Embed Size (px)
Citation preview

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/228698976
PKR:aNMRperspective
ArticleinProgressinNuclearMagneticResonanceSpectroscopy·August2007
DOI:10.1016/j.pnmrs.2007.07.001
CITATIONS
2
READS
52
5authors,including:
Someoftheauthorsofthispublicationarealsoworkingontheserelatedprojects:
DynamiclightscatteringtostudyhomogeneityofbiomacromoleculesViewproject
NetrinandDependenceReceptorViewproject
DarrinALindhout
NGMBiopharmaceuticals,Inc.
21PUBLICATIONS478CITATIONS
SEEPROFILE
SeanA.McKenna
UniversityofManitoba
46PUBLICATIONS1,139CITATIONS
SEEPROFILE
ColinEcheverríaAitken
NationalInstitutesofHealth
32PUBLICATIONS1,071CITATIONS
SEEPROFILE
AllcontentfollowingthispagewasuploadedbyDarrinALindhouton27August2014.
Theuserhasrequestedenhancementofthedownloadedfile.Allin-textreferencesunderlinedinblueareaddedtotheoriginaldocument
andarelinkedtopublicationsonResearchGate,lettingyouaccessandreadthemimmediately.

PKR: A NMR perspective
Darrin A. Lindhout a, Sean A. McKenna a, Colin Echeverrıa Aitken a, Corey W. Liu b,Joseph D. Puglisi a,b,*
a Department of Structural Biology, Stanford University School of Medicine, Stanford, CA 94305-5126, USAb Stanford Magnetic Resonance Laboratory, Stanford University School of Medicine, Stanford, CA 94305-5126, USA
Received 30 May 2007Available online 18 July 2007
Keywords: NMR; PKR; eIF2a; Mechanism; Structure
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2002. PKR is a modular system of independently folded domains . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2013. NMR solution studies of the N-terminal region of PKR1–169: dsRBD1/2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
3.1. Solution structure and dynamics of dsRBD1/2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2013.2. Statistical comparison of dsRBD1/2 with other solved RNA-binding domains . . . . . . . . . . . . . . . . . . . . . . . . . . 202
4. NMR solution studies of the central region of PKR169–251: inter-domain linker . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2035. NMR solution studies of the C-terminal region of PKR252–551: kinase domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.1. The kinase domain crystal structure demonstrates eIF2a binding site and dimer interface contacts . . . . . . . . . . . . 2035.2. NMR chemical shift assignments of the kinase domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2035.3. NMR assignments of kinase domain cannot verify protomer orientations observed in the crystal structure . . . . . . 203
6. Model of latent form PKR: dynamically tuned auto-inhibition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2046.1. NMR titration of isolated dsRBD1/2 domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2046.2. NMR titration of isolated kinase domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
7. Determining choice of RNA ligand for solution NMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2057.1. Limitations of protein!RNA structure by NMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2057.2. Molecular interactions within solved dsRBD!dsRNA complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2057.3. Two types of PKR regulators: RNA activators and inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2067.4. High-affinity dsRNA interactions requires simultaneous binding of both dsRBD1 and dsRBD2. . . . . . . . . . . . . . 2067.5. Choice of dsRNA ligand for NMR structural studies of PKR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 207
8. Specific recognition of dsRBD1/2 by activator TAR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2088.1. Thermodynamic and mutagenesis analysis of TAR!dsRBD1/2 interaction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2088.2. Chemical shift perturbation analysis of TAR!dsRBD1/2 interaction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
9. Specific recognition of dsRBD1/2 by inhibitors VAI and EBERI . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2099.1. Thermodynamic, autophosphorylation, and truncation analysis
of VAI!dsRBD1/2 and EBERI
!dsRBD1/2 interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2099.2. Chemical shift perturbation analysis of VAI
!dsRBD1/2 and EBERI!dsRBD1/2 interactions . . . . . . . . . . . . . . . . . 209
9.3. Regions distal to the dsRNA!dsRBD1/2 binding interface are critical for PKR inhibition . . . . . . . . . . . . . . . . . . 21010. NMR of latent PKR suggests an extended confirmation in solution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
10.1. NMR analysis of PKR. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21010.2. Correlation of extended and auto-inhibition hypotheses of latent PKR. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
0079-6565/$ - see front matter ! 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.pnmrs.2007.07.001
* Corresponding author. Address: Department of Structural Biology, Stanford University School of Medicine, Stanford, CA 94305-5126, USA. Tel.: +1650 498 4397; fax: +1 650 723 8464.
E-mail address: [email protected] (J.D. Puglisi).
www.elsevier.com/locate/pnmrs
Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

11. TAR binding to PKR induces conformational changes within dsRBD1/2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21112. Activated, phosphorylated PKR reveals a dimeric conformation in solution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211
12.1. Activated PKR demonstrates chemical shift perturbations localized within the kinase domain . . . . . . . . . . . . . . 21112.2. Dynamic communication of the inter-domain linker . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21212.3. Resonance assignments for dsRBD1/2 in activated PKR are unperturbed . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212
13. Current model of PKR activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21314. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
1. Introduction
Double-stranded RNA-dependent protein kinase (PKR)is an integral part of the cellular response to viral infection[1–3]. The presence of both intracellular and extracellularviral particles stimulates the body’s innate immuneresponse, resulting in the production of interferon and theup-regulation of PKR within the cell [4,5]. PKR is a cellularsensor of viral contamination that binds to highly struc-tured double-stranded RNA (dsRNA) elements within theviral genome [1,6–9]; binding initiates an autophosphoryla-tion reaction whereby PKR becomes phosphorylated(PKR-P), which increases its a!nity for the a-subunit ofeukaryotic initiation factor 2 (eIF2a) [10–13] (Fig. 1a).eIF2 is a heterotrimeric GTPase that binds to initiatorMet-tRNA in the GTP-bound form and delivers Met-tRNA to the 40S subunit of the ribosome during translationassembly in a GTP-dependent fashion [14,15]. PKR-P phos-phorylates the a-subunit at position S51 of eIF2 (eIF2a-P),which compromises the ability of the eFI2 to undergoGDP/GTP exchange in the presence of exchange factoreIF2B [13,16]. Resultantly, eIF2a-P remains in a GDP-bound state, is unable to bind to Met-tRNA, and initiatesa down-regulation of translation [14]. Therefore, PKR-
mediated modulation of translation hampers the ability ofthe invading viral particle to replicate using cellular transla-tion machinery, and allows su!cient time for the body tobegin an immune response. Additionally, PKR is alsoknown to play a role in other cellular processes such asinduction of apoptosis, cellular growth, immune response,the activation of transcription factors such as IRF-1 andNF-jB, and cancers such as melanoma and leukemia[9,17–33].
PKR is a 551-residue enzyme consisting of two indepen-dently folded domains connected by a long, flexiblelinker (Fig. 1b). The N-terminal domain contains two con-served 70-residue dsRNA-binding domains (dsRBD1 anddsRBD2) connected by a short 20-residue linker that com-prise the first 170 N-terminal residues (dsRBD1/2). ThedsRBD motif is characterized by a conserved a–b–b–b–afold consisting of two a-helices packed along one face of a3-stranded b-sheet, positioning multiple basic residues fordsRNA-mediated contacts [34–37]. In order to achievehigh-a!nity interactions between PKR and dsRNA, bothdsRBDs of PKR are required for binding, as well as specificlength and structural requirements of the viral dsRNA[7,8,38,39]. The C-terminal domain consists of an arch-typ-ical 299-residue Ser/Thr protein kinase with a standard
Fig. 1. The role of PKR in translation initiation. (a) eIF2 ternary complex (a, b, c) is responsible for the delivery of initiator Met-tRNA to ribosomalassembly in a GTP-dependent fashion. PKR senses viral dsRNA, undergoes autophosphorylation, and then performs phosphorylation on eIF2a,inhibiting guanine nucleotide exchange in the presence of eIF2B. Resultantly, the active pool of GTP-bound eIF2 ternary complex decreases, and there is adown-regulation of translation initiation. (b) PKR contains two N-terminal dsRBDs (dsRBD1 and dsRBD2) and a C-terminal kinase domain connectedby a long central linker. Position of residues critical in autophosphorylation catalysis (T446 and T451) and in ATP coordination (K296) are as indicated.
200 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

catalytic loop (L410–I420), Mg2+ binding and coordinationsite (K426–S438), activation loop (L439–G450), and aP + 1 substrate recognition loop (T451–I460). The activa-tion/P + 1 loops contain two conserved Thr residues(T446/T451) that are phosphorylated during dsRNA-med-iated autophosphorylation catalysis and are critical for theability to perform phosphor transfer on end substrateeIF2a. The N- and C-terminal domains are connected bya 80-residue linker rich in Ser and Thr, which may propa-gate the dsRNA activation signal to the kinase domainfrom the dsRBD1/2 domain.
In recent years, nuclear magnetic resonance (NMR) hasbecome a commonly used method for pursuing structuralstudies of large biomacromolecules. NMR is an excellentalternative to crystallization as it is able to overcome flex-ibility and dynamic issues sometimes inherent with crystal-lography. New strategies and technical advances such ashigher magnetic field strength (900 MHz), segmental iso-tope labeling (13C, 15N and/or 2H) and pulse sequencedevelopment (Transverse Relaxation Optimized Spectros-copY – TROSY, Residual Dipolar Coupling – RDC) haveenabled NMR solution studies of protein and RNA sys-tems of 100 kDa and larger [40–49]. Recent examplesinclude the dsRNA structural determination of domain IIwithin the 100 kDa internal ribosome entry site from hep-atitis C virus, and the solution derived global fold of the82 kDa enzyme malate synthase G from Escherichia coli[50–56]. In addition to structural determination, NMR isan excellent diagnostic tool in which parameters such asdynamic motion, conformational changes, binding interac-tion(s) via chemical shift perturbation(s), dissociation con-stants, internal motions, and thermodynamics of bindingcan be measured [57–62].
The purpose of this review is to provide a comprehen-sive perspective examining the role that NMR has playedon our current understandings of PKR, and how NMRhas aided in relating structure to biological function.PKR is a large 66 kDa biomacromolecule whose inherentdynamic flexibility has hindered structural studies by crys-tallography. PKR proves to be an excellent test case thatsupports the viability for the use of NMR in larger macro-molecular structural settings. This review will examine therelationship between NMR and PKR from a largely struc-tural perspective; beginning with initial structural studies ofthe isolated dsRBD1/2 and kinase domains, and buildingtowards more complex systems of the full-length proteinincluding an 85 kDa PKR!dsRNA complex.
2. PKR is a modular system of independently folded domains
PKR is a compartmentalized enzyme consisting of a 80-residue inter-domain linker region between the N-terminaldsRBDs and the C-terminal kinase domain. Subsequenttruncation analysis of the separate domains demonstratedindividually folded domains capable of high level recombi-nant protein expression in a bacterial source, retainingboth RNA binding and kinase activities endemic to the
full-length protein [8,10,11]. Mutagenesis analyses werealso very helpful in producing properly folded, yet catalyt-ically dead mutants (i.e. K296R), which have proved to behelpful in trapping PKR conformational state(s) for struc-tural studies [63,64].
The use of NMR as a PKR structural and biochemicalinvestigation technique is an enticing choice due to themodularity of the protein, as all three domains possess suf-ficient solubility and maintain monomeric behavior at con-centrations required for NMR. The following four sectionswill therefore focus on NMR solution studies of the indi-vidual, isolated domains within PKR, and the conclusionsthat can be derived from these studies.
3. NMR solution studies of the N-terminal region ofPKR1–169: dsRBD1/2
PKR contains two tandem copies of the highly con-served dsRBD motif connected by a short 20-residue lin-ker, which compose the first 170 N-terminal residues ofthe enzyme (19 kDa). To date, there has been more than20 functionally distinct proteins arising from both cellularand viral sources, which contain one or multiple copiesof the dsRBD motif, and can be dispersed N-, centraland/or C-terminally throughout the protein [65]. The con-served dsRBD motif generally spans between 65 and 73 res-idues and follows a a–b–b–b–a fold where two a-helicespack against a 3-stranded anti-parallel b-sheet. The lengthbetween adjacent dsRBDs is protein dependent, howeverthe distance generally does not exceed 100 residues [65].
3.1. Solution structure and dynamics of dsRBD1/2
The NMR solution structure of the N-terminal regionof PKR1–174 was first reported by Nanduri et al. in 1998,which showed a well-resolved 1H,15N HSQC spectrumgiven by two well-defined dsRBD motifs existing as adumb-bell shape, connected by a flexible 20-residue lin-ker region [34,66]. For purposes of this review, we haverepeated the 1H,15N HSQC spectrum of 15N-PKR1–169 at800 MHz, to demonstrate the excellent spectral disper-sion of dsRBD1/2 in solution (Fig. 2a). The two dsRBDmotifs share a 27% sequence identity (50% sequencehomology) and exhibit very similar tertiary folds with areported backbone RMSD of 2 A [34] (Fig. 2b). The cen-tral hydrophobic core was shown to contain highly con-served residues responsible for stabilizing the conservedfold. The authors exploited 20 multi-dimensional hetero-nuclear experiments to overcome chemical shift degener-acy and obtain unambiguous assignments of medium andlong-range NOEs, producing "15 NOE restraints perresidue (2580 NOE restraints and 3215 total experimentalrestraints) [34,66]. However, despite the abundance oflong-distance NOE restraints (ji–jj > 5 A), the authorswere unable to detect any contacts between dsRBD1and dsRBD2, indicating that the dsRBD motifs tumbleindependently of one another in solution. This inherent
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 201

flexibility between the two domains was confirmed using het-eronuclear 1H{15N}-nOeand 13Ca/13Cb secondary chemicalshifts [34,67–69]. Solution dynamics of dsRBD1/2 werefurther investigated in 2006 by Barnwal et al. using 13C-methyl relaxation techniques [70–75]. These measurementsdemonstrated that the small anisotropy inflicted by theflexible linker on the dumb-bell shaped molecule plays anegligible role in the individual methyl dynamics of therespective dsRBD motifs and that the hydrophobic coreof dsRBD1 is more tightly packed than that of dsRBD2[76].
3.2. Statistical comparison of dsRBD1/2 with other solvedRNA-binding domains
To demonstrate the conserved nature of the dsRBDfold between protein families, we have compared theisolated dsRBD1 and dsRBD2 domains of PKR with
other solved structures of dsRBD motifs (Table 1).Included in our analysis are the NMR solution struc-tures of the third dsRBD from Drosophila staufen pro-tein (Staufen), a dsRBD from Rnt1 endoribonuclease(Rnt1p), a dsRBD from the PKR protein activatorPACT (PACT), and two dsRBDs from the adenosinedeaminase rat ADAR (ADAR1 and ADAR2) [34,77–79]. The comparison of the calculated RMSD valuesof each respective dsRBD motif demonstrates the stabil-ity of the a–b–b–b–a fold within biology and the con-servation of the structural fold through evolution ofRNA-binding protein families. Interestingly, when com-paring the reported ‘tighter’ packed hydrophobic coreof dsRBD1 within PKR with other measured dsRBDmotifs, we did not observe significant RMSD deviationswhen compared with the ‘looser’ packed dsRBD2 [76].This observation seems to suggest that the relative back-bone conformations between dsRBD1 and dsRBD2 are
Table 1RMSD di"erences between solved dsRBD motifs
dsRBD1a dsRBD2b Staufenc Rnt1pd PACTe ADAR1f ADAR2g
dsRBD1 0.00 1.55 1.27 1.59 2.19 2.32 3.74dsRBD2 1.55 0.00 1.33 1.93 1.57 2.21 3.29
All values are derived from backbone (C CA N) superimposition using software VMD v1.8.5 [115] and are reported in Angstroms (A). The best conformerwithin each respective NMR ensemble was used (as specified in each pdb file), except for Staufen dsRBD3 (1STU) where the first conformer was used.a dsRBD1 uses residues Phe10 to Lys77 in the calculation from 1QU6 [34].b dsRBD2 uses residues Tyr101 to Glu168 in the calculation from 1QU6 [34].c Staufen uses residues Pro1 to Leu68 in the calculation from 1STU [77].d Rnt1p uses residues Ala370 to Leu438 in the calculation from 1T4N [78].e PACT uses residues Pro10 to Ala78 in the calculation from 1DIX (unpublished).f ADAR1 uses residues Leu5 to Phe72 in the calculation from 2B7T [79].g ADAR2 uses residues Lys4 to Leu71 in the calculation from 2B7V [79].
Fig. 2. NMR solution structure of the N-terminal domain of PKR (1–174). (a) 1H,15N HSQC spectrum of PKR 1–169 at 800 MHz. (b) Superimpositionof all 20 conformers of the solution structure of dsRBD1/2 reveals the flexible nature of the linker region (1QU6.pdb) [34]. CA atom superimposition of (c)residues 10–80 of dsRBD1 and (d) residues 100–160 of dsRBD2 demonstrate the conserved a–b–b–b–a fold of each respective domain. Figure wasgenerated using Pymol v0.98 for Mac OSX [116].
202 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

not greatly a"ected by side-chain hydrophobic packingconsiderations within the core(s) of the domains.
4. NMR solution studies of the central region of PKR169–251:inter-domain linker
The central 80-residue linker region (PKR169–251,8.5 kDa) that connects the dsRBD1/2 and kinase domainscontains a disproportionately high number of serine resi-dues (22 out of 80) and is negatively charged at physiolog-ical conditions (estimated pI = 4.08). The published 1H,15NHSQC spectrum of the linker region displayed poor spec-tral dispersion with the majority of the cross-peaks over-lapping in the unstructured region (1HN " 8.0–8.5 ppm),confirming earlier hypotheses that the linker region isunstructured in solution [80]. Regardless of structural fold,the linker may play some form of electrostatic role in thefull-length protein, potentially in the coordination ofdsRBD1/2 with dsRNA, the propagation of the activatingsignal to the kinase domain for autophosphorylation andthe subsequent release of dsRNA upon activation. Addi-tionally, the high Ser/Thr content (35%) makes this regionan attractive substrate for potential PKR regulation byboth itself and other cellular kinases.
5. NMR solution studies of the C-terminal region ofPKR252–551: kinase domain
The C-terminus of PKR252–551 consists of a 35 kDaglobular Ser/Thr kinase domain comprising 299 residues,which is centrally involved in PKR autophosphorylation.The kinase domain can be described as a bi-lobed enzymecontaining a b-barrel N-lobe (residues 252–370) and a lar-ger a-helical C-lobe (residues 371–551), such that the activesite is sandwiched in a cleft between the two lobes. The N-lobe is proposed to be involved in PKR self-recognition/dimerization and the C-lobe involved in substrate recogni-tion [10].
5.1. The kinase domain crystal structure demonstrates eIF2abinding site and dimer interface contacts
The published crystal structure of the kinase domain incomplex with the a-subunit of eIF2 by Dar et al. in 2005was a critical breakthrough in the molecular understandingof PKR fold, substrate recognition, and the ability to per-form phosphor transfer with substrate eIF2a [10]. The crys-tal structure of the phosphorylated form of the kinasedomain at residue T446 (pT446) revealed two importantpacking interactions occurring with PKR: the eIF2a bind-ing interface on the C-terminal lobe of the kinase domain,and the PKR!PKR homodimer interface within the N-ter-minal lobe of the kinase domain. The eIF2a substrate bind-ing face required for PKR binding involves residues withinthe P + 1 loop (T451–R453) and additional regions (E379,R382, T487–K493, and T496), and defined the spatial ori-entation of substrate required for phosphor transfer on S51
of eIF2a by the PKR active site. The PKR!PKR packingwas shown to be localized to the N-terminal lobe of thekinase domain and revealed a network of salt bridge(R262–D266) and an H-bonding triad (D289–Y293–Y323) interactions involved in dimer formation, demon-strating the potential importance of electrostatics in PKRself-association and activity [11]. Strangely, the PKR dimerin the crystal lattice oriented the active site of each protomerin an arrangement that suggested cis-autophosphorylation,opposing the currently accepted trans-autophosphorylationmodel [81]. How protein!protein dimerization occurswithin the full-length protein awaits further study.
5.2. NMR chemical shift assignments of the kinase domain
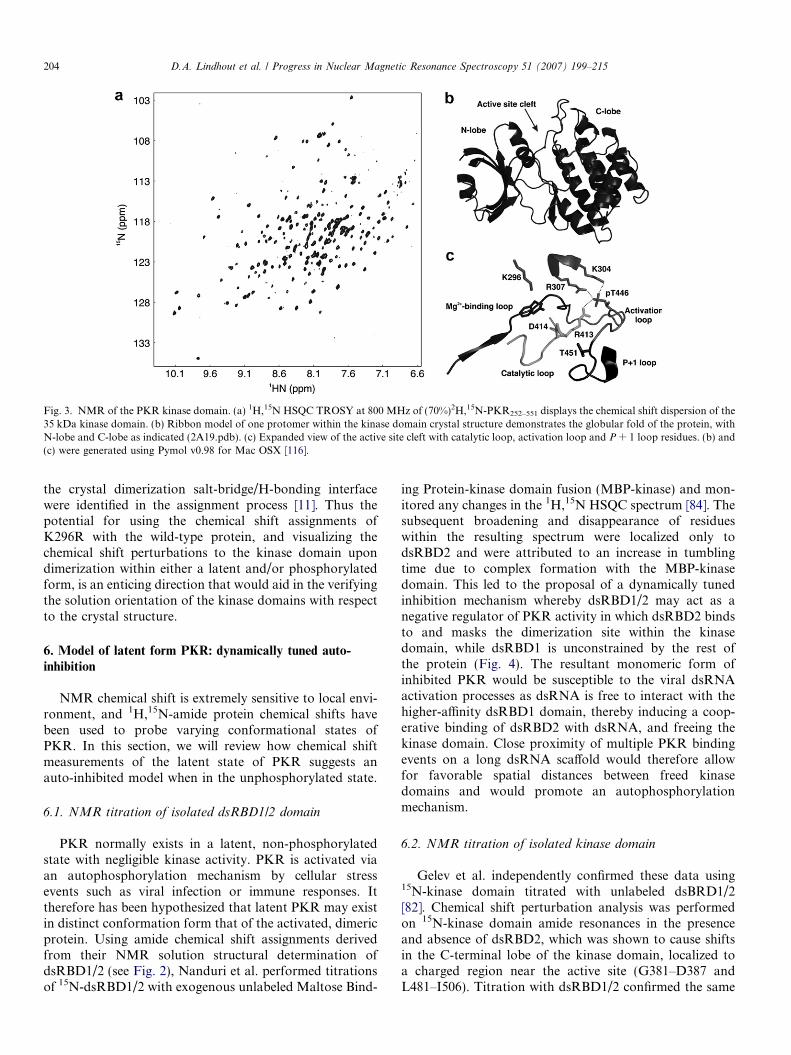
The NMR assignments for the protein backbone of thekinase domain were reported in 2006 by Gelev et al. [82],using a combination of 2H,15N and/or 13C labeling strate-gies. For purposes of this review, we have repeated a1H,15N HSQC TROSY spectrum of 2H,15N-kinase domainat 800 MHz to demonstrate the excellent spectral disper-sion of this domain in solution (Fig. 3a). The authors usedthe catalytically dead mutant K296R to ensure that thekinase domain is present in solution as a homogeneousunphosphorylated monomer for ease in NMR assignment.Normally, the K296 side-chain extends into the ATP-bind-ing pocket and is important in phosphate coordination(Fig. 3b and c), however the conserved mutation to Arginterrupts this coordination; inhibiting phosphate transferwithout compromising ATP binding [83], hence limitingunwanted phosphorylation during recombinant proteinexpression and suppressing dimer a!nity. Additionally,the phosphorylation of T446 is critical in kinase activityas pT446 stabilizes and rigidifies the activation loop(L439–G450) via a hydrogen-bonding network with neigh-boring residues K304, R307, and R413, allowing for theproper active site orientation to perform phosphor trans-fer, as inferred from the reported kinase domain crystalstructure [10] (Fig. 3c).
NMR analysis of the kinase domain revealed a well-resolved 1H,15N HSQC TROSY spectrum, allowing for theassignment of 75% of all HN, N, Ca, and Cb resonances.There were two distinct localized areas that constituted thebulk of unassigned residues; the acidic, serine-rich conservedDYDPEXSXXSXXS region (P334–S357), and the activa-tion and P + 1 substrate recognition loops (R445–E458).
5.3. NMR assignments of kinase domain cannot verifyprotomer orientations observed in the crystal structure
Although, the accomplishment of assigning a 35 kDaprotein in solution is significant, the use of the K296Rmutant precluded the ability to directly visualize theN-terminal lobe dimerization interface and to examinethe orientation of the kinase domain protomers (cis-auto-phosphorylation) reported in the crystal structure. Indeed,4 of the 5 residues (R262, D266, Y293, Y323) involved in
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 203
the crystal dimerization salt-bridge/H-bonding interfacewere identified in the assignment process [11]. Thus thepotential for using the chemical shift assignments ofK296R with the wild-type protein, and visualizing thechemical shift perturbations to the kinase domain upondimerization within either a latent and/or phosphorylatedform, is an enticing direction that would aid in the verifyingthe solution orientation of the kinase domains with respectto the crystal structure.
6. Model of latent form PKR: dynamically tuned auto-inhibition
NMR chemical shift is extremely sensitive to local envi-ronment, and 1H,15N-amide protein chemical shifts havebeen used to probe varying conformational states ofPKR. In this section, we will review how chemical shiftmeasurements of the latent state of PKR suggests anauto-inhibited model when in the unphosphorylated state.
6.1. NMR titration of isolated dsRBD1/2 domain
PKR normally exists in a latent, non-phosphorylatedstate with negligible kinase activity. PKR is activated viaan autophosphorylation mechanism by cellular stressevents such as viral infection or immune responses. Ittherefore has been hypothesized that latent PKR may existin distinct conformation form that of the activated, dimericprotein. Using amide chemical shift assignments derivedfrom their NMR solution structural determination ofdsRBD1/2 (see Fig. 2), Nanduri et al. performed titrationsof 15N-dsRBD1/2 with exogenous unlabeled Maltose Bind-
ing Protein-kinase domain fusion (MBP-kinase) and mon-itored any changes in the 1H,15N HSQC spectrum [84]. Thesubsequent broadening and disappearance of residueswithin the resulting spectrum were localized only todsRBD2 and were attributed to an increase in tumblingtime due to complex formation with the MBP-kinasedomain. This led to the proposal of a dynamically tunedinhibition mechanism whereby dsRBD1/2 may act as anegative regulator of PKR activity in which dsRBD2 bindsto and masks the dimerization site within the kinasedomain, while dsRBD1 is unconstrained by the rest ofthe protein (Fig. 4). The resultant monomeric form ofinhibited PKR would be susceptible to the viral dsRNAactivation processes as dsRNA is free to interact with thehigher-a!nity dsRBD1 domain, thereby inducing a coop-erative binding of dsRBD2 with dsRNA, and freeing thekinase domain. Close proximity of multiple PKR bindingevents on a long dsRNA sca"old would therefore allowfor favorable spatial distances between freed kinasedomains and would promote an autophosphorylationmechanism.
6.2. NMR titration of isolated kinase domain
Gelev et al. independently confirmed these data using15N-kinase domain titrated with unlabeled dsBRD1/2[82]. Chemical shift perturbation analysis was performedon 15N-kinase domain amide resonances in the presenceand absence of dsRBD2, which was shown to cause shiftsin the C-terminal lobe of the kinase domain, localized toa charged region near the active site (G381–D387 andL481–I506). Titration with dsRBD1/2 confirmed the same
Fig. 3. NMR of the PKR kinase domain. (a) 1H,15N HSQC TROSY at 800 MHz of (70%)2H,15N-PKR252–551 displays the chemical shift dispersion of the35 kDa kinase domain. (b) Ribbon model of one protomer within the kinase domain crystal structure demonstrates the globular fold of the protein, withN-lobe and C-lobe as indicated (2A19.pdb). (c) Expanded view of the active site cleft with catalytic loop, activation loop and P + 1 loop residues. (b) and(c) were generated using Pymol v0.98 for Mac OSX [116].
204 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

region of shifts that corresponded to dsRBD2, howeverthere were additional smaller changes within the N-termi-nal lobe of the kinase domain indicating that the entiredsRBD1/2 is capable of interacting with the kinasedomain; a system in which the dsRBD1!N-lobe interactionis of lower a!nity than that of the dsRBD2!C-lobe interac-tion. Subsequent mutagenesis analysis of dsRBD1/2 andthe kinase domain confirmed that the two domains arecapable of interacting along one face of the kinase domainencompassing the active site, C-lobe (helices aD, aE, aG),and the N-lobe (sheets b3, b-barrel, the hinge region of b7–b8).
7. Determining choice of RNA ligand for solution NMR
Having reviewed the NMR reports of the isolateddomains, we shall now review dsRNA ligands of PKRand investigate length, sequence, size, and specificityrequirements for protein!RNA interactions. We shall lookto three solved structures of dsRBD!dsRNA complexes togain both structural and potential size restrictions involvedwith dsRBD recognition of a dsRNA sca"old, and attemptto correlate this in choosing an appropriate dsRNA ligandfor NMR studies of PKR.
7.1. Limitations of protein!RNA structure by NMR
The past decade has witnessed a dramatic rise in bothpulse sequence sophistication and magnetic field strength,which has now raised the upper echelons of NMR proteinstructural size limits to nearly 100 kDa [50–56]. It thusmight be tempting to assume that structural determinationof large RNA molecules has also witnessed this same5-fold increase in size limitations [85]. However, a quickscan of solved NMR structures within the protein data-bank (circa Dec2006) yields 332 RNAs compared to5095 proteins. This large disparity in the quantity of struc-tures is a direct indicator of the di!culties inherent withsolution NMR of large RNA molecules. RNA containsonly four nucleotide bases as compared to 20 naturalamino acids for protein, which yields a lower degree ofpossible chemical shift dispersion. The relative abundance
of observable protons within RNA is also significantlyreduced when compared with that of proteins on a perDalton basis, yielding the potential for a lower numberof structural restraints. Additionally, dsRNA hasincreased NMR line widths due to the anisotropic natureof the helix and the slower tumbling rates associated withthis behavior. Finally, may RNAs form elongated struc-tures with few long-range NOEs of the type that defineprotein fold. Thus at present, the upper workable sizelimit for NMR studies of RNA is approximately25 kDa, or 75 nucleotide bases.
7.2. Molecular interactions within solved dsRBD!dsRNAcomplexes
In choosing an appropriate dsRNA ligand for NMRstudies with PKR, careful consideration must be given tolength, thermodynamic and chemical stability, aggregationstate, and binding a!nity with PKR. To gain structuralinsights, we can look to three examples of solved structuresof various dsRBDs in complex with dsRNA; a 1.9 A X-raystructure of the 2nd dsRBD (Xlrbpa-2) from Xenopus laevisin complex with model RNA, an NMR structure of the 3rddsRBD (Staufen) from Drosophila in complex with a syn-thetic RNA stem-loop, and a NMR structure of thedsRBD (Rnt1p) from Saccharomyces cerevisiae in complexwith the 5 0 terminal hairpin of one of its small nucleolarRNA substrates; the snR47 precursor [35–37]. In all threeexamples, the RNAs adopt an A-form RNA helical struc-ture and present similar binding interfaces for dsRBD-mediated interactions (Fig. 5).
In the Xlrbpa-2 complex (Fig. 5b), the dsRNA bindinginterface spans 16 bp encompassing one full turn of thehelix with three distinct regions involved in the interaction[36]. Specifically, there is an interaction of the N-terminalXlrbpa-2 a-helix (helix a1) with the RNA minor groove,an interaction of the C-terminal a-helix (helix a2) acrossthe RNA major groove, and an interaction of the loopbetween b-strands 1 and 2 (loop 2) with a successiveRNA minor groove. The Staufen dsRBD3 structure incomplex with a 30mer RNA stem-loop (Fig. 5c) displayssimilar binding characteristics, spanning 12 bp with
Fig. 4. Model of PKR auto-inhibition. The latent form of PKR exists in an auto-inhibited conformation in which dsRBD2 binds to the kinase domain asa negative repressor. Upon viral infection, the dsRBD1/2 domains undergo cooperative binding to dsRNA, allowing the freed kinase domain to undergoan autophosphorylation reaction, yielding the active form of the PKR.
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 205

interactions between the RNA minor groove and loop 2,the minor groove with the loop between b-strand 3 andhelix a2 (loop 4), and with the single-stranded loop thatcaps the RNA helix with helix a1 [37]. The Rnt1p dsRBDin complex with a 32mer RNA hairpin spans one entireface of the RNA helix, encompassing 13 bp plus theAGAA tetraloop, and makes similar RNA-mediated con-tacts as observed in the previous two structures (Fig. 5d)[35]. Protein side-chains insert into two minor groovesand a major groove in three regions, where helix a1 packsalong the RNA minor groove, helix a2 and loop 4 contactthe sugar–phosphate backbone of the major groove, andloop 2 contacts the successive minor groove. Rnt1p alsopossesses a third C-terminal a-helix (helix a3) not directlyinvolved in RNA binding, however it possesses a hydro-phobic face that when packed against helix a1 and the loopbetween helix a1 and b-strand 1 may stabilize the a–b–b–b–a structural fold.
The majority of the RNA!dsRBD interactions centeraround regions (outlined above) in which the dsRBD motifbinds to one face of the RNA helix, are stabilized predom-inantly by hydrogen bonds between specific chargeddsRBD side-chain moieties and the 2 0-OH groups of thehelical dsRNA [86]. Mutation of specific dsRBD residuesimplicated in RNA binding have been shown to decreaseRNA a!nity for various dsRBD motifs [37,87]. The high-lighted structural studies suggest that dsRBD-mediatedbinding to RNA is not sequence specific, but rather is moredependent on secondary structural elements within theindividual RNA helices. The RNA length dependence ofa single dsRBD binding appears to require 13 bp or longer,encompassing a minimum of one full turn of RNA helix, inwhich two consecutive minor grooves and one majorgroove are exposed [86]. Additionally, the dsRBD struc-ture remains largely unaltered in the RNA-bound form[37]. Importantly, the solved structures of the Stau-fen!30mer (17.7 kDa) and the Rnt1p!32mer (19.7 kDa)complexes demonstrate the feasibility of using NMR as astructural determination tool in large, RNA!proteincomplexes.
7.3. Two types of PKR regulators: RNA activators andinhibitors
The interaction between dsRNAs and PKR represents acrucial host cellular defense against viral infection, in whichdsRNA sca"olds that promote PKR autophosphorylationare referred to as activators. The ability of the host-cell torecognize specific elements of virally encoded dsRNAs istherefore a first step in understanding the mechanism ofPKR activation. Alternatively, certain viruses can circum-vent PKR function by the production of virally encodedtranscripts that bind to and inactivate PKR, which makeup the inhibitor class of dsRNA ligands.
7.4. High-a!nity dsRNA interactions requires simultaneousbinding of both dsRBD1 and dsRBD2
Prior biochemical analysis has demonstrated that RNA-mediated PKR autophosphorylation requires the simulta-neous binding of both dsRBD1 and dsRBD2 motifs todsRNA [88–90]. A longer dsRNA sca"old than thosedescribed in Fig. 5 is therefore required as the RNA mole-cule must be in excess of one full helical turn to allowappropriate surface area for dsRBD1/2 binding. Indeed,Ucci et al. recently demonstrated dsRNA length depen-dence on dsRBD1/2 binding using a combination analyti-cal centrifugation, circular dichroism and NMR, tomeasure the e"ects of synthetic dsRNA sequences rangingfrom 15 to 45 bp [39]. For shorter dsRNA sequences suchas 20 bp, resonance perturbations within the 1H,15N HSQCwere localized predominantly within dsRBD1, and only asthe dsRNA sca"old was lengthened to 40 bp was cross-peak exchange broadening within dsRBD2 observed. Thereported binding stoichiometries suggested a bindingmodel in which dsRBD1 plays the dominant role indsRNA complexation and is able to bind to multiplegrooves of the helical dsRNA sca"old, and that bothdsRBD1 and dsRBD2 domains are required for high-a!n-ity interactions with dsRNA sequences of lengths greaterthan 30 bp.
Fig. 5. Structures of solved dsRBD!dsRNA complexes. (a) dsRBD1 of PKR (1–78) displaying relative orientation of helices and loops involved in RNArecognition (1QU6.pdb) [34]. (b) Ribbon model of the 1.9 A crystal structure of Xlrbpa-2 in complex with a self-complimentary RNA 10mer (1DI2.pdb)[36]. (c) The lowest energy conformer from the NMR ensemble of Staufen dsRBD3 in complex with a RNA 30mer stem-loop (1EKZ.pdb) [37]. (d) Thelowest energy conformer from the NMR ensemble of Rnt1p in complex with a RNA 32mer snR47 hairpin precursor (1T4L.pdb) [35]. Figure wasgenerated using Pymol v0.98 for Mac OSX [116].
206 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

7.5. Choice of dsRNA ligand for NMR structural studies ofPKR
In the past, PKR autophosphorylation biochemicalanalysis has tended to revolve around the usage of the syn-thetic dsRNA polymer polyinosinic–polycytidylic acid(poly I!C) as a potent activator of autophosphorylation[29,91,92]. Although, an excellent substrate for PKR func-tional studies, poly I!C is not an appropriate molecule forstudies of PKR!dsRNA interactions by NMR due to itsheterogeneity in solution and large molecular-weight(P33 · 103 kDa). Therefore, more appropriate choicesfor NMR and mechanistic studies of a RNA!PKR complexare biological ligands, which include activator RNAs suchas the 3 0-UTR of a-tropomyosin, the internal ribosome
entry site (IRES) of hepatitis C virus (HCV), interferon-cmRNA, human hepatitis d agent RNA, the TAR domainof the human immunodeficiency virus (HIV) HIV-1, andinhibitor RNAs such as adenovirus VAI RNA, andEpstein–Barr virus EBER-1 and -2 RNAs [7,38,92–101].
Most viral regulators of PKR activity are distorteddsRNA helices, possessing structural features beyond sim-ple duplex, such as bulges and hairpin loops (Fig. 6) [102–105]. Some of these RNAs are well beyond the size limita-tions previously described and thus are poor choices forNMR structural studies. The TAR, VAI, and EBERI
domains are excellent RNA substrates due to their smallsize, relative stability, monomeric behavior at concentra-tions needed for NMR (>100 lM), and ease of productionby current in vitro transcription methodologies [106].
Fig. 6. Examples of biological PKR viral ligands. Predicted secondary structural elements of viral dsRNAs demonstrate position of helices, bulges, andhairpin loops required for PKR binding: (a) the internal ribosome entry site (IRES) of hepatitis C virus, (b) Adenovirus – VAI, (c) Ebstein–Barr virus –EBERI, and (d) the TAR domain of human immunodeficiency virus (HIV). The HCV IRES domain is too large of a viral substrate to monitor interactionwith PKR by NMR, however smaller domains such as VAI, EBERI, and TAR are more suitable candidates for NMR studies due to their smaller sizes,and highly structured folds.
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 207

The subsequent areas of this review will therefore focussolely on NMR experiments conducted on PKR in thelatent state, the phosphorylated state (PKR-P), and inthe presence of inhibiting/activating biological viral RNAligands EBERI, VAI, and TAR. These will provide aframework for the characterization of biochemical require-ments of physiological ligands that participate in the PKRautophosphorylation mechanism.
8. Specific recognition of dsRBD1/2 by activator TAR
The HIV-1 TAR domain (TAR), located at the 5 0 end ofall spliced and unspliced HIV-1 mRNAs, contains thebinding site of the Tat viral protein [107]. Tat is thetrans-activator of the HIV-1 long terminal repeat region,and is controlled via acetylation by Tat-associated histoneacetyltransferase [108]. It consists of a highly folded stem-bulge-loop that binds the cellular protein Tat-cyclin T1-CDK9 complex, which has been shown to mediate tran-scriptional elongation via recruitment of RNA polymeraseII [109,110]. TAR is a targeted domain of the cellular anti-viral response where PKR directly binds to and mediates, adown-regulation of translation.
8.1. Thermodynamic and mutagenesis analysis ofTAR!dsRBD1/2 interaction
Using the TAR domain as a model ligand, Kim et al. cor-relatedRNArecognitionwithPKRactivation by comparingbinding thermodynamics and chemical shift perturbationsinvolved in TAR!dsRBD1/2 association [8]. The authorsdemonstrated that TAR interactions with dsRBD1/2 canbe described as amodel in which oneTARmolecule containstwo binding sites: one for each of the dsRBD domainspresent within PKR. This binding event contains adissociation constant of 70 nM with a 1:1 stoichiometry, inwhich both dsRBD1 and dsRBD2 are required for thehigh-a!nity interaction and determined the thermodynamicparameters involved in this interaction (DH0 = #11.17 kcal/mol, DG0 = #9.88 kcal/mol, and DS0 = #4.26 cal/mol K).Indeed, subsequent binding titrations with either individualdsRBD domain gave 15-fold (dsRBD1) and 1175-fold(dsRBD2) weaker TAR!dsRBD a!nities. Additional analy-sis regarding the sequence specifics of the TARRNA associ-ation with dsRBD1/2 demonstrated that TAR structuralvariants such as bulge-deletions, a UUCG tetraloop varia-tion and a truncation of 17 nucleotides all resulted in signif-icantly weaker a!nities. These results demonstrate thatdsRBD!RNA interactions are directly dependent on second-ary structural elements of the RNA sca"old such as bulges,loops, and length dependence.
8.2. Chemical shift perturbation analysis of TAR!dsRBD1/2interaction
NMR analysis of larger molecular-weight pro-tein!RNA complexes involving more than one isolated
dsRBD motif and a RNA ligand require additional con-siderations for acquisition of high quality spectral data.Indeed, the 40 kDa complex of dsRBD1/2 domains ofPKR with the 57-nucleotide dsRNA TAR elementrequires higher levels of NMR sophistication to over-come molecular-weight obstacles such as chemical shiftoverlap and redundancy, peak broadening, and anisot-ropy. Site-specific labeling strategies of both protein res-idues and RNA nucleotide base, TROSY-based pulsesequences and increased magnetic field strengths havetherefore been developed to allow for high-resolutiondata acquisition for complex NMR systems such as theTAR!dsRBD1/2 complex [40–49].
Using a combination of unlabeled, 2H, 13C, and/or 15Nlabeling strategies, the NMR chemical shift assignments ofboth dsRBD1/2 and TAR were assigned in the free mono-meric form, and in a 40 kDa TAR!dsRBD1/2 complexundergoing slow-exchange. For purposes of this review,we have repeated the 1H,15N HSQC (800 MHz) and 1D1H NMR (500 MHz) of 15N-dsRBD1/2 in the presenceand absence of 1H TAR to demonstrate the high qualityof the spectral dispersion. Both the protein 1H,15N HSQCand 1D 1H NMR RNA imino spectra of dsRBD1/2 andTAR experienced large chemical shift perturbations uponassociation and were shown to be localized to specificregions of each molecule using chemical shift mappingtechniques (see Fig. 7). Within TAR RNA, imino protonresonance of U10, G11, G12, U13, U14, G21, U38, andG43 were perturbed upon dsRBD1/2 association, indicat-ing that dsRBD1/2 binding occurs within distinct, yet spa-tially distant regions of the TAR RNA helix (Fig. 7c andd). For both dsRBDs, the residues that underwent the larg-est chemical shift perturbations were localized to helices a1and a2 (Fig. 7a and b), which correlates well with the pre-vious described solved NMR dsRBD!dsRNA structures[35–37], in which loop 2 makes contacts with the secondminor dsRNA groove, which is 10–13 bp distance fromthe first recognized minor groove by helix a1, encompass-ing a full turn of the helix (see Fig. 2). However, loop 2of dsRBD1/2 was largely perturbed only in dsRBD1, indi-cating that dsRBD1 interacts with the TAR RNA, whereasdsRBD2 is involved in TAR RNA recognition to a lesserextent. This result was supported by ITC titration analysis,which demonstrated that TAR RNA has a greater a!nityfor isolated dsRBD1 when compared to that of dsRBD2.These results suggest that the association of dsRBD1/2with TAR RNA exists in a slightly di"erent structural scaf-fold than those previously reported for individual dsRBDs,which is in close agreement with that reported by Ucciet al. for dsRNA sca"olds in excess of 40 bp [39]. Thishypothesis is supported by the observation of large chem-ical shift perturbations in the 20-residue linker betweendsRBD1 and dsRBD2 domains, indicating that these lin-ker residues maybe more than just a tether between thetwo dsRBD domains and may play a structural role inboth the recognition and coordination of the TAR RNAligand.
208 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

9. Specific recognition of dsRBD1/2 by inhibitors VAI andEBERI
The ability of dsRBD1/2 to sense and recognize specificdsRNAs was continued and extended using inhibitor RNAsVAI andEBERI in our laboratory byMcKenna et al. in 2006[7]. Both VAI and EBERI are virally encoded dsRNAs thatbind to and inhibit PKR autophosphorylation, allowingviral replication to continue [97,111]. They are approxi-mately equal size ("150 bp) and form roughly the same sec-ondary structural fold containing a central stem-loop (CS),apical stem-loop (AS), and distal stem (DS), although basecomposition is varied (see Fig. 6b and c).
9.1. Thermodynamic, autophosphorylation, and truncationanalysis of VAI
!dsRBD1/2 and EBERI!dsRBD1/2
interactions
Correlation of PKR inactivation with RNA recognitionwas demonstrated using [c-32P]ATP incorporation, wherethe addition of VAI was shown to attenuate the phosphory-lation incorporation within full-length PKR by activatorTAR. ITC and native gel-shift analysis of the recognitionof dsRBD1/2 with either VAI or EBERI demonstrated sim-
ilar thermodynamic and binding a!nities as that reportedfor the activator TAR, indicating a similar mode of bindingfor both activator and inhibitor RNAs (VAI –KD = 76 nM,DHo = #11.2 kcal/mol, DGo = #9.9 kcal/mol, DSo = #4.4cal/mol K and EBERI – KD = 72 nM, DHo = #11.4 kcal/mol,DGo = #9.9 kcal/mol,DSo = #4.9 cal/mol K) [7]. Sub-sequent truncation analysis of the inhibitor RNAs demon-strated that the high-a!nity dsRBD1/2 interactions werelocalized to VAI-AS (Fig. 6b – domain II) and EBERI-CS(Fig. 6c – domain III).
Various base-pairs and bulges within VAI-AS andEBERI-CS were shown to be highly important in thedsRBD1/2 interaction, demonstrating that high-a!nitydsRNA recognition is critical in regards to both sequencespecific and secondary structural elements, as observedfor the TAR RNA [7].
9.2. Chemical shift perturbation analysis of VAI!dsRBD1/2
and EBERI!dsRBD1/2 interactions
NMR chemical shift perturbation analysis using 1H,15N-dsRBD1/2 amide and 1H-imino RNA was employed toverify the binding interaction suggested by ITC and trunca-tion analysis. The 1H,15NHSQCTROSY spectra of 1H,15N-
Fig. 7. NMR of a TAR!dsRBD1/2 complex. (a) 1H,15N HSQC TROSY spectral overlay of 2H15N-dsRBD1/2 in the absence (black) and presence (grey) ofunlabeled TAR dsRNA at 800 MHz. (b) Ribbon diagram of dsRBD1/2 (1QU6.pdb), in which amide resonances undergoing chemical shift perturbationsgreater than the average (Dd > 0.19 ppm) are colored in black (Dd = [(DH)2 + (0.25DN)2]1/2). (c) 1D 1N NMR proton imino resonances of TAR dsRNA inthe absence (top) and presence (bottom) of dsRBD1/2. (d) Nucleotides within TAR dsRNA that are significantly perturbed upon dsRBD1/2 binding arecolored in grey and displayed within the dsRNA secondary structure. (b) was generated using Pymol v0.98 for Mac OSX [116].
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 209

dsRBD1/2 in the presence of either VAI-AS or EBERI-CSdisplayed peak shift perturbations within the same reso-nances as that observed for TAR activator (see Fig. 7a) [7].Using amide chemical shift mapping on the surface of thesolved structure of dsRBD1/2, it was shown that perturba-tions within theRNA-binding domains is localized to helicesa1 and a2, loop regions, and the 20-residue linker, regardlessof whether activator (TAR) or inhibitor (VAI-AS or EBERI-CS) was used (see Fig. 7b). Thus, dsRBDs interact in a sim-ilar manner with activator and inhibitor binding sites.
9.3. Regions distal to the dsRNA!dsRBD1/2 bindinginterface are critical for PKR inhibition
Although, it is reported that a single stem-loop for eachof the dsRNA inhibitors tested is responsible for high-a!n-ity interactions with dsRBD1/2 (VAI-AS and EBERI-CS),the full-length inhibitor is required for PKR autophospho-rylation inhibition. When the inhibitory region distal to thehigh-a!nity dsRNA binding stem was removed, the distalstem behaved in a similar manner to the activator TAR [7].This demonstrates that although activators such as TARand inhibitors such as VAI and EBERI have a similar modeof binding to PKR, the inhibitors possess regions distal tothe binding stem-loops that contribute to the inhibition.This conclusion is supported by mutagenesis of distalregions within VAI as mutations in the central stem(VAI-CS – Fig. 6b domain III) are generally not well toler-ated [100,112,113].
10. NMR of latent PKR suggests an extended confirmationin solution
Having covered NMR of the isolated domains, and het-erogeneous dsRNA!dsRBD1/2 complexes, we shall nowreview more complex systems of PKR involving the full-length protein in the free form (PKR), complexed todsRNA (PKR!RNA), and in the phosphorylated form(PKR-P).
10.1. NMR analysis of PKR
NMR analysis of the full-length 66 kDa enzyme wasrecently investigated in our laboratory in 2006 byMcKennaet al. using a 2H,15N-labeling strategy to maximize TROSYcross-peak intensities at 800 MHz [80]. Using the unphos-phorylated wild-type PKR, we demonstrated that PKRdisplays a surprisingly well-resolved spectrum and behavesas a monomer in solution, as confirmed by size exclusionchromatography and dynamic light scattering (DLS). Forpurposes of this review, we have repeated the 1H,15NHSQCTROSY of (70%)2H,15N-PKR at 800 MHz to demonstratethe high quality of spectral dispersion. The 1H,15N HSQCTROSY spectrum revealed a near-perfect superimpositionwith those of either the isolated dsRBD1/2 (see Fig. 2a)and the kinase domain (see Fig. 3a) previously reported,and were used as a starting guide for amide chemical shift
assignments [34,66,82]. In total, 81% of the resonances inthe RNA-binding domains (137 out of 169) and 27% of theresonances in the kinase domain (82 out of 299) wereunambiguously assigned in the full-length protein (Fig. 8).The 80-residue linker region between dsRBD1/2 and thekinase domains was unassigned as it is apparently unstruc-tured in solution and present with the bulk signal in theoverlapping region of the spectrum (1HN " 7.5–8.5 ppm).Interestingly, the near-perfect superimposition of cross-peaks between dsRBD1/2 and/or kinase domain with PKRindicates that the isolated domains behave in a similarmanner as that observed in the full-length protein.
10.2. Correlation of extended and auto-inhibition hypothesesof latent PKR
The auto-inhibition model predicts noticeable chemicalshift perturbations to the full-length protein if there wassome form of interaction occurring between the dsRBD2domain and the kinase domain [84]. This apparent lackof chemical shift perturbations suggests that PKR is inan extended conformation in the latent form where theRNA-binding and kinase domains do not interact. How-ever, the lack of observable perturbations within the full-length protein might be due to the dynamic nature of theauto-inhibitory interaction, and as such we were unableto visualize the interaction under the conditions used.Additionally, the 80-residue inter-domain linker wasunresolved in the full-length protein and may play a rolein the modulation of a!nity of the respective domainsfor one another. Although currently unresolved, it isintriguing to propose that the self-modulation of PKRdomains and corresponding a!nities may be dependenton electrostatic interactions involved between the basic
Fig. 8. 1H,15N HSQC TROSY of PKR at 800 MHz. The excellent spectraldispersion of (70%)2H,15N-PKR allowed for a large number of unambig-uous assignments of the 66 kDa full-length protein.
210 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

RNA-binding domains (estimated pI = 9.42), the acidicinter-domain linker (pI = 4.08), and the basic kinasedomain (pI = 9.11).
11. TAR binding to PKR induces conformational changeswithin dsRBD1/2
We next monitored any potential conformationalchanges occurring within PKR during complexation withRNA in solution by using 15N-amide chemical shift per-turbations in the presence of the 19 kDa viral 57-nucleo-tide dsRNA TAR domain (Fig. 9a). The resultant85 kDa complex revealed a 1H,15N HSQC TROSY spec-trum that was well resolved and could be assigned usingthe methods previously described for the unligandedPKR. Confirmation of a 1:1 complex was confirmed usingDLS, gel filtration, and native gel-shift analyses. Perturba-tions of 15N-amide resonances within the TAR!PKR com-plex indicated that binding occurs solely in the dsRBD1/2domains, with chemical shifts of the dsRBDs within PKRclosely matching those previously reported for the isolated40 kDa TAR!dsRBD1/2 complex (see Fig. 7a). The con-formational changes occurring upon TAR complexationare therefore believed to be the same in the TAR!PKRcomplex as that reported for the TAR!dsRBD1/2 com-plex. The relative lack of chemical shift perturbationswithin the kinase domain in the presence of TAR indi-
cates that the kinase domain does not directly interactwith the TAR!dsRBD1/2 complex in the full-length pro-tein, and that the inter-residue linker region may beimportant in the propagation of the activation signal fromthe dsRBDs to that of the kinase domain.
For purposes of this review, we also performed the anal-ogous experiment of measuring the 1D imino 1H NMRchemical shift perturbations to the TAR domain upon asso-ciation with PKR at 500 MHz. The TAR imino proton per-turbations were identical in both the isolated 40 kDaTAR!dsRBD1/2 complex andwithin the 85 kDaTAR!PKRcomplex, confirming our hypothesis that a covalently boundkinase domain tethered to the dsRBD1/2 domains does notcontribute to further chemical shift changes with the TARimino protons (Fig. 9b). The only noticeable di"erencesobserved is increased line widths in the TAR!PKR complexdue to increased tumbling times of the larger complex whencompared to the TAR!dsRBD1/2 complex [8].
12. Activated, phosphorylated PKR reveals a dimericconformation in solution
12.1. Activated PKR demonstrates chemical shiftperturbations localized within the kinase domain
To investigate any conformational changes occurringwithin PKR upon activation, we further investigated the
Fig. 9. TAR binding to PKR induces perturbations localized solely within the dsRBD1/2 domains. (a) Selected views of the 1H,15N HSQC TROSY at800 MHz of (70%)2H,15N-PKR in the absence (black) and presence (grey) of an equimolar addition of TAR domain, demonstrating that only resonancesin the dsRBD1/2 domains are perturbed upon complexation. (b) 1D 1H NMR imino spectra of a 40 kDa TAR!dsRBD1/2 complex (top), and as a 85 kDaTAR!PKR complex (bottom). The lack of further 1H NMR TAR imino chemical shift perturbations in the presence of a covalently bound kinase domainindicates that the kinase domain is not involved in dsRNA complexation.
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 211

purified, RNA-free form of the protein using NMR. Forpurposes of this review, we repeated a 1H,15N HSQC TRO-SY spectrum at 800 MHz of phosphorylated, activatedPKR-P that displayed a spectrum with extensive peakbroadening with significant chemical shift perturbationswithin the kinase domain resonances [80]. We attributedbroadening to an equilibrium between monomeric anddimeric, phosphorylated forms of the protein in solution(Fig. 10). The formation of dimeric form of PKR-P wasindependently confirmed using gel filtration and DLSmethodologies [80]. We were able to attain 54% and 16%unambiguous spectral assignments for the backbone reso-nances of the dsRBD1/2 and kinase domains, respectively.The spectrum is suggestive of a single phosphorylationstate of the protein as resonance doubling was notobserved, which suggests a fast exchange time scalebetween molecular species in solution.
Backbone amide groups that exhibited the largest chem-ical shift perturbations within PKR-P were localized tothree distinct surface exposed regions of the kinase domain;around the kinase active site centered at K296 (the b6–b7loop (R413, D414, K416, S418), b7 (I420,F421), b7–b8(loop (T425)), b8 (Q427, I430), and the loop C-terminalto b8 (D432)), around the N-terminal b-barrel (b1 (L272,G274), b1–b2 loop (S275, G276, G277, G279), b2 (F282,K285), b2–b3 loop (H286, R287), b3 (I294, V295), b4(N324, G325, C326, G329), and b5 (Q365)), and theregions proximal to the eIF2a binding site (aD (E375,R381), aD–aE loop (G383), aE (L390, Q397, G401,D403, H406, K408) and aF–aH loop (V484)). These datasuggest how changes within the kinase domains N-terminal
lobe dimerization site, the active site, and the eIF2a bind-ing sites may be occurring. Chemical shift perturbationsmay reflect conformation changes and phosphorylationstate within the kinase domain, and PKR!PKR bimolecu-lar interactions.
12.2. Dynamic communication of the inter-domain linker
Our NMR data strongly suggest that the inter-domainlinker region is mainly unstructured within PKR, howevera functional role of this linker in the autophosphorylationprocess has recently been demonstrated [80]. We have pre-viously confirmed that PKR is able to undergo dsRNA-independent high-concentration autophosphorylation atconcentrations of PKR > 5 lM [80]. Using a truncationconstruct lacking the RNA-binding domains and com-posed of only the inter-domain linker and the kinasedomain (PKR169–551), we confirmed this same dsRNA-independent activation phenomenon [80]. Interestingly,when repeating the experiment with the isolated kinasedomain (PKR252–551) we observed near-basal levels of acti-vation over the same concentration range, demonstratingthat high-concentration autophosphorylation is dependenton the presence of the linker region. Therefore, the inter-domain linker may mediate the dynamic communicationbetween kinase and RNA-binding domains of PKR.
12.3. Resonance assignments for dsRBD1/2 in activatedPKR are unperturbed
Backbone amide resonances corresponding to thedsRBD1/2 domains within PKR-P were largely unper-turbed, indicating that the RNA-binding domains areconformationally unaltered in the phosphorylated formof the protein, and behave as independently tumblingdomains as they appear in the latent state. We also dem-onstrated via NMR chemical shift mapping, DLS andnative gel-shift analysis that PKR-P releases dsRNAupon activation, and in the case of TAR binding,dsRNA a!nity is greatly decreased in the phosphorylatedstate. dsRNA a!nity is likely not compromised due toconformational changes or modification (i.e. phosphory-lation state) within the dsRBD1/2 domains, but rathermay be caused by conformational changes occurring bydimerization of the kinase domain. This can lead to thefollowing 2-part hypothesis regarding dsRNA releaseand the observed decrease in dsRNA a!nity followingactivation; phosphorylation state within PKR-P may illi-cit an unfavorable steric conformation within the full-length protein that would compromise dsRNA binding,or that an unfavorable electrostatic imbalance due tophosphate negative charge buildup on the surface ofthe kinase domain may decrease dsRNA!dsRBD1/2 a!n-ity due to charge repulsion. The electrostatics hypothesisis supported by a report that polyanionic agent heparin isa potent activator of PKR in the absence of dsRNA[114].
Fig. 10. The activated form of PKR is a dimer in solution. 1H,15N HSQCTROSY spectra acquired at 800 MHz of latent (black) and phosphory-lated (grey) forms of PKR. The increased line broadening of the activatedform of PKR displays monomer/dimer equilibrium in solution.
212 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

13. Current model of PKR activation
The use of NMR as a tool in the elucidation of an acti-vation mechanism for PKR has been critical in the charac-terization of the isolated domains, mapping of high-a!nitydsRNA interactions with dsRBD1/2, and of the phosphor-ylated form of PKR. Supplemented with other biochemicalresults, the various reaction steps involved in the activationprocess can be delineated from a rich resource of NMRinvestigations.
A generalized mechanism of PKR activation can there-fore be generated which incorporates much of the NMRdata presented within this review and represents an intrigu-ing view into the innate anti-viral response to cellular infec-tion (Fig. 11). We propose that PKR exists in a latent statethat is monomeric and extended, and acts as a cellular sen-sor of dsRNA genomic components of a viral invading par-ticle. High-a!nity interactions of the viral dsRNA with theN-terminal dsRBD1/2 domains are dependent not only ondsRNA length, but specific secondary structural elementssuch as nucleotide base-pairing, stem-loop compositionand bulges within the dsRNA sca"old. Upon complexa-tion, the C-terminal kinase domain is not directly involvedin any dsRNA!PKR-mediated contacts, such that dsRNArecognition is localized within the dsRBD1/2 domains.There is then a proposed conformational change in whichthe central inter-domain linker domain may rearrangeand allow for a weak dimeric association of dsRNA!PKRcomplexes via protein!protein interactions between adja-cent N-terminal lobes of the respective kinase domainsand/or the inter-domain linkers. Once properly positionedrelative to one another, the active sites are positioned in afavorable spatial orientation, which overcomes the ener-getic barrier required for a trans-autophosphorylationcatalysis. This step marks the rate-limiting step of the acti-vation mechanism. Once phosphorylated, the dsRNAligand is ejected and PKR-P self-a!nity greatly increases,and may proceed to trans-phosphorylate additional PKRmolecules in a cascade fashion, which rapidly increasesthe cellular pool of PKR-P. The resultant high level ofPKR-P can then perform e!cient phosphor transfer on
S51 of substrate eIF2a, e"ectively halting translation, andthereby slowing viral proliferation within the cell.
14. Conclusion
We have explored the role of NMR as a structural toolin elucidating the mechanism of autophosphorylationwithin the 66 kDa enzyme PKR. The modularity of PKRcoupled with the favorable spectral dispersion and cross-peak signal to noise of the domains involved within theindividual states of the activation mechanism have beenshown to be an excellent proof of concept argument inthe pursuit of studying high molecular-weight complexesin excess of 80 kDa using NMR as both a structural anddiagnostic tool. Indeed, the in vitro use of NMR has beena cornerstone in the growing understanding regarding themechanism of activation of PKR in a physiological,in vivo setting.
Acknowledgements
We thank M. Margaris for assistance, and other mem-bers of the Puglisi laboratory for their help and advice.D.A. Lindhout and S.A. McKenna are each supportedby Alberta Heritage Foundation for Medical Research fel-lowships, and S.A. McKenna is additionally supported bya Canadian Institutes of Health Research fellowship. Sup-ported by NIH AI47365 and GM 078346.
References
[1] M.J. Gale Jr., M.G. Katze, Pharmacol. Ther. 78 (1998) 29.[2] M.J. Gale Jr., M.J. Korth, M.G. Katze, Clin. Diagn. Virol. 10 (1998)
157.[3] J.L. Cole, Trends Biochem. Sci. 32 (2007) 57.[4] L. Malmgaard, J. Interferon Cytokine Res. 24 (2004) 439.[5] C.E. Samuel, Clin. Microbiol. Rev. 14 (2001) 778.[6] R.K. Maitra, N.A. McMillan, S. Desai, J. McSwiggen, A.G.
Hovanessian, G. Sen, B.R. Williams, R.H. Silverman, Virology204 (1994) 823.
[7] S.A. McKenna, I. Kim, C.W. Liu, J.D. Puglisi, J. Mol. Biol. 358(2006) 1270.
[8] I. Kim, C.W. Liu, J.D. Puglisi, J. Mol. Biol. 358 (2006) 430.
Fig. 11. Proposed molecular framework involved in PKR activation. The latent form of the protein consists of the dsRBDs (R1 and R2), the inter-domainlinker (L) and the kinase domain (K). The R1 and R2 domains sense, and bind to helical regions of activating dsRNA molecules, which may lead to aconformational change in which an enhanced bimolecular interaction occurs between two PKR molecules via the K and/or L domains. In thisconformation, autophosphorylation occurs and there is composite dsRNA release and activation competency of PKR. Activated PKR may then feedbackto trans-phosphorylated remaining latent PKR, or may proceed to phosphorylate the cellular pool of the a-subunit of eIF2, thereby initiating a down-regulation of translation.
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 213

[9] S. Balachandran, P.C. Roberts, L.E. Brown, H. Truong, A.K.Pattnaik, D.R. Archer, G.N. Barber, Immunity 13 (2000) 129.
[10] A.C. Dar, T.E. Dever, F. Sicheri, Cell 122 (2005) 887.[11] M. Dey, C. Cao, A.C. Dar, T. Tamura, K. Ozato, F. Sicheri, T.E.
Dever, Cell 122 (2005) 901.[12] D.R. Taylor, S.B. Lee, P.R. Romano, D.R. Marshak, A.G.
Hinnebusch, M. Esteban, M.B. Mathews, Mol. Cell. Biol. 16(1996) 6295.
[13] S.S. Taylor, N.M. Haste, G. Ghosh, Cell 122 (2005) 823.[14] C.G. Proud, Semin. Cell Dev. Biol. 16 (2005) 3.[15] M. Jaramillo, K. Browning, T.E. Dever, S. Blum, H. Trachsel, W.C.
Merrick, J.M. Ravel, N. Sonenberg, Biochim. Biophys. Acta 1050(1990) 134.
[16] G.D. Pavitt, Biochem. Soc. Trans. 33 (2005) 1487.[17] R. Jagus, B. Joshi, G.N. Barber, Int. J. Biochem. Cell. Biol. 31
(1999) 123.[18] A.R. Cuddihy, A.H. Wong, N.W. Tam, S. Li, A.E. Koromilas,
Oncogene 18 (1999) 2690.[19] A.R. Cuddihy, S. Li, N.W. Tam, A.H. Wong, Y. Taya, N.
Abraham, J.C. Bell, A.E. Koromilas, Mol. Cell. Biol. 19 (1999) 2475.[20] S.D. Der, Y.L. Yang, C. Weissmann, B.R. Williams, Proc. Natl.
Acad. Sci. USA 94 (1997) 3279.[21] S. Guerra, L.A. Lopez-Fernandez, M.A. Garcia, A. Zaballos, M.
Esteban, J. Biol. Chem. 281 (2006) 18734.[22] B.K. Cheung, D.C. Lee, J.C. Li, Y.L. Lau, A.S. Lau, J. Immunol.
175 (2005) 7218.[23] J.C. Li, D.C. Lee, B.K. Cheung, A.S. Lau, FEBS Lett. 579 (2005)
3055.[24] D. Scheuner, M. Gromeier, M.V. Davies, A.J. Dorner, B. Song,
R.V. Patel, E.J. Wimmer, R.E. McLendon, R.J. Kaufman, Virology317 (2003) 263.
[25] F. Weber, V. Wagner, N. Kessler, O. Haller, J. Interferon CytokineRes. 26 (2006) 1.
[26] Y.L. Yang, L.F. Reis, J. Pavlovic, A. Aguzzi, R. Schafer, A. Kumar,B.R. Williams, M. Aguet, C. Weissmann, EMBO J. 14 (1995) 6095.
[27] M. Benkirane, C. Neuveut, R.F. Chun, S.M. Smith, C.E. Samuel, A.Gatignol, K.T. Jeang, EMBO J. 16 (1997) 611.
[28] M. Zamanian-Daryoush, S.D. Der, B.R. Williams, Oncogene 18(1999) 315.
[29] J.M. Murad, L.G. Tone, L.R. de Souza, F.L. De Lucca, Blood CellsMol. Dis. 34 (2005) 1.
[30] J.M. Murad, L.R. de Souza, F.L. De Lucca, Blood Cells Mol. Dis.(2006).
[31] A. Kumar, J. Haque, J. Lacoste, J. Hiscott, B.R. Williams, Proc.Natl. Acad. Sci. USA 91 (1994) 6288.
[32] A. Maran, R.K. Maitra, A. Kumar, B. Dong, W. Xiao, G. Li, B.R.Williams, P.F. Torrence, R.H. Silverman, Science 265 (1994) 789.
[33] A. Kumar, Y.L. Yang, V. Flati, S. Der, S. Kadereit, A. Deb, J.Haque, L. Reis, C. Weissmann, B.R. Williams, EMBO J. 16 (1997)406.
[34] S. Nanduri, B.W. Carpick, Y. Yang, B.R. Williams, J. Qin, EMBOJ. 17 (1998) 5458.
[35] H. Wu, A. Henras, G. Chanfreau, J. Feigon, Proc. Natl. Acad. Sci.USA 101 (2004) 8307.
[36] J.M. Ryter, S.C. Schultz, EMBO J. 17 (1998) 7505.[37] A. Ramos, S. Grunert, J. Adams, D.R. Micklem, M.R. Proctor, S.
Freund, M. Bycroft, D. St Johnston, G. Varani, EMBO J. 19 (2000)997.
[38] R.J. Spanggord, M. Vuyisich, P.A. Beal, Biochemistry 41 (2002)4511.
[39] J.W. Ucci, Y. Kobayashi, G. Choi, A.T. Alexandrescu, J.L. Cole,Biochemistry 46 (2007) 55.
[40] V. Tugarinov, L.E. Kay, J. Biomol. NMR 28 (2004) 165.[41] V. Tugarinov, L.E. Kay, Chembiochem 6 (2005) 1567.[42] V. Tugarinov, P.M. Hwang, L.E. Kay, Annu. Rev. Biochem. 73
(2004) 107.[43] K. Pervushin, R. Riek, G. Wider, K. Wuthrich, Proc. Natl. Acad.
Sci. USA 94 (1997) 12366.
[44] M. Salzmann, K. Pervushin, G. Wider, H. Senn, K. Wuthrich, Proc.Natl. Acad. Sci. USA 95 (1998) 13585.
[45] R. Riek, G. Wider, K. Pervushin, K. Wuthrich, Proc. Natl. Acad.Sci. USA 96 (1999) 4918.
[46] P.J. Lukavsky, J.D. Puglisi, Methods 25 (2001) 316.[47] G.M. Clore, A.M. Gronenborn, Trends Biotechnol. 16 (1998) 22.[48] G.M. Clore, A.M. Gronenborn, Curr. Opin. Chem. Biol. 2 (1998)
564.[49] H. Zhou, A. Vermeulen, F.M. Jucker, A. Pardi, Biopolymers 52
(1999) 168.[50] V. Tugarinov, W.Y. Choy, V.Y. Orekhov, L.E. Kay, Proc. Natl.
Acad. Sci. USA 102 (2005) 622.[51] V. Tugarinov, L.E. Kay, Biochemistry 44 (2005) 15970.[52] V. Tugarinov, L.E. Kay, J. Am. Chem. Soc. 125 (2003) 13868.[53] V. Tugarinov, R. Muhandiram, A. Ayed, L.E. Kay, J. Am. Chem.
Soc. 124 (2002) 10025.[54] P.J. Lukavsky, I. Kim, G.A. Otto, J.D. Puglisi, Nat. Struct. Biol. 10
(2003) 1033.[55] J.D. Puglisi, I. Kim, P. Lukavsky, G. Otto, A. Lancaster, P. Sarnow,
Nucleic Acids Res. Suppl. (2001) 263.[56] I. Kim, P.J. Lukavsky, J.D. Puglisi, J. Am. Chem. Soc. 124 (2002)
9338.[57] D.A. Lindhout, M.X. Li, D. Schieve, B.D. Sykes, Biochemistry 41
(2002) 7267.[58] D.A. Lindhout, B.D. Sykes, J. Biol. Chem. 278 (2003) 27024.[59] S. McKenna, T. Moraes, L. Pastushok, C. Ptak, W. Xiao, L.
Spyracopoulos, M.J. Ellison, J. Biol. Chem. 278 (2003) 13151.[60] S. McKenna, J. Hu, T. Moraes, W. Xiao, M.J. Ellison, L.
Spyracopoulos, Biochemistry 42 (2003) 7922.[61] L. Spyracopoulos, Protein Pept. Lett. 12 (2005) 235.[62] L. Spyracopoulos, M.J. Lewis, L.F. Saltibus, Biochemistry 44 (2005)
8770.[63] D.C. Thomis, C.E. Samuel, J. Virol. 67 (1993) 7695.[64] D.C. Thomis, C.E. Samuel, J. Virol. 69 (1995) 5195.[65] L.R. Saunders, G.N. Barber, FASEB J. 17 (2003) 961.[66] S. Nanduri, B. Carpick, Y. Yang, B.R. Williams, J. Qin, J. Biomol.
NMR 12 (1998) 349.[67] D.S. Wishart, B.D. Sykes, J. Biomol. NMR 4 (1994) 171.[68] D.S. Wishart, B.D. Sykes, Met. Enzymol. 239 (1994) 363.[69] D.S. Wishart, B.D. Sykes, F.M. Richards, J. Mol. Biol. 222 (1991)
311.[70] H. Frauenfelder, S.G. Sligar, P.G. Wolynes, Science 254 (1991) 1598.[71] M. Karplus, J.A. McCammon, Annu. Rev. Biochem. 52 (1983) 263.[72] L.E. Kay, Nat. Struct. Biol. 5 (1998) 513.[73] S.M. Gagne, S. Tsuda, L. Spyracopoulos, L.E. Kay, B.D. Sykes, J.
Mol. Biol. 278 (1998) 667.[74] G.D. Henry, J.H. Weiner, B.D. Sykes, Biochemistry 25 (1986) 590.[75] D.M. LeMaster, Biochemistry 35 (1996) 14876.[76] R.P. Barnwal, T.R. Chaudhuri, S. Nanduri, J. Qin, K.V. Chary,
Proteins 62 (2006) 501.[77] M. Bycroft, S. Grunert, A.G. Murzin, M. Proctor, D. St Johnston,
EMBO J. 14 (1995) 3563.[78] N. Leulliot, S. Quevillon-Cheruel, M. Graille, H. van Tilbeurgh,
T.C. Leeper, K.S. Godin, T.E. Edwards, S.T. Sigurdsson, N.Rozenkrants, R.J. Nagel, M. Ares, G. Varani, EMBO J. 23 (2004)2468.
[79] R. Stefl, M. Xu, L. Skrisovska, R.B. Emeson, F.H. Allain, Structure14 (2006) 345.
[80] S.A. McKenna, D.A. Lindhout, I. Kim, C.W. Liu, V.M. Gelev, G.Wagner, J.D. Puglisi, J. Biol. Chem. 282 (2007) 11474.
[81] S. Wu, R.J. Kaufman, J. Biol. Chem. 272 (1997) 1291.[82] V. Gelev, H. Aktas, A. Marintchev, T. Ito, D. Frueh, M. Hemond,
D. Rovnyak, M. Debus, S. Hyberts, A. Usheva, J. Halperin, G.Wagner, J. Mol. Biol. 364 (2006) 352.
[83] A.C. Carrera, K. Alexandrov, T.M. Roberts, Proc. Natl. Acad. Sci.USA 90 (1993) 442.
[84] S. Nanduri, F. Rahman, B.R. Williams, J. Qin, EMBO J. 19 (2000)5567.
214 D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215

[85] A.G. Tzakos, C.R. Grace, P.J. Lukavsky, R. Riek, Annu. Rev.Biophys. Biomol. Struct. 35 (2006) 319.
[86] P.C. Bevilacqua, T.R. Cech, Biochemistry 35 (1996) 9983.[87] R.C. Patel, P. Stanton, G.C. Sen, J. Biol. Chem. 271 (1996) 25657.[88] B. Tian, P.C. Bevilacqua, A. Diegelman-Parente, M.B. Mathews,
Nat. Rev. Mol. Cell. Biol. 5 (2004) 1013.[89] B. Tian, M.B. Mathews, J. Biol. Chem. 276 (2001) 9936.[90] F. Zhang, P.R. Romano, T. Nagamura-Inoue, B. Tian, T.E. Dever,
M.B.Mathews,K.Ozato,A.G.Hinnebusch, J. Biol.Chem. 276 (2001)24946.
[91] A. Sudhakar, A. Ramachandran, S. Ghosh, S.E. Hasnain, R.J.Kaufman, K.V. Ramaiah, Biochemistry 39 (2000) 12929.
[92] S. Nekhai, A. Kumar, D.P. Bottaro, R. Petryshyn, Virology 222(1996) 193.
[93] J. Vyas, A. Elia, M.J. Clemens, RNA 9 (2003) 858.[94] J.M. Nussbaum, S. Gunnery, M.B. Mathews, Nucleic Acids Res. 30
(2002) 1205.[95] S. Davis, J.C. Watson, Proc. Natl. Acad. Sci. USA 93 (1996) 508.[96] R. Kaempfer, Cell Res. 16 (2006) 148.[97] P.A. Clarke, M. Schwemmle, J. Schickinger, K. Hilse, M.J. Clemens,
Nucleic Acids Res. 19 (1991) 243.[98] T.V. Sharp, M. Schwemmle, I. Je"rey, K. Laing, H. Mellor, C.G.
Proud, K. Hilse, M.J. Clemens, Nucleic Acids Res. 21 (1993) 4483.[99] G.D. Ghadge, S. Swaminathan, M.G. Katze, B. Thimmapaya, Proc.
Natl. Acad. Sci. USA 88 (1991) 7140.[100] K.H. Mellits, M. Kostura, M.B. Mathews, Cell 61 (1990) 843.
[101] H.D. Robertson, L. Manche, M.B. Mathews, J. Virol. 70 (1996)5611.
[102] P.C. Bevilacqua, C.X. George, C.E. Samuel, T.R. Cech, Biochem-istry 37 (1998) 6303.
[103] K.H. Mellits, M.B. Mathews, EMBO J. 7 (1988) 2849.[104] K.H. Mellits, T. Pe’ery, M.B. Mathews, J. Virol. 66 (1992) 2369.[105] T. Pe’ery, K.H. Mellits, M.B. Mathews, J. Virol. 67 (1993) 3534.[106] P.J. Lukavsky, J.D. Puglisi, RNA 10 (2004) 889.[107] J. Karn, J. Mol. Biol. 293 (1999) 235.[108] R.E. Kiernan, C. Vanhulle, L. Schiltz, E. Adam, H. Xiao, F.
Maudoux, C. Calomme, A. Burny, Y. Nakatani, K.T. Jeang, M.Benkirane, C. Van Lint, EMBO J. 18 (1999) 6106.
[109] A. Daher, M. Longuet, D. Dorin, F. Bois, E. Segeral, S. Bannwarth,P.L. Battisti, D.F. Purcell, R. Benarous, C. Vaquero, E.F. Meurs, A.Gatignol, J. Biol. Chem. 276 (2001) 33899.
[110] M. Stevens, E. De Clercq, J. Balzarini, Med. Res. Rev. 26 (2006) 595.[111] J. Kitajewski, R.J. Schneider, B. Safer, S.M. Munemitsu, C.E.
Samuel, B. Thimmappaya, T. Shenk, Cell 45 (1986) 195.[112] G.D. Ghadge, P. Malhotra, M.R. Furtado, R. Dhar, B. Thimma-
paya, J. Virol. 68 (1994) 4137.[113] A. Rahman, P. Malhotra, R. Dhar, T. Kewalramani, B. Thimma-
paya, J. Virol. 69 (1995) 4299.[114] S. Fasciano, B. Hutchins, I. Handy, R.C. Patel, FEBS J. 272 (2005)
1425.[115] W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graph. 14 (1996) 33.[116] W.L. DeLano, DeLano Scientific, San Carlos, CA, USA (2002).
D.A. Lindhout et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 51 (2007) 199–215 215
View publication statsView publication stats





























