathromboglobulin as markersof thrombogenesis and platelet activation in atrialfibrillation. Effects of introducing ultra-low-dose warfarinand aspirin. Circulation 1996;94:425–31.
[7] Minamino T, Kitakaze M, Sanada S, Asanuama H, Kurotobi T,Koretsune Y, et al. Increased expression of Pselectin onplatelets is a risk factor for silent cerebral infarction inpatients with atrial fibrillation: role of nitric oxide. Circu-lation 1998;98:1721–7.
[8] Yamauchi K, Furui H, Taniguchi N, Sotobata I. Plasma beta-thromboglobulin and platelet factor 4 concentrations inpatients with atrial fibrillation. Jpn Heart J 1986;27:481–7.
[9] Furui H, Taniguchi N, Yamauchi K, Sotobata I, Saito H,Inagaki H. Effects of treadmill exercise on platelet function,blood coagulability and fibrinolytic activity in patients withatrial fibrillation. Jpn Heart J 1987;28:177–84.
[10] Heppell RM, Berkin KE, McLenachan JM, Davies JA. Haemo-static and haemodynamic abnormalities associated with leftatrial thrombosis in nonrheumatic atrial fibrillation. Heart1997;77:407–11.
[11] Conway DSG, Pearce LA, Chin BSP, Hart RG, Lip GY. Plasmavon Willebrand factor and soluble Pselectin as indices ofendothelial damage and platelet activation in 1321 patientswith nonvalvular atrial fibrillation: relationship to strokerisk factors. Circulation 2002;106:1962–7.
[12] Kamath S, Blann AD, Caine GJ, Gurney D, Chin BSP, Lip GYH.Platelet P-selectin levels in relation to plasma soluble P-selectin and beta-thromboglobulin levels in atrial fibrilla-tion. Stroke 2002;33:1237–42.

629Platelet adhesion in atrial fibrillation
[13] Caine GJ, Nadar SK, Stonelake P, Lip GYH, Blann AD. Plateletadhesion in breast cancer. Platelet adhesion in breastcancer: development and application of a novel assay.Blood Coagul Fibrinolysis 2004;15:513–8.
[14] Nadar SK, Caine GJ, Blann AD, Lip GY. Platelet adhesion inhypertension: application of a novel assay of plateletadhesion. Ann Med 2005;37:55–60.
[15] Gage BF, Waterman AD, Shanon W, Boechler M, Rich MW,Radford MJ. Validation of clinical classification schemes forpredicting stroke: results from the National Registry ofAtrial Fibrillation. JAMA 2001;285:2864–70.
[16] ACC/AHA/ESC 2006 guidelines for the management ofpatients with atrial fibrillation-executive summary:a report of the American College of Cardiology/AmericanHeart Association Task Force on practice guidelines and theEuropean Society of Cardiology Committee for PracticeGuidelines (Writing Committee to Revise the 2001 Guide-lines for the Management of Patients with Atrial Fibrilla-tion). Eur Heart J 2006;27:1979–2030.
[17] Nieswandt B, Watson SP. Platelet–collagen interaction:is GPVI the central receptor? Blood 2003;102:449–61.
[18] Niiya K, Hodson E, Bader R, Byers-Ward V, Koziol JA, Plow EF,Ruggeri ZM. Increased surface expression of the membraneglycoprotein IIb/IIIa complex induced by platelet activa-tion. Relationship to the binding of fibrinogen and plateletaggregation. Blood 1987;70:475–83.
[19] Geho DH, Smith Jr WI, Liotta LA, Roberts DD. Fibrinectin-based masking molecule blocks platelet adhesion. Biocon-jug Chem 2003;14:703–6.
[20] Tuszynski GP, Smith M, Rothman VL, Capuzzi DM, Joseph RR,Katz J, et al. Thrombospondin levels in patients withmalignancy. Thromb Haemost 1992;67:607–11.
[21] Ruggeri ZM, De Marco L, Gatti L, Bader R, Montgomery RR.Platelets havemore than one binding site for vonWillebrandfactor. J Clin Invest 1983;72:1–12.
[22] Coller BS, Peerschke EI, Scudder LE, Sullivan CA. Studieswith a murine monoclonal antibody that abolishes ristoce-tin-induced binding of von Willebrand factor to platelets:additional evidence in support of GPIb as a platelet receptorfor von Willebrand factor. Blood 1983;61:99–110.
[23] Merten M, Thiagarajan P. P-selectin expression on plateletsdetermine size and stability of platelet aggregates. Circu-lation 2000;102:1931–1936.
[24] Andrioli G, Ortolani R, Fontana L, Gaino S, Bellavite P, LechiC, et al. Study of platelet adhesion in patients withuncomplicated hypertension. J Hypertens 1996;14:1215–21.
[25] Massberg S, Brand K,Gruner S, Page S,Muller E, Muller I, et al.
A critical role of platelet adhesion in the initiation ofatherosclerotic lesion formation. J Exp Med 2002;196:887–96.
[26] Asakura H, Hifumi S, Jokaji H, Saito M, Kumabashiri I, UotaniC, et al. Prothrombin fragment F1+2 and thrombin-antithrombin III complex are useful markers of hypercoag-ulable state in atrial fibrillation. Blood Coagul Fibrinolysis1992;3:469–73.
[27] Kumagai K, Fukunami M, Ohmori M, Kitabatake A, Kamada T,Hoki N. Increased intracardiovascular clotting in patients withchronic atrial fibrillation. J Am Coll Cardiol 1990;16:377–80.
[28] Lip GYH, Lowe GDO, Rumley A, Dunn FG. Increased markersof thrombogenesis in chronic atrial fibrillation: effects ofwarfarin treatment. Br Heart J 1995;73:527–33.
[29] Inoue H, Nozawa T, Okumura K, Jong-Dae L, Shimizu A, YanoK. Prothrombotic activity is increased in patients withnonvalvular atrial fibrillation and risk factors for embolism.Chest 2004;126:687–92.
[30] Tan KT, Lip GYH. The assessment of platelet activation inantiplatelet drug development. Curr Med Chem 2005;12:3117–25.
[31] Kamath S, Chin BS, Blann AD, Lip GYH. A study of plateletactivation in paroxysmal, persistent and permanent atrialfibrillation. Blood Coagul Fibrinolysis 2002;13:627–36.
[32] Kamath S, Blann AD, Chin BS, Lanza F, Aleil B, Cazenave JP,et al. A study of platelet activation in atrial fibrillation andthe effects of antithrombotic therapy. Eur Heart J 2002;23:1788–95.
[33] Lip GYH, Edwards SJ. Stroke prevention with aspirin,warfarin and ximelagatran in patients with non-valvularatrial fibrillation: a systematic review and meta-analysis.Thromb Res 2006;118:321–33.
[34] Goette A, Ittenson A, Hoffmanns P, Reek S, Hartung W,Klein H, et al. Increased expression of P-selectin inpatients with chronic atrial fibrillation. Pacing ClinElectrophysiol 2000;23:1872–5.
[35] Antithrombotic Trialists’ Collaboration. Collaborative anal-ysis of randomised trials of anti platelet therapy forprevention of death, myocardial infarction, and stroke inhigh risk patients. BMJ 2002;324:71–86.
[36] ACTIVE Writing Group on behalf of the ACTIVE Investiga-tors; Connolly S, Pogue J, Hart R, Pfeffer M, Hohnloser S,Pfeffer M, et al. Clopidogrel plus aspirin versus oralanticoagulation for atrial fibrillation in the atrial fibrilla-tion clopidogrel trial with irbesartan for prevention ofvascular events (ACTIVE W): a randomised controlledtrial. Lancet 2006;367(9526):1903–12.