Embed Size (px)
Citation preview

www.elsevier.com/locate/chemphys
Chemical Physics 320 (2006) 75–83
Photoinduced intramolecular charge transfer (ICT) reactionin trans-methyl p-(dimethylamino) cinnamate: A combined
fluorescence measurement and quantum chemical calculations
Amrita Chakraborty a, Samiran Kar b,1, Nikhil Guchhait a,*
a Department of Chemistry, University of Calcutta, 92, A. P. C. Road, Kolkata 700009, Indiab Department of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India
Received 7 April 2005; accepted 22 June 2005Available online 18 August 2005
Abstract
The photophysical behaviour of trans-methyl p-(dimethylamino) cinnamate (t-MDMAC) donor–acceptor system has been inves-tigated by steady-state absorption and emission spectroscopy and quantum chemical calculations. The molecule t-MDMAC showsan emission from the locally excited state in non-polar solvents. In addition to weak local emission, a strong solvent dependent redshifted fluorescence in polar aprotic solvents is attributed to highly polar intramolecular charge transfer state. However, the forma-tion of hydrogen-bonded clusters with polar protic solvents has been suggested from a linear correlation between the observed redshifted fluorescence band maxima with hydrogen bonding parameters (a). Calculations by ab initio and density functional theoryshow that the lone pair electron at nitrogen center is out of plane of the benzene ring in the global minimum ground state structure.In the gas phase, a potential energy surface along the twist coordinate at the donor (–NMe2) and acceptor (–CH = CHCOOMe)sites shows stabilization of S1 state and destabilization S2 and S0 states. A similar potential energy calculation along the twist coor-dinate in acetonitrile solvent using non-equilibrium polarized continuum model also shows more stabilization of S1 state relative toother states and supports solvent dependent red shifted emission properties. In all types of calculations it is found that the nitrogenlone pair is delocalized over the benzene ring in the global minimum ground state and is localized on the nitrogen centre at the 90�twisted configuration. The S1 energy state stabilization along the twist coordinate at the donor site and localized nitrogen lone pairat the perpendicular configuration support well the observed dual fluorescence in terms of proposed twisted intramolecular chargetransfer (TICT) model.� 2005 Elsevier B.V. All rights reserved.
Keywords: trans-Methyl p-(dimethylamino) cinnamate; TICT; Density functional theory; Absorption; Fluorescence
1. Introduction
The photoinduced charge transfer reaction betweentwo distinct parts of a molecule (intramolecular) or be-
0301-0104/$ - see front matter � 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.chemphys.2005.06.031
* Corresponding author. Tel.: +91 33 2350 8386; fax: +91 33 23519755.
E-mail address: [email protected] (N. Guchhait).1 Present Address: CHEMGEN Pharma International, Dr. Sie-
mens Street, Bolck GP, Sect. V, Salt Lake City, Kolkata 700 091,India.
tween two molecules (intermolecular) generates a chargetransfer (CT) state having geometry and properties verydifferent from its ground and locally excited states. In-tra- or intermolecular charge transfer reactions are themost encountered photochemical reaction for the lastfour decades as photoinduced CT reactions play a fun-damental role in photosynthesis in green plants, visionand is used as a molecular probe to investigate the crit-ical miceller concentration, degree of water penetrationinto the surfactant aggregate and local polarity ofthe microenvironment of the binding sites of proteins.

76 A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83
Since the first observation of dual fluorescence fromdimethylaminobenzonitrile (DMABN) by Lippertet al. [1], the photophysics and photochemistry of simi-lar donor–acceptor molecules is a matter of debate formany years [2,3]. It is well established that the propertiesof DMABN molecule in the excited state are differentfrom its ground state as the charge distribution is chan-ged after excitation. The electronic structure and specialexcited state properties of DMABN was establishedfrom experimental evidences and subsequently theoreti-cal work complemented the experimental results [4–8].Various models have been proposed to account the dualfluorescence of DMABN [2,3,9–11]. Till date, it is foundthat, out of all models, twisted intramolecular chargetransfer (TICT) model is the most acceptable one forexplaining dual fluorescence from DMABN and itsanalogous molecules [8,12].
In spite of a large number of investigation have beenperformed for DMABN and analogous molecules byexperimentally and theoretically, neither the mecha-nism of the ICT reaction nor the structure of theICT state is presently known with any certainty. Re-search is going on from condensed phase to molecularjet to get a better understanding of photoinduced ICTreactions with different newer donor–acceptor systems[13,14]. Very recently, photoinduced ICT reactions havebeen studied by absorption and emission spectroscopyfor p-(dimethylamino) cinnamaldehyde [15,16], ethylp-(dimethylamino) cinnamate [17] and p-(dimethylamino)cinnamic acid [18]. All the above mentioned moleculesare very similar to that of DMABN except one unsatu-rated bond is connected in between the donor andacceptor sites. In all cases the observed dual fluorescencewas explained by TICT model. Only a semiempiricalAM1 calculation predicts S1 and S2 states crossing alongthe twist coordinate at the NMe2 site for p-(dimethyla-mino) cinnamaldehyde and for p-(dimethylamino)cinnamic acid. But concrete theoretical supports forthe observed experimental phenomena for all thesemolecules are also lacking. It is expected that ab initiocalculations may provide better understanding on pho-toinduced ICT process for the above molecules as wasperformed for DMABN and its analogous. Keeping thisin mind, we have synthesized t-MDMAC and investi-gated photoinduced ICT processes in condensed phaseby steady-state absorption and emission spectroscopy.The polarity and hydrogen bonding ability dependenceemission have been studied for the excited charge state.Although the molecule is similar to that of the abovestudied molecules, but the acceptor group in t-MDMACis different. The presence of unsaturated bond betweenthe donor and acceptor provides an extra flexibility tothe molecule. Also we have performed ab initio and den-sity function theory (DFT) level of calculations in thegas and solvated states to provide a better understand-ing between the experimental results with the theoretical
one and try to find out the mechanism of excited stateCT process in t-MDMAC.
2. Experimental
(trans)-Methyl p-(dimethylamino) cinnamate (t-MDMAC) was prepared from p-(dimethylamino)benzaldehyde by standard procedure. In brief, 4-dimeth-ylaminobenzaldehyde and methyl (triphenylphosphor-axylidene) acetate in dry dichloromethane were stirredat room temperature for 36 h. After the solvent wasremoved over vacuum, the crude compound was puri-fied by silica gel column chromatography and repeatedcrystallization with minimum amount of methanol toget t-MDMAC, 1H NMR (400 MHz, CHCl3): d 3.04(S, 6H, –N–CH3), 3.79 (S, 3H, OMe), 6.23 (d,J = 21.2 Hz, 1H), 6.73 (d, J = 10.8 Hz, 2H), 7.43 (d,J = 11.6 Hz), 7.63 (d, J = 20.8 Hz, 1H).
O
O
NCH3
CH3
CH3 7
8
1
6
2 3
4
5
9
13
14
15
10
11
Structure of t-methyl p-(dimethylamino) cinnnamate (t-MDMAC)
The steady-state absorption and emission spectra indifferent spectroscopic grade solvents were recordedusing a Hitachi (UV/Vis) and Perkin Elmer (Model50B) spectrophotometer, respectively. The concentra-tion of (t-MDMAC) was maintained to 10�5 M in differ-ent spectroscopic grade solvents.
All quantum chemical calculations were performedby Gaussian 03 software [19]. Structural calculationshave been done at Hartee-Fock (HF) level and also atdensity functional theory (DFT) with B3LYP hybridfunctional and 6-31++G(d,p) and 6-311++G(d,p) basissets [20–22]. The excited states� potential energy surfaces(PES) along the twist coordinates were performed bytime dependent density functional theory (TDDFT)with same functional and basis set [23]. Validation ofusing TDDFT in calculating excitation energies forcharge transfer excited state of such conjugated systemwas extremely discussed by Jodicke and Luthi [24,25].All excited state calculations are limited to single pointcalculations. We have defined twisting of donor oracceptor as the reaction coordinate and freeze all otherdegrees of freedoms. Recently, Hass et al. [26] per-formed relaxed calculation by CASSCF method andshowed that relaxation of the benzene ring could pro-vide a better mapping of twisting surfaces for ICT reac-tion. However, our limited computational facility doesnot allow us to perform such type of relaxed calculation.

A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83 77
Solvation calculations were performed using timedependent polarized continuum model (TD-PCM)[27,28].
3. Results and discussion
The absorption spectra of trans-methyl p-(dimethyla-mino) cinnamate (t-MDMAC) (�1 · 10�5 M solution)in polar and non-polar solvents are shown in Fig. 1.As seen from the absorption spectra, t-MDMAC exhib-its absorption bands at 314 nm and at �360 nm. Com-pared to similar molecular systems it is assumed thatonly a single species is present in the ground state[15,17,18]. These two bands are assigned to the transi-tion from ground state to Lb (S2) and La (S1) states of(t-MDMAC), respectively, as was assigned in the similar
300 320 340 360 380 4000.00
0.25
0.50
0.75
1.00
5
42
3
6
1
Abs
(a.
u.)
Wavelength (nm)
Fig. 1. Absorption spectra of t-MDMAC at room temperature indifferent solvents: (1) n-hexane; (2) THF; (3) dioxane; (4) ACN; (5)EtOH and (6) water.
Table 1Observed parameters from the absorption and emission spectra of t-MDMA
Solvent Df kabs (nm) kflu
Water 0.319 314 474362 396
Acetonitrile (ACN) 0.304 314 458362 392
Ethanol (EtOH) 0.288 314 453361 391
Cyclohexane 0.002 314 390345
Tetrahydrofuran (THF) 0.207 314 427360
Hexane 0.016 314 392345
Dioxane 0.020 314 418360
Carbontetrachloride (CCl4) 0.005 314 399354
a Dm(cm�1) = mabs (cm�1) � mflu (cm�1).
type of molecules [15,18]. The maximum of the �360 nmabsorption band slightly blue shifted in the non-polarsolvent.
To study the excited state properties of t-MDMAC,the excitation and emission spectra of t-MDMAC havebeen measured in different solvents and the results arepresented in Table 1. The emission spectra of t-MD-MAC (kext = 350 nm) in different polar and non-polarsolvents are shown in Fig. 2. In non-polar solvent(cyclohexane or n-hexane), upon 350 nm excitation,t-MDMAC shows an emission band at 390 nm, whichis ascribed to locally excited (LE) state emission. Whereas the same 350 nm excitation in polar solvents exhibitsdual fluorescence, weak higher energy emission at390 nm arising from the locally excited (LE) stateand the red-shifted strong emission at �450 nm arisingfrom the CT state. The fluorescence quantum yields for
C in different solvents
(nm) mabs (cm�1) mflu (cm�1) Dm (cm�1)
27,624 21,097 652725,253
27,624 21,834 579025,510
27,701 22,075 562625,575
28,986 25,641 3345
27,778 23,419 4359
28,986 25,510 3475
27,778 23,923 3854
28,249 25,063 3185
400 450 500 5500
10
20
30
40
50
2
3
6
4
51F
luor
esce
nce
Inte
nsit
y (a
.u.)
Wavelength (nm)
Fig. 2. Fluorescence emission spectra of t-MDMAC at room temper-ature in different solvents: (1) hexane; (2) THF; (3) dioxane; (4) ACN;(5) EtOH and (6) water.
a

400 450 5000
20
40
60
80
5
1
Flu
ores
cenc
e In
tens
ity
(a.u
.)
Wavelength (nm)
Fig. 4. Fluorescence emission spectra of t-MDMAC in cyclohexaneand ethanol mixture: (1) cyclohexane; (2) cyclohexane + 2% EtOH; (3)cyclohexane + 6% EtOH; (4) cyclohexane + 10% EtOH and (5)cyclohexane + 20% EtOH.
78 A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83
the red shifted CT band is very high compared to theLE band (Table 2). The maxima of the LE band arefound to be independent of solvent polarity, but thefluorescence emission spectrum of the CT band showsa strong solvent dependency (Fig. 2). This large Stokesshifted emission band is progressively red-shifted withincreasing the polarity of the solvents. This could beinterpreted by assuming that the polarity dependentemission originates from the charge transfer fromdonor (–NMe2) to acceptor part in the excited state.Therefore, the emissive state has higher dipole momentthan the neutral ground state. The polarity dependentStokes shifted emission is due to different extents ofstabilization of the photo generated charge species inthe excited state. As can be seen in Fig. 3, the excita-tion spectra are solvent independent and the observedexcitation bands correspond well with the absorptionspectra (Fig. 1). This clearly states that the moleculeexists as single species in the ground state. The emis-sion spectra of t-MDMAC in cyclohexane ethanolmixed solvent are shown in Fig. 4. With increasing eth-anol concentration the intensity of CT band (453 nm)increases with the decrease of LE emission and a clearisoemissive point is observed. This clearly points out
Table 2Quantum yields of LE and CT states of t-MDMAC in differentsolvents at room temperature and 77 K
Solvent a Quantum yields
LE state CT state
Cyclohexane – 4.70 · 10�3 –Acetonitrile – 5.50 · 10�4 2.7 · 10�2
Methanol 0.98 5.28 · 10�4 4.8 · 10�2
Water 1.1 4.16 · 10�4 9.0 · 10�3
Methylcyclohexane (77 K) – 6.0 · 10�2 –Ethanol (77 K) – 7.0 · 10�2 –
a is the index of solvent hydrogen bond donor ability.
250 300 350 4000
40
80
120
160
14
3
2Inte
nsit
y (a
.u.)
Wavelength (nm)
Fig. 3. Excitation spectra of t-MDMAC at room temperature indifferent solvents: (1) hexane; (2) ACN; (3) EtOH and (4) water.
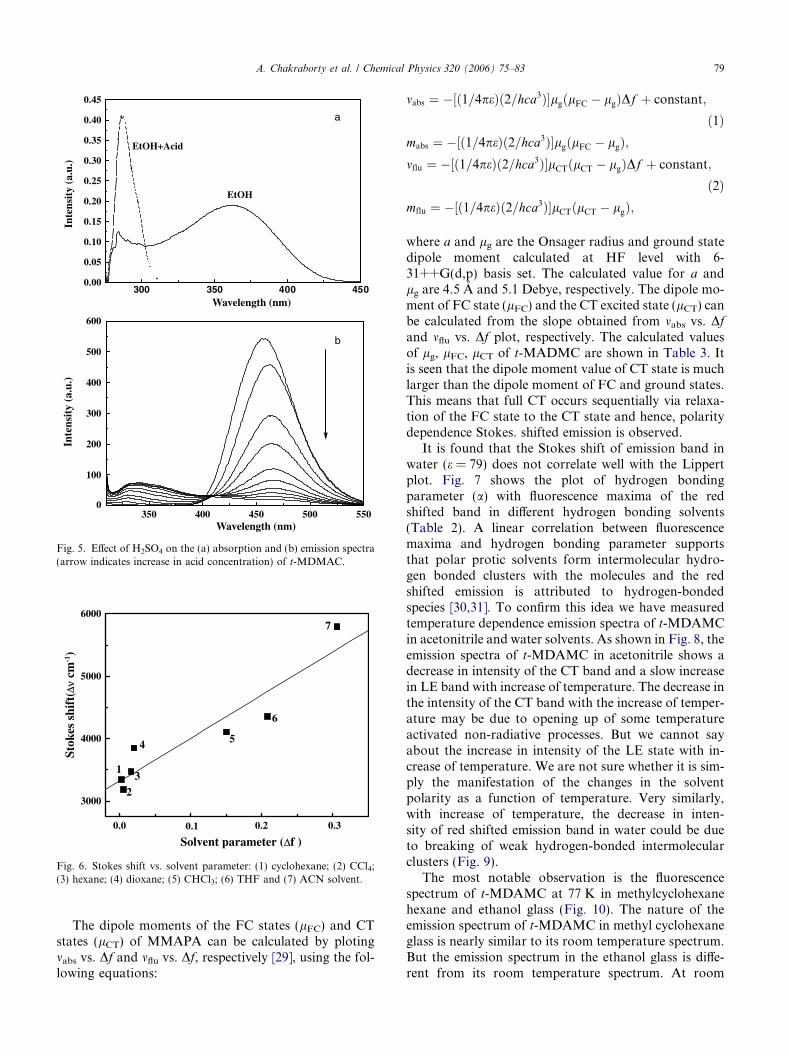
that the CT state is arising from the LE state. A chargetransfer in the excited state may result from the trans-fer of electron upon excitation from the donor (–NMe2)to the acceptor (–CH@CHCO2Me) connected throughbenzene ring. That means that the lone pair of nitrogenin the donor is transfer to the acceptor to form ahighly polar CT state. To proof this, we have measuredabsorption and emission spectra of t-MDMAC in pres-ence of acid and are shown in Fig. 5. On addition of2N H2SO4 acid, a new absorption band appears at300 nm with the disappearance of 360 nm band. TheH+ ions of acid try to bind with the nitrogen lone pairof the donor. The absorption band at 300 nm is basi-cally arising from the normal p–p* transition of simplebenzene type of chromophore. In the emission spectra(Fig. 5(b)), upon increase in acid concentration, thecharge transfer band intensity is decreased with theincrease of LE emission of the protonated species. Asthe H+ ion binds to the nitrogen lone pair, the lonepair electron of nitrogen is not available for the gene-ration of excited CT state. Therefore, red shiftedemission (450 nm) corresponding to CT emission disap-pears in the emission spectra.
The Stokes shifts (Dm in cm�1), calculated from themaxima of the absorption and fluorescence spectra,are plotted against solvent parameter (Df) and areshown in Fig. 6 (Lippert plot). The solvent parameterDf basically [29] describes solvent polarity and polarisi-bility by the following equation:
Df ¼ ½ðe� 1=2eþ 1Þ� � ½ðg2 � 1=2g2 þ 1Þ�;
where e and g are the dielectric constant and refractiveindex of the medium, respectively. As seen in Fig. 6, alinear dependency of Stokes shifts (Dm in cm�1) with sol-vent parameter (Df) indicates high dipole moment of thered shifted emissive species.
300 350 400 4500.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
EtOH+Acid
EtOH
a
Inte
nsit
y (a
.u.)
Wavelength (nm)
350 400 450 500 5500
100
200
300
400
500
600
b
Inte
nsit
y (a
.u.)
Wavelength (nm)
Fig. 5. Effect of H2SO4 on the (a) absorption and (b) emission spectra(arrow indicates increase in acid concentration) of t-MDMAC.
0.0 0.1 0.2 0.3
3000
4000
5000
60007
6
54
32
1
Stok
es s
hift
(∆ν
cm-1)
Solvent parameter (∆f )
Fig. 6. Stokes shift vs. solvent parameter: (1) cyclohexane; (2) CCl4;(3) hexane; (4) dioxane; (5) CHCl3; (6) THF and (7) ACN solvent.
A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83 79
The dipole moments of the FC states (lFC) and CTstates (lCT) of MMAPA can be calculated by plotingmabs vs. Df and mflu vs. Df, respectively [29], using the fol-lowing equations:
mabs ¼ �½ð1=4peÞð2=hca3Þ�lgðlFC � lgÞDf þ constant;
ð1Þmabs ¼ �½ð1=4peÞð2=hca3Þ�lgðlFC � lgÞ;mflu ¼ �½ð1=4peÞð2=hca3Þ�lCTðlCT � lgÞDf þ constant;
ð2Þmflu ¼ �½ð1=4peÞð2=hca3Þ�lCTðlCT � lgÞ;
where a and lg are the Onsager radius and ground statedipole moment calculated at HF level with 6-31++G(d,p) basis set. The calculated value for a andlg are 4.5 A and 5.1 Debye, respectively. The dipole mo-ment of FC state (lFC) and the CT excited state (lCT) canbe calculated from the slope obtained from mabs vs. Dfand mflu vs. Df plot, respectively. The calculated valuesof lg, lFC, lCT of t-MADMC are shown in Table 3. Itis seen that the dipole moment value of CT state is muchlarger than the dipole moment of FC and ground states.This means that full CT occurs sequentially via relaxa-tion of the FC state to the CT state and hence, polaritydependence Stokes. shifted emission is observed.
It is found that the Stokes shift of emission band inwater (e = 79) does not correlate well with the Lippertplot. Fig. 7 shows the plot of hydrogen bondingparameter (a) with fluorescence maxima of the redshifted band in different hydrogen bonding solvents(Table 2). A linear correlation between fluorescencemaxima and hydrogen bonding parameter supportsthat polar protic solvents form intermolecular hydro-gen bonded clusters with the molecules and the redshifted emission is attributed to hydrogen-bondedspecies [30,31]. To confirm this idea we have measuredtemperature dependence emission spectra of t-MDAMCin acetonitrile and water solvents. As shown in Fig. 8, theemission spectra of t-MDAMC in acetonitrile shows adecrease in intensity of the CT band and a slow increasein LE band with increase of temperature. The decrease inthe intensity of the CT band with the increase of temper-ature may be due to opening up of some temperatureactivated non-radiative processes. But we cannot sayabout the increase in intensity of the LE state with in-crease of temperature. We are not sure whether it is sim-ply the manifestation of the changes in the solventpolarity as a function of temperature. Very similarly,with increase of temperature, the decrease in inten-sity of red shifted emission band in water could be dueto breaking of weak hydrogen-bonded intermolecularclusters (Fig. 9).
The most notable observation is the fluorescencespectrum of t-MDAMC at 77 K in methylcyclohexanehexane and ethanol glass (Fig. 10). The nature of theemission spectrum of t-MDAMC in methyl cyclohexaneglass is nearly similar to its room temperature spectrum.But the emission spectrum in the ethanol glass is diffe-rent from its room temperature spectrum. At room

0.7 0.8 0.9 1.021000
21500
22000
225004
3
2
1
ν (c
m-1
)
Hydrogen bonding parameter (α )
Fig. 7. Plot of hydrogen bonding parameter (a) vs. fluorescence bandposition of t-MDMAC: (1) water; (2) methanol; (3) ethanol and (4)isopropanol.
350 400 450 500 5500
10
20
30
40
50
1
4
1
4Inte
nsit
y (a
.u.)
Wavelength (nm)
Fig. 8. Temperature dependent fluorescence emission spectra oft-MDMAC in ACN solvent at (1) 283 K; (2) 293 K; (3) 303 K and(4) 313 K.
400 450 500 5500
1
2
3
4
5
6
7
8
4
1
Flu
ores
cenc
e In
tens
ity
(a.u
.)
Wavelength (nm)
Fig. 9. Temperature dependent fluorescence emission spectra oft-MDMAC in water at: (1) 283 K; (2)293 K; (3)303 K and (4)313 K.
400 450 500 5500
200
400
600
Inte
nsit
y(a.
u.)
Wavelength(nm)
Fig. 10. Fluorescence spectra of t-MDMAC in (-d-) methylcyclohex-ane and (-h-) EtOH glass at 77 K and in (–j–) EtOH at roomtemperature.
Table 3Calculated nitrogen out-of-plane dihedral angle and dipole moments of t-MDMAC at different level of calculations
Method/basis set Dihedral angle atthe nitrogen centre(\C8–N7–C1–C6)
Calculated groundstate dipole moment(Debye)
FC state dipolemoment (Debye)
CT state dipolemoment (Debye)
HF/6-31++G(d,p) 13.209 5.1 6.3 12.4HF/6-311++G(d,p) 13.161 4.5 5.9 12.1B3LYP/6-311++G(d,p) 7.048 5.6 6.7 12.7B3LYP/6-31++G(d,p) 6.901 5.5 6.6 12.6
80 A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83
temperature, t-MDAMC shows two emission bands inethanol solvent. But in ethanol glass, t-MDAMC showsonly LE state emission band as observed in non-polarmethyl cyclohexane solvent. The properties of the sol-vent such as polarity, polarizability and viscosity canchange with the decrease of temperature. High viscosityat low temperature glass may also hindered twisting mo-tion for the relaxation path from FC to CT state. This
observation may support that twisting is hindered inthe rigid glass matrix and hence, photoinduced ICT pro-cess did not occur. In addition it is found that the quan-tum yields for the LE state increases in the lowtemperature glass matrix as charge transfer processand/or any other deactivated channel are stopped.
To interpret the excited state CT character of t-MADMC, structural calculations have been carried

Table 4Ground state optimized geometrical parameters of t-MDMAC at DFTlevel with B3LYP functional and 6-311++G(d,p) basis set
Bond Calculatedvalues (A)
Bond angle/aihedral angle
Calculatedvalues (�)
RC1–C2 1.421 \C9–N7–C8 118.738RC2–C3 1.386 \C9–N7–C1 199.965RC3–C4 1.410 \C3–C4–C10 123.946RC1–N7 1.381 \O13–C12–O14 122.506RN7–C9 1.456 \C8–N7–C1–C6 7.048RC4–C10 1.455 \C3–C4–C10–C11 �0.299RC10–C11 1.352 \C11–C12–O13-O14 �179.991RC11–C12 1.470RC12–O13 1.221RC12–O14 1.363
A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83 81
out for the ground state at ab initio (HF) and DFT lev-els of theory. Calculated parameters are summarized inTable 4. It is seen that in all levels of calculation, in theglobal minimum structure, the nitrogen lone pair is outof plane of the benzene ring plane i.e. not parallel to the
0 20 40 60 80 100
- 671. 68
- 671. 56
- 671. 54
- 671. 52 b S2
S1
S03.64 eV
3.23 eV
Ene
rgy
(Har
tree
)
Twist Angle (Degree )
0 20 40 60 80 100 120-671.69
-671.68
-671.67
-671.58
-671.55
-671.52a
S2
S1
S0
2.77 eV
3.6 eV
Ene
rgy
(Har
tree
)
Twist angle (Degree)
Fig. 11. Potential energy surface of t-MDMAC for the ground andfirst two excited states along the twisting coordinate at the (a) donorside (–NMe2) and (b) acceptor side (CH@CHCOOMe) using TDDFTmethod with B3LYP functional and 6-311++G(d,p) basis set.
benzene p-orbitals. All calculations show a pyramidalstructure at the nitrogen center in the ground state. Po-tential energy surfaces for the ground and two other ex-cited states along the twist coordinate at TDDFT levelare shown in Fig. 11. As seen in Fig. 11, the energyalong the twist coordinate increases for the groundand second excited states. However, stabilization alongthe twist coordinate is observed for the first excited state(S1). It is also seen that the twisting along the donor siteshows more stabilized CT state than along the acceptorcoordinate and energetically twisting along donor isfavorable than acceptor site. Similar calculation byPCM model (Fig. 12) shows a stabilization of the CTstate in acetonitrile solvent. Calculated PES in acetoni-trile also shows a solvent dependency of the red shiftedemission band. Fig. 13 shows the picture of HOMO andLUMO molecular orbital for the global minimum andthe CT states. As seen in Fig. 13, the shape of the nitro-gen lone pair orbital at the 90� configuration is localizedcompared to that of the delocalized minimum energystate. This indicates that upon photo excitation it maybe possible to transfer charge from the nitrogen lonepair to the acceptor group at the 90� configuration inthe S1 state, but not at the ground state.
0 20 40 60 80 100
-671.70
-671.60
-671.58
-671.56
-671.54
-671.52
3.35 eV
2.97 eV
b S2
S1
S0
Ene
rgy
(Har
tree
)
Twist angle (Degree)
0 20 40 60 80 100 120
-671.706
-671.685
-671.580
-671.545a S
2
S1
S0
2.58 eV
3.36 eV
Ene
rgy
(Har
tree
)
Twist Angle (Degree)
Fig. 12. Potential energy surface of t-MDMAC for the ground andfirst two excited states along the twisting coordinate at the (a) donorside (–NMe2) and (b) acceptor side (CH@CHCOOMe) in ACN solventusing TDDFT method with B3LYP functional and 6-311++G(d,p)basis set.

Fig. 13. Molecular orbital representation of t-MDMAC: (a) HOMO of ground state; (b) LUMO of ground state; (c) HOMO of 90� twistedconformer and (d) LUMO of 90� twisted conformer.
82 A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83
4. Summary
We have synthesized methyl p-(dimethylamino) cin-namate and its photoinduced intramolecular chargetransfer reaction in different solvents has been studiedby steady state absorption and emission spectroscopy.The effect of pH, temperature variation, solvent pola-rity, and emission measurement at 77 K in glass matrixestablish well the existence photoinduced intramolecu-lar charge transfer reaction mechanism by twistedintramolecular charge transfer model. In addition,the ground state structural calculations at ab initoand DFT levels show that the nitrogen lone pair isout-of-plane of the benezene p-orbitals. Calculateddipole moment for the CT states is found to be 12.6Debye and is responsible for the observed red shiftedsolvent dependence ICT emission. A PES calculationalong the twist coordinate predicts S1 state stabiliza-tion both in the gas and condensed (acetonitrile sol-vent) phase. Molecular orbital pictures indicate thatthe lone pair electron is localized at the nitrogen centerat the 90� twisted configurations and is responsible forthe photoinduced charge transfer process in the excitedS1 state.
Acknowledgements
This work has been supported by a grant form DST,India (Grant no. SR/S1/PC-01/2003). NG would like toacknowledge Prof. Mihir Chowdhury of IACS, Kolk-ata, for fruitful discussion. The authors would like tothank Dr. S.N. Bhattacharyya, Dr. S.S.Z. Adnan, Dr.S. Chakraborty and Dr. Swapan Chakrabarti for theirconstant encouragement throughout this work. The
authors also thank Professor Sanjib Ghosh of Presi-dency College, Chemistry Department for making thelow temperature measurements available for them. Theauthors express their sincere thanks to the reviewersfor constructive suggestions.
References
[1] E. Lippert, W. Luder, F. Moll, W. Nagele, H. Boos, H. Prigge, I.Seibold-Blankenstein, Angew. Chem. 73 (1961) 695.
[2] K. Rotkiewicz, K.H. Grellmann, Z.R. Grabowski, Chem. Phys.Lett. 19 (1973) 315.
[3] K.A. Zachariasse, Th. Von der Haar, A. Hebecker, U. Leinhos,W. Kuhnle, Pure Appl. Chem. 65 (1993) 1765.
[4] O. Krauss, B. Brutschy, Chem. Phys. Lett. 350 (2001) 427.[5] A.L. Sobolewski, W. Sudholt, W. Domcke, J. Phys. Chem. A 102
(1998) 2716.[6] H. Saigusa, E. Iwase, M. Nishimura, J. Phys. Chem. A 107 (2003)
3759.[7] D. Rappoport, F. Furche, J. Am. Chem. Soc. 126 (2004) 1277.[8] A.B.J. Parusel, W. Rettig, W. Sudholt, J. Phys. Chem. A 106
(2002) 804.[9] A. Kohn, C. Hattig, J. Am. Chem. Soc. 12 (2004) 67399.
[10] A.S. Sobolewski, W. Domcke, Chem. Phys. Lett. 250 (1996)428.
[11] K.A. Zachariasse, Chem. Phys. Lett. 320 (2000) 8.[12] A.D. Gorse, M. Pesquer, J. Phys. Chem. 99 (1995) 4039.[13] D. Gregory, D.P. Scholes, I.R. Gould, Chem. Phys. Lett. 266
(1997) 521.[14] H. Ishikawa, M. Sugiyama, Y. Shimanuki, Y. Tajima, W. Setaka,
M. Kira, N. Mikami, J. Phys. Chem. A 107 (2003) 10781.[15] P.R. Bangal, S. Chakravorti, J. Photochem. Photobio. A: Chem-
istry 139 (2001) 5.[16] S. Panja, P. Chowdhury, S. Chakravorti, Chem. Phys. Lett. 368
(2003) 654, and references therein.[17] R. Das, D. Guha, S. Mirta, S. Kar, S. Lahiri, S. Mukherjee,
J. Phys. Chem. A 101 (1997) 4042.[18] P.R. Bangal, S. Chakravorti, J. Photochem. Photobio. A: Chem-
istry 116 (1998) 191.

A. Chakraborty et al. / Chemical Physics 320 (2006) 75–83 83
[19] Gaussian 03, Revision B.03, M.J. Frisch et al., Gaussian, Inc.,Pittsburgh PA, 2003.
[20] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[21] M.J. Frisch, J.A. Pople, J.S. Binkley, J. Chem. Phys. 80 (1984)
3265.[22] T. Clark, J. Chandrasekhar, G.W. Spitznagel, P.V.R. Schleyer, J.
Comp. Chem. 4 (1983) 294.[23] X.U. Duan, X.Y. Li, R.X. He, X.M. Cheng, J. Chem. Phys. 122
(2005) 084314.[24] C.J. Jodicke, H.P. Luthi, J. Am. Chem. Soc. 125 (2003) 252.
[25] C. Jamorski, J.B. Foresman, C. Thilgen, H.P. Luthi, J. Chem.Phys. 116 (2002) 8761.
[26] S. Zilberg, Y. Hass, J. Phys. Chem. A 106 (2002) 1.[27] M. Cossi, V. Barone, J. Chem. Phys. 115 (2001) 4708.[28] R. Cammi, B. Mennucci, J. Tomasi, J. Phys. Chem. A 104 (2000)
5631.[29] F.D. Lewis, W. Weigel, J. Phys. Chem. A 104 (2000) 8146.[30] R.W. Taft, M.J. Kamlet, J. Am. Chem. Soc. 98 (1976)
2886.[31] A.C. Testa, J. Lumin. 50 (1991) 243.

















![Bis[4-(dimethylamino)pyridinium] octaaquachloridolanthanum ...journals.iucr.org/e/issues/2012/11/00/su2504/su2504.pdfBis[4-(dimethylamino)pyridinium] octaaquachloridolanthanum(III)](https://img.pdfslide.us/doc/110x75/5e0610443af6f93e3057972f/bis4-dimethylaminopyridinium-octaaquachloridolanthanum-4-dimethylaminopyridinium.jpg)










