Embed Size (px)
Citation preview

Polyhedron Vol. IO, No. 16. pp. ‘939-1949, ‘99’ 0277-5387/91 s3.O!z!+.OO
Printed in Great Britain Perganlon Press plc
PHOSPHOLE COMPLEXES OF GOLD(III) HALIDES : SYNTHESIS, STRUCTURE, ELECTROCHEMISTRY AND
LIGAND REDISTRIBUTION REACTIONS
SAEED ATTAR and JOHN H. NELSON*
Department of Chemistry, University of Nevada, Reno, NV 89557, U.S.A.
and
WILLIAM H. BEARDEN
JEOL NMR Application Laboratory, Peabody, MA 91960, U.S.A.
and
NATHANIEL W. ALCOCK
Department of Chemistry, University of Warwick, Coventry CV4 7AL, U.K.
and
LJILJANA SOLUJIC’ and EMIL B. MILOSAVLJEVIC’
Institute of Chemistry, Faculty of Sciences, University of Belgrade, 11001 Belgrade, Yugoslavia
(Received 26 October 1990 ; accepted 3 May 1991)
Abstract-The complexes LAuCl, [L = I-phenyl-3,4-dimethylphosphole(RMPP), l- phenyldibenzophosphole(DBP) and triphenylphosphine], LAuBr, (L = DBP, Ph3P) and LAuClBr, (L = DBP, Ph3P) were prepared and characterized by physical properties, cyclic voltammetry, far IR, 3’P{‘H} and CP/MAS 3’P{ ‘H) NMR spectroscopy. Ligand redistribution reactions of the types : LAuCl+ L’AuBr G= L’ AuCl -I- LAuBr ; LAuCl 3 + LAuBr 3~LAuC1,Br3_.; LAuC13+L’AuBr 3~LAuC1,Br3_,+L’AuCl,Br3_,,; and LAu Cl + L’AuBr 3 e LAuBr + L’AuCl f L’AuBr -I- LAuCl,Br,_, + L’AuCl,Br,_, all occur rapidly and have been studied by 3’P{ ‘H} NMR spectroscopy. These reactions all appear to proceed by way of halide-bridged intermediates. The solid state identities of LAuClBr, were established by a combination of far IR and CP/MAS “P{ ‘H} NMR spectroscopies and single crystal X-ray crystallography for Ph3PAuBrzC1. The R,PAuBr,Cl complexes are substitutionally disordered, probably with two-thirds bromine and one-third chlorine in each site. Ph3PAuBr,C1 crystallized in the monoclinic space group P2,/c with a = 9.086(2), b = 11.410(2), c = 18.734(4) A, b = 95.01”(2), and Z = 4. The structure was solved by least-squares methods with RF = 0.074 for 1919 unique reflections with Z/o(Z) 2 3.0. The gold atom has nearly regular square-planar coordination with Au-P = 2.314(6) A” and Au-X = 2.399(3), 2.413(3) and 2.415(4) A”.
Solid-state nuclear magnetic resonance spec- the utility of CP/MAS ’ 'P( ‘H} NMR spectroscopy troscopy is attracting increasing interest in inor- for the solid-state characterization of phosphines,2 ganic chemistry. ’ We and others have investigated phosphine-oxides, -sulphides and -selenides;3 man-
ganese, molybdenum and tungsten ;4 iron, ruth- * Author to whom correspondence should be addressed. enium and osmium;’ rhodium,6*7 nickel,* palla-
1939

1940 S. ATTAR et al.
dium, ‘-’ ’ platinum,p~‘0~‘2~’ 3 copper, ‘b25 silver,26~27 gold,28,2p cadmium3 O and mercury3 ’ phosphine complexes. These studies have shown that the 3’P chemical shift is very environmentally dependent and provides information concerning the phos- phorus and metal site symmetries and the overall geometries of the complexes. The magnitudes of ‘J(CuP) and ‘J(AgP) allow determination of metal coordination numbers and metal-phosphorus bond distances. For the palladium, platinum, cadmium and mercury complexes, the magnitude of ‘J(PP) is a function of the PMP bond angles. Thus, for some complexes their { 3’P ‘H) CP/MAS spectra can provide details of solid-state structures similar to those available from the more difficult to obtain EXAFS32,33 measurements.
Gold(II1) phosphine complexes of the type R3 PAuX3 are prepared by oxidative addition of the appropriate halogen, XZ, to the gold(I) complexes R,PAuX. Previous workers3”37 have found that when the oxidative additions (reactions 1 and 2) were studied, highly crystalline homogeneous samples were obtained. In each case elemental analyses were consistent with the R3PAuXX’, stoichiometries. NMR studies of these substances in solution36*38 showed that halide redistribution” reactions occurred rapidly to produce all six of the possible compounds: R3PAuC13, cis- and trans- R,PAuCl,Br, cis- and trans-R,PAuClBr, and R,PAuBr,. These authors discussed the probable solid state identities suggesting that a mixture of R,PAuCl, and R3PAuBr3,37 pure R3PA~XX234’35 or a mixture of all six species36 was present in the solid state. They also discussed the mechanism36,38 of the halide redistribution. We have conducted a thorough study of the complexes of DMPP, DBP (see Struct. 1) and Ph3P by solution 3’P{‘H) and CP/MAS 3’P{‘H} NMR, electrochemistry, far IR spectroscopy and X-ray crystallography to obtain a further insight into the solid state identities and ligand redistribution reactions.
R,PAuCl+ Br, + R,PAuBr,Cl (1)
R,PAuBr + Cl, ---+ R,PAuBrCl, (2)
m, I
EXPERIMENTAL
(a) Reagents andphysical measurements
All chemicals were reagent grade and were used as received or synthesized as described below. All
solvents, when necessary, were dried by standard procedures and stored over Linde 4 8, molecular sieves. All reactions involving phosphines or phos- pholes were conducted under a nitrogen atmos- phere. R3PA~X,2p,34 1 -phenyl-3,4-dimethylphos- phole4’(DMPP) and I-phenyldibenzophosphole4’ (DBP) were prepared by literature methods.
Elemental analyses were performed by Galbraith Laboratories, Knoxville, TN. Melting points were determined on a Meltemp apparatus and are uncor- rected. IR spectra were recorded on a Perkin-Elmer 1800 FT-IR instrument as polyethylene pellets. The solution 3’P{ ‘H) NMR spectra were recorded at 40.26 MHz on a JEOL FX-100 spectrometer in the FT mode. Typical acquisition conditions were 1 and 5 kHz spectral windows, acquisition times of 4 and 0.8 s, recycle time of 5 s ; typically 5 to 10 k tran- sients were collected with proton noise decoupling and 2 Hz line broadening was applied. A JEOL NM 5471 controller was used for temperature control and temperatures were measured with a calibrated platinum resistance thermometer. Chemical shifts were referenced to an external sample of PPh3 in CDC13 (6 = -6 ppm) and converted to an 85% H3P04 reference with a positive value being down- field of the respective reference. Cross-polarization magic-angle-spinning (CP/MAS) 3’P{ ‘H) NMR spectra were obtained on a JEOL GX-270 wide bore spectrometer operating at 6.43 T (3’P at 109.25 MHz) using a 40 kHz sweep width, recycle delay time of 6 s, and a proton-decoupling field of 10 G. No line broadening was applied. Between 200 and 300 mg of the compound was spun at 4 kHz in Delrin or Kel-F rotors. All the CP/MAS chemical shifts were referenced to an external sample of Ph,P (6 = -6.0 ppm). The uncertainties in chemical shifts are estimated to be f0.5 ppm. Cyclic vol- tammetry was performed as previously described.42
(b) Syntheses LAuCl,
The three LAuCl, (L = DBP, Ph3P and DMPP) complexes were prepared by the same procedure. The following is exemplary. Chlorine gas was slowly bubbled through a CHC13 (20 cm’) solution con- taining 0.25 g (0.51 mmol) DBPAuCl while the solution was magnetically stirred. The originally colourless solution turned bright yellow within a few seconds. This yellow solution was filtered, the solution volume was reduced to a few cm3 by rotary evaporation, n-hexane was slowly added and the mixture was cooled to 0°C. The precipitate was isolated by filtration, washed with anhydrous diethyl ether, and vacuum dried to yield 0.24 g (82.8%) of orange crystals, m.p. 119-121°C.

Phosphole complexes of gold(III) halides 1941
Found: C, 38.5; H, 2.5. Calc. for ClgH13AuC13P: theses, and were treated as Br, which gave satis- C, 38.4; H, 2.3. Ph3PAuC13,37 yellow plates (58%), factory refinement. Substitution of Br by Cl cannot m.p. 179-181°C. DMPP AuCl,, yellow powder be excluded, and the relatively high thermal par- (98%) m.p. 121-122°C. All three LAuCl, ameters of the Br atoms could be consistent with (L = DBP, Ph3P, DMPP) complexes are photo- this [Ueq of Br (trans to Br) 0.0727 (10) (mean), of chemically and thermally unstable, decomposing Br (trans to P) 0.0846 (15)]. This possibility is not even in the absence of light to black solids. The testable by direct refinement because of the cor- DMPP complex decomposes in 2 weeks and the relation of thermal parameters and occupancy ; ran- Ph,P complex decomposes in approx. 5 weeks. dom substitution would be equivalent to 0.83 Br
LAuBr,. The LAuBr, (L = DBP, Ph3P) com- at each site. Anisotropic temperature factors plexes were prepared in the same manner. The were used for all non-hydrogen atoms. Hydrogen following is exemplary. To a solution containing atoms were given fixed isotropic temperature fac- 0.17 g (0.31 mmol) DBPAuBr in 20 cm3 CHC13 was tors, u = 0.07 A’ and were inserted at calculated added 1 .O cm3 of 0.34 M Br2 in CHCl,. The resulting positions and not refined. Final refinement was dark red solution was magnetically stirred at am- by cascaded least-squares methods [minimizing bient temperature for 3 h. The solution was then &(Fo-Fe)*]. The largest positive and negative filtered, the filtrate reduced in volume to a few cm3 peaks on final difference Fourier syntheses were by rotary evaporation, anhydrous diethyl ether was of height (e A- 3, + 2 and -4 in the vicinity of slowly added and the mixture was cooled to 0°C. the Au atom. A weighting scheme of the form The precipitate was isolated by filtration, washed 1/(a2(J’)+gF2) with g = 0.0026 was used and with anhydrous diethyl ether, and vacuum dried to shown to be satisfactory by a weight analysis. Three yield 0.15 g (68.2%) of burgundy-red crystals, m.p. standard reflections monitored every 200 reflections 121-123°C. Found: C, 30.95; H, 2.17. Calc. for showed no regular changes during data collection. ClsH,,AuBr3P: C, 31.0; H, 1.9. Ph3PAuBr3,37 Final RF = 0.074 and RWF = 0.078. This rather high brick-red plates (92%) m.p. 149-150°C. Attempted final figure is likely to result principally from preparation of (DMPP)AuBr, by the same pro- approximations in measurement of the platy cedure resulted in the formation of a black oil. The crystal, leading to an imprecise absorption correc- 3’P{‘H} NMR spectrum of this oil suggested the tion, and the weakly diffracting crystals. Computing addition of Br, to the diene system of DMPP.40*43 was with the SHEXTL system44 on a Data General
LAuClBr,. The LAuClBr, (L = DBP, Ph,P) DG30 computer. Scattering factors were taken complexes were prepared in the same manner as from ref. 45. ClsHl =,AuBr*Cl, M = 654.33, mono- for the LAuBr3 complexes except that the starting clinic, a = 9.086(2), b = 11.410(2), c = 18.734(4) A, material was LAuCl. DBPAuClBr, (84.2%), bur- fl = 95.01(2)“, U = 1934.8 A3, space group P2,/c, gundy-red crystals, m.p. 128-130°C. Found: C, Z = 4, D, = 2.25 g cmp3, burgundy-red plates, 32.8; H, 2.1. Calc. for ClgH13AuBr2ClP: C, 33.1 ; p(Mo-K,) = 138.1 cm-‘, 1= 0.71069 A, F(OOO) = H, 2.0%. Ph3PAuC1Br, (52.1%) burgundy-red 1288. crystals, m.p. 162-164°C. Found : C, 32.7 ; H, 2.5. Calc. for C,*H, SAuBr,C1 : C, 33.0, H, 2.3%.
RESULTS AND DISCUSSION
(c) Crystal structure analyses (1) Oxidative additions to R3PAuX complexes
A 0.04 x 0.10 x 0.40 mm burgundy-red lath of Two-coordinate 1Celectron d1 O LAuX com- Ph,PAuBr$l was selected from among the crystals plexes may be expected to readily undergo oxidative formed by the reaction of Ph,PAuCI with Br2 in addition reactions46 to form four-coordinate 16- CHC1,/Et20 in stoichiometric quantities. They electron d* LAuX, complexes. Neither DBPAuCl were found to be only weakly diffracting. Intensity nor Ph,PAuCl react with CH,I, BrCN, ICN or data were taken with a Nicolet P2, diffractometer ClCH2CN. In each case the R,PAuCl complex was in the 8-28 mode and were corrected for Lorentz, quantitatively recovered. However, the colourless polarization and absorption effects, the last by DBPAuCl, Ph,PAuCl and DMPPAuCl complexes the Gaussian method. Systematic absences (h01, each reacted with Cl* in CHC13 at ambient tem- 1 # 2n ; OkO, k # 2n) indicated the P2,lc space perature to give the corresponding yellow LAu group. The heavy atoms were located by Patterson Cl3 complexes. All three complexes are photo- techniques, and the light atoms were then found on chemically and thermally unstable, eventually successive Fourier syntheses. The three heavy Au decomposing into black solids. The relative rates of ligand atoms were of equal height in Fourier syn- decomposition are : DMPPAuC13 > DBPAuC13 >

1942 S. ATTAR et al.
Ph3PAuCI,, which is the order of decreasing ease of oxidation of the respective phosphorus donors. The colourless DBPAuBr and Ph,PAuBr com- plexes reacted with Br, in CHC13 at ambient tem- perature to give the photochemically and thermally stable burgundy-red DBPAuBr, and Ph,PAuBr, complexes. For DMPPAuBr, Br2 in CHC13 not only oxidized gold(I) to gold(II1) but also bro- minated the carbon-carbon double bonds43 of DMPP leading to a complex mixture of 20 phos- phorus containing species as a black oil. The colour- less DBPAuI and Ph,AuI when reacted with I2 in CHCl, at ambient temperature formed black oils that gave single “P ‘H) NMR chemical shifts in {
CDC13 at 6 = 31.8 ppm (DBP) and 38.4 ppm (Ph3P), which are identicalz9 to those of DBPAuI and Ph,PAuI, respectively. This is consistent with the observations that gold(II1) complexes are reduced to gold(I) complexes by iodide.47
Both DBPAuCl and Ph,PAuCl reacted with Br2
in CHCl, at ambient temperature to form bur- gundy-red homogeneous appearing crystals of the corresponding LAuClBr, complexes, whose stoi- chiometry was evidenced by elemental analyses and these substances have narrow melting point ranges. However, DBPAuI and Ph,PAuI reacted with Br, to form DBPAuBr, and Ph3PAuBr3, respectively, and DBPAuBr and Ph,PAuBr reacted with Cl, to form DBPAuCl, and Ph3PAuC13, respectively.
(2) Halide redistribution reactions
Although the elemental analyses and melting point data for the LAuClBr, complexes would indi- cate that their solid state identity is as formulated, their 3’P{ ‘H} NMR spectra in CDC13 solution at 30°C [Fig. l(a)] show the presence of more than one species in solution. For both complexes six phosphorus-containing species are present in solu- tion. These species result from halide redistribu- tion39 reactions of the LAuClBr* complexes. The 3’P{ ‘H} NMR resonances are assigned as fol- lows : DBPAuCl, (27.9 ppm), cis-DBPAuCl,Br (24.2 ppm), trans-DBPAuCl,Br (22.3 ppm), trans- DBPAuBr2Cl (19.9 ppm), cis-DBPAuBr,Cl (19.6 ppm) and DBPAuBr3 (16.4 ppm). These assign- ments are based upon the chemical shifts of solu- tions of pure DBPAuCl, (28.3 ppm) and DBPAuBr, (16.8 ppm) in CDC13 at 30°C. We reason that the greater the tram influence4’-SO of the halide, the weaker the tram Au-P bond and the more upfield the 3’P chemical shift. Thus, we expect the “P chemical shift of cis-DBPAuCl,Br to be upfield of that of DBPAuCl, but downfield of that of trans-DBPAuCl,Br. Also, if the halide re-
(a)
28 26 24 22 20 16
pm
Fig. 1. 40.26 MHz “P{ ‘H} NMR spectra in CDCl, at 30°C for (a) DBPAuClBr, and (b) an equimolar mixture of DBPAuCl, and DBPAuBr,. Assignments as follows : DBPAuCl, (27.9 ppm), cis-DBPAuCl,Br (24.2 ppm), trans-DBPAuCl,Br (22.3 ppm), trans-DBPAuBr,Cl (19.9 ppm), cis-DBPAuBr,Cl (19.6 ppm), DBPAuBr,
(16.4 ppm).
distribution reaction is nearly statistical as is often found for such reactions,39 then the DBPAuBr*Cl complexes should be more abundant than the DBPAuBrCl, complexes and their corresponding 3 ‘P resonances should be more intense. The 3’P{ ‘H} resonances for a CDC13 solution of Ph,PAuBr&l at 30°C were similarly assigned: Ph3PAuC13 (42.9 ppm), cis-Ph,PAuCl,Br (38.3 ppm), trans-Ph3PAuClZBr (38.1 ppm), trans- Ph,PAuBrCl (34.8 ppm), cis-Ph,PAuBr,Cl (33.0 ppm) and Ph,PAuBr, (30.5 ppm).
Heaton and Kelsey3’ similarly observed by 3’P NMR spectroscopy that reaction of Cl2 with Et,PAuBr in CHC13 at 25°C resulted in the rapid formation of all six Et,PAuCl,Br,_, species but their assignments as to the relative chemical shifts of Et3PAuClZBr and Et,PAuClBr, differ from ours. They based their assignments on the systematic trends observed upon cis-substitution of chloride by bromide in mixed chloride/bromide platinum(IV) complexes. We believe that our assignments of the relative chemical shifts are more internally con- sistent than theirs and they are also consistent with the following observation. Figure l(b) shows the

Phosphole complexes of gold(III) halides 1943
3 ‘P{ ‘H} NMR spectrum of a solution prepared from equimolar amounts of DBPAuCl, and DBPAuBr, in CDC13 immediately after mixing at 30°C. The same six species are present in this solu- tion indicating that the halide redistribution reac- tion is rapid. For a solution of DBPAuClBr, the Br/Cl ratio is 2 : 1 but in a solution containing equi- molar amounts of DBPAuCl, and DBPAuBr, the Br/Cl ratio is 1 : 1. Hence, the relative amounts of DBPAuCl,Br to DBPAuClBr, species in the latter solution should increase relative to those in the former. The ratio of the integrated intensities of the resonances ascribed’ to DBPAuCl,Br complexes to those ascribed to the DBPAu ClBr4 complexes is 0.5 in Fig. l(a) and 1.0 in Fig. l(b), consistent with the relative molar ratio of the halides in the two solutions.
(3) The mechanisms of halide redistribution
The halide redistributions of LAuBr,Cl com- plexes could proceed by two alternative mech- anisms : (1) catalysis by LAuC~~~ (reactions 3-6) ; or (2) simple halide redistribution (reaction 7).
LAuCl+ Br2 - LAuClBr 2 (3)
LAuClBr* + LAuCl- LAuCl,Br + LAuBr (4)
LAuBr + Br2 ------+ LAuBr 3 (9
LAuCl + LAuCl, Br - LAuCl 3 + LAuBr (6)
2LAuBr$l _
- LAuClsBr + LAuBrs
Table 1. Electrochemical properties of some gold(I) complexes
Complex Ep (V vs Fc+/Fc)
Au’ + Au”’ Au’ + Au0 Others
(DBP)AuCl 1.07 h 1.27 (DBP)AuBr 1.03 h 1.20 (Ph,P)AuCl 1.13 - 2.46 (Ph,P)AuBr 1.14 -2.52
’ Measured by cyclic voltammetry (v = 200 mV s- ‘) in CH,Cl, solution at a platinum working electrode, with 0.1 M tetrabutylammonium perchlorate as supporting electrolyte.
‘Very near the cathodic limit of the solvent/electrolyte/ electrode system used.
‘Oxidation. The current for this process is less than that for the Au’ + Au”’ oxidation. The two values (1.27 and 1.20 V) are close enough so that ligand (DBP) oxi- dation may be suggested. DPP (Differential Pulse Polar- ography) results agree very well with CVs.
than on the oxidation potential of LAuX. But none of the effects on the redox potentials are very large.
For the sequence of reactions (3-6) to occur as redox reactions, the potentials for reactions (4) and (6) must be greater than zero. Although the reduction potentials of LAuClBr, and LAuCl,Br are not available, we may approximate them by the appropriate weighted averages of the potentials for the LAuCl, and LAuB, complexes.53 Thus, the redox potential for Ph,PAuClBr, + 2e- -+ Ph,P
Table 2. Electrochemical properties of some gold(II1) complexes
Reactions (3-6), were first suggested by Puddephatt and Thompson36 on the basis of similarities with platinum(I1) catalysed halide redistributions of pla- tinum(IV) complexes. These reactions imply that the LAuCl,Br and LAuClBr, complexes are better oxidants than Br, or that they react with the gold(I) complexes faster than Br,. We have therefore exam- ined the electrochemical behaviour of the R,PAuX and R,PAuX, complexes most of which have not previously been reported. 5 ’
The redox potentials, determined by cyclic vol- tammetry in CHzClz solution are given in Tables 1 and 2. All cyclic voltammograms were quasi-revers- ible. 52 As can be seen, the phosphine generally has a smaller effect on the reduction potential of LAuX,
(7) Complex
Ep (V vs Fc+/Fc) Au”’ + Au’ Au’ + Au0 Others
(DBP)AuCl, (DBP)AuBr, (Ph,P)AuCI, (Ph,P)AuBr,
- 0.49 h 1.2Wd -0.37 h l.l7’,d -0.49 -2.55 e -0.31 -2.54 1.34’
p Measured by cyclic voltammetry (v = 200 mV s- ‘) in CH2Clz solution at a platinum working electrode, with 0.1 M tetrabutylammonium perchlorate as supporting electrolyte.
“Very near the cathodic limit of the solvent/electro- lyte/electrode system used.
‘Oxidation. The current for this process is less than that for other processes; see ref. 51.
“The closeness of these values may indicate DBP oxi- dation.
‘Near the anodic limit.

1944 S. ATTAR et al.
AuBr+Br- +Cll is estimated to be -0.37 V vs Fc/Fc+ or -0.03 V vs NHE as Fc/Fc+ is 0.40 vs NHE. The redox potentials4 for Br, + 2e- Z$ 2Br is 1.087 V. Thus, Ph,PAuClBr, is a much poorer oxidant than Brz. While this argument suggests that the gold(I) catalysed mechanism for halide scrambling is not likely to compete with oxidation by Br2 it does not necessarily establish that a redox reaction between a gold(I) and a gold (III) species cannot take place. To test the latter possi- bility, a series of ( 31P ‘H} NMR experiments were performed on CDC13 solutions containing equimolar mixtures of LAuX and L’AuX,. Since the halides on both complexes are the same, no halide redistribution takes place. The 3’P{‘H) NMR spectrum of one such mixture (DBPAuCl plus Ph,PAuCl,) is shown in Fig. 2. All four poss- ible species viz Ph,PAuCl (32.4 ppm), DBPAuCl (24.0 ppm), Ph3PAuC1, (42.8 ppm) and DBPAuCl, (27.9 ppm) are present in this solution. The same four species were found in the same relative amounts in a solution prepared from equimolar amounts of DBPAuCl, and Ph,PAuCI. Similarly, CDCl, solutions of DBPAuBr plus Ph,PAuBr, and DBPAuBr3 plus Ph3PAuBr each showed the pres- ence of Ph,PAuBr (34.5 ppm), Ph,PAuBr, (30.5 ppm), DBPAuBr (26.6 ppm) and DBPAuBr, (16.3 ppm) in the same relative amounts. These reactions could be the result of phosphine redistribution3’ or of redox reactions. To ascertain if redox reactions are possible, we can crudely estimate55 the electro- chemical potential for one of these reactions. Con- sider the following half reactions (8 and 9) :
Ph3PAuC13 + 2e- - Ph,PAuC1+2Cl-
E” = -0.49V (8)
DBPAuCl+ 2Cl- ---+ DBPAuCl, + 2e-
E0 = +0.49v. (9)
Thus, for reaction (10) E” g O.OV and from
Ph3PAuC13 +DBPAuCl I DBPAuCl 3
+Ph,AuCl, (10)
E” = -0.0295 log KS, we predict I& r 1 and the redox reaction (10) is thermodynamically feasible. Reaction (10) was found to occur rapidly and &s = 0.76, as measured by 31P( ‘H) NMR spectro- scopy at 30°C in CDC13.
This result poses the interesting question as to what would be the products of reactions (11) and (12)?
Ph,PAuCl+ PhsPAuBr3 L
DBPAuBr +Ph3PAuC13 L.
(11)
(12)
In both these cases, halide redistribution as well as a redox reaction are conceptually possible. The results of 3’P(‘H} NMR assessments of these reac- tions are shown in Figs 3 and 4. As can be seen, complete ligand scrambling occurs in both cases. The assignments given in these figures are based upon comparisons with the “P chemical shifts of the pure LAuX and LAuX 3 complexes and the pre- vious arguments for the LAuBrCl, and LAuBr$l complexes. The same equilibria were observed to occur for CDC13 solutions containing equimolar amounts of Ph,PAuBr plus Ph,PAuCl, and DBP AuCl plus DBPAuBr,.
Since the two possible pathways of halide redis- tribution (reactions 3-6 or 7) would lead to the same products, both pathways might be operating in the absence of added halogen. This postulation is sup- ported by the results of an additional series of 3 ‘P ‘H} NMR experiments on equimolar mixtures ( of LAuX and L’AuX3’. One such example was shown in Fig. 4 where complete ligand redis-
Fig. 2. 40.26 MHz “P{ ‘H} NMR spectrum of an equimolar mixture of DBPAuCl and Ph3PAuC13 in CDC13 at 30°C. Assignments as follows : Ph,PAuCl, (42.8 ppm), Ph,PAuCl(32.4 ppm), DBPAuCl3
(27.9 ppm), DBPAuCl(24.0 ppm).

Phosphole complexes of gold(II1) halides 1945
1....,....,....a
40 35 30
PPm
Fig. 3. 40.26 MHz 3’P{ ‘H} NMR spectrum of an equi- molar mixture of Ph,PAuCl and Ph,PAuBr, in CDC13 at 30°C. Assignments as follows : cis-Ph,PAuCl,Br (38.3 ppm), trans-Ph,PAuCl,Br (37.9 ppm), Ph,PAuBr (34.5 ppm), cis-Ph,PAuClBr, (32.9 ppm), Ph,PAuCl
(32.4 ppm), Ph,PAuBr, (30.4 ppm).
tribution is evident. Ph,PAuBr plus DBPAuCl,, PhrPAuCl plus DBPAuBr,, and DBPAuCl plus Ph,PAuBr, mixtures gave similar results. In each case 12-14 species with the same chemical shifts as given in Fig. 5 and with intensities varying in direct proportion to the varying ligand ratios were observed. In no case were the anticipated 16 reson- ances observed because of coincidentally equal chemical shifts for some of the species.
Furthermore, complete ligand redistribution was observed for a solution containing an equimolar
Fig. 4. 40.26 MHz 3’P{ ‘H) NMR spectrum of an equi- molar mixture of DBPAuBr and Ph,PAuCl, in CDCl, at 30°C. Assignments as follows : Ph3PAuCl, (42.7 ppm), cis-Ph3PAuClzBr (38.1 ppm), trans-Ph,PAuCl,Br (37.8 ppm), Ph,PAuBr (34.5 ppm), cis-Ph,PAuClBr* (32.8 ppm), Ph3PAuCl(32.4ppm), DBPAuCl, (27.8 ppm), cis-DBPAuCl,Br (24.1 ppm), DBPAuCl (23.9 ppm), trans-DBPAuCl,Br (22.1 ppm), trans-DBPAuClBr, (19.5 ppm), cis-DBPAuClBr, (19.5 ppm), DBPAuBr,
(16.2 ppm).
ratio of Ph,PAuCI, and DBPAuBr3 (Fig. 5). The same species were also found in the same relative amounts for an equimolar mixture of Ph,PAuBr, and DBPAuCI,.
Ligand redistribution also occurs among gold(I) complexes [reaction (13), Fig. 61. As is evident by the line widths and temperature dependence of this spectrum, the redistributions among gold(I) species are more rapid than those among gold(II1) species. Note also that the rate of halide redis-
n=o-3
1....m....1....,....,_...,..~., #....I 45 40 35 30 25 20 15
PPm
Fig. 5.40.26 MHz 3’P{ ‘H} NMR spectrum of an equimolar mixture of Ph,PAuCl, and DBPAuBr, in CDCl, at 30°C. Assignments as follows: Ph,PAuCIS (42.9 ppm), cis-Ph3PAuClZBr (38.3 ppm), truns-Ph,PAuCl,Br (38.0 ppm), truns-Ph,PAuClBr, (34.7 ppm), cis-Ph,PAuClBr, (32.9 ppm), Ph,PAuBr, (30.5 ppm), DBPAuCl, (27.9 ppm), cis-DBPAuCl,Br (24.3 ppm), trans-DBPAuCl,Br
(22.3 ppm), truns-DBPAuClBr, (19.9 ppm), cis-DBPAuClBr, (19.6 ppm), DBPAuBr, (16.5 ppm).

1946 S. ATTAR et al.
,...a’.‘1.,‘1-.‘*-* 36 34 32 30 28 26 24 22
wm
Fig. 6.40.26 MHz 3’P( ‘H} NMR spectra of an equimolar mixture of Ph,PAuCl and DBPAuBr in CDCI, at (a) 30°C and (b) - 13°C. Assignments as follows : Ph,P AuBr (34.3 ppm), Ph,PAuCI (32.2 ppm), DBPAuBr
(26.2 ppm), DBPAuCl (23.6 ppm).
tribution is faster for DBPAuX than for Ph,PAuX species, suggesting that DBP has a greater trans- effect than Ph?P. The same result was observed for an equimolar- mixture of Ph,PAuBr and
AuCl. DBP
Ph,PAuCl+ DBPAuBr # Ph,PAuBr
+ DBPAuCl
We believe that all of these equilibrium tribution reactions involve halide-bridged
(13)
redis- inter-
mediates, 39 similar to that shown in reaction (7). No ionic species such as [(Ph,P)(DBP)Au]+ or [(Ph,P)(DBP)AuX,]+ were ever observed in any of these solutions. Addition of Ph3P or DBP to solutions containing LAuX3 (L = PhjP, DBP ; X = Cl, Br) resulted in immediate redox reactions [see eq. (14)]. These results suggest that very little, if any, free Ph,P or DBP is present during the course of the ligand redistribution reactions. The kinetics and mechanism56 of the redistribution reactions of pentacoordinate complexes of the type [Au(N - N) (CN),X](N - N is a l,lO-phenanthroline ; X = Cl, Br) have been described. Anion redistribution of Ph,PAuCzCPh complexes have also been described. j7
Ph,P+DBPAuCl,- Ph3PC12+DBPAuCl (14)
(4) The solid state identity of LAuClBr,
The isolated products of the reactions of Ph,PAuCl and DBPAuCl with Br2 appear to be homogeneous crystalline substances with narrow melting point ranges and elemental analyses agree- ing with the LAuClBr, composition. However, their 3’P{ ‘H} NMR spectra in CDCl, at 30°C show the presence of all six LAuCl,Br,_, species. Mann and Purdie34 suggested that only trans-Et3PAuXX’2 is present in the solid state. Williamson and Baird3’ suggested that a mixture of cis- and trans-LAu ClBr2(L = Ph3P, Et3P) is present in the solid state. Puddephatt and Thompson36 suggested that all six LAuCl,Br,_, species are present and McAuliffe et al. 37 suggested that the solid state products are mix- tures of LAuCl, and LAuBr, in appropriate pro- portions. The simplest interpretation of the far IR spectroscopic data (Table 3) is that LAuBr, and LAuC13 are present in a 2 : 1 ratio. This interpre- tation rests on the fact that the two LAuClBr, (L = DBP, Ph,P) complexes show v(AuC1) and v(AuBr) vibrations at essentially the same fre- quencies as those of LAuC13 and LAuBr,. If the mixed halide species were present in the solid state the v(AuC1) and v(AuBr) vibrations for them would be expected to occur at different frequencies, as has been found for HgX,, HgXX’, HgX2’ systems.” The CP/MAS 3’P{ ‘H} NMR spectra (Fig. 7) of the two LAuClBr, complexes each show two major resonances in a 2 : 1 intensity ratio (L = Ph3P, 41 .O
* -10 -
wm
100 90 80 70 60 50 40 30 20 IO 0 -10 -20 -30
pm
Fig. 7. 109.25 MHz CP/MAS 3’P{‘H) NMR spectra of (a) DBPAuClBr, and (b) Ph,PAuClBr,. SSB are spin-
ning sidebands.

Phosphole complexes of gold(II1) halides
Table 3. Gold(III)-halogen stretching frequencies” for the LAuX, and LAuXX’, complexesh
Complex v(X -Au-X) (asymm.) v(Au-X) (truns to L)
(DBP)AuC13 366 318 (DBP)AuBr, 260 219 (BBP)AuClBr, Cl: 365 322
Br: 261 224 (Ph,P)AuCl, 366, (371)‘(361) 307, (31 l)d (300) (PhjP)AuBr, 261, (264)’ (258) 225, (222)d (215) (Ph,P)AuClBr, Cl : 366, (347)d 310, (313)d(301)
Br : 259, (261)d (255) 220, (216)d
a v(Au-X) in cm- ‘. ’ Recorded as polyethylene pellets. ‘Refers to the asymmetric (higher energy) as opposed to symmetric (lower
energy) stretch of the X-Au-X unit. dFrom ref. 35, recorded as Nujol mulls mounted between polyethylene
discs. ‘From ref. 37, as Nujol mulls.
1947
and 34.4 ppm ; L = DBP, 36.9 and 3 I. 1 ppm). These compare with the CP/MAS 3’P{ ‘H) NMR spectra of pure Ph,PAuCl, (46.9 ppm), Ph,PAuBr, (36.2 ppm), DBPAuCl, (42.2 ppm) and DBPAuBr3 (30.5 ppm). Each of these resonances have line widths (Av(l/2) -480 Hz or -4.4 ppm). We have pre- viously shown that the CP/MAS 3’P chemical shift is very environmentally dependent.9s’0*‘2,29 If Ph,PAuCl,Br,_, existed in the solid state with ran- domly disordered halides, we might expect to see three “P chemical shifts spanning a range of about 8 ppm. These three resonances would represent Ph,PAuBr, (29.6%), cis-Ph3PAuBr2C1 (29.6%) plus trans-Ph,PAuBr,Cl (14.8%) and cis-Ph3 AuCl,Br (14.8%). Given the large CP/MAS line widths together with their lower relative abundances resonances for transPh,PAuCl,Br (7.4%) and Ph,PAuCl, (3.7%) would probably not be observed (see Fig. 1 for comparison). If only truns-Ph,PAu Br,Cl was present, only one 3’P chemical shift would be observed. Finally, if the solid were a mix- ture of cis- and truns-Ph,PAuBr,Cl or a mixture of Ph,PAuCl, and Ph,PAuBr,, then two resonances should be observed. In the latter case their relative intensities would be 2: 1. The observation of three resonances (two major and a minor shoulder) in the CP/MAS 3’P NMR spectra (Fig. 7) suggests that these solids probably contain randomly disordered halides.
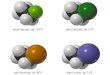
As further evidence, the X-ray crystal structure of Ph3PAuBr2C1 (Fig. 8) was obtained. No inhomo- geneity was observable in the crystalline sample. Microscopic observation shows that crystals of Ph,PAuBr,Cl are medium-red and those of
Ph,PAuBr, are deeper red and that they have different morphologies ; they show similar platy morphologies, though the first species gives almost rectangular plates, while crystals of the tribromide are generally diamond-shaped. For Ph,PAuBr,Cl the three halides were almost identical in electron density, implying approximately equal substitution of Br by Cl at each site. Bond distances and angles are given in Table 4. Ph3PAuBrzC1 is a nearly reg- ular square-planar complex. The two Au-X bonds tram to each other [2.399(3) and 2.413(3) A] are essentially the same length as the Au-X bond tram to Ph3P [2.415(4) A]. The corresponding distances
u- d Br(3)
Fig. 8. View of Ph,PAuBr,Cl showing the atom labelling scheme; 50% probability ellipsoids. Atoms labelled Br are considered to be substitutionally disordered with
approximately 213 Br and l/3 at each site.

1948 S. ATTAR et al.
Table 4. Selected structural parameters for [(Ph,P)AuBr,Cl]
Bond lengths (A)
Au-Br( 1) Au-Br(3)
P(l)-C(1) P(l)-C(31)
2.399(3) 2.413(3) 1.794(22) 1.823(26)
Au-Br(2) Au-P( 1) P( l)-C(21) Ph : C-C(ave)
2.415(4) 2.314(6) 1.825(22) 1.391(46)
Bond angles (“)
Br( I)-Au-Br(2) Br(2)-Au-Br(3) Br(2)-Au-P( 1) Au-P( I)-C( 11) c(11)-P(1)-c(21) C( 1 I)-P( l)-C(3 1) Ph : C-C-C(ave)
88.9(l) 91.1(l)
177.2(2) 112.7(7) 102.0(9) 111.1(11) 120.0(30)
Br( I)-Au-Br(3) Br( l)-Au-P( 1) Br(3)-Au-P( 1) Au-P( l)-C(21) Au-P( l)-C(3 1) C(21)-P(l)--C(31)
177.3(l) 92.6(2) 87.3(2)
116.0(7) 104.1(8) lll.l(ll)
with approximately 2/3 Br and l/3 Cl at each ‘Note : the atoms labelled Br are considered to be substitutionally disordered
I site.
in Ph3PAuC1359 [2.273(4), 2.282(4) and 2.347(4) A] 10. and Et3PAuBr360 [2.407(l), 2.416(l) and 2.468(l) A] show that in these complexeS the bond trans to 11. the phosphine is lengthened because of the high trans influence4&” of the phosphine. The fact that 12.
the Au-X bond truns to Ph3P in Ph,PAuBr,Cl is not lengthened to a similar extent suggests greater
l3 ’
substitution of Cl for Br in this site, which is l4 ’ consistent with its rather higher apparent thermal
parameter.
15. Acknowledgements-The financial support of the UNR Research Advisory Board and the donors of the Pet- 16. roleum Research Fund, administered by the American Chemical Socity is gratefully acknowledged. 17.
J. A. Rahn, D. J. O’Donnell, A. R. Palmer and J. H. Nelson, Znorg. Chem. 1989, 28, 2631. G. Bodenhausen, J. A. Deli, A. Clements and P. S. Pregosin, Inorg. Chim. Acta 1983,77, L17. J. A. Rahn, L. Baltusis and J. H. Nelson, Znorg. Chem. 1990,29,750. L. Bemi, H. C. Clark, J. A. Davies, C. A. Fyfe and R. E. Wasylishen, J. Am. Chem. Sot. 1982,104,438. G. A. Bowmaker, J. D. Cotton, P. C. Healy, J. D. Kildea, S. D. Silong, B. W. Skelton and A. H. White, Znorg. Chem. 1989,28, 1462. D. A. Edwards and R. Richards, Spectrochim. Acta
1978,34A, 167. G. A. Bowmaker and D. A. Rogers, J. Chem. Sot.,
Dalton Trans. 1984, 1249. G. A. Bowmaker, J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai and A. H. White, J.
Chem. Sot., Dalton Trans. 1987, 1089. G. A. Bowmaker, L. M. Englehardt, P. C. Healy, J. D. Kildea, R. I. Papasergio and A. H. White, Znorg. Chem. 1987, 26, 3533. E. M. Menger and W. S. Veeman, J. Magn. Reson.
1982,46, 257. P. F. Barron, J. C. Dyason, L. M. Engelhardt, P. C. Healy and A. H. White, Znorg. Chem. 1984,23,3766. J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai, V. A. Patrick and A. H. White, J.
Chem. Sot., Dalton Trans. 1985, 831. P. E. Barron, J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai, V. A. Patrick and A. H. White, J. Chem. Sot., Dalton Trans. 1987, 1099. P. F. Barron, J. C. Dyason, L. M. Engelhardt, P. C. Healy and A. H. White, Aust. J. Chem. 1985,38,261. S. Attar, G. A. Bowmaker, N. W. Alcock, J. S. Frye, W. H. Bearden and J. H. Nelson, Znorg. Chem. 199 1, 30.
NOTES AND REFERENCES 18.
1. 2.
3.
4.
5.
6.
7.
8.
9.
N. J. Clayden, Chem. Ser. 1988,28,211. G. H. Penner and R. E. Wasylishen, Can. J. Chem. 1989,67, 1909. J. B. Robert and L. Wiesenfeld, Molec. Phys. 1981, 44,319. E. Lindner, R. Fawzi, H. A. Mayer, K. Eichle and K. Pohmer, Znorg. Chem. 199 1,30, 1102. A. J. Catty, C. A. Fyfe, M. Lettinga, S. Johnson and L. H. Randall, Znorg. Chem. 1989,28,4120. J. W. Diesveld, E. M. Menger, H. T. Edzes and W. S. Veeman, J. Am. Chem. Sot. 1980,102,7935. A. Naito, D. L. Sastry and C. A. McDowell, Chem. Phys. Lett. 1985, 115, 19. R. Berm, R. Mynott, I. Topalovic and F. Scott, Organometallics 1989, 8, 2299. J. H. Nelson, J. A. Rahn and W. H. Bearden, Znorg. Chem. 1987,26,2192.
19.
20.
21.
22.
23.
24.

Phosphole complexes of gold(II1) halides
25. G. A. Bowmaker, A. Camus, P. C. Healy, B. W. Skelton and A. H. White, Znorg. Chem. 1989, 28, 3883.
26. P. F. Barron, J. C. Dyason, P. C. Healy, L. M. Engelhardt, B. W. Skelton and A. H. White, J. Chem. Sot., Dalton Trans. 1986, 1965.
27. S. Attar, N. W. Alcock, G. A. Bowmaker, J. S. Frye, W. H. Bearden and J. H. Nelson, Znorg. Chem. 1991, 30.
complexes see : R. G. Pearson and P. E. Figdore, J. Am. Chem. Sot. 1980, 102, 1541; A. Tanaki and J. K. Kochi, J. Chem. Sot., Dalton Trans. 1973, 2620 ; A. Johnson and R. J. Puddephatt, J. Organo- met. Chem. 1975,85,115.
47. L. I. Elding and L. H. Skibsted, Znorg. Chem. 1986, 25,4084.
28. P. F. Barron, L. M. Engelhardt, P. C. Healy, J. Oddy and A. H. White, Aust. J. Chem. 1987,40, 1545.
29. S. Attar, W. H. Bearden, N. W. Alcock, E. C. Alyea and J. H. Nelson, Znorg. Chem. 1990, 29,425.
30. J. M. Kessler, J. H. Reeder, R. Vat, C. Yeung, J. H. Nelson, J. S. Frye and N. W. Alcock, Mag. Reson. Chem. 1991,29.
48. T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev. 1973,10, 335.
49. M. M. Gofman and V. I. Nefedov, Znorg. Chim. Acta 1978,28, 1.
50. J. K. Burdett and T. A. Albright, Znorg. Chem. 1979, ‘is, 2112.
31. T. Allman and R. E. Lenkinski, Znorg. Chem. 1986, 25, 3202.
32. E. A. V. Ebsworth, D. W. H. Rankin and S. Cradock, Structural Methods in Inorganic Chemistry, Ch. 1 and 8. Blackwell Scientific Publications, Oxford (1987).
33. B. K. Teo and D. C. Joy, EXAFS Spectroscopy, Techniques and Applications. Plenum, New York (1981).
51. J. E. Anderson, S. M. Sawtelle and C. E. McAndrews, Znorg. Chem. 1990,29,2627. [Have just shown that electrochemical oxidation of Ph,PAuCl in CH,Cl, under similar conditions is quasi-revers- ible (E,, = 1.22 V vs Fc/Fc+) and proceeds by an ECE(C) process, see Electrochemical Methods (Edited by A. J. Bard and L. R. Faulkner). John Wiley & Sons, New York, 1980, pages vii-xvi for a list of abbreviations.]
34. F. G. Mann and D. Purdie, J. Chem. Sot. 1940,1235. 35. D. R. Williamson and M. C. Baird, J. Znorg. Nucl.
Chem. 1972,34,3393. 36. R. J. Puddephatt andP. J. Thompson, J. Chem. Sot.,
Dalton Trans. 1975, 1810. 37. C. A. McAuliffe, R. V. Parish and P. D. Randall, J.
Chem. Sot., Dalton Trans. 1979, 1730. 38. B. T. Heaton and R. J. Kelsey, Znorg. Nucl. Chem.
Lett. 1975, 11, 363. 39. J. A. Rahn, M. S. Holt and J. H. Nelson, Polyhedron
1989, 8, 897. 40. A. Breque, F. Mathey and P. Savignac, Synthesis
1981,983.
52. P. H. Rieger, Electrochemistry, p. 322 ff. Prentice- Hall, Englewood Cliffs, New Jersey (1987).
53. Ligand additivity effects on the redox potentials of transition metal coordination and organometallic compounds have been widely observed. See for example: P. Lemoine, M. Gross, P. Braunstein, F. Mathey, D. Deschamps and J. H. Nelson, Organo- metallics 1984, 3, 1303 ; D. Riley and J. Lyon III, J. Chem. Sot., Dalton Trans. 1991, 157 ; A. B. P. Lever, Znorg. Chem. 1990, 29, 1271. The subject has also been treated theoretically: B. E. Bursten, J. Am. Chem. Sot. 1982,104, 1299.
54. J. E. Huheey, Inorganic Chemistry, Principles of Structure and Reactivity 3rd edn, p. A50. Harper and Row, New York (1983).
41. S. Affandi, R. L. Green, B. T. Hsieh, M. S. Holt, J. H. Nelson and E. C. Alyea, Synth. React. Znorg. Met.-Org. Chem. 1987, 17, 307.
42. E. B. Milosavljevic’, Lj. Solujic’, D. W. Krassowski and J. H. Nelson, J. Organomet. Chem. 1988, 352, 177.
55. See reference 43, page 334. 56. L. Catalini, M. Giampaolo, G. Paolucci, B. Pitteri
and M. L. Tobe, Znorg. Chem. 1987,26,2450. 57. R. J. Cross and M. F. Davidson, J. Chem. Sot.,
Dalton Trans. 1986,411.
43. F. Mercier, S. Holland and F. Mathey, J. Organomet. Chem. 1986,316,271.
58. R. L. Ammlung and T. B. Brill, Znorg. Chim. Acta 1974,11,201.
44. G. M. Sheldrick, SZZEXTL User Manual. Nicolet, Madison, Wisconsin (1983).
45. International Tables for X-ray Crystallography, Vol. IV. Kynoch, Birmingham (1974).
46. For oxidative additions of alkyl halides to gold(I)
59. G. Bandoli, D. A. Clemente, G. Marangoni and L. Cattalini, J. Chem. Sot., Dalton Trans. 1973, 886.
60. D. S. Eggleston, D. F. Chodosh, D. T. Hill and G. R. Girard, Acta Cryst. 1984, C40, 1357.
1949