Embed Size (px)
Citation preview

J. Appl. Toxicol. 2009; 29: 629–637 Published in 2009 by John Wiley & Sons, Ltd.
Research Article
Received 30 March 2009, Revised 29 May 2009, Accepted 1 June 2009, Published online in Wiley InterScience: 23 July 2009
(www.interscience.wiley.com) DOI 10.1002/jat.1455
Physiologically based pharmacokinetic modeling of cyclotrimethylenetrinitramine in male ratsKannan Krishnan,a Lee C.B.Crouse,b Matthew A. Bazar,b Michael A. Majorb and Gunda Reddyb*
ABSTRACT: A physiologically based pharmacokinetic (PBPK) model for simulating the kinetics of cyclotrimethylene trinitra-mine (RDX) in male rats was developed. The model consisted of fi ve compartments interconnected by systemic circulation. The tissue uptake of RDX was described as a perfusion-limited process whereas hepatic clearance and gastrointestinal absorp-tion were described as fi rst-order processes. The physiological parameters for the rat were obtained from the literature whereas the tissue : blood partition coeffi cients were estimated on the basis of the tissue and blood composition as well as the lipophilicity characteristics of RDX (logP = 0.87). The tissue : blood partition coeffi cients (brain, 1.4; muscle, 1; fat, 7.55; liver, 1.2) obtained with this algorithmic approach were used without any adjustment, since a focused in vitro study indicated that the relative concentration of RDX in whole blood and plasma is about 1 : 1. An initial estimate of metabolic clearance of RDX (2.2 h-1 kg-1) was obtained by fi tting PBPK model simulations to the data on plasma kinetics in rats administered 5.5 mg kg-1 i.v. The rat PBPK model without any further change in parameter values adequately simulated the blood kinetic data for RDX at much lower doses (0.77 and 1.04 mg kg-1 i.v.), collected in this study. The same model, with the incorporation of a fi rst order oral absorption rate constant (Ka 0.75 h-1), reproduced the blood kinetics of RDX in rats receiving a single gavage dose of 1.53 or 2.02 mg kg-1. Additionally, the model simulated the plasma and blood kinetics of orally administered RDX at a higher dose (100 mg kg-1) or lower doses (0.2 or 1.24 mg kg-1) in male rats. Overall, the rat PBPK model for RDX with its parameters adequately simulates the blood and plasma kinetic data, obtained following i.v. doses ranging from 0.77 to 5.5 mg kg-1 as well as oral doses ranging from 0.2 to 100 mg kg-1. Published in 2009 by John Wiley & Sons, Ltd.
! Supporting information may be found in the online version of this article.
Keywords: RDX; PBPK model; pharmacokinetics
*Correspondence to: G. Reddy , US Army Center for Health Promotion and Preventive Medicine, 5158 Blackhawk Road, Aberdeen Proving Ground, MD 21010–5483, USA.Email: [email protected]
aDSEST, Université de Montreal, Montreal, Quebec, Canada
bDirectorate of Toxicology, U.S. Army Center for Health Promotion and Preventive Medicine, Aberdeen Proving Ground, MD, USA
This article is a US Government work and is in the public domain in the USA.
INTRODUCTION
Cyclotrimethylenetrinitramine (RDX, Royal Demolition Explosive), a six-member heterocyclic compound, has been used as an explosive in military munition formulations since World War II. General physicochemical properties, toxicological data and car-cinogenicity bioassays for RDX in rats and mice have been reviewed by various authors (McLellan et al., 1992; ATSDR, 1995; Parker et al., 2006). The neurotoxic eff ects of RDX have been reported in animals and humans (ATSDR, 1995). The acute oral LD50 values of RDX in mice and rats range from 59 to 97 mg kg−1 and from 71 to 300 mg kg−1 respectively (see McLellan et al., 1992). RDX did not produce cancer in two chronic studies in rats (Levine et al., 1983; Hart, 1977), but did produce hepatocellular carcinoma in only female B6C3F1 mice (Lish et al., 1984). Genotoxicity studies of RDX revealed that it was not genotoxic in vitro or in vivo test systems (Reddy et al., 2005). Only a few studies have reported the metabolite profi les and the parent chemical concentrations in laboratory animals dosed with RDX (Schneider et al.1977; Major et al., 2007). However, the available pharmaco-kinetic data on this substance have not yet been used to develop a physiologically based pharmacokinetic (PBPK) model.
PBPK models, increasingly fi nding application in risk assess-ments, are mathematical representations of the absorption, dis-
tribution, metabolism and excretion of chemicals in biota. These models are developed on the basis of the interrelationships among mechanistic determinants of the pharmacokinetic pro-cesses (Krishnan and Andersen, 2001). The PBPK models, once developed and validated, are useful in predicting the internal dose of the toxic moiety of chemicals (Clewell et al., 2002; US EPA, 2006). The use of internal dose represents a scientifi cally sound approach for evaluating dose–response relationships and con-ducting health risk assessments (reviewed in Lipscomb and Ohanian 2007). By facilitating the prediction of internal doses, the PBPK models enable the interspecies, route-to-route and exposure scenario extrapolations essential for human health risk assessment based on animal data (Clewell and Andersen, 1985;
629

K. Krishnan et al.
www.interscience.wiley.com/journal/jat Published in 2009 by John Wiley & Sons, Ltd. J. Appl. Toxicol. 2009; 29: 629–637
Krishnan and Andersen 2001; Lipscomb and Ohanian, 2007; Thompson et al., 2008).
A thorough examination of the published literature indicates that there are no PBPK models for RDX in any species. The present study was therefore undertaken to develop a PBPK model in the rat by estimating key parameters related to absorption, distribu-tion and clearance of RDX. Input parameters for the development of PBPK models, such as tissue : blood partition coeffi cients and metabolic clearance values, have not been reported in the litera-ture. Further, there are only limited data on the oral bioavailabil-ity and pharmacokinetics of RDX in the rat (Schneider et al., 1977; Major et al. 2007; Crouse et al., 2008). Even though animal-alter-native algorithms are available to predict tissue : blood partition coeffi cients of organic chemicals (Poulin and Krishnan, 1995), they do not readily facilitate the evaluation of the potential dif-ferential partitioning of chemicals into erythrocytes and pla-sma. Since RDX concentrations in pharmacokinetic studies are reported as either plasma or whole blood concentrations, the present study evaluated the distribution of RDX in whole blood and plasma in vitro. Further, focused, low-dose pharmacokinetic studies by the i.v. and oral routes were conducted to facilitate the evaluation of the rat PBPK model for RDX.
EXPERIMENTALChemicals
Neat RDX was procured from Dr William M. Koppes, Naval Surface Warfare Center, Indian Head, MD, USA. The purity of the RDX was further con-fi rmed to be 99.9% by gas chromatographic method by Mr Michael Hable, Directorate of Laboratory Sciences, US Army CHPPM (APG, MD, USA).
In vitro Evaluation of the Relative Concentration of RDX in Whole Blood and Plasma
Blood collection
Whole blood samples were obtained from three 20-month-old male F344 rats used as controls in an experimental study. The animals were anes-thetized with isofl urane, the blood was drawn via cardiac puncture and the animals were immediately euthanized via carbon dioxide. The indi-vidual blood samples were initially placed in 1.3 ml Sarstedt EDTA KE microtubes containing 1.6 mg EDTA anti-coagulant. The samples were then composited in a 15 ml polystyrene BD Falcon tube for a total volume of 14 ml, and placed on a blood rocker.
RDX spiking and replicate preparation
Three RDX spiked blood replicates and a control were prepared. Aliquots of 3 ml of whole blood were placed in four untreated polypropylene vials. Thirty microliters of 100 ppm RDX solution (10 mg RDX dl−1 water) was individually added to three of the whole blood vials. The theoretical target concentration was 1 µg RDX ml−1 (0.001 µg µl−1) of blood. The RDX solution was maintained on a hot plate at 37 °C with a stir bar prior to amending the blood. Thirty microliters of water was added to the fourth vial for the control. All four vials were returned to the rocker.
RDX sampling and complete blood count
At 10, 30, 60, and 90 min after spiking the blood, 100 µl aliquots were taken from each vial and placed in fl asks with 100 ml of deionized water for RDX analysis. Water was used specifi cally to hemolyze the erythro-cytes. Blood samples of 350 µl were also taken and centrifuged in a Stat-Spin Veterinary centrifuge to obtain plasma for RDX analysis. A 100 µl aliquot of plasma was then similarly placed in fl asks with 100 ml of water.
Following aliquot collection, a complete blood count was performed on the remaining blood in each replicate with an Abbott Cell-Dyn 3700 Hematology Analyzer (Abbott Laboratories, Abbott Park, IL, USA).
Statistical analysis and calculation of blood partitioning
All statistical analyses were performed with SPSS© for Windows, Release 15.0.0 (SPSS, Chicago, IL, USA). Repeated measures analysis of variance was performed to assess whether the RDX concentrations in the whole blood and plasma changed over time. Total erythrocytes and hematocrit were also analyzed for changes over time to determine whether hemo-lysis of cells was occurring. A two-tailed paired samples t-test was per-formed to determine whether the calculated RDX plasma and cellular percentages were statistically diff erent. Statistical signifi cance was defi ned at the P < 0.05 level. Hematocrit data from the complete blood count was used as a surrogate for packed cell volume to determine the apportioning of RDX between the cellular component of the blood and plasma on a per volume basis. A 1% buff y coat was assumed for calcula-tions. A mass adjustment was also included based on the specifi c gravity of whole blood (1.05) and plasma (1.02) (Trudnowski and Rico, 1974).
Evaluation of the Oral and Intravenous Pharmacokinetics of RDX in Rats
Animals
Adult male Sprague–Dawley rats (approximately 380–420 g b.w.) were obtained from Charles River Laboratories Inc., Raleigh, NC, USA. All rats were implanted with femoral artery catheters at Charles River prior to shipment. The attending veterinarian exam-ined the animals and found them to be in acceptable health. Because of the relatively short patency period of the catheters, the rats were only quarantined for 12 h prior to study initiation. All rats were maintained at a room temperature between 64 and 79˚F and relative humidity 30–70%, under a 12 h light/dark cycle. With the exception of an 8 h fasting cycle (4 h prior to dosing and 4 h post-dosing), a certifi ed pesticide-free rodent chow (Harlan Teklad®, 8728C Certifi ed Rodent Diet, Madison, WI, USA) and drinking water were available ad libitum. Rats were housed indi-vidually in suspended polycarbonate boxes with Harlan Sani-Chip® bedding (P.J. Murphy Forest Products Corp. Montville, NJ, USA). Each rat was uniquely identifi ed by number using cage cards. The animal husbandry was performed in animal facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Research was conducted under an IACUC-approved animal use protocol, and was compliant with Animal Welfare Regulations and requirements set forth in The Guide for the Care and Use of Research Animals (NRC, 1996).
Dosing solutions
Dosing occurred on two separate days to accommodate the short inter-vals between blood sampling. Therefore, fresh dosing solutions were prepared for each day of dosing. The fi rst solution was prepared by mixing 20 mg dry RDX in 100 ml deionized water. The mixture was warmed to 38 °C (body temperature of rat) and mixed with a magnetic stir bar to get RDX into solution. The second dosing solution was pre-pared by mixing 21.2 mg dry RDX in 100 ml deionized water since the concentration of the fi rst solution proved to be lower than expected. Each dosing solution was mixed for approximately one hour once the temperature reached 38 °C and a single sample was taken from the middle of the solution for analysis.
Oral dosing
Rats were dosed orally with RDX solutions via gavage using a 16 gage × 2 inch gavage tube. Oral doses administered were calculated based on a
630

Plarmacokinetic modeling of cyclotrimethylenetrinitramine
J. Appl. Toxicol. 2009; 29: 629–637 Published in 2009 by John Wiley & Sons, Ltd. www.interscience.wiley.com/journal/jat
10 ml kg−1 dosing volume. Doses were calculated from body weights taken 1 h prior to dosing. A total of 10 rats were dosed orally with the RDX/deionized water solution and blood samples were collected at dif-ferent times up to 10 h and analyzed as described below.
I.v. dosing
Rats were dosed intravenously with the RDX solutions directly into the implanted femoral artery catheter using a 3 ml syringe equipped with a 21 gage Luer stub adaptor. Intravenous doses were calculated based on a 5 ml kg−1 dosing volume in water solutions and dosed via catheters to each rat. Doses were calculated from body weights taken 1 h prior to dosing. The target dose was 1.0 mg kg−1 with actual doses calculated from analytical concentrations of the dosing solutions. A total of 10 rats were dosed by the i.v. route and blood samples were collected at diff er-ent times up to 10 h and analyzed as described below.
Based on the analytical concentrations of the dosing solutions (153 and 207 mg l−1) and the body weights of the rats, intravenously dosed rats in the fi rst round of dosing (n = 5) received 0.77 mg kg−1 of RDX and orally dosed rats received 1.53 mg kg−1. Intravenously dosed rats in the second round of dosing (n = 5) received 1.04 mg kg−1 of RDX and orally dosed rats received 2.07 mg kg−1.
Blood sampling and analysis
Blood sampling was performed via the pre-implanted femoral artery catheter of each rat at 0.5, 1, 2, 4, 6, 8, and 10 h post-dosing. A 100 µl sample of blood was taken from the animals at each sample time using a 21 gage Luer stub adapter attached to a 1 ml syringe. The sampling procedure remained the same for each rat. The stainless steel catheter plug was removed using hemostats and the lock solution was removed from the catheter using a syringe. A 100 µl blood sample was taken from each rat. The catheter was fl ushed with 0.2 ml sterile saline. Finally, 0.07 ml heparinized saline (500 IU heparin/1 ml fi nal solution in saline) was injected into the catheter so that it would remain patent for the next sampling time. The catheter plug was placed back into the tubing and the area was cleaned with an alcohol swab before insertion back into the wound clip. Blood samples collected at diff erent time points were placed (100 µl) directly in a volumetric fl ask containing 100 ml of water and placed in the refrigerator prior to analysis.
The blood samples were analyzed by the Explosives Analysis Team according to methods described by Bishop et al. (2003). Blood samples (100 µl) were added to water (100 ml) as soon as collected and placed in the refrigerator and extracted with 1 ml of isoamyl acetate (anhydrous, ≥99%) and aliquots of isoamyl acetate extractions were analyzed by gas chromatography equipped with electron capture detector. The detection limit of the analytical method was 0.10 µg l−1 water (Bishop et al., 2003).
Modeling
Development of RDX PBPK model in the rat
A PBPK model for RDX in the rat was constructed on the basis of its known physicochemical and toxicological properties. Since RDX is not stored to a signifi cant extent in the skeletal/structural components of the body, 91% of the body weight of rat was represented by the tissue com-partments included in the models (100% body weight − 9% skeletal/structural component weight) (Krishnan and Andersen, 2001). The tissue compartments included in the PBPK model were: liver, fat, brain, slowly perfused tissues and richly perfused tissues (Fig. 1). Each tissue compart-ment in the RDX PBPK model was described with a mass balance diff er-ential equation (MBDE) that consisted of a series of clearance terms (Ramsey and Andersen, 1984). The perfusion-limited tissue uptake of RDX in blood was described according to Fick’s law of simple diff usion, which states that the fl ux of a chemical is proportional to its concentration gradient. Metabolism in liver was described as a fi rst-order process. Accordingly, hepatic metabolism was described using a fi rst order rate constant (Kfc; h−1 kg−1) and computed as Kfc × volume of liver × free con-centration of RDX in the liver. Values specifi c to test animals were obtained
by multiplying Kfc by body weight−0.33. Oral absorption was also described as a fi rst order process, and the absorbed RDX was introduced into the liver compartment (Krishnan and Andersen, 2001). The equations consti-tuting the RDX PBPK were written as a program and solved using Berkeley Madonna® (version 8.0, University of California, Berkeley, CA, USA).
For the rat PBPK model for RDX, four sets of input parameter values were needed, the tissue volumes, tissue blood fl ow rates, partition coef-fi cients and metabolism constants. The fi rst two, tissue volumes and blood fl ows, are species-specifi c, and were obtained from the literature (Timchalk et al., 2002) (Table 1). The latter two, tissue partition coeffi cients and metabolic constants, are compound-specifi c and needed to be determined for RDX. These parameters have never been evaluated for RDX. The tissue : blood partition coeffi cients (Pt) were therefore estimated on the basis of the n-octanol : water partition coeffi cient (Po:w) of RDX as well as the composition of rat tissues and blood in terms of neutral lipids, phospholipids and water as follows (Poulin and Krishnan, 1995; Poulin and Theil, 2000):
PP F FP F Ft
o:w nlet wet
o:w nleb web
=×( ) +×( ) +
where Fnlet = fractional volume of neutral lipid equivalents in tissue, cal-culated as the sum of neutral lipids plus 0.3 × phospholipid content, Fwet = fractional volume of water equivalents in tissue, calculated as the sum of tissue water content plus 0.7 × phospholipid content, Fnleb = fractional volume of neutral lipid equivalents in blood, calculated as the sum of neutral lipids plus 0.3 × phospholipid content, and Fweb = fractional volume of water equivalents in blood, calculated as the sum of water content plus 0.7 × phospholipid content.
The fraction of neutral-lipid equivalents and water-equivalent compo-nents in tissues and blood were obtained mainly from Poulin and Krishnan (1996) whereas the log Pow value for RDX (0.87) was obtained from ATSDR (1995). The fi rst-order metabolic rate for RDX was estimated by fi tting model simulations to the time-course data on RDX kinetics in rats administered 5.5 mg kg−1 i.v. (Schneider et al., 1977).
Evaluation of the rat RDX PBPK model
The rat PBPK model was then evaluated for its performance by simulating the following experiments:
• a low dose experiment in the rat in which 0.77 or 1.04 mg kg−1 RDX was administered by i.v. route (described in the preceding section);
• a high dose experiment in the rat in which 100 mg kg−1 RDX was admin-istered by gavage and plasma concentrations were determined up to 24 h (Schneider et al., 1977);
BLOOD
BRAIN
FAT
SLOWLY PERFUSED TISSUES
LIVER
i.v. Dose
G.I. Tract
Oral Dose Metabolism
RICHLY PERFUSED TISSUES
Figure 1. Conceptual representation of the PBPK model for simulating uptake and disposition of RDX in rats.
631

K. Krishnan et al.
www.interscience.wiley.com/journal/jat Published in 2009 by John Wiley & Sons, Ltd. J. Appl. Toxicol. 2009; 29: 629–637
• a low dose experiment in which 1.53 or 2.07 mg RDX kg−1 b.w. was given in water by gavage to rats and blood RDX concentrations mea-sured during 10 h post-dosing (described in the preceding section); and
• an oral bioavailability study in which groups of rats (n = 11) were dosed with neat RDX in gelatine capsules at the dose levels of 0.2 or 1.24 mg kg−1 b.w., and blood concentrations collected for up to 12 h post-dosing (Crouse et al., 2008).
RESULTS
In the in vitro study, at 10 min following incubation, the whole blood concentration of RDX ranged from 0.43 to 0.67 µg ml−1 whereas the plasma concentrations were between 0.50 and 0.57 µg ml−1. At the 30 min interval, the whole blood ranged from 0.59 to 0.68 µg ml−1 compared with 0.52 to 0.59 µg ml−1 in the plasma. Similar data were also obtained at 60 min (whole blood, 0.58–0.62 µg ml−1 vs plasma, 0.53–0.60 µg ml−1) as well as 90 min (whole blood, 0.65–0.68 µg ml−1 vs plasma, 0.49–0.62 µg ml−1) after incubation. Taken together, these data indi-cate that the whole blood and plasma concentrations of RDX are comparable. The overall average concentration of RDX in eryth-rocytes (0.54 ± 0.02) and plasma (0.46 ± 002) was approximately 1 : 1. Statistical analyses indicated no signifi cant diff erence in RDX concentrations during 90 min in blood, erythrocyte or plasma. Therefore, the tissue : blood partition coeffi cients estimated per Poulin and Krishnan (1995) were used in the PBPK model, for
comparing with whole blood or plasma concentrations of RDX reported in experimental studies.
The tissue : blood partition coeffi cients for RDX, predicted using the algorithmic approach of Poulin and Krishnan (1996), ranged from 1 to 7.55 for the various tissues (brain, 1.4; muscle, 1; fat, 7.55; liver, 1.2). Incorporating these tissue : blood partition coeffi cients along with the rat physiological parameters (Table 1), simulations of the blood and plasma kinetics of RDX in rats receiving 5.5 mg kg−1 i.v. were obtained (data from Schneider et al., 1977). As shown in Fig. 2(A), the experimental data from Schneider et al. (1977) were contained within the series of model simulations obtained using various values of fi rst order metabolic rate constant (Kfc) ranging from 2 to 100 h−1 kg−1. With increasing values of Kfc (5–100 h−1 kg−1), the PBPK model simulations moved further away from the experimental data points [Fig. 2(A)]. Subsequently, a single value of Kfc was determined by visual fi tting of model simulations to experimental data of Schneider et al. (1977) [Fig. 2(B); Kfc = 2.2 h−1 kg−1]. The rat PBPK model without any further change in parameter values adequately sim-ulated the kinetics of RDX at much lower doses evaluated in the present study, fi rst for the i.v. route and then for the oral route (data in Table 2). Figure 3 shows the comparison of rat PBPK model simulations with experimental data obtained in this study following a single i.v. dose of 0.77 or 1.04 mg kg−1.
The PBPK model, with the incorporation of a Ka value, also adequately simulated the kinetics of RDX in rats receiving a single gavage dose of 100 mg kg−1 (Schneider et al., 1977) (Ka, 0.0075 h−1;
Table 1. List of parameters used in the rat PBPK model for RDX
Parameter type Compartment Value Units
Volumea
Blood 6 % b.w.Brain 1.2 % b.w.Fat 7 % b.w.Liver 4 % b.w.Richly perfused 4 % b.w.Slowly perfused b % b.w.
Blood fl ow ratea
Brain 3 % cardiac outputFat 9 % cardiac outputLiver 25 % cardiac outputSlowly perfused 20 % cardiac outputRichly perfused c % cardiac output
Partition coeffi cientsd
Brain 1.4 l blood l−1 tissueFat 7.55 l blood l−1 tissueLiver 1.2 l blood l−1 tissueRichly perfused e l blood l−1 tissueSlowly perfused 1 l blood l−1 tissue
Cardiac output 15 l h−1 kg−1 b.w.aPhysiological parameters were obtained from Timchalk et al. (2002); cardiac output was calculated for individual animals as a function of body weight0.74.bCalculated as body weight minus skeletal weight and the volumes of other compartments.cCalculated as cardiac output minus the blood fl ow to all other compartments.dComposition data from Poulin and Theil (2000) (brain), Poulin and Krishnan (1995) (blood), and Poulin and Krishnan (1996) (other tissues).eSet equal to that of the brain.
632

Plarmacokinetic modeling of cyclotrimethylenetrinitramine
J. Appl. Toxicol. 2009; 29: 629–637 Published in 2009 by John Wiley & Sons, Ltd. www.interscience.wiley.com/journal/jat
A
121086420
100
10
1
0.1
0.01
0.001
1e-4
1e-5
Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
B
1
10
100
0 2 4 6 8 10 12Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
Figure 2. Estimation of the rate of metabolism of RDX in rats. PBPK model simulations (solid lines) were obtained using varying values of metabolic rate constant (A, 0.1–100 h−1 kg−1; B, 2.2 h−1 kg−1). Experimental data (symbols; mean ± SE, n = 10) were obtained from Schneider et al. (1977).
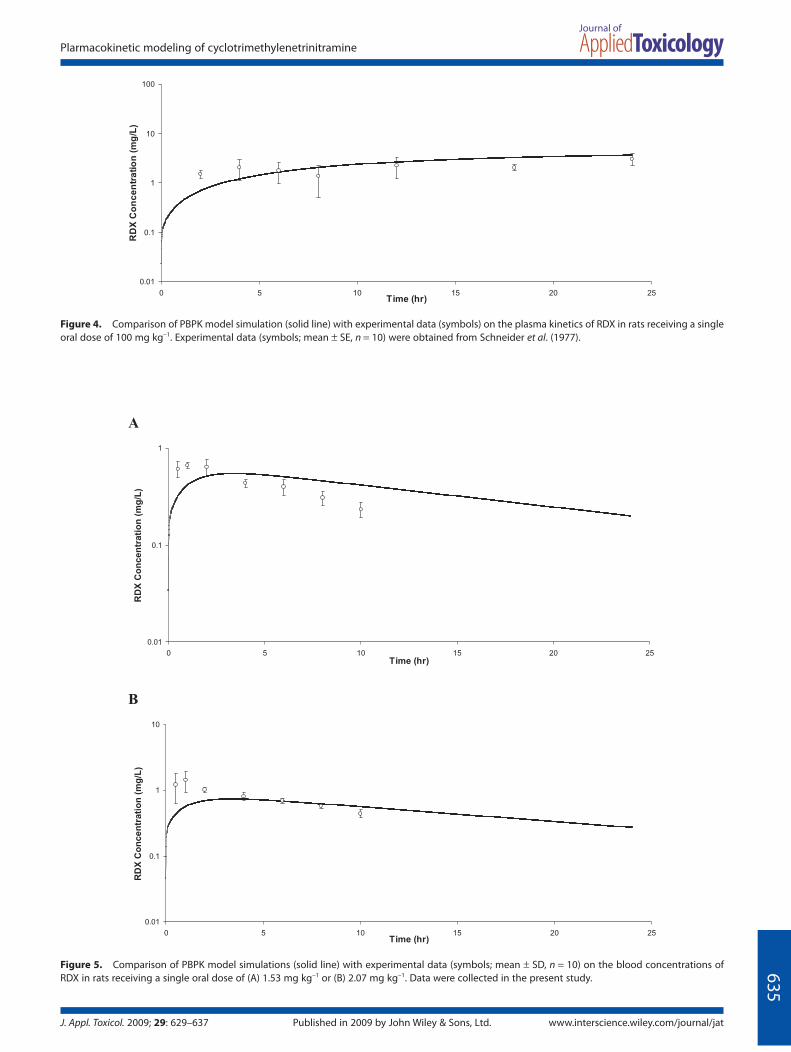
Fig. 4) or lower doses of 1.53 or 2.07 mg kg−1 used in the present study [Ka, 0.75 h−1; Fig. 5(A, B)]. Figure 6 shows that, using the same Ka value as well as other model parameter values, the model could simulate the kinetics of neat RDX (given via gelatine capsules) at even lower doses (0.2 or 1.24 mg kg−1) as part of a recent bioavailability study in male rats (Crouse et al., 2008).
DISCUSSION
The metabolism and pharmacokinetics of RDX has been investi-gated in several studies (reviewed in McLellan et al., 1992; ATSDR, 1995; Parker et al., 2006). Schneider et al. (1977) reported a volume of distribution of 2.18 l kg−1 and a half-life of 6.32 min based on very limited blood kinetic data collected following a single i.v. dose in rats. Apart from these scant data analyses, no
pharmacokinetic parameters or models for RDX have been reported in the literature. The present study developed, for the fi rst time, a PBPK model for RDX in the rat. This initial PBPK model for RDX, based on the perfusion-limitation of RDX uptake by tissues, adequately simulated the whole body kinetics or more specifi cally blood and plasma kinetics of RDX in the rat. In this regard, the model was also applied for high dose to low dose and route to route (i.v. to oral) extrapolations of the blood and plasma kinetics of RDX – and compared with such data collected in this and previous studies.
In order to develop the RDX PBPK model, physiological, physi-cochemical and biochemical parameters were required. The physiological parameters for rats were obtained from published literature (Timchalk et al., 2002; Table 1) whereas the tissue : blood partition coeffi cients were determined on the basis of rat tissue and blood composition data as well as the log Po:w value for RDX.
633

K. Krishnan et al.
www.interscience.wiley.com/journal/jat Published in 2009 by John Wiley & Sons, Ltd. J. Appl. Toxicol. 2009; 29: 629–637
Table 2. Time-course of blood RDX concentration in rats receiving a single intrave-nous (i.v.) or oral dose
Route Dose (mg kg−1)Blood concentration (mg l−1)a
0.5 h 1 h 2 h 4 h 6 h 8 h 10 h
Intravenous 0.767 1.84 0.924 0.602 0.412 0.294 0.238 0.160.493 0.179 0.081 0.015 0.026 0.022 0.007
Intravenous 1.038 1.618 1.06 0.786 0.504 0.44 0.312 0.2340.394 0.23 0.069 0.081 0.117 0.058 0.047
Oral 1.53 0.608 0.666 0.636 0.434 0.398 0.308 0.2340.116 0.047 0.132 0.04 0.077 0.053 0.043
Oral 2.07 1.21 1.432 1.022 0.814 0.696 0.582 0.4420.573 0.484 0.092 0.113 0.053 0.061 0.06
aAverage values of fi ve rats ± SD.
A
0.01
0.1
1
10
100
0 2 4 6 8 10 12 14 16Time (hr)
RD
X C
on
cen
tra
tio
n (
mg
/L)
B
0.01
0.1
1
10
100
0 2 4 6 8 10 12 14 16Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
Figure 3. Comparison of PBPK model simulation (solid lines) with experimental data (open circles; mean ± SD, n = 5) on the blood concentration of RDX in rats receiving a single dose of 0.77 mg kg−1 (A) or 1.04 mg kg−1 (B) by the intravenous route. Data were obtained in the present study.
634
Plarmacokinetic modeling of cyclotrimethylenetrinitramine
J. Appl. Toxicol. 2009; 29: 629–637 Published in 2009 by John Wiley & Sons, Ltd. www.interscience.wiley.com/journal/jat
0.01
0.1
1
10
100
5202510150Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
Figure 4. Comparison of PBPK model simulation (solid line) with experimental data (symbols) on the plasma kinetics of RDX in rats receiving a single oral dose of 100 mg kg−1. Experimental data (symbols; mean ± SE, n = 10) were obtained from Schneider et al. (1977).
A
0.01
0.1
1
5202510150Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
B
0.01
0.1
1
10
5202510150Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
Figure 5. Comparison of PBPK model simulations (solid line) with experimental data (symbols; mean ± SD, n = 10) on the blood concentrations of RDX in rats receiving a single oral dose of (A) 1.53 mg kg−1 or (B) 2.07 mg kg−1. Data were collected in the present study.
635

K. Krishnan et al.
www.interscience.wiley.com/journal/jat Published in 2009 by John Wiley & Sons, Ltd. J. Appl. Toxicol. 2009; 29: 629–637
A
2520151050
1
0.1
0.01
0.001
Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
B
2520151050
1
0.1
0.01
Time (hr)
RD
X C
on
cen
trat
ion
(m
g/L
)
Figure 6. Comparison of PBPK model simulation (solid lines) with experimental data (open circles) on the blood concentration of RDX in rats receiving a single dose of 0.2 mg kg−1 (A) or 1.24 mg kg−1 by the oral route. Data were obtained from Crouse et al. (2008).
The algorithm based on lipid and water solubility for predicting tissue : blood partition coeffi cients in the rat has been success-fully done in the past for several chemicals that are fairly lipo-soluble or hydrosoluble (Krishnan and Andersen, 2001). The tissue : blood partition coeffi cients for RDX are likely to be quite similar to tissue : plasma partition coeffi cients as indicated by similar concentrations found in whole blood and plasma in the in vitro distribution studies. In these studies, the target concen-tration of RDX was lower than the nominal concentrations. It is not known as to whether binding of RDX to the vials occurred or whether there was any metabolism of RDX within the samples. The impact, if any, of the anesthetic used in this study (isofl uor-ane) on the behavior of RDX in blood is not known and this concern might be applicable to in vivo as well as in vitro studies. The primary benefi t of the in vitro approach for evaluating the relative concentrations of RDX in whole blood and plasma is that it is not aff ected by the fi rst-pass metabolism that would occur in an in-vivo study, and provides a certain level of confi dence in
using the tissue : blood partition coeffi cients obtained with the animal-replacement algorithms in the in vivo PBPK models.
The maximal velocity, Michaelis parameter or the intrinsic clearance of RDX in vivo is not reported in the literature. Therefore, the present study evaluated the hepatic metabolism parameter (as a fi rst order rate constant, Kfc) using the limited in vivo i.v. kinetic data in rats. This parameter refl ects the rate of metabolism in the liver; however, it may also refl ect unknown non-hepatic clearance processes. The Kfc was determined initially in rats based on visual fi tting of model simulations to kinetic data reported by Schneider et al. (1977) for a single i.v. dose of 5.5 mg kg−1. The plasma kinetics and plasma half-life reported by Schneider et al. (1977) are essentially reproduced by the present model. Using the same value for Kfc (i.e. 2.2 h−1 kg−1), the model adequately simulated the elimination kinetic curves obtained following lower i.v. doses (0.2 and 2.04 mg kg−1) evaluated in the present study as well as by Crouse et al. (2008). Similarly, the pharmaco-kinetics of RDX following a single oral dose of 100 mg kg−1
636

Plarmacokinetic modeling of cyclotrimethylenetrinitramine
J. Appl. Toxicol. 2009; 29: 629–637 Published in 2009 by John Wiley & Sons, Ltd. www.interscience.wiley.com/journal/jat
(Schneider et al., 1977) was also adequately simulated, indicating the absence of metabolic saturation in the dose range investi-gated in this study. The metabolism of RDX has been reported in vitro to be mediated by CYP2B4, a phenobarbital-inducible enzyme (Bhushan et al., 2003). This isozyme is thought to exhibit a relatively higher Km (compared with CYP2E1, for example) for a number of substrates, indicating that metabolic saturation is likely to occur only at very high doses.
Overall, the rat PBPK model for RDX described in this article (i) consistently simulates the blood and plasma kinetic data follow-ing the i.v. and oral routes available in the literature as well as those collected in the present study; (ii) suggests that the perfu-sion limitation description (of tissue uptake) can yield adequate simulation of plasma and blood kinetics of RDX in rats; and (iii) indicates that the metabolism of RDX is likely to be a fi rst-order process in the dose range investigated in this study (0.2–100 mg kg−1 oral dose; 0.2–5.5 mg kg−1 i.v.). This initial model can serve as a basis to develop PBPK descriptions for predicting phar-macokinetics of RDX in other species as well as for investigating the potential pharmacokinetic basis of species diff erences in tox-icity and carcinogenicity of RDX.
SUPPORTING INFORMATION
Supporting information can be found in the online version of this article.
REFERENCESATSDR. 1995. Toxicological Profi le for RDX. US Department of Health and
Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA.
Bhushan B, Trott S, Spain JC, Halasz A, Paquet L,Hawari J. 2003. Biotransformation of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) by a rabbit liver cytochrome P450: insight into the mechanism of RDX biodegradation by Rhodococcus sp. strain DN22. Appl. Environ. Microbiol. 69: 1347–1351.
Bishop RW, Hable MA, OliverCG, Valis RJ. 2003. The USACHPPM gas chromatographic procedure for the analysis of waters and soils for energetic and related compounds. J. Chromatogr. Sci. 41: 73–79.
Clewell HJ III, Andersen ME. 1985. Risk assessment extrapolations and physiological modeling. Toxicol. Ind. Health 1: 111–131.
Clewell HJ III, Andersen ME, Barton HA. 2002. A consistent approach for the application of pharmacokinetic modeling in cancer and noncan-cer risk assessment. Environ. Health Perspect. 110: 85–93.
Crouse LCB, Michie MW, Major MA, Leach GJ, Reddy G. 2008. Oral bioavail-ability of cyclotrimethylenetrinitramine (RDX) from contaminated site soils in rats. Int. J. Toxicol. 27: 317–322.
Hart ER 1976. Two Year Feeding Study in Rats. Litton Bionetics Inc.: Kensington, MD. Offi ce of Navel Research Contract number N00014–73-C-0162. ADA 040161.
Krishnan K, Andersen ME 2001. Physiologically based pharmacokinetic modeling in toxicology. In Principles and Methods of Toxicology, Hayes AW (ed.). Taylor & Francis: Philadelphia, PA; 193–241.
Levine BS, Furedi-Machacek EM, Rac VS, Gordon DE, Lish PM. 1983. Determination of the Chronic Mammalian Toxicological Eff ects of RDX: Twenty-four Month Chronic Toxicity/carcinogenicity Study of Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in Fisher 344 Rat. Phase V, Vol. 1. Contract no. DAMD 17–79-C-9161. IIT Research Institute: Chicago, Illinois. ADA 160774. US Army Medical Research and Development Command: Fort Detrick, MD.
Lipscomb JC, Ohanian EV. 2007. Toxicokinetics and Risk Assessment. Informa Healthcare: New York.
Lish PM, Levine BS, Furedi-Machacek EM, Sagartz EM, Rac VS. 1984. Determination of the Chronic Mammalian Toxicological Eff ects of RDX: Twenty-four Month Chronic Toxicity/carcinogenicity Study of Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in B6C3F1 Hybrid Mouse. Phase VI, Vol 1. Contract no. DAMD 1–79-C-9161. IIT Reserach Institute: Chicago, Illinois. ADA160774. U.S. ArmyMedical research and development Command: Fort Detrick, MD.
Major MA,Reddy G, Berge MA, Patzer SS, Li AC, Gohdes M. 2007. Metabo-lite profi ling of [14C]hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in Yucatan miniature pigs. J. Toxicol. Environ. Health A 70: 1191–202.
McLellan WL, Hartley WR, Brower ME. 1992. Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX). In Drinking Water Health Advisory: Munitions, Roberts WC, Hartley WR (eds). Lewis:, Boca Raton, FL; 133–180.
NRC. 1996. Guide for the Care and Use of Laboratory Animals. Institute of Laboratory Animal: Resources commission on Life sciences, National Research Council. National Academy Press: Washington DC.
Parker GA, Reddy G, Major MA. 2006. Reevaluation of a twenty-four-month chronic toxicity/carcinogenicity study of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in the B6C3F1 hybrid mouse. Int. J. Toxicol. 25: 373–378.
Poulin P, Krishnan K. 1995. An algorithm for predicting tissue: blood parti-tion coeffi cients of organic chemicals from n-octanol: water partition coeffi cient data. J. Toxicol. Environ. Health. 46: 117–129.
Poulin P, Krishnan K. 1996. Molecular structure-based prediction of the partition coeffi cients of organic chemicals for physiological pharma-cokinetic models. Toxicol. Meth. 6: 117–137.
Poulin P, Theil FP. 2000. A priori prediction of tissue : plasma parti-tion coeffi cients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J. Pharm. Sci. 89: 16–35.
Ramsey JC, Andersen ME. 1984. A physiologically based description of the inhalation pharmacokinetics of styrene in rats and humans. Toxicol. Appl. Pharmacol. 73: 159–175.
Reddy G, Erexson GL, Cifone MA, Major MA, Leach GJ. 2005. Genotoxicity assessment of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX). Int. J. Toxicol. 24: 427–438.
Schneider NR, Bradley SL, Andersen M E. 1977. Toxicology of cyclotri-methylenetrinitramine: distribution and metabolism in the rat and the miniature swine. Toxicol. Appl. Pharmacol. 39: 531–541.
Thompson CM, Sonawane B, Barton HA, DeWoskin RS, Lipscomb JC, Schlosser P, Chiu WA, Krishnan K. 2008. Approaches for applications of physiologically based pharmacokinetic models in risk assessment. J. Toxicol. Environ. Health B Crit. Rev. 11: 519–547.
Timchalk C, Nolan RJ, Mendrala AL, Dittenber DA, Brzak KA, Mattsson JL. 2002. A physiologically based pharmacokinetic and pharmacody-namic (PBPK/PD) model for the organophosphate insecticide chlor-pyrifos in rats and humans. Toxicol. Sci. 66: 34–53.
Trudnowski RJ, Rico RC. 1974. Specifi c gravity of blood and plasma at 4 and 37 °C. Clin. Chem. 20: 615–616.
US EPA. 2006. Approaches for the application of physiologically based pharmacokinetic (PBPK) models and supporting data in risk assess-ment. EPA/600/R-05/043F. 2006. National Center for Environmental Assessment, Offi ce of Research and Development, US Environmental Protection Agency: Washington, DC.
637





























