Embed Size (px)
Citation preview

Photosensitized (electron transfer) carbon-carbon bond cleavage of radical cations: the 2-phenylethyl ether and acetal systems1
DONALD R. A R N O L D ~ AND LAURIE J. LAMONT Department of Chemistry, Dalhousie Universiry, Halifax, N.S., Canada B3H 453
Received April 7, 1989
DONALD R. ARNOLD and LAURIE J. LAMONT. Can. J. Chem. 67,2119 (1989). The scope of the photosensitized (electron transfer) carbon-carbon bond cleavage involving radical cations has been defined
for 2-phenylethyl ethers and acetals. The thresholds for reactivity of the monophenylethyl and gem-diphenylethyl derivatives are compared. While the radical cation of methyl 2,2-diphenylethyl ether (7) cleaves to give ultimately diphenylmethane (2) and dimethoxymethane (8), the radical cation of methyl 2-phenylethyl ether (9) was stable under these conditions. In contrast to the lack of reactivity of the radical cation of 9, the radical cations of methyl 2-phenyl-2-propyl ether ( l l ) , methyl 2-phenylcyclopentyl ether (13), and 2-phenylmethyl-l,3-dioxolane (16) cleave. Cleavage in the monophenylethyl series is limited to formation of a carbocation at least as stable as the secondary a-oxyalkyl or di-a-oxyalkyl. The basis for predicting this type of reactivity of radical cations is defined. The rate of carbon-carbon bond cleavage is increased by increasing the oxidation potential of the molecule, by decreasing the carbon-carbon bond strength, and (or) by decreasing the oxidation potential of that fragment that will become the carbocation. The results obtained from the reactions of 2-diphenylmethyl-l,3-dioxolane (14) and 2-phenylmethyl-1,3-dioxolane (16) cast doubt on the published oxidation potential for the 1,3-dioxolan-2-yl radical.
Key words: photochemistry, radical cation, electron transfer, bond cleavage, radical.
DONALD R. ARNOLD et LAURIE J. LAMONT. Can. J. Chem. 67,2119 (1989). On a dCfini le champ d'application du clivage, photosensibilis6 (par transfert Clectronique) et impliquant des cations radicaux,
de la liaison carbone-carbone d'tthers et d'acCtals de la strie 2-vhCnvlCthvle. On a comvare les seuils de rtactivitt des dCrivCs monophCnylCthylts et gem-diphCnyltthylts. Alors que le cation'radical d; 1-mtthoxy-i,2-diphCnyltthane (7) subit un clivage conduisant Cventuellement au diphCnylmCthane (2) et au dimtthoxymtthane (8), le cation radical du mCthoxy-1-2-phtnylCthane (9) est stable dans ces conditions. En opposition au fait que le cation radical du composC 9 n'est pas reactif, les cations radicaux du 2-mCthoxy-2-phCnylpropane ( l l ) , du I-mtthoxy-2-phtnylcyclopentane (13) et du 2-phCnylmtthy1-1,3-dioxolane (16) se clivent. Le clivage dans la sCrie des composCs monophtnylCthylts est limit6 2 la formation d'un cation au moins aussi stable que les a-oxyalkyles ou di-a-oxyalkyles secondaires. On a dCfini les bases permettant de pridire ce type de reactivitt des cations radicaux. Les vitesses de clivage de la liaison carbone-carbone augmentent lorsqu'on augmente le potentiel d'oxydation de la molCcule, lorsqu'on diminue la force de la liaison carbone-carbone et (ou) lorsqu'on diminue le potentiel d'oxydation du fragment qui deviendra le carbocation. Les rtsultats obtenus lors des rtactions des 2,2-diphCnylmtthyl-l,3-dioxolane (14) et 2-phtnylmtthyl-l,3-dioxolane (16) permettent de mettre en doute les rCsultats qui ont CtC publits relativement au potentiel d'oxydation du radical 1,3-dioxolan-2-yle.
Mots clks : photochimie, cation radical, transfert d'tlectron, clivage de la liaison, radical. [Traduit par la revue]
Introduction Reaction 1
A frequently observed reaction of organic radical cations is hv, Sen., 80°C Ph2CH-CHPh2 Ph2CH2 + CH30CHPh2
cleavage of a carbon-carbon bond to give a carbon-centered CH,CN, CH30H radical and a carbocation fragment. This process accounts for 1 2 3 many of the fragments in the mass spectrum of an organic molecule. The relative efficiencv of the various modes of Sens.: 174-Dicyan0benzene (4)
cleavage of the radical cation, in the gas phase, are predictable and relatively well understood. In marked contrast, when the radical cation is generated in solution by photosensitization (electron transfer), cleavage is much less common; in fact, it is not yet possible to predict when this reaction will occur (3). Other reactions of the radical cation often occur instead. It is now clear that for the carbon-carbon bond cleavage process to compete effectively with these other reactions the nascent fragments, both radical and carbocation, must be relatively stable (lb).
The reactivity of the radical cations of several compounds having in common the gem-diphenyl moiety has been studied. Even though carbon-carbon bond cleavage of the radical cation of these compounds would give the highly delocalized diphen- ylalkyl radical, not all react in this way. The photosensitized (electron transfer) reactivity of 1,1,2,2-tetraphenylethane (1) is
particularly interesting; the efficiency of the cleavage process is temperature dependent in this case. Essentially no cleavage of 1 occurred upon irradiation of an acetonitrile-methanol solution at 10°C, with 1,4-dicyanobenzene (4) as the electron-accepting photosensitizer; however, at 80°C, cleavage was relatively efficient and an excellent yield of diphenylmethane (2) and methyl diphenylmethyl ether (3) was obtained (reaction 1) (1 b). The energy of carbon-carbon bond cleavage of this radical cation is clearly at the threshold for reactivity.
The proposed mechanism for this reaction is shown in Scheme 1 (1 6). The temperature dependence of the reaction of the radical cation of 1 was attributed to the cleavage step (step 4) and an estimate of E, = 7.2 kcal mol-' was obtained from the slope of an Arrhenius plot (log (relative efficiency) versus temp-'). This estimate for the bond dissociation energy of the central bond in the radical cation of 1 is in fair agreement with -
qh i s is part 22 of the series ~ ~ d i ~ ~ l in photochemistry, the value (9.3 kcal mol- ') calculated from thermochemical data Preceding publications in these series are refs. 1 and 2. (Fig. l a ) ( lb).
2 ~ u t h o r to whom correspondence may be addressed. Some similar 1,l-diphenylethyl derivatives are stable to
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

CAN. J . CHEM. VOL. 67, 1989
P h 2 C H + + P h C H 2'
FIG. 1. Thennochemical cycles useful for estimating the bond dissociation energy of the radical cations of (a) 1,1,2,2-tetraphenylethane (1); (b) 1,1,2-triphenylethane (6); (c) 1,2-diphenylethane (5). (Data taken from Table 2, and refs. I b, 5, and 6.)
1. Sensitizer (A) hl) A*
2. Ph2CH -R + A* + P ~ , C H - R ~ + A'
3. P ~ ~ C H - R ~ +A' d Ph2CH-R+A
4. P ~ ~ C H -R+ d Ph2CH - + R+
5. R+ + CH30H d R-0CH3 + H+
6. Ph2CH. +A' + Ph2CH-+ A
7. Ph2CH- + CH30H d Ph2CH2 + CH30-
8. P~,CH-R+ d Ph2C(.)-R + H+
9. Ph2C(.)-R + A' ---+ Ph2C(-)-R + A
10. Ph2C(-)-R + H+ ---+ Ph2CH-R
SCHEME 1. The mechanism for the photosensitized (electron trans- fer) carbon-carbon bond cleavage.
irradiation under these conditions, even at the higher tempera- ture (80°C). Apparently, with these unreactive derivatives cleavage cannot compete with other reactions, such as deactiva- tion by back electron transfer (step 3) or deprotonation (step 8). The carbon-carbon bond dissociation energy of the radical cation must be too high. An understanding of the factors that influence the thermodynamics, and more importantly the kinetics, of step 4 will be required to be able to predict whether cleavage of any given radical cation will occur.3
The main structural features that influence the thermody- namics of step 4 have been identified (Fig. 1) (1 b). To decrease the bond dissociation energy of the radical cation, one must increase the oxidation potential of the molecule, decrease the carbon-carbon bond dissociation energy, and (or) decrease the oxidation potential of that fragment that will become the carbocation.
The reactivity of 1,1,2,2-tetraphenylethane (1) gives an indication of the threshold carbon-carbon bond energy required for cleavage to occur; it is about 10 kcal mol-'. One of our goals is to be able to predict generally, on the basis of the structure of the radical cation, when this cleavage reaction will occur. We have therefore extended our study to the monophenylethyl analogues in an effort to find the threshold for cleavage in this series.
There are conflicting reports in the literature regarding the reactivity of the radical cation of the parent member of this series, 1,2-diphenylethane (5) (7). We find that 5 is stable to irradiation under conditions which cause cleavage of 1 (reaction 2); this is consistent with most of the reported behaviour (7). It is
Reaction 2
hv, Sen., 80°C PhCH2-CH2Ph Noreaction
CH3CN, CH30H
3 ~ f the reaction of the radical with the carbocation, to give the radical also clear, from consideration of the thennochemical cycle (Fig.
cation, has little or no activation energy, the activation energy for the lc), that the bond dissociation energy for 5 is significantly
cleavage of the radical cation will be the bond dissociation energy. This greater (29.3 kcal mol-l) than that for may be the case in the gas phase (4); however, solvent reorganization at Can the bond dissociation energy of the radical cation of the transition state must require an activation energy, particularly for monophenyl analogues be decreased, by appropriate substitu- solutions in polar solvents. tion, to the point where cleavage will occur? For example, will
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

ARNOLD AND LAMONT 2121
Reaction 3
hv, Sen., 80°C Ph2CH-CH2Ph P No reaction
CH3CN, CH30H
the radical cation of 1 , l ,2-triphenylethane (6) cleave? While the oxidation potentials of 5 and 6 are essentially the same (Table 2), the central carbon-carbon bond in 6 is weaker than that in 5 (5a), and the oxidation potential of the diphenylmethyl radical (0.35 V (6))4 is appreciably less than that of the benzyl radical (0.73 V (6)). Both of these factors tend to favour cleavage of 6 relative to 5 (Fig. 1 b). But will the combined effect, which lowers the bond dissociation energy of the radical cation of 6 to 16.7 kcal mol-', be large enough?
Methyl 2,2-diphenylethyl ether (7) is readily cleaved under these conditions, even at 10°C (reaction 4) (8). The data
Reaction 4
hv, Sen., 10°C Ph2CH-CH20CH3 -b Ph2CH2 +
CH3CN, CH30H rn
required to complete the thermochemical cycle for the cleavage of 7 are not available; however, the significantly lower oxidation potential of the a-oxymethyl radical (-0.24 V (6)) relative to the diphenylmethyl radical tends to favour cleavage. Substitution of the a-oxymethyl group for the benzylic group in 5 makes an even greater incremental difference in the carbon- carbon bond energy. Will methyl 2-phenylethyl ether (9) cleave under these conditions?
Reaction 5
hv, Sen., 80°C PhCH2- CH20CH3 No reaction
CH3CN, CH 30H
9 The radical cations of methyl 1 , l -diphenyl-2-propyl and
methyl 1-phenyl-2-propyl ethers (10 and 11) are even more likely to cleave. The a-~x~carbocat ion from these derivatives is secondary; the oxidation potential of this secondary radical (-0.45 V (6)) is significantly less than that of the analogous primary a-oxymethyl radical.
The radical cation of methyl 2,2-diphenylcyclopentyl ether (12) cleaves to give the 1,5-radical cation under these conditions ( la) . Would the radical cation of the monophenyl derivative, methyl 2-phenylcyclopentyl ether (13), cleave?
The effect of the single oxygen in 7 and 9-13 is understood; while the radical a to ether oxygen is stabilized by interaction with the nonbonding pair of electrons, the a-oxycarbocation is stabilized even more. This is evident from the relatively low oxidation potential of the radical. The effect of the two oxygens in the 1,3-dioxolan-2-yl system can also be predicted. Both the radical and the carbocation should be stabilized by more than the stabilization provided .by only one adjacent oxygen, although the effect is not additive (9). However, this reasoning is not consistent with the measured oxidation potential of the 1,3-diox- olan-2-yl radical (0.3 1 V (6)), which is significantly greater than that of the a-oxymethyl radical. It will therefore be interesting to know if the radical cations of the 2-diphenylmethyl-l,3-diox-
4 ~ 1 1 of the potentials mentioned in this paper are relative to the saturated calomel electrode (SCE).
olanes (14 and 15) and the 2-phenylmethyl-1,3-dioxolanes (16 and 17) (Chart 1) cleave under these conditions. The reactivity of these compounds will provide another indication of the oxidation potential of the 1,3-dioxolan-2-yl radical.
12 13 cis and trans
14 R = H 16 R = H 15 R = C H 3 17 R = C H 3
The specific objective of this work was to determine the reactivity of the radical cations of 2,2-diphenylethyl and 2-phenylethyl ethers and acetals upon photosensitization (elec- tron transfer). The ultimate goal is to provide a firm basis for predicting reactivity in this type of cleavage reaction in general.
Results The compounds of interest are shown in Chart 1; all except
methyl 1,l-diphenyl-2-propyl ether (10) were known com- pounds, prepared by standard methods, and characterized by spectroscopic techniques ('H nmr, ms). The new compound (lo), prepared in two steps from 1,l-diphenyl-2-propanone, was fully characterized (ir, 'H nrnr, 13C n m , ms, and elemental analysis).
The methyl cis- and trans-2-phenylcyclopentyl ethers (13) and methyl 1-phenyl-2-propyl ether (11) were obtained by the photosensitized (electron transfer) anti-Markovnikov addition of methanol to the corresponding alkenes (10). The ratio of isomers obtained from 1 -phenylcyclopentene (cis-13/trans-13 = 0.4) was similar to that reported (0.6, ref. lob). These isomers were separated by flash column chromatography.
The irradiation procedure was similar to that used in the previous study (1 b), so the results can be compared directly with those reported. Solutions of the ether or acetal and the photosensitizer, 1,4-dicyanobenzene (4), in acetonitrile-meth- an01 (3:1), were degassed by nitrogen ebullition and then were irradiated through a Pyrex filter using a medium-pressure mercury vapour lamp. The irradiation vessel was kept at a constant temperature, 10°C or 80°C, by acirculating water bath.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

CAN. J. CHEM. VOL. 67, 1989
TABLE 1. Summary of results of irradiations"
Reaction Compound Product(s) (yield, %)
Diphenylmethane (2); methyl diphenylmethyl ether (3)
No reactiond
No reactiond
4"vb Methyl 2,2-diphenylethyl ether (7) (2); dimethoxylmethane (8)
5' Methyl 2-phenylethyl ether (9) No reactiond
6" Methyl 1 , 1 -diphenyl-2-propyl ether (10) (2) (70); 1,l-dimethoxyethane (18); 1,1,2,2-tetraphenylethane (1) (10)
7" Methyl 1-phenyl-2-propyl ether (11) Toluene (19) (25); (5) (15); (4-cyanopheny1)phenylmethane (20) (25)
8 " ~ ~ Methyl 2,2-diphenylcyclopentyl ether (12) 53-Diphenylpentanal dimethylacetal (21) (60)
9" Methyl cis and trans-2-phenylcyclopentyl 5-Phenylpentanal dimethylacetal (22);f ether (13) 5-(4-cyanopheny1)-5-phenylpentanal
dimethylacetal (23)g
loe 2-Diphenylmethyl- l,3-dioxolane (14) (2) (90); (1) (10); 2-methoxy- l,3-dioxolane (24)
"A solution of the compound (0.05 M) and 1,4-dicyanobenzene (4) (0.05 M) in acetonitrile-methanol (3:l) at 10°C was irradiated through a Pyrex filter with a medium-pressure mercury vapor lamp. The usual irradiation period was ca. 20 h.
*Reference l b. 'The temperature of the irradiation was 80°C; there was little or no reaction upon irradiation at 10°C. dTrace amounts of the expected cleavage products were detected by gc/ms. eReference l a . lThe acetal 21 was the only volatile product detected at low conversion; the yield was not determined. The reaction
becomes inefficient and the product mixture becomes complex during irradiation. 8Tentative structural assignment based upon retention time and mass spectrum (gc/ms).
Progress of the reaction was followed by capillary column gas chromatography with a mass selective detector (gclms). The products, including minor amounts of compounds resulting from competing reactions or secondary photolysis, were readily detected. The results of the irradiations are illustrated in reactions 1-13, and are summarized in Table 1. The reported yields are based upon starting material consumed, at greater than 85% conversion, and purified product isolated by column chromatography. The yield of toluene (19) was determined by
gas chromatography using a calibrated flame ionization detector (gclfid).
Most of the products are well-known compounds, character- ized here by spectroscopic techniques and by direct comparison with authentic samples.
The (4-cyanopheny1)phenylmethane (20), isolated by column chromatography, was identified by mass spectrum, 'H nmr, and comparison with reported melting point. The ortho ester, 2-methoxy-2-methyl-l,3-dioxolane (25), was hydrolyzed to
Reaction 7
hv, Sen., 10°C PhCH2-CH(CH3)0CH3 - CH3CH(OCH3),+ PhCH3 + Ph(4-NC-C6H4)CH2 + PhCH2CH2Ph
CH3CN, CH30H 11 18 19 20 5
Reaction 9
?CH3
hv, Sen., 10°C Ph d - PhCH2(CH2)3CH(OCH3)2 + PhCH(CH2)3CH(OCH3)2 CH3CN, CH30H I 13 cis or trans
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.
ARNOLD AND LAMONT
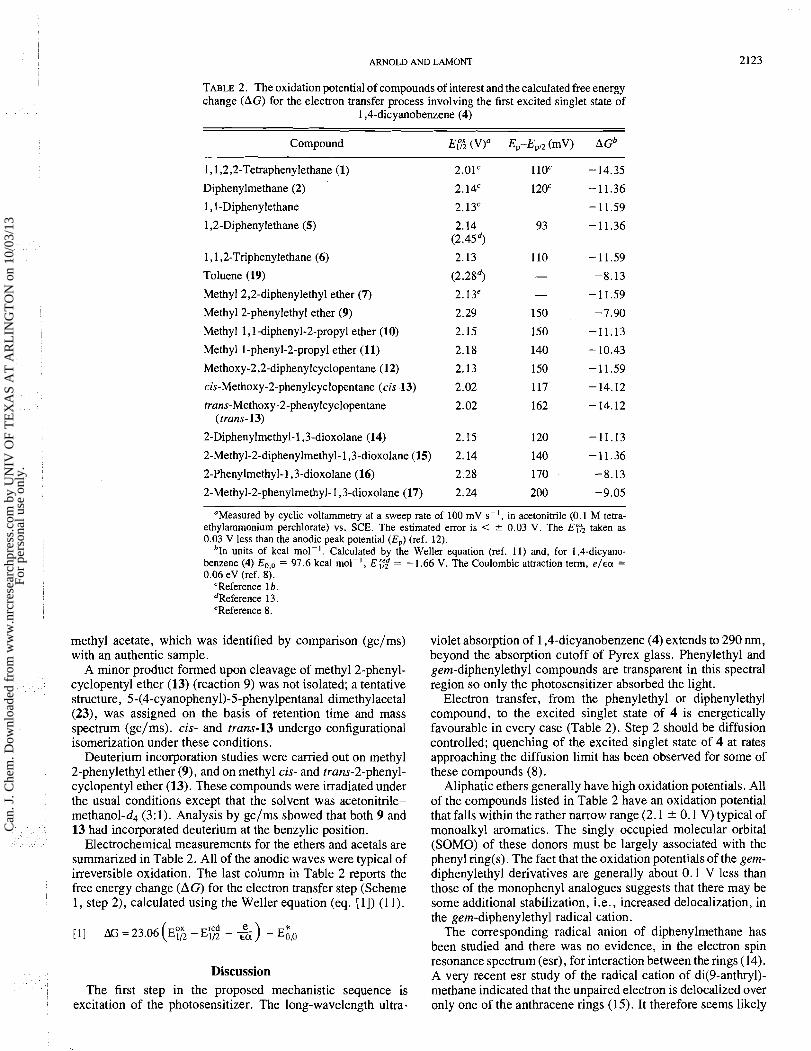
TABLE 2. The oxidation potential of compounds of interest and the calculated free energy change (AG) for the electron transfer process involving the first excited singlet state of
1,4-dicyanobenzene (4)
Compound E14; (Vy Ep-EPI2 (mV) AGb
1,1,2,2-Tetraphenylethane (1)
Diphenylmethane (2)
1,l-Diphenylethane
1,2-Diphenylethane (5)
1,1,2-Triphenylethane (6) 2.13 110 -11.59
Toluene (19) (2.2Sd) - -8.13
Methyl 2,2-diphenylethyl ether (7) 2.13' - -11.59
Methyl Zphenylethyl ether (9) 2.29 150 -7.90
Methyl 1 , l -diphenyl-2-propyl ether (10) 2.15 150 -11.13
Methyl 1-phenyl-2-propyl ether (11) 2.18 140 -10.43
Methoxy-2,2-diphenylcyclopentane (12) 2.13 150 -11.59
cis-Methoxy-2-phenylcyclopentane (cis-13) 2.02 117 - 14.12
"Measured by cyclic voltammetry at a sweep rate of 100 mV s-I, in acetonitrile (0.1 M tetra- ethylammonium perchlorate) vs. SCE. The estimated error is < * 0.03 V. The E r 2 taken as 0.03 V less than the anodic peak potential (E,) (ref. 12).
*In units of kcal mol-I. Calculated by the Weller equation (ref. 11) and, for 1,4-dicyano- benzene (4) Eo,, = 97.6 kcal mol-', E i7;d = - 1.66 V. The Coulombic attraction term, e / ~ a = 0.06 eV (ref. 8).
'Reference 1 b . dReference 13. 'Reference 8.
methyl acetate, which was identified by comparison (gc/ms) with an authentic sample.
A minor product formed upon cleavage of methyl 2-phenyl- cyclopentyl ether (13) (reaction 9) was not isolated; a tentative structure, 5-(4-cyanopheny1)-5-phenylpentanal dimethylacetal (23), was assigned on the basis of retention time and mass spectrum (gc/ms). cis- and trans-13 undergo configurational isomerization under these conditions.
Deuterium incorporation studies were carried out on methyl 2-phenylethyl ether (9), and on methyl cis- and trans-2-phenyl- cyclopentyl ether (13). These compounds were irradiated under the usual conditions except that the solvent was acetonitrile- methanol-d4 (3:l). Analysis by gc/ms showed that both 9 and 13 had incorporated deuterium at the benzylic position.
Electrochemical measurements for the ethers and acetals are summarized in Table 2. All of the anodic waves were typical of irreversible oxidation. The last column in Table 2 reports the free energy change (AG) for the electron transfer step (Scheme 1, step 2), calculated using the Weller equation (eq. [I]) (1 1).
Discussion The first step in the proposed mechanistic sequence is
excitation of the photosensitizer. The long-wavelength ultra-
violet absorption of 1,4-dicyanobenzene (4) extends to 290 nm, beyond the absorption cutoff of Pyrex glass. Phenylethyl and gem-diphenylethyl compounds are transparent in this spectral region so only the photosensitizer absorbed the light.
Elect~on transfer, from the phenylethyl or diphenylethyl compound, to the excited singlet state of 4 is energetically favourable in every case (Table 2). Step 2 should be diffusion controlled; quenching of the excited singlet state of 4 at rates approaching the diffusion limit has been observed for some of these compounds (8).
Aliphatic ethers generally have high oxidation potentials. All of the compounds listed in Table 2 have an oxidation potential that falls within the rather narrow range (2.1 ? 0.1 V) typical of monoalkyl aromatics. The singly occupied molecular orbital (SOMO) of these donors must be largely associated with the phenyl ring(s). The fact that the oxidation potentials of the gem- diphenylethyl derivatives are generally about 0.1 V less than those of the monophenyl analogues suggests that there may be some additional stabilization, i.e., increased delocalization, in the gem-diphenylethyl radical cation.
The corresponding radical anion of diphenylmethane has been studied and there was no evidence, in the electron spin resonance spectrum (esr), for interaction between the rings (14). A very recent esr study of the radical cation of di(9-anthry1)- methane indicated that the unpaired electron is delocalized over only one of the anthracene rings (15). It therefore seems likely
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

2124 CAN. 1. CHEM. \
that any interaction between the two phenyl rings in the radical cation of these gem-diphenylethyl derivatives is small.
One factor that is known to inhibit bond cleavage of a radical cation is lack of overlap of the adjacent carbon-carbon bond with the SOMO (1 a). All of the compounds studied here can attain conformations that allow such overlap, hence this is not a limiting factor.
Back electron transfer from the radical anion of the sensitizer to the radical cation of the donor, step 3, is the major competing reaction. This process is energetically very favourable (ca. -85 kcal mol-') for all of the donors studied; the rate should be nearly the same in every case. Back electron transfer from the monophenylethyl compounds might, in fact, be somewhat slower than for the gem-diphenylethyl compounds because the process is exothermic enough to fall within the inverted-Marcus region (16).
The contrast in reactivity between the 1,1,2,2-tetraphenyl- ethane (1) and 1,2-diphenylethane (5) continues with 1,1,2-tri- phenylethane (6); no cleavage was observed (reaction 3). This is perhaps not surprising in view of the fact that 1 is on the threshold for reactivity. The bond strength of the radical cation of 6. estimated to be 16.7 kcal mol-' from the thermochemical cyclk (Fig. 1 b), is too great for cleavage to compete with back electron transfer, even at the higher temperature (80°C).
Cleavage of the radical cation of methyl 2,2-diphenylethyl ether (4) occurs at 10°C. Reaction 4 is more favourable than reaction 1; nevertheless, the radical cation of the monophenyl- ethyl analog, methyl Zphenylethyl ether (9), does not cleave, even at 80°C (reaction 5). Apparently, the bond dissociation energy of the radical cation of 9 is too large for cleavage to compete.
When the irradiation of 9 is carried out in acetonitrile-meth- anol-d4, deuterium is incorporated at the benzylic position. The acidity of a benzylic hydrogen is greatly increased upon oxidation to the radical cation (1 7). Deprotonation, followed by back electron transfer from the sensitizer radical anion to the resulting radical and reprotonation (or redeuteration), provides a mechanism for deuterium exchange (steps 8- 10).
Progressing through the series of monophenylethyl deriva- tives of decreasing bond strength, not until methyl l-phenyl-2- propyl ether ( l l ) , where the a-oxyalkyl radical is secondary, does the radical cation of a monophenylethyl derivative cleave. Both the gem-diphenyl-2-propyl and the monophenyl ethers (10 and 11) cleave upon photosensitization (electron transfer) at 10°C (reactions 6 and 7).
The reaction mixtures involving 11, and all of the other monophenylethyl derivatives that cleave, are more complex than in the case of the analogous gem-diphenylethyl derivatives; (4-cyanopheny1)phenylmethane (20) is a major product in these reactions. A key step accounting for the formation of diphenyl- methane (2) from the gem-diphenylethyl derivatives is the reduction of the diphenylmethyl radical by the sensitizer radical anion (step 6). Since the reduction potential of 1,4-dicyanoben- zene (4) (- 1.66 V) is significantly greater (more negative) than that of the diphenylmethyl radical (- 1.14 V (6)), reduction of the radical is favourable. Reduction of the benzyl radical (- 1.45 V (6)) and a-phenethyl radical (- 1.60 V (6)) is less favourable. Formation of (4-cyanopheny1)phenylmethane (20) may result from coupling of the benzyl radical and the 1,4- dicyanobenzene radical anion at the ips0 position followed by loss of cyanide ion. This coupling of the radical and radical anion may become more important as reduction of the radical becomes less favourable. Steric hindrance may also inhibit the
coupling reaction involving the more hindered and more delocalized diphenylmethyl radical.
The formation of the 1,s-radical cation upon cleavage of the radical cation of methyl 2,2-diphenylcyclopentyl ether (12) has been reported (reaction 8) ( la) . In view of the fact that the acyclic~monophenyl derivative (11) cleaves, it is now not surprising that the monophenylcyclopenty1 ether (13) also cleaves (reaction 9). The observations that cis- and trans-13 are equilibrated under these conditions and that deuterium is incorporated at the benzylic position, when the irradiation is carried out in acetonitrile-methanol-d4, leads to the conclusion that deprotonation-reprotonation (steps 8- 10) may be competi- tive with cleavage in this case. Isomerization could also result from reversible carbon-carbon bond cleavage; there was no evidence for cyclization of the 1,s-radical cation into the terminal phenyl ring.
In addition to the expected cleavage product, 5-phenylphen- tanal dimethylacetal (22), a minor product having a retention time and mass spectrum (gc/ms) consistent with 5-(4-cyano- pheny1)-5-phenylpentanal dimethylacetal(23) was also detected (not isolated). This product would arise upon coupling of the benzylic radical with the radical anion of 1,4-dicyanobenzene (4) at the ips0 position followed by loss of cyanide ion, the same mechanism as that invoked for the formation of (4-cyanopheny1)- phenylmethane (20).
All four of the dioxolanes studied (14-17) undergo photosen- sitized (electron transfer) cleavage. Cleavage of the radical cation of 16, the derivative with the strongest carbon-carbon bond, is particularly interesting. If the reported oxidation potential of the 1,3-dioxolan-2-yl radical (0.31 V (6)) were correct, cleavage of 16 would be less favourable than cleavage of 9, which does not cleave. This, and other evidence vide infra, leads us to question the reported oxidation potentials of the 1,3-dioxolan-2-yl radicals.
The cleavage process (step 4) yields a carbocation and a radical fragment. We have suggested that the direction of this process, that is, which fragment becomes the radical and which becomes the carbocation, is a reflection of the relative oxidation potentials of the two fragment radicals. That fragment radical with the lower oxidation potential becomes the carbocation (1 b, c). In all of the cases involving cleavage of the radical cation of ethers, the radical bearing an a oxygen has an oxidation potential lower than either the benzylic or the diphenylmethyl radical; hence the products observed in reactions 4 and 6-9 are consistent with this hypothesis. There is, however, a quantita- tive discrepancy between calculated and observed product ratios in the diphenylmethyl-dioxolane (14) case.
Reaction 10
Ph2CH 0 hv, Sen., 10°C X 1 - Ph2CH2 + C H 3 0 ~ 0 ] H 0 CH3CN,CH30H H 0
14 2 24 Reaction 13
hv, Sen., 10°C - PhCH3 + CH30~01 P ~ ~ ~ ~ ~ l CH3CN, CHI OH H3C 0
17 19 25
Ph(4NC- C6H4)CH2
20
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

ARNOLD AND LAMONT 2125
When the difference between the oxidation potentials of the two possible radicals is small (ca. 0.1 V), cleavage is less regioselective and all four possible products are obtained. The product ratio in such a case reflects the differences in energy of the two possible modes of separation of the radical and cation, according to eq. [2] (18). Since the difference in the reported
hv, Sen., (A) R-R' -< RH + R10CH3
CH3CN, CH30H R'H + ROCH3
[R'H] [ROCH3] r=- - - [RH] [R10CH3]
values for the oxidation potential of the diphenylmethyl radical (0.35 V) and the 1,3-dioxolan-2-yl radical (0.31 V) is only 0.04 V, we would expect some (ca. 5%) methyl diphenylmethyl ether (3) from reaction 10; however, none was observed. Less than 1 % might have gone undetected (gc/fid), which means the difference in oxidation potential must be >0.10 V. If we accept the oxidation potential of the diphenylmethyl radical, this places an upper limit on the oxidation potential of the 1,3-dioxolan-2-yl radical of <0.25 V, 0.06 V less than the reported value (6). Work in progress will resolve this di~crepancy.~
Experimental General information
The 'H nmr spectra were recorded on a Nicolet Instruments Corp. narrow bore 360-MHz spectrometer and are reported in parts per million to high frequency from tetramethylsilane. Infrared spectra were recorded on a Perkin Elmer 283B ir spectrometer and are recorded in wavenumbers. Gas chromatography with mass selective detection (gc/ms) was done on a Hewlett Packard 5890A gc/5970 mass selective detector interfaced with a Hewlett Packard 9816 microcomputer. The column used was a 25 m x 0.2 mm 5% phenyl methyl silicone on fused silica with a film thickness of 0.25 pm. Gas chromatography with flame ionization detection (gc/fid) was carried out on a Hewlett Packard 5890. The column used was a 25 m x 0.25 mm OV-1701 fused silica column. Preparative gas chromatography was carried out on an Aerograph A-700 gc, fitted with a thermal conductivity detector, using a 6 ft x 314 in. 10% SE-30 on Chromosorb 60180 W column. Melting points were taken on a Thermolyne melting point apparatus and are corrected. Combustion analysis was done by Canadian Microanalytical Service Limited, New Westminster, B.C.
Electrochemical measurements Oxidation potentials were obtained by cyclic voltammetry using a
three-electrode cell, which has been described previously (19). The working electrode was a platinum sphere (1 mm diameter) and the counter electrode was a platinum wire. The reference electrode was saturated calomel (SCE). The electrolyte was 0.1 M tetraethylammo- nium perchlorate (TEAP) in acetonitrile. Substrate concentrations were typically 1 mM.
The electrochemical measurements are summarized in Table 2. The peak potentials were measured using a 100 mV/s sweep rate. No cathodic wave was detected following the anodic sweep. The half- wave potentials were taken as 0.03 V before the anodic peak potentials (12).
po he oxidation potential of the di(4-methylpheny1)methyl radical is 0.188 V (1 8). The product ratio from the cleavage of the radical cation of 2-(di-(4-methylpheny1)methyl)-1,3-dioxolane will allow a more accurate measure of the oxidation potential of the 1,3-dioxolan-2-yl radical if it is greater than 0.1 V.
Irradiations Irradiations were done in Pyrex tubes using a Canadian General
Electric Co. 1-kW medium-pressure mercury vapour lamp (CGE H 1000 DX 36-15) with a quartz cooling jacket immersed in a constant temperature bath held at either 10°C or 80°C. Samples were degassed by bubbling nitrogen through the solution for 5 min prior to irradiation.
Materials Solvents were distilled prior to use. Acetonitrile and 1,4-dicyano-
benzene (4) (Aldrich Chem. Co.) were purified as previously described (lb). Diphenylacetaldehyde, 1,l-diphenyl-2-propanone, 2-phenyl- methyl-l,3-dioxolane (16), 1,2-diphenylethane (S), 2-phenylethanol, 5-phenylpentanol, 1-phenyl-2-propanone, 1,1,2-triphenylethylene, P- methylstyrene, and phenyl-2-propanone were obtained from Aldrich Chem. Co.
Preparation of compounds Preparation of 1 , I ,2-triphenylethane (6) 1,1,2-Triphenylethane (6) was obtained by reduction of 1,1,2-tri-
phenylethylene using H2(g) over 10% Pd on charcoal in hexanes at 170 Torr (1 Torr = 133.3 Pa). The reduction was complete after 2 days of stirring at room temperature. The product was purified by recrystalliza- tion using hexanes to yield pure 6 (mp 52-54°C (lit. (20) mp 54.4"C)) in 90% yield; 'H nmr (360 MHz) in CDC13: 7.12 (m, 15H), 4.23(t, lH, J = 7.8 Hz), 3.36 (d, 2H, J = 7.8 Hz) (in agreement with that reported (2 1 )) .
Preparation of methyl 2-phenylethyl ether (9 ) Dimethyl sulphate (1 1.7 mL, 0.12 mol) was added dropwise to a
stirred solution of 2-phenylethanol(5 g, 4.1 X mol) and NaH (3 g, 0.13 mol) in anhydrous diethyl ether. The reaction mixture was refluxed for 20 h under nitrogen and was quenched by addition of water. The aqueous layer was extracted with diethyl ether. The combined organic extract was washed with concentrated NH40H, then with saturated NaCl solution, dried over magnesium sulphate, and the solvent evaporated to give the crude product. Column chromatography on silica gel (Kieselgel 60) using methylene chloride - hexanes as eluant gave the ether (9) in 60% yield; 'H nmr (360 MHz) in CDC13: 7.19-7.31 (m, 5H), 3.60(t, 2H, J = 7.2 Hz), 3.36(s, 3H), 2.89(t, 2H, J = 7.2 Hz) (in agreement with that reported (22)).
Preparation of methyl 1 ,I-diphenyl-2-propyl ether (10) Methyl 1,l-diphenyl-2-propyl ether (10) was obtained through a
two-step synthesis from 1,l-diphenyl-2-propanone in 55% overall yield. The 1,l-diphenyl-2-pentanone was reduced, using LiAlH4, to the corresponding alcohol (mp 62-63°C (lit. (23) mp 63-64"C), which was recrystallized from hexanes. The ether was prepared from this alcohol using NaH and dimethyl sulphate as described above for the preparation of 9. Pure 10 (mp 31-32°C) was obtained by column chromatography on silica gel (Kieselgel60) using methylene chloride and hexanes as eluants; ir (PE 283B) neat: 3070(m), 3030(s), 2980(s), 2930(s), 2900(s), 1600(s), 1500(s), 1455(s), 1375(m), 1340(w), 1140(s), 1090(s), 755(s), 745(s), 700(s); 'H nmr(360 MHz) in CDC13: 7.20(m, 10H),4.01 (m,lH),3.80(d, l H , J = 3 . 9 H z ) , 3 . 2 2 ( ~ , 3 H ) , 1.08 (d, 3H, J = 6.1 Hz); I3C nmr (90 MHz) in CDC13: 142.7, 129.3, 127.5, 125.2, 78.8 (d), 58.6 (d), 56.6 (t), 17.6 (q ) . Anal. calcd. for Cl6HI80: C 84.91, H 8.02; found: C 84.90, H 8.05.
Preparation of methyl I-phenyl-2-propyl ether (11) (24) Methyl 1-phenyl-2-propyl ether (11) was obtained by the photo-
chemical method (lob). P-Methyl styrene was irradiated at 10°C in the presence of an electron-accepting sensitizer, 1,4-dicyanobenzene (4), in acetonitrile-methanol (3: 1) solution. The pure ether 11 was obtained in 40% yield by column chromatography on silica gel (Kieselgel60) with methylene chloride and hexanes as eluant; 'H nmr (360 MHz) in CDCl3:7.16-7.27 (m,5H),3.51 (m, lH),3.30(s, 3H),2.93(dd, lH, 'J= -14.0Hz, J=5.99Hz),2.62(dd, 1 H , 2 ~ = -14.0Hz,J=6.74 Hz), l . lO(d, 3H, J = 6.15 Hz); 13~nmr(90MHz)inCDC13: 138.9, 129.3, 128.1, 125.9, 78.0, 56.1,42.7, 18.8.
Preparation of methyl cis- and trans-2-phenylcyclopentyl ether (cis- and trans-13)
The methyl cis- and trans-2-phenylcyclopentyl ethers were synthes-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

2126 CAN. J. CHEM. VOL. 67, 1989
ized from I-phenylcyclopentene using the photochemical method (lob).
Bromobenzene (7.8 g, 0.05 mol) was added dropwise to magnesium (1.2 g, 0.05 mol) in anhydrous diethyl ether. Once the magnesium was consumed, the reaction mixture was cooled and cyclopentanone (3.14 g, 0.0375 mol) was added dropwise. The reaction mixture was poured into a concentrated hydrochloric acid - ice mixture, which was then extracted with ether (3 X). The organic layers were combined, washed with 5% NaHC03 solution, and then with water. The extract was then dried with magnesium sulphate and the solvent was evaporated. Pure I-phenylcyclopentene was obtained in 73% yield by flash column chromatography using hexanes as eluant.
A solution of 1-phenylcyclopentene (173 mg, 1.2 x mol) and 4 (124 mg, 9.7 x mol) in acetonitrile-methanol (30 mL, 3:l) was irradiated at 10°C for 24 h. Flash column chromatography of the photolysate, using methylene chloride - hexanes (1:l) as eluant, separated the isomeric ethers, cis- (17%) and trans-13 (38%), at 97% conversion.
cis-13: 'H nmr (360 MHz) in CDC13: 7.19-7.33 (m, 5H), 3.82 (ddd, lH,J=4.8Hz,J=4.3Hz,J=2.1Hz),3.05(s,3H),2.99(ddd,lH, J = 4 . 8 H z , J = 7 . 9 4 H z , J = 11.06Hz),2.01 (m,2H), 1.92(m,2H), 1.88 (m, 2H).
trans-13: 'H nmr (360 MHz) in CDC13: 7.18-7.31 (m, 5H), 3.78 (ddd, IH, J = 3.15Hz, J = 5 . 5 8 H z , J=6.0Hz),3.25(s,3H),3.03 (ddd, IH, J = 6.0Hz, J = 6.5 Hz, J = 7.0Hz), 1.65-2.15 (m, 6H).
Preparation of 2-diphenylmethyl-l,3-dioxolane (14) 2-Diphenylmethyl-l,3-dioxolane (14) was prepared from diphenyl-
acetaldehyde using chlorotrimethylsilane and ethylene glycol (25). Pure 14 (mp 74-76°C) was obtained, following recrystallization from methanol, in 95% yield; 'H nmr (360 MHz) in CDC13: 7.19-7.38 (m, IOH), 5.55 (d, 2H, J = 5.01 Hz), 4.22 (d, 2H, J = 5.01 Hz), 3.78-3.86 (m, 4H) (in agreement with the reported spectrum (26)).
Preparation of 2-diphenylmethyl-2-methyl-1,3-dioxolane (15) (27) 2-Diphenylmethyl-2-methyl-1,3-dioxolane (15) was prepared from
1,l-diphenyl-2-propanone by the reported procedure (25). Pure 15 (mp 86-87°C) was obtained in 95% yield following recrystallization from methanol; 'H nmr (360 MHz) in CDCI3: 7.48-7.50 (m, 4H), 7.18- 7.28 (m, 6H), 4.22 (s, IH), 3.79 (m, 2H), 3.35 (m, 2H), 1.36 (s, 3H); I3C nmr (90MHz) inCDC13: 140.7 (s) 129.9, 127.9, 126.3, 11 1.3 (s), 65.0 (t), 61.0 (d), 25.3 (q).
Preparation of 2-phenylmethyl-I ,3-dioxolane (16) (28) 2-Phenylmethyl-1,3-dioxolane (16) was prepared from I -phenyl-2-
propanone by the reported procedure (25). Compound 16 was puri- fied by column chromatography on silica gel (Kieselgel 60) using methylene chloride - hexanes as eluant. Pure 16 was obtained in 80% yield; 'H nmr (360 MHz) in CDC13: 7.22-7.28 (m, 5H), 3.90 (m, 2H), 3.76 (m, 2H), 2.92 (s, 2H), 1.3 1 (s, 3H).
Irradiations Irradiation of l,2-diphenylethane (5) A solution of 5 (83 mg, 4.4 x mol) and 4 (68 mg, 5.3 X
mol) in acetonitrile-methanol (3:l) was irradiated at both 10°C and 80°C. Trace amounts of toluene (19), the expected product from carbon-carbon bond cleavage, were observed (gclms) after irradia- tion for 20 h at 1 O°C and after 15 h at 80°C.
Irradiation of 1 , I ,2-triphenylethane (6) A solution of 6 (157 mg, 6.1 x mol) and 4 (96 mg, 7.5 x
mol) in acetonitrile-methanol (20 mL, 3:l) was irradiated at 80°C for 30 h. Trace amounts of diphenylmethane (2), 1 ,Zdiphenylethane (5), and methyl diphenylmethyl ether (3) were detected (gclms).
Irradiation of methyl 2-phenylethyl ether (9) A solution of 9 (40 mg, 2.9 x lop4 mol) and 1,4-dicyanobenzene (4)
(15 mg, 1.2 x mol) in acetonitrile-methanol (2 mL, 3:l) was irradiated at 10°C for 25 h. Only trace amounts of toluene (19) and (4-cyanopheny1)phenylmethane (20), expected cleavage products, were detected (gclfid).
A solution of 9 (25 mg, 1.8 X mol) and 4 (24 mg, 1.9 x mol) in acetonitrile-methanol (2 mL, 3:l) was irradiated at 80°C for 15 h. Only trace amounts of 19 and 20 were detected (gclfid).
A solution of 9 (20 mg, 1.5 X mol) and 1,4-dicyanobenzene (4) (I I mg, 8.6 X mol) in acetonitrile-methanol-d4 (2 mL, 3:l) was irradiated at 10°C for 24 h. Analysis (gclms) of the photolysate, using selected ion monitoring of the molecular ion and the benzylic fragment (mlz = 91-95), showed that deuterium had been incorporated (9%, one atom deuterium) at the benzylic position.
Irradiation of methyl I , I -diphenyl-2-propyl ether (10) A solution of 10 (600 mg, 2.66 X loe3 mol) and 4 (340 mg, 2.66 x
mol) in acetonitrile-methanol (20 mL, 3:l) was irradiated for 23 h. Diphenylmethane (2) and 1 ,I ,2,2-tetraphenylethane (1) were isolated by flash column chromatography of the photolysate on silica gel (Baker Analyzed) using hexanes and methylene chloride as eluant. After 90% conversion, 2 (70%) and 1 (10%) were isolated.
Irradiation of methyl I -phenyl-2-propyl ether (11) A solution of 11 (15 mg, 1 .O X rnol), 4 (22 mg, 1.7 X
rnol), and cyclododecane (12 mg, 7.0 X mol) in acetonitrile- methanol (2 mL, 3: 1) was irradiated for 20 h. The major products of the irradiation were toluene (19), 1,2-diphenylethane ( 9 , and (4-cyano- pheny1)phenylmethane (20). Product analysis was done using gc/fid with cyclododecane as an internal calibration standard. The percent yields at 98% conversion were as follows: 19, 25%; 5, 15%; and 20, 25%. The yields for 5 and 20 were confirmed by product isolation following column chromatography.
Irradiation of methyl cis- and trans-2-phenylcyclopentyl ether (cis- and trans-13)
A solution of cis- and trans-13 (ca. 1:2) (52 mg, 3.0 X mol) and 4 (30 mg, 2.3 X mol) in acetonitrile-methanol (2 mL, 3:l) was irradiated at 10°C. After 9 h of irradiation, the only product detected (gclms) was 5-phenylpentanal dimethylacetal(22), identified by comparison (gclms, retention time, and fragmentation pattern) with an authentic sample. After prolonged irradiation, an additional product was detected (gclms), to which the structure 5-(4-cyanopheny1)-5- phenylpentanal dimethylacetal (23) was tentatively assigned on the basis of the fragmentation pattern: 277, 245, 218, 230, 192, 165, 103 (the molecular ion was neither expected nor observed).
Authentic 23 was synthesized by a two-step process from the corresponding alcohol. 5-Phenylpentanol was oxidized, using chro- mium trioxide/pyridine, to 5-phenylpentanal in 88% yield by the standard procedure (29). Chlorotrimethylsilane (1 .O8 g, 0.01 mol) was added dropwise to a stirred solution of 5-phenylpentanal in methanol. The mixture was stirred overnight at room temperature; it was then cooled in ice, and 50 mL aqueous NaHC03 (5%) was added. The mixture was extracted (3x) with ether, and the organic layers were combined, washed with brine solution, and then dried over magnesium sulphate. The acetal23 was purified by flash column chromatography on silica gel, eluting with methylene chloride - hexanes (7525). The yield of pure 23 from the aldehyde was 50%; 'H nmr (360 MHz) in CDC13: 7.15-7.28 (m, 5H), 4.34 (t), IH, J = 5.72 Hz), 3.29 (s, 6H), 2.61 (t, 2H, J = 7.73 Hz), 1.62 (m, 4H), 1.39 (m, 2H); I3C nmr (90 MHz): 142.5, 128.3, 125.9, 125.6, 104.4, 52.6, 35.9, 32.4, 31.3, 24.3.
A solution of cis- and trans-13 (ca. 1:2) (20 mg, 1.1 X mol) and 4 (24 mg, 1.9 X 1 mol) in acetonitrile-methanol-d4 (2 mL, 3: 1) was irradiated at 10°C for 24 h. Analysis (gclms, using selective ion monitoring) of the photolysate showed that deuterium (2%, one atom deuterium) was incorporated, at the benzylic position, in both isomers.
A solution of methyl cis- and trans-2-phenylcyclopentyl ether (13) in the ratio cis:trans = 1 5 4 (12mg, 6.8 X mol) and4 (18 mg, 1.4 x mol) was irradiated at 10°C. The ratio of cis to trans isomers was monitored by integrated peak areas on the gclfid. After 45 h of irradiation, the ratio was 1:37.
Irradiation of 2-diphenylmethyl-I ,3-dioxolane (14) A solution of 14 (443 mg, 1.85 x mol) and 4 (273 mg, 2.13 x
mol) in acetonitrile-methanol (20 mL, 3: 1 ratio) was irradiated at 10°C and monitored (gclfid). The products from the irradiation, 2-methoxy-1,3-dioxolane (24), diphenylmethane (2), and 1,1,2,2- tetraphenylethane (I), were identified by comparison (gclms, retention times, and fragmentation pattern) with those of authentic samples. After 23 hof irradiation, diphenylmethane (2) and 1 ,I ,2,2-tetraphenyl-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.

ARNOLD AND LAMONT 2127
ethane (1) were isolated by flash column chromatography in 90 and 10% yield at 75% conversion.
Irradiation of 2-diphenylmethyl-2-methyl-l,3-dioxolane (15) A solution of 15 (441 mg, 1.74 X mol) and 4 (289 mg, 2.26 X
mol) in acetonitrile-methanol (20 mL, 3: 1) was irradiated at 10°C for 23 h. Progress of the reaction was monitored (gclfid). The products of the irradiation were 2-methoxy-2-methyl-l,3-dioxolane (25), di- phenylmethane (2), and 1,1,2,2-tetraphenylethane (1). Diphenyl- methane (2) and 1,1,2,2-tetraphenylethane (1) were identified by comparison (gc/ms, retention times, and fragmentation pattern) with authentic samples. The identity of the ortho ester 25 was confirmed by hydrolysis to methyl acetate, as indicated by comparison (gc/ms) with an authentic sample. The hydrolysis was accomplished by refluxing the crude reaction mixture with 20 mL of 15% aqueous HCl for 1 h. The products, diphenylmethane (2) (85%) and 1,1,2,2-tetraphenylethane (1) (lo%), were isolated by flash column chromatography after 85% conversion of 15.
Irradiation of 2-phenylmethyl-1,3-dioxolane (16) A solution of 16 (18 mg, 1.1 X rnol), 4 (14 mg, 1.1 x
rnol), and cyclododecane (16. 5 mg, 9.8 X lop5 mol) in acetonitrile- methanol (2 mL, 3:l) was irradiated at 10°C for 20 h. The major products of the irradiation were toluene (19), 2-methoxy-1,3-dioxolane (24), 1,2-dipheny lethane (5), and (4-cyanopheny1)phenylmethane (20). Toluene (19), 2-methoxy-l,3-dioxolane (24), and 1,2-diphenylethane (5) were identified by comparison of retention times and fragmentation pattern (gc/ms) with authentic samples. The (4-cyanopheny1)phenyl- methane (20) was isolated by column chromatography and identified by comparison ('H nrnr and melting point) with reported data. (4-Cyano- pheny1)phenylmethane (mp 44-46°C (lit. (14) mp 49-50°C)); 'H nrnr (360 MHz) in CDC13: 7.14-7.56 (m, 9H), 4.02 (s, 2H). Gas chromatography/fid was used to calculate the percent yields of the products at 85% conversion; cyclododecane was used as an internal standard. The yields were toluene (19) 40%, 1,2-diphenylethane (5) lo%, and (4-cyanopheny1)phenylmethane (20) 20%. The estimated error in these values is 25%. The values for 5 and 20 were confirmed by isolation using column chromatography on silica gel (Kieselgel60) eluting with methylene chloride and hexanes.
Irradiation of 2-phenylmethyl-2-methyl-] ,3-droxolane (1 7) A solution of 17 (22 mg, 1.2 X lop4 rnol), 4 (20 mg; 1.6 x
rnol), and cyclododecane (14 mg, 8.3 X mol) in acetonitrile- methanol (2 mL, 3:l) was irradiated at 10°C for 20 h. The major products of the photolysis were toluene (19), 2-methoxy-2-methyl- 1,3- dioxolane (25), 1,2-diphenylethane (5), and (4-cyanopheny1)phenyl- methane (20). Product analysis was done using gc/fid with cyclo- dodecane as an internal standard. The yields of 19 ,5 , and 20 were 35, 5, and 20% respectively, at 95% conversion. The yields of 5 and 20 were confirmed by isolation using column chromatography as described above for the reaction of 16.
Acknowledgments Acknowledgment is made to the Donors of the Petroleum
Research Fund, administered by the American Chemical Society, and to the Natural Sciences and Engineering Research Council of Canada for support of this research. We thank Dr. D. L. Hooper and the Atlantic Region Magnetic Resonance Centre at Dalhousie University for the 360 MHz nmr spectra and for help in the preparation of the manuscript.
4. (a) H. M. ROSENSTOCK, K. DRAXL, B. W. STEINER, and J. T. HERRON. J. Phys. Chem. Ref. Data, 6 (1977) Suppl. No. 1 (1977); (b) I. HOWE and D. H. WILLIAMS. J. Am. Chem. Soc. 91, 7137 (1969); (c) F. W. MCLAFFERTY, T. WACHS, C. LIFSHITZ, G. INNORTA, and P. IRVING. J. Am. Chem. Soc. 92, 6867 (1970).
5. (a) M. J. MANKA, R. L. BROWN, and S. E. STEIN. J. Phys. Chem. 89,5421 (1985); (b) A. C. BUCHANAN nI , T. D. J. DUNSTAN, E. C. DOUGLAS, and M. L. POUTSMA. J. Am. Chem. Soc. 108,7703 (1986).
6. (a) D. D. M. WAYNER, J. J. DANNENBERG, and D. GRILLER. Chem. Phys. Lett. 131, 189 (1986); (b) D. D. M. WAYNER, D. J. MCPHEE, and D. GRILLER. J. Am. Chem. Soc. 110, 132 (1988).
7. (a) H. F. DAVIS, P. K. DAS, L. W. REICHEL, and G. W. GRIFFIN. J. Am. Chem. Soc. 106,6968 (1984); (b) L. W. REICHEL, G. W. GRIFFIN, A. J. MULLER, P. K. DAS, andS. N. EGE. Can. J. Chem. 62,424(1984); (c) A. ALBINI, E. FASANI, and M. MELLA. J. Am. Chem. Soc. 108.41 19 (1986); (d) A. ALBINI, E. FASANI, and E. MONTESSORO. Z. Naturforsch. 29B, 1409 (1984); (e) A. ALBINI and S. SPRETI. J. Chem. Soc. Perkin Trans. 2, 1175 (1987); (f) E. BACIOCCHI and R. RUZZICONI. J. Chem. Soc. Chem. Commun. 445 (1984).
8. D. R. ARNOLD and A. J. MAROULIS. J. Am. Chem. Soc. 98,5931 (1976).
9. (a) R. H. MARTIN, F. W. LAMPE, and R. W. TAFT. J. Am. Chem. Soc. 88,1353 (1966); (b) L. R. C. BARCLAY, J. LUSZTYK, andK. U. INGOLD. J. Am. Chem. Soc. 106, 1793 (1984); (c) V. MALATESTA and K. U. INGOLD. J. Am. Chem. Soc. 103, 609 (1981); (d) D. J. PASTO, R. KRASNANSKY, and C. ZERCHER. J. Org. Chem. 52, 3062 (1987).
10. (a) R. A. NEUNTEUFEL and D. R. ARNOLD. J. Am. Chem. Soc. 95,4080 (1973); (b) Y. SHIGEMITSU and D. R. ARNOLD. J. Chem. Soc. Chem. Comrnun. 407 (1975).
11. D. REHM and A. WELLER. Isr. J. Chem. 8,259 (1970). 12. R. S. NICHOLSON and I. SHAIN. Anal. Chem. 36,706 (1964); 37,
178 (1965). 13. (a) K. K. BARNES and C. K. MANN. J. Org. Chem. 32, 1474
(1967); (b) E. S. WSH and N. C. YANG. J. Am. Chem. Soc. 85, 2124 (1963).
14. J. D. YOUNG, G. R. STEVENSON, and N. L. BAULD. J . Am. Chem. Soc. 94, 8790 (1972).
15. A. TERAHARA, H. 0.-NISHIGUCHI, N. HIROTA, H. HIGUCHI, and S. MISUMI. J. Phys. Chem. 90,4958 (1986).
16. (a) R. A. MARCUS. Disc. Faraday Soc. 29, 21 (1960); (b) I. R. GOULD and S. FARID. J. Am. Chem. Soc. 110,7883 (1988).
17. (a) A. M. DE P. NICHOLAS and D. R. ARNOLD. Can. J. Chem. 60, 2165 (1982); (b) F. G. BORDWELL and J.-P. CHENG. J. Am. Chem. Soc. 111, 1792 (1989).
18. (a) R. POPIELARZ. Ph.D. Thesis, Dalhousie (1989); (b) R. POPIELARZ and D. R. ARNOLD. Submitted for publication.
19. D. D. M. WAYNER and D. R. ARNOLD. Can. J. Chem. 63, 871 (1985).
20. A. KLAGES and S. HEILMANN. Ber. 37, 1447 (1904). 21. I. GRANOTH, Y. SEGALL, H. LEADER, and R. ALKABETS. J. Org.
Chem. 41, 3682 (1976). 22. The Sadtler Handbook of Proton NMR Spectra. 1978. Compound
number 1484, p. 654. 23. F. A. ABD ELHAFEZ and D. J. CRAM. J. Am. Chem. Soc. 75,339
(1953). 24. S. UEMURA and S. FUKUZAWA. J. Chem. Soc. Perkin Trans. 1.
1. (a) D. R. ARNOLD, B. J. FAHIE., L. J. LAMONT, J. WIERZCHOW- 471 (1985).
SKI, and K. M. YOUNG. can. J. them. 65, 2734 (1987); (b) A. 25. T. H. CHAN, M. A. BROOK, and T. CHALY. Synthesis, 203
OKAMOTO, M. S. SNOW, and D. R. ARNOLD. Tetrahedron, 42, (1983).
6175 (1986); (c) A. OKAMOTO and D. R. ARNOLD. Can. J. Chem. 26' B. L. JENsEN and R. E' COUNSELL' Can' J ' 51, 3820
63, 2340 (1985). (1973). 2. D. R. ARNOLD and S. A. MINES. Can. J , Chem. 67,689 (1989). 27. V. I. STENBERG and D. A. KUBIK. J. Org. Chem. 39, 2815 (1974).
3. (a) X. CI and D. G. WHITTEN. J. Am. Chem. Soc. 109, 7215 28. I. M. TAKAUS and W. C. J. Org. 437 1952
(1987); (b) A. ALBINI and M. MELLA. Tetrahedron, 42, 6219 (1978).
(1986); (c) T. GOTOH, M. KATO, M. YAMAMOTO, and Y. 29. R. RATcLIFFE and R. RODEHORST. J. 0%. Chem. 35, 4000
NISHUIMA. J. Chem. Soc. Chem. Comrnun. 90 (1981); (d) D. M. (1970). I CAMAIONI and J. A. FRANZ. J. Org. Chem. 49, 1607 (1984).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F T
EX
AS
AT
AR
LIN
GT
ON
on
10/0
3/13
For
pers
onal
use
onl
y.



![Carfentanil · RISK ASSESSMENTS 28 Carfentanil Report on the risk assessment of methyl 1-(2-phenylethyl)-4-[phenyl(propanoyl) amino]piperidine-4-carboxylate in the framework of the](https://img.pdfslide.us/doc/110x75/5f8b296f33927873be52150b/carfentanil-risk-assessments-28-carfentanil-report-on-the-risk-assessment-of-methyl.jpg)

























