Embed Size (px)
Citation preview

Indian Journal of Biotechnology
Vol 15, October 2016, pp 495-506
Phylogenetic characterization of novel cathelicidin from Indian water buffalo
Shahid Hussain1, Chandra Sekhar Mukhopadhyay
1*, B V Sunil Kumar
1 and Simarjeet Kaur
2
1School of Animal Biotechnology, Post Graduate Institute of Veterinary Education and Research and 2Department of Animal Genetics and Breeding, College of Veterinary Sciences
Guru Angad Dev Veterinary and Animal Sciences University, Ludhiana 141 004, India
Received 5 May 2015; revised 17 November 2015; accepted 24 November 2015
The bubaline cathelicidins have been an important area of research because of possibility of exploring novel
antimicrobial peptides (AMPs) in the buffaloes, which are comparatively more sturdy animals and are better adapted to
Indian climate as compared to the crossbred cattle. The rationale behind the current study was to in silico characterize the
cloned bubaline cathelicidin3 (Cath3) peptide and to study the evolution of bubaline caths. Multiple sequence alignment of
the bubaline Cath3 (from this study) and homologous peptide sequences revealed an insertion of 6 amino acids in the
cathelin domain of the bubaline Cath3 peptide when compared with that of cattle. Biocomputational analyses of the Cath3
coding sequence (cds) as well as the amino acid sequences (using MEGA6 software & Datamonkey server) indicated that
different types and transcript variants of cathelicidin varied considerably within the same species, indicating the role of
natural selection during the evolution of caths. The cathelicidin cds belonging to divergent species were analyzed using
different models like SLAC, FEL and REL (Datamonkey server). It is concluded from the REL model that bubaline
cathelicidin3 antimicrobial peptides have undergone episodic positive selection (in upto 36 codons) conferring selective
advantage in evolution of the peptide.
Keywords: Buffalo, cathelicidin, evolution, immunity, selection
Introduction The role of antimicrobial peptides (AMPs) in the
immune system of eukaryotes has opened up new
research areas directed towards their possible
exploitation in improving disease resistance in
domestic animals and humans. The immune system is
persistently evolving among species, given its
necessity to act against the unceasing evolution of
pathogenic microbes for acquiring resistance. Genes
functioning in host-pathogen interaction may be the
targets of directional or balancing selection more
often than the genes which are no way related to the
health and immunity of the living being1,2
.
AMP family is widely distributed among
eukaryotes3,4
. These AMPs are evolutionarily ancient
component of the innate immune system and are the
fundamental defensive weapons of multicellular
organisms5. AMPs show diversity in both ways, i.e.,
inter species diversity vis-à-vis within species
variations among higher organism in their spectrum of
activity and structure6. Mammals have two major
categories of AMPs, defensins and cathelicidins. The
precursors of cathelicidins are divided into a highly
conserved N-terminal pre-proregion, which includes
the conserved signal sequence and the cathelin domain
as well as the divergent C-terminal antimicrobial
domain. Proteolytic cleavage of the peptide at a stretch
of specific amino acid residues by specific processing
enzymes releases the antimicrobial domain from the
whole peptide7-9
. Antimicrobial nature of these peptides
is attributed to their positive charge and their ability to
fold into amphipathic structure around a lipid bilayer
membrane10
.
The emergence of multiple drug resistance has led to
the need for identifying novel and effective antimicrobial
agents. Given the wide continuum of activity of naturally
occurring AMPs towards different pathogens including
multi-drug resistant bacteria and fungi, these peptides are
being studied as new anti-microbial agents11
. The
indigenous buffaloes are known for disease tolerance as
compared to crossbred cattle12
. The current study could
give an insight towards the use of cathelicidin in the
development of novel antimicrobial drugs. Presently, few
cathelicidins of bubaline origin have been identified and
no work on its evolution with respect to their Bovidae
counterparts has been reported. The present study aims to
unveil the evolutionary perspective of cathelicidin
peptides of the bubaline species.
___________
*Author for correspondence:
Mobile: +91-161-2414023

INDIAN J BIOTECHNOL, OCTOBER 2016
496
Materials and Methods Peripheral blood samples were aseptically collected
in sterile, 50 mL centrifuge tubes using 0.5 M EDTA as
an anticoagulant. Blood samples were collected from
adult dairy buffalo housed at the Dairy Farm,
Department of Animal Genetics and Breeding, Guru
Angad Dev Veterinary and Animal Sciences University,
Ludhiana, Punjab, India. The experiment was approved
by the Institutional Animal Ethics Committee.
Isolation of total RNA
Peripheral blood mononuclear cells (PBMCs) were
extracted from whole blood using HiSep based gradient
centrifugation. The isolated PBMCs were resuspended in
1 mL of Trizol reagent (Invitrogen, USA) and the total
RNA was isolated from the PBMCs using 1× RNA-lysis
buffer (ammonium chloride, 0.83 g; ammonium
bicarbonate, 0.1 g; EDTA 0.5 M, 20 µL and DEPC
treated water (0.1%) up to 100 mL). The concentration
and integrity of total RNA was checked by Nanodrop
spectrophotometer (Thermo Scientific, USA). Total RNA
samples having absorbance ratio (260/280) between
2.0 and 2.1 were subjected to first strand cDNA synthesis,
using RevertAid cDNA synthesis Kit (Fermentas, USA)
according to manufacturer’s instruction.
Cloning of Cathelicidin Coding Sequence
The primers targeting cathelicidin3 coding
sequence (cds) were designed from published
nucleotide sequence of taurine cathelicidin3 (Cath3)
mRNA (NCBI acc. no. NM_174001.1). Two pairs of
the primers were designed from overlapping
fragments using online primer designing tool Primer3
(CAT3F: 5′-GACCATGGAGACCCAGAGG-3′,
CAT3R: 5′-CCAGGAGGCGGTAGAGATTA-3′;
Cat1F: 5′-TGAGCGGTCCTCAGAAGCTA-3′;
Cat1R: 5′-CCTTGATGTTGGCGTTCCCA-3′). PCR
was carried out in a final volume of 100 µL
[containing cDNA 400 ng, primer pairs 0.4 µM each,
1× PCR-buffer, MgCl2 25 mM, 2 units Taq DNA
polymerase (Invitrogen, USA)] in a thermal cycler
(ABI, USA). The annealing temperature of 53°C and
55°C were standardized for CAT1 and CAT3 primer-
pairs, respectively. The amplicon was checked for
quality by horizontal agarose gel (2%)
electrophoresis. The purified PCR products were
ligated into pGEMT easy cloning vector (Promega,
USA) and transformed into Escherichia coli TOP10
strain as per standard protocol given in by Sambrook
and Russel13
. Recombinant clones were screened
according to blue-white selection method on
Luria-Bertani (LB) agar plate containing ampicillin
(100 µg/mL) + X-gal (40 mg/mL) + IPTG (100 mM).
Recombinant white colonies were picked up, cultured in
LB broth and subjected to plasmid isolation13
using the
GeneJET™ Plasmid Miniprep Kit (Thermo Scientific,
USA). The presence of insert was confirmed by
restriction endonuclease digestion, using EcoRI. The
isolated plasmids were also confirmed by custom
sequencing from the Department of Biochemistry,
University of Delhi, New Delhi, India.
Sequence Analysis
Processing and Submission of Sequence Data
The electropherogram of the cds (forward and
reverse) of cathelicidin were displayed, quality
checked and analyzed using BioEdit sequence
alignment editor, Version 7.2.314
. The open reading
frame was deduced and two overlapping sequences
(421 & 169 bp nucleotides) were merged to obtain a
single complete coding sequence (612 bp), which was
submitted to DDBJ, Japan (http://getentry.ddbj.nig.
ac.jp/getentry/na/AB918736/) (Getentry Acc. No.
AB918736.2). The open reading frame of Cath3 was
translated in silico using Expasy “Translate” tool
(http://web.expasy.org/translate/).
Blast Analyses
The final cDNA sequence was subjected to NCBI
BLASTn15
(http://blast.ncbi.nlm.nih.gov/) and a total
of 84 full length cathelicidin cds belonging to different
cathelicidin variants, from divergent species were
retrieved from GenBank. The nucleotide and
corresponding amino acid sequences of different
cathelicidin types and transcript variants were arranged
in FASTA format and maintained in separate text files.
Multiple Sequence Alignment (MSA)
The amino acid sequences belonging to cathelicidin
types of different species were aligned using Multiple
Alignment Fast Fourier Transform (MAFFT)
(http://www.ebi.ac.uk/Tools/msa/mafft/) online server.
The overview of the alignment of all the 83 peptide
sequences was obtained to depict the specific
conserved and variable regions of the whole peptide.
The bubaline cath variants as well as other ruminant
caths were also subjected to MSA for the conserved
pre-proregion and the variable AMP region separately
to study the extent of conservativeness of the sequence.
Phylogenetic Analysis
The deduced amino acid sequences of cathelicidin
variants of divergent species were first subjected to

HUSSAIN et al: EVOLUTIONARY ANALYSES OF BUBALINE CATHELICIDIN3
497
selection of best evolutionary model, followed by
multiple sequence alignment and construction of
phylogenetic tree using maximum likelihood (ML)
method16
(MEGA 6 software)17
, with 1000 bootstrap
resampling18
. The maximum likelihood method
utilizes optimality criterion to apply an explicit model
of evolution to phylogenetic tree construction16
.
Jones-Taylor-Thornton substitution model19
with
5 discrete Gamma categories and complete deletion of
missing data were selected as parameters. Tree
inference was drawn using nearest neighbor
interchange heuristic method. Evolutionary Divergence Estimation
The MEGA6 software17
was used for calculating
the evolutionary divergence (variance estimation by
100 bootstrap replications, Jones-Taylor-Thornton
substitution model19
, Gamma distribution with 5 rate
categories and homogeneity among lineages), amino
acid composition, disparity index to estimate the
homogeneity of substitution pattern (with complete
deletion of missing data and 100 Monte Carlo
replications) using the amino acid sequences. The
heatmap plots were generated using WGCNA
module20
of R program (Version 3.0.2) from the
matrix of evolutionary divergence.
Selection Pressure on Coding Sequences
The type of selection pressure operating on the cath
codons was determined using tests of neutrality
[Z-test of selection, using modified Nei-Gojobori
method (Jukes-Cantor)21
with transition/transversion
ratio of 2 and complete deletion of missing data] by
MEGA6 software17
. The codon based test of neutrality
compares synonymous and non-synonymous
substitution rates in protein-coding genes, and
considers a non-synonymous rate elevated above the
synonymous rate as evidence for Darwinian selection
for detecting adaptive molecular evolution22
. The
inferred number of synonymous (s) and
nonsynonymous (n) substitutions vis-à-vis the
estimated numbers of synonymous (S) and
nonsynonymous (N) sites were calculated using the
joint maximum likelihood reconstructions of ancestral
states according to a Muse-Gaut model23
of codon
substitution and Felsenstein 1981 model16
of
nucleotide substitution (data not shown). The variance
of the difference between the ‘N’ and ‘S’ was
computed with bootstrapping (500 replicates) using
the Nei-Gojobori method21
. The evolutionary
divergence between sequences was also estimated to
obtain the base substitution per site (using the
composite maximum likelihood method16
).
Analyzing Positive Selection Sites
A total of 33 complete cds were selected, which
were representing all the type variants of cathelicidin
of taurine, bubaline, ovine and caprine species and
one representative sequence each from wild Bactrian
camel, swine, killer whale, fresh-water dolphin and
minke whale (as outgroup). The specific codons that
have undergone positive selection were determined
by sequence analysis using Datamonkey
(www.datamonkey.org/) server24
applying the
statistical methods: Single Likelihood Ancestor
Counting (SLAC), Fixed Effects Likelihood (FEL)
and Random Effects Likelihood (REL)25
. At the
beginning, a model selection was run to test all of the
203 time-reversible models. A hierarchical testing
amalgamated with nested LRT tests with AIC
selection is done (http://www.datamonkey.org/help/
models.php) by the server to obtain a single "best-
fitting" rate matrix. The significance level was
separate for each of the models (SLAC, FEL &
REL)26
. Branch-site REL test26
for episodic
diversifying selection was done to undermine the
variation in the rate of evolution along both branches
and sites simultaneously (http://www.datamonkey.org/
help/index.php). This analysis enables us to determine
the lineages on which a subset of sites has evolved
under positive selection, without requiring prior
knowledge about which lineages are of interest.
Results and Discussion
Sequencing of Clones and Analysis
The clones of the overlapping partial sequences of
cathelcidin3, namely, CAT3 (169 bp) and CAT1 (463 bp)
(Fig. 1) were custom sequenced and the final
cathelicidin-3 (Cath3) cds of the Indian water buffalo
(Bubalus bubalis) was deduced. The functional
characterization of bubaline cathelicidin-3 has already
been done in our laboratory32
. Evolutionary Divergence of Antimicrobial Peptides
The lowest Bayesian information content (BIC)
score indicates the best model for analyzing the
substitution pattern of the coding sequences. In the
current study, the Jones-Taylor-Thornton (JTT)+
Gamma (G) was found to be the best model with the
lowest BIC score (8304.3). In this model, the Gamma
distribution (+G) adjusts the non-uniformity of
evolutionary rates among the sites of the codons19
.

INDIAN J BIOTECHNOL, OCTOBER 2016
498
MSA
MSA was done (using MAFFT27
) for divergent
species (represented as an overview of MSA in Fig. 2)
and also for bubaline cathelicidin types and transcript
variants (Figs 3 & 4) as well as different cathelicidin
types of the ruminant-species (Figs 5 & 6). The
pre-proregion of the full-length cathelicidin peptide is
conserved among the variants belonging to divergent
species, while the antimicrobial domain was highly
divergent among the cathelicidin types within same
vis-à-vis different species. The novel Cath3, obtained
from this study, presented an insertion of five amino
acids in the pre-proregion, which was absent in all
other ruminant species incorporated in the study (Fig. 5).
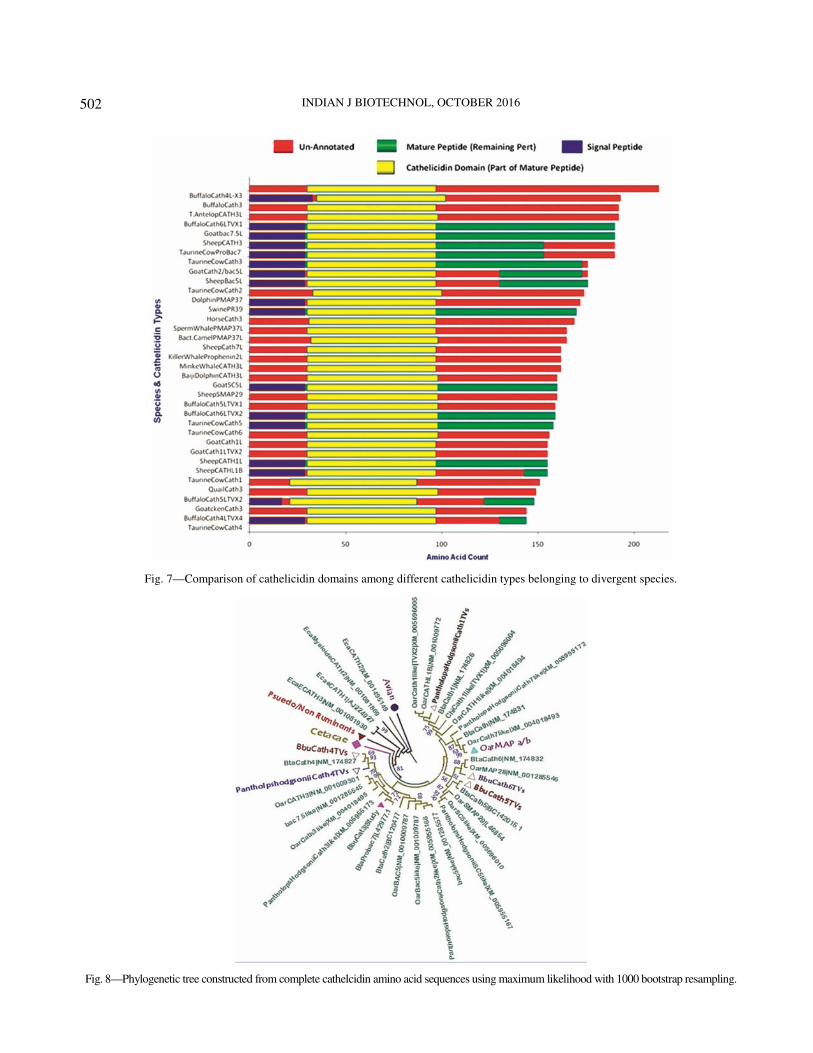
The structural annotations of the reported
cathelicidin peptides of divergent species and types
were collected from NCBI Protein database. The
functional domains of three divergent cathelicidin
peptides have been diagrammatically represented in
Fig. 7, which indicates that the length of the whole
cathelicidin peptide varies considerably among the
types within the same species. However, the length of
the signal peptide and the cathelicidin domain is
nearly same among the cathelicidin variants among
different species. There is much variation in the
mature peptide (harboring the AMP domain) among
different types of species-specific cathelicidin peptide.
Phylogenetic Analysis
The phylogenetic tree (Fig. 8) depicts that the
mammalian cathelicidins are clustering together,
while they are distinctly separate from that of the
aquatic and avian species. The branches representing
the Aves, Cetacea as well as the pseudoruminants
have been compressed to single nodes each, in order
to improve the resolution of the tree. Within the
mammalian species, the primates have formed a
separate clad from the non-primate, hoofed species.
This indicates that cathelicidin AMPs might have
undergone natural selection in accordance with the
requirement of environment and species-specific
needs to defend against microbes. The tree, in general,
demonstrates that different cathelicidin types
belonging to the same species have clustered in
different clads due to the difference in sequences.
Furthermore, some transcript variants (Cath5
transcript variants X1 and X2 of B. bubalis) fall in the
same node, while some (bubaline Cath4 transcript
variants X1, X2 & X3, X4) are not clustered within a
single node. Similar observations have also been
made with cathelicidin types and transcript variants of
other species like poultry with differences in
cathelicidin types of different breeds (not shown in
this phylogenetic tree). In certain cases, however,
different cathelicidin types from the same breed
(Fowlicidin1 and 3 from Brisson breed of chicken)
formed part of the same node. In porcine species,
some cathelicidin types like Protegrin2, 3 and also
Protegrin1, 4 formed part of the same node. The
cathelicidin types or variants belonging to the same
species present high bootstrapping values (>90% in
some cases), indicating the higher consistency of the
given data and taxonomical bipartitioning16
. Bootstrap
Fig. 1—Restriction digestion of CAT3 and CAT1 plasmids with restriction enzyme EcoR1 to release the inserts of defined sizes (169 & 463 bp).

HU
SS
AIN
et al: E
VO
LU
TIO
NA
RY
AN
AL
YS
ES
OF
BU
BA
LIN
E C
AT
HE
LIC
IDIN
3
49
9
Fig. 3—Multiple sequence alignment (using MAFFT) of the conserved pre-proregion of different transcript variants of the bubaline cathelicidin variants.
Fig. 2—Depiction of the overview of the multiple sequence alignment (using MAFFT online server) of cathelicidin peptide sequences of all species under study.

IND
IAN
J BIO
TE
CH
NO
L, O
CT
OB
ER
201
6
500
Fig. 4—Multiple sequence alignment (using MAFFT) of the variable AMP region of the transcript variants of the bubaline cathelicidin variants.
Fig. 5—Multiple sequence alignment (using MAFFT) of the conserved pre-proregion of ruminant cathelicidins.

HUSSAIN et al: EVOLUTIONARY ANALYSES OF BUBALINE CATHELICIDIN3
501
values being indicative of the stability of branching
pattern, not the accuracy of the constructed tree.
Similar studies by Tomasinsig and Zanetti28
on the
evolution of bovine cathelicidins showed that most
sequences do not cluster monophyletically within
Bovidae, i.e., these sequences are more closely related
between than within species, suggesting the existence
of common ancestor genes before the divergence of
bovids from other cloven-footed animals like caprines
and ovines. Conversely, all porcine family genes
cluster monophyletically. This suggests that the
cathelicidin family genes presently observed in pigs
and bovids might have originated by independent
rounds of duplications and subsequent diversification
after the separation of Suidae and Bovidae28
.
Evolutionary Divergence of Cathelicidin Types and Transcript
Variants
The evolutionary divergence measures the departure of
sequences between two or more species with respect to
certain parameter. Here, we have compared the
evolutionary divergence of cathelicidins types and
transcript variants of different species. The analyses for
estimating the evolutionary divergence between amino
acid sequences were conducted using the Jones-Taylor-
Thornton matrix-based model18
and then Poisson’s
correction model, using MEGA6 software17
. The results
showed that the number of amino acid substitutions per
site was the minimum for a pair of sequences belonging
to the same clad, as is evident from the phylogenetic
tree. The evolutionary divergence estimates among
ruminant species like Pantholops hodgsonii (Tibetian
wild antelope), Ovis aries (sheep) and Bos taurus (cattle)
ranged 0.130-0.196. Gallus gallus (domestic chicken)
Fowlicidin and Tibetian antelope showed maximum
evolutionary divergence (heatmap plot; Fig. 9). The least
evolutionary divergent cathelicidin homologs belong to
the same families (dark blue). Intermediary evolutionary
divergence (light red, light blue & white) was noted in
case of porcine and equine cathelicidin types. A sample
dendrogram and heatmap of the evolutionary divergence
values among the cathelicidin peptide sequences of the
ruminant species was also generated for more clear
visualization of the evolutionary divergence (Fig. 10).
Ruminant cathelicidin transcript variants of same species
formed one node, indicating the minuscule variation of
evolutionary divergence. Furthermore, ruminant
cathelicidin variants were part of a larger clad implying
towards interspecies relatedness of ruminant
cathelicidins. In general, the sample dendrogram results
are comparable to that of evolutionary divergence
estimates.
Amino Acid Composition
The amino acid composition of bubaline
cathelicidin (Cath3) was compared with a closely related
Fig
. 6
—M
ult
iple
seq
uen
ce a
lig
nm
ent
(usi
ng
MA
FF
T)
of
the
var
iab
le A
MP
reg
ion
of
rum
inan
t ca
thel
icid
ins.
INDIAN J BIOTECHNOL, OCTOBER 2016
502
Fig. 7—Comparison of cathelicidin domains among different cathelicidin types belonging to divergent species.
Fig. 8—Phylogenetic tree constructed from complete cathelcidin amino acid sequences using maximum likelihood with 1000 bootstrap resampling.

HUSSAIN et al: EVOLUTIONARY ANALYSES OF BUBALINE CATHELICIDIN3
503
Fig. 9—Evolutionary divergence heatmap: Relative distance among transcript variants vis-à-vis species specific cathelicidin.
Fig. 10—Sample dendrogram and evolutionary divergence of ruminant-specific cathelicidin types and transcript variants.

INDIAN J BIOTECHNOL, OCTOBER 2016
504
species (taurine cattle, Cath4) and one distant species
(Chicken, Fowlicidin3). The differences in amino
acids among the cathelicidins of selected species have
been depicted in Fig. 11. Proline (17.09%) and
leucine (15.02%) were in maximum proportion in
bubaline cathelicidin3.
Homogeneity of Substitution Patterns between Sequences
Disparity index is used to measure the apparent
difference in evolutionary patterns for a pair of
sequences. A common assumption in comparative
sequence analysis is that the sequences have evolved
with the same pattern of nucleotide substitution
(homogeneity of the evolutionary process)29
. In our
study, disparity index (DI) was estimated for the
target amino acid of various species with respect to
that of the bubaline cathelicidin peptide (Cath3) and
was compared. Different cathelicidin types in cattle
and non-ruminant species, i.e., swine and equines
species, showed significant disparity indicating that
during evolution substitution pattern varied among
species with respect to buffalo. This implies that the
branching pattern obtained in the phylogenetic tree
could be misleading, as the assumption of
homogeneity of substitution pattern has not been met.
This limitation was further studied using “branch-site
REL analysis” (BSR)26
of the Datamonkey online
server (http://www.datamonkey.org/)25
. The BSR
method analyzes the episodic diversifying selection of
divergent homologous coding sequences. In this
analysis, in total 14 branches (or nodes) were detected
(p<0.05) to undergo episodic diversifying selection
(Fig. 12). This test estimates the ratio (ω) of
nonsynonymous (β) to synonymous (α) substitution
rates along with a lineage of interest, as a measure of
selective pressure. The branches of the tree in this
figure have different colours which signify the
strength of selection. Blue colour is indicative of
purifying selection (ω=0), black indicating neutral
selection (ω=1) and red colour implying
diversifying/positive selection (ω > 5). The variation
in thickness corresponds to the proportion of sites
undergoing harmonic diversifying selection. In this
case, three bubaline cathelicidins, namely,
XM_006065192|BbuCath6like|TVX1, XM_006065186|
BbuCath2like|TVX3 and XM_006040871|BbuCath
Fig. 11—Amino acid composition of cathelicidin peptides
belonging to buffalo (Cath3), taurine cow (Cath4) and chicken
(Fowlicidin3).
Fig. 12—Branch-site REL analysis of ruminant cathelicidin-types
and single, representative cathelcidin from other species. The red
oval shapes encircle the ruminant cathelicidins undergoing
episodic diversifying selection. The green coloured rectangle
indicates the bubaline Cath3 sequence of the present study.

HUSSAIN et al: EVOLUTIONARY ANALYSES OF BUBALINE CATHELICIDIN3
505
5like|TVX2, are shown to be undergoing harmonic
diversifying selection, with the taurine BC142015.1|
BtaCath5 also falling in this category. This suggests that
the ruminant cathelicidin sequences have been evolved
under heterogeneity of substitution pattern. Similar
statistical analysis has previously been attempted by
Singh and coworkers30
in the evolutionary analysis of
dicer.
Estimation of Selection Pressure for Various Codons
Maximum likelihood method is the codon
substitution model that was used to estimate the biases
introduced in nucleotide substitution and branch
length16
. Selection pressure on different codons of
cathelicidin types and variants was done using online
server Datamonkey (http://www. datamonkey.org/)25
.
Selection pressure on a codon was determined by
comparing the rates of synonymous (s) substitution per
synonymous site (dS) and non-synonymous (n)
substitution per non-synonymous site (dN). The value of
the test statistic dN-dS objectifies whether the codon
has undergone positive, negative or neutral selection.
Three different models, single likelihood ancestor
counting (SLAC), random effects likelihood (REL) and
fixed effects likelihood (FEL) were used to analyse26
the
data. SLAC yielded the minimum number of false
positive and false negative results, whereas REL model
detected more number of positively and negatively
selected codons compared to SLAC. In general, SLAC
is considered as the most conservative method, while
REL is the least intensive27-32
. SLAC model showed that
overall there were three positively selected codon sites
(23, 24 & 34), while REL showed as many as 36
positively selected codons (Figs 13A & B; Fig. 14). The
intermediary intensive FEL model showed 8 positively
selected codons (23, 24, 48, 50, 154, 162, 165 & 182).
The SLAC method for the cathelicidin sequences under
study presented somewhat uniform distribution of
positively selected codons based on the dN-dS values in
the conserved cathelin and the highly evolved
antimicrobial domain (Fig. 13A). On the other hand,
based on the REL method positive selected codons were
more dispersed towards the right of the graph, which
represented the antimicrobial domain (Fig. 14). The
REL method, in this case, gave a more factual picture of
positive selection of cathelicidin types and transcript
variants, as the incorporated input sequences were
divergent due to addition of the outgroup sequences in
the analysis.
The result from REL analysis clearly indicates that
almost all the positively selected sites are localized
within the highly variable, functional AMP domain of
cathelicidin. This signifies that the various
cathelicidin variants/types in the species under study
have a specific role to play in conferring immunity and
thereby fitness of the organism. Similarly, in buffalo,
although a very limited number of cathelicidin types
have been experimentally reported33
, the transcript
variants might have some specific roles. In a similar
type of work on ovine cathelicidin variants 1 and 2,
Dhaliwal et al34
reported that positive selection has
played an important role in the evolution of cathelicidin
variants among animal species.
Conclusion
Evidence of positive selection was seen in the
AMP, cathelicidin in the species that were included in
the study. The position of some of the positively
selected codons indicates that pathogens exert most of
Fig. 13 (A & B)—Graphical representation of the dN-dS test
statistic versus the codon positions obtained from SLAC (A) and
REL (B) analyses of cathelicidin cds.
Fig. 14—Positions of the positively selected codons of
cathelicidin peptides detected by REL analysis.

INDIAN J BIOTECHNOL, OCTOBER 2016
506
the selective pressures that lead to the changes observed.
Crystallographic studies would be helpful for assessing
the functional relevance of the cathelicidin.
Acknowledgement
The authors thankfully acknowledge the financial
assistance provided by the Department of Agriculture
and Cooperation, Ministry of Agriculture, Government
of India, New Delhi under Rashtriya Krishi Vikas
Yojana for conducting the present research work.
References 1 Hamblin M T, Thompson E E & Di Rienzo A, Complex
signautres of natural selection at the duffy blood group locus, Am
J Hum Genet, 70 (2002) 369-383.
2 Vallender E J & Lahn B T, Positive selection on the human
genome, Hum Mol Genet, 13 (2004) R245-254.
3 Martin E, Ganz T & Lehrer R I, Defensins and other endogenous
peptide antibiotics of vertebrates, J Leukoc Biol, 58 (1995) 128-
136.
4 White S H, Wimley W C & Selsted M E, Structure, function,
and membrane integration of defensins, Curr Opin Struct Biol, 5
(1995) 521-527.
5 Ganz T, Antimicrobial polypeptides in host defense of
respiratory tract, J Clin Invest, 109 (2002) 693-697.
6 Zanetti M, Gennaro R, Skerlavaj B, Tomasinsig L & Circo R,
Cathelicidin peptides as candidates for a novel class of
antimicrobials, Curr Pharm Des, 8 (2002) 779-793.
7 Bals R, Wang X, Zasloff M & Wilson J M, The peptide
antibiotic LL-37/hCAP-18 is expressed in epithelia of human
lung where it has broad antimicrobial activity at airway surface,
Proc Natl Acad Sci USA, 95 (1998) 9541-9546.
8 Frohm M, Agerbeth B, Ahangari G, Sthle-Backdahl M, Liden S
et al, The expression of gene coding for antibacterial peptide LL-
37 is induced in human keratinocytes during inflammatory
disorders, J Biol Chem, 272 (1997) 15258-15263.
9 Gutsmann T, Hagge S O, Larrick J W, Seydel U & Wiese A,
Interaction of CAP18 derived peptides with membrane made from
endotoxins or phospholipids, Biophys J, 80 (2001) 2935-2945.
10 Shinnar A E, Butler K L &Park H J. Cathelicidin family of
antimicrobial peptides: Proteolytic processing and protease
resistance, Bioorg Chem, 31 (2003) 425-436.
11 Hsu C H, Chen C, Jou M L, Lee A Y, Lin Y C et al, Structural
and DNA binding studies on the bovine antimicrobial peptide,
indolicidin: Evidence for multiple conformations involved in
binding to membranes and DNA, Nucleic Acids Res, 33 (2005)
4053-4046.
12 Dua K, Comparative disease susceptibility of cattle and buffalo
in Punjab (India), in Proce 10th Int Symp Vet Epidemiol Econ,
2003. [Available at: www.sciquest.org.nz].
13 Sambrook J M & Russell W D, Molecular cloning: A laboratory
manual, 3rd edn, edited by M R Green & J Sambrook (Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New
York, USA) 2001.
14 Hall T A, BioEdit: A user-friendly biological sequence
alignment editor and analysis program for Windows 95/98/NT,
Nucleic Acids Symp Ser, 41 (1999) 95-98.
15 Altschul S F, Gish W, Miller W, Myers E W & Lipman D J,
Basic local alignment search tool, J Mol Biol, 215 (1990)
403-410.
16 Felsenstein J, Evolutionary trees from DNA sequences: A
maximum likelihood approach, J Mol Evol, 17 (1981) 368-376
17 Tamura K, Stecher G, Peterson D, Filipski A & Kumar S,
MEGA6: Molecular evolutionary genetics analysis, version 6.0,
Mol Biol Evol, 30 (2013) 2725-2729.
18 Hedges S B, The number of replications needed for accurate
estimation of bootstrap P value in phyllogenetic studies,
Mol Biol Evol, 9 (1992) 366-369.
19 Jones D T, Taylor W R & Thornton J M, The rapid generation
of mutation data matrices from protein sequences, Comput Appl
Biosci, 8 (1992) 275-282.
20 Zhang B & Horvath S, A general framework for weighted gene
co-expression network analysis, Stat Appl Genet Mol Biol, 4
(2005) 1.
21 Nei M & Gojobori T, Simple methods for estimating the
numbers of synonymous and nonsynonymous nucleotide
substitutions, Mol Biol Evol, 3 (1986) 418-26.
22 Yang Z & Bielawski J P, Statistical methods for detecting
molecular adaptation, Trends Ecol Evol, 15 (2000) 496-503.
23 Muse S V & Gaut B S, A likelihood approach for comparing
synonymous and non synonymous nucleotide substitutions, with
applications to chloroplast genome, Mol Biol Evol, 11 (1994)
715-724.
24 Pond S L & Frost S D, Datamonkey: Rapid detection of
selective pressure on individual sites of codon alignments,
Bioinformatics, 21 (2005) 2531-2533.
25 Kosakovsky P S L & Frost S D, Not so different after all: A
comparison of methods for detecting amino acid sites under
selection, Mol Biol Evol, 22 (2005) 1208-1222.
26 Pond S L K, Murrell B, Fourment M, Frost S D W, Delport W et
al, A random effects branch-site model for detecting episodic
diversifying selection, Mol Biol Evol, 28 (2011) 3033-3043.
27 Katoh K, Kuma K, Toh H & Miyata T, MAFFT version 5:
Improvement in accuracy of multiple sequence alignment,
Nucleic Acids Res, 33 (2005) 511-518.
28 Tomasinsig L & Zanetti M, The cathelcidins structure, function
and evolution, Curr Protein Pept Sci, 6 (2005) 23-34.
29 Kumar S & Gadagkar S R, Disparity Index: A simple statistic to
measure and test the homogeneity of substitution patterns
between molecular sequences, Genetics, 158 (2001) 1321-1327.
30 Singh J, Mukhopadhyay C S, Arora J S & Kaur S,
Biocomputational characterization and evolutionary analysis of
bubaline dicer1 enzyme, Asian-Aust J Anim Sci, 28 (2015) 876-887.
31 Bhardwaj R, Mukhopadhyay C S, Deka D, Verma R,
Dubey P P & Arora J S, Biocomputational analysis of
evolutionary relationship between toll-like receptor and
nucleotide-binding oligomerization domain-like receptors genes,
Vet World, 9 (2016) 1218-1228.
32 Bhardwaj R, Brah G S, Arora J, Kaur S, Mukhopadhyay C S,
Cloning and molecular characterization of toll-like receptor 4
(TLR-4) gene in Indian water buffalo (Bubalus bubalis), Indian
J Anim Sci, 85 (2015) 19-24
33 Hussain S, Mukhopadhyay C S, Kumar B V S & Arora J S,
Functional characterization of bubaline recombinant-
cathelicidin, Proc Natl Acad Sci India, Sect B Biol Sci, 85 (2015)
965-969.
34 Dhaliwal K K, Arora J S, Mukhopadhyay C S & Dubey P P, In
silico characterization of functional divergence of two
cathelicidin variants in Indian sheep, Evol Bioinform Online, 1
(2015) 189-196.





























