Embed Size (px)
Citation preview

Pharmacological Characterization of a Novel NonpeptideAntagonist Radioligand, (�)-N-[2-Methyl-4-methoxyphenyl]-1-(1-(methoxymethyl) propyl)-6-methyl-1H-1,2,3-triazolo[4,5-c]pyridin-4-amine ([3H]SN003) for Corticotropin-ReleasingFactor1 Receptors
GE ZHANG, NING HUANG, YU-WEN LI, XIAOPING QI, ANNE P. MARSHALL, XIAO-XIN YAN, GERALDINE HILL,CYNTHIA ROMINGER, SHIMOGA R. PRAKASH, RAJAGOPAL BAKTHAVATCHALAM, DAVID H. ROMINGER,PAUL J. GILLIGAN, and ROBERT ZACZEK
CNS Diseases Research (G.Z., N.H., Y.-W.L., X.Q., A.P.M., X.-X.Y., G.H., C.R., D.H.R., R.Z.), Drug Metabolism (S.R.P.), and Chemical andPhysical Sciences (R.B., P.J.G.), the Bristol-Myers Squibb Pharmaceuticals Research Institute, Wilmington, Delaware
Received October 29, 2002; accepted January 03, 2003
ABSTRACTThe in vitro pharmacological profile of a novel small moleculecorticotropin-releasing factor 1 (CRF1) receptor antagonist, (�)-N-[2-methyl-4-methoxyphenyl]-1-(1-(methoxymethyl)propyl)-6-methyl-1H-1,2,3-triazolo[4,5-c]pyridin-4-amine (SN003), andthe characteristics of its radioligand ([3H]SN003) are described.SN003 has high affinity and selectivity for CRF1 receptors ex-pressed in rat cortex, pituitary, and recombinant HEK293EBNA(HEK293e) cells with respective radiolabeled ovine CRF([125I]oCRF) binding Ki values of 2.5, 7.9, and 6.8 nM. SN003was shown to be a CRF1 receptor antagonist inasmuch as itinhibited CRF-induced cAMP accumulation in humanCRF1HEK293e cells and CRF-stimulated adrenocorticotropinhormone release from rat pituitary cells without agonist activi-ties. Significant decreases in the Bmax of [125I]oCRF binding bySN003 suggest that this antagonist is not simply competitive.To further explore the interaction of SN003 with the CRF1receptors, [3H]SN003 binding to rat cortex and human
CRF1HEK293e cell membranes was characterized and shownto be reversible and saturable, with KD values of 4.8 and 4.6nM, and Bmax values of 0.142 and 7.42 pmol/mg protein, re-spectively. The association and dissociation rate constants of[3H]SN003 (k�1 0.292 nM�1 min�1 and k�1 0.992 � 10�2
min�1) were also assessed using human CRF1HEK293e cellmembranes, giving an equilibrium dissociation constant of 3.4nM. Moreover, [3H]SN003 binding displayed a single affinitystate and insensitivity to 5�-guanylylimidodiphosphate, consis-tent with characteristics of antagonist binding. Incomplete in-hibition of [3H]SN003 binding by CRF peptides also suggeststhat SN003 is not simply competitive with CRF at CRF1 recep-tors. The distribution of [3H]SN003 binding sites was consistentwith the expression pattern of CRF1 receptors in rat brainregions. Small molecule CRF1 antagonist radioligands like[3H]SN003 should enable a better understanding of small mol-ecule interactions with the CRF1 receptor.
Corticotropin-releasing factor (CRF) was first isolated fromovine hypothalamus (Vale et al., 1981) and identified as a keysecretagogue for ACTH release from the anterior pituitary.
In the past 10 years, considerable progress has been made inunderstanding the physiological and potential pathologicalroles of the CRF system. In addition to its endocrine role inthe regulation of the hypothalamic-pituitary-adrenal axis inresponse to stress, CRF appears to be implicated in a varietyof other central and peripheral functions including arousal,anxiety-like behaviors, learning and memory, feeding, im-
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
DOI: 10.1124/jpet.102.046128.
ABBREVIATIONS: CRF, corticotropin-releasing factor; ACTH, adrenocorticotropin hormone; CP-154,526, butyl-[2,5-dimethyl-7-(2,4,6-trimethyl-phenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]ethylamine; SN003, (�)-N-[2-methyl-4-methoxyphenyl]-1-(1-(methoxymethyl) propyl)-6-methyl-1H-1,2,3-triazolo[4,5-c] pyridin-4-amine; NBI 27914, 5-chloro-N-(cyclopropylmethyl)-2-methyl-N-propyl-N�-(2,4,6-trichlorophenyl)-4,6-pyrimidinediaminehydrochloride; SSR125543A, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-meth-yl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride; hrCRF, human/rat corticotropin-releasing factor; oCRF, ovine corticotropin-releasing factor;�-helical CRF, �-helical corticotropin-releasing factor9 – 41; D-PheCRF, [D-Phe12,Nle21,38,Ca-MeLeu37]-CRF12– 41; Gpp(NH)p, 5�-guanylylimidodiphosphate; HEK, human embryonic kidney; PBS, phosphate-buffered saline; DMP696, 4-(1,3-dimethoxyprop-2-ylamino)-2,7-dimethyl-8-(2,4-dichlorophenyl)-pyrazolo[1,5-a]-1,3,5-triazine; DMP904, 4-(3-pentylamino)-2,7-dimethyl-8-(2-methyl-4-methoxyphenyl)-pyrazolo-[1,5-a]-pyrimidine; R-121919, 3-[6-dimethylamino)-4-methyl-pyrid-3-yl]-2,5-dimethyl-N,N-dipropyl-pyrazolo[2,3-a]pyrimidin-7-amine.
0022-3565/03/3051-57–69$7.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 305, No. 1Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 46128/1052158JPET 305:57–69, 2003 Printed in U.S.A.
57
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from

mune, and autonomic functions (Owens and Nemeroff, 1991;De Souza and Grigoriadis, 1998; Gilligan et al., 2000; Daut-zenberg and Hauger 2002).
The entire spectrum of CRF peptide effects is mediatedthrough two known receptor subtypes, CRF1 and CRF2.Both receptor subtypes belong to the class B family of Gprotein-coupled receptors that include receptors for secre-tin and parathyroid hormone, among others (review Daut-zenberg and Hauger, 2002). Despite their high degree ofsequence homology and their common coupling through Gs
proteins to cAMP signaling, CRF1 and CRF2 receptorsdiffer markedly from each other in pharmacological prop-erties and anatomic distribution (DeSouza et al., 1998;Gilligan et al., 2000; Dautzenberg and Hauger, 2002).CRF1 receptors are widely distributed in the central ner-vous system. There exist three splice variants of the CRF2
receptor (CRF2�, CRF2�, and CRF2�) with distinct ana-tomic localization. CRF2� receptors are primarily locatedin discrete rat brain areas such as lateral septum, andCRF2� receptors are located in rat choroid plexus, heart,lung, and skeletal muscle. CRF1 and CRF2 receptors areactivated by several related peptides identified from vari-ous species. These include CRF, sauvagine, urotensin, andthe urocortins, including recently identified urocortin IIand III (Lewis et al., 2001; Reyes et al., 2001), whichdisplay differential affinity for CRF1 and CRF2 receptors.
Studies of the neuronal circuitry mediating fear andanxiety states (Davis, 1992) suggest that both CRF1 andCRF2 receptors located in differential brain areas may beinvolved in the regulation of various stress-induced behav-iors, albeit the relative importance of CRF2 receptors isless clear (Lewis et al., 2001; Reyes et al., 2001; Bakshi etal., 2002). Clinical findings support the hypothesis thatdysfunction of the CRF system is involved in certain neu-ropsychiatric disorders such as anxiety and depression(Gilligan et al., 2000; Keck and Holsboer, 2001). Numerousanimal studies using CRF ligands and genetically alteredmice provide strong evidence for the role of CRF1 receptorsin the coordination of the behavioral response to stress andin stress-related psychiatric disorders (Gilligan et al.,2000; Bakshi et al., 2002; Dautzenberg and Hauger, 2002).
In recent years there has been much emphasis on devel-oping orally active, nonpeptidic CRF1 antagonists to eval-uate the putative role of CRF1 receptors in psychopathol-ogy and to test their potential as novel therapeutic agents.Schulz et al. (1996) were the first to report a pyrazolopy-rimidine CRF1 antagonist, CP-154,526, with high affinityfor the CRF1 receptor and anxiolytic activity in rats. Ad-ditional CRF1 antagonists, such as antalarmin, NBI27914, DMP696, R121919, and SSR125543A, also exhibitCRF1 antagonist properties in vitro and in vivo (He et al.,2000; McCarthy et al., 1999; Habib et al., 2000; Griebel etal., 2002; Gully et al., 2002; Heinrichs et al., 2002; Maciaget al., 2002; McElroy et al., 2002). Although a variety of theiodine-125-labeled peptides such as [125I]ovine CRF havebeen extensively employed in previous studies of CRF1
receptors, the use of a small molecule CRF1 antagonistradioligand as a tool would permit investigation of inter-actions between CRF and small molecule antagonists atCRF1 receptors and allow direct mapping of the smallmolecule binding sites in discrete brain regions.
The aim of the present studies is to describe the in vitro
pharmacological properties of tritiated (�)-N-[2-methyl-4-methoxyphenyl]-1-(1-(methoxymethyl)propyl)-6-methyl-1H-1,2,3-triazolo[4,5-c]pyridin-4-amine ([3H]SN003), a small-molecule radioligand for rat and human CRF1 receptors. Thebinding characteristics of [3H]SN003 were profiled in ratcortical and human CRF1 cell membranes, and the specificityand anatomic distribution of [3H]SN003 binding sites in ratbrain were illustrated by brain section phosphorimaging. Thein vitro pharmacological profile of the unlabeled SN003 li-gand was also studied. Parts of these studies were previouslypresented in abstract form (Li et al., 2001; Zhang et al.,2001). This is the first report identifying a small moleculeantagonist radioligand specifically labeling CRF1 receptorsin brain tissues and slices. This nonpeptide radioligand as atool provides an opportunity to further understand the inter-actions of CRF and small molecule antagonists with CRF1
receptors.
Materials and MethodsAnimals. Male Sprague-Dawley rats weighing 250 to 350 g
were obtained from Charles River Laboratories, Inc. (Wilmington,MA). They were housed two per cage in a room with controlledillumination, humidity, and temperature. Food and water wereprovided ad libitum. All animal studies were conducted in accor-dance with the National Institutes of Health Guide for the Careand Use of Laboratory Animals. Protocols were approved by theCommittee on Animal Care and Use of the Bristol-Myers SquibbCompany.
Materials. SN003 was obtained by directed synthesis efforts(Bakthavatchalam et al., 1997) as were other small-moleculeCRF1 antagonists, DMP696 (He et al., 2000) and DMP904 (Gilli-gan et al., 2000). SN003, DMP696, DMP904, and CP-154,526 weresynthesized by the Department of Chemical and Physical Sci-ences, Bristol-Myers Squibb Company. The chemical structures ofthe small molecule CRF1 receptor antagonists are shown in Fig. 1.CRF-related peptides, human/rat CRF (hrCRF), ovine CRF(oCRF), sauvagine, urocortin I (human), urocortin I (rat), �-helical
Fig. 1. Chemical structures of SN003 and other small molecule CRF1receptor antagonists. A, SN003 [(�)-N-[2-methyl-4-methoxyphenyl]-1-(1-(methoxymethyl) propyl)-6-methyl-1H-1,2,3-triazolo[4,5-c] pyridin-4-amine]; B, DMP696, DMP904, and CP-154,526.
58 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from

CRF9 – 41 (�-helical CRF), and [D-Phe12,Nle21,38,Ca-MeLeu37]-CRF12–41
(D-PheCRF), were purchased from American Peptide Co., Inc. (Sunny-vale, CA), Bachem California (Torrance, CA), and Peninsula Labora-tories (Merseyside, UK). [125I]oCRF and [125I]sauvagine (specificactivities, 2200 Ci/mmol) were purchased from PerkinElmer LifeSciences (Boston, MA). Gpp(NH)p (5�-guanylylimidodiphosphate)and other standard reagents were purchased from Sigma-Aldrich(St. Louis, MO) and Invitrogen (Carlsbad, CA).
Synthesis of [3H]SN003. [3H]SN003 was labeled by the radioli-gand synthesis facility in the drug metabolism group of the Bristol-Myers Squibb Company (former DuPont Pharmaceuticals Compa-ny).
Compound 1 (ST613) was prepared from Compound 2a (SN003) bydemethylation with sodium thiomethoxide in dimethylformamide (Fig.1A). To a solution of compound 1 in dimethylformamide, potassiumcarbonate was added and the mixture was stirred. Tritium-labeledmethyl iodide (250 mCi, 80 Ci/mmol) in toluene was transferred to thisreaction mixture, followed by rinsing, extraction, and separation bychromatography on a reversed-phase column (Vydac Protein-peptideC18 Semiprep column). Fractions containing the product were lyophi-lized to provide radiochemically pure compound 2 ([3H]SN003, purity�99%). [3H]SN003 was dissolved and stored as an ethanolic solution(1mCi/ml; specific activity 74 Ci/mmol).
Cell Culture of HEK293e Cells Expressing Human CRF Re-ceptors. Full-length human cDNAs for human CRF1 and CRF2� re-ceptors were subcloned into plasmids and transfected intoHEK293EBNA (HEK293e) cells (Invitrogen) using lipofectamine (In-vitrogen). The details of the plasmid construct were described in previ-ous studies (Horlick et al., 1997; Kostich et al., 1998). HEK293e cellsstably expressing human CRF1 or CRF2� receptors were grown inDulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovineserum at 37°C in a humid environment (5% CO2) for 10 days. The cellswere then adapted to spinner culture for bulk processing. Cells wereharvested, washed in phosphate-buffered saline (PBS), and counted;and the cell pellet (containing approximately 1 � 108 HEK293e cells)was stored at –80°C until use.
Peptide Radioligand Binding to Homogenates. For bind-ing assays, the total particulate fraction of rat frontal cortex andpituitary tissues or cell pellets was prepared as a crude membranesource expressing CRF1 or CRF2� receptors. Frozen tissues or cellpellets were thawed on ice and homogenized in tissue buffer(containing 50 mM HEPES, 10 mM MgCl2, 2 mM EGTA, and 1�g/ml each of aprotonin, leupeptin, and pepstatin, pH 7.0 at 23°C)using a Brinkmann Instruments (Westbury, NY) Polytron (PT-10,setting 6 for 10 s). The homogenate was centrifuged at 48,000g for12 min, and the resulting pellet was washed by resuspension andcentrifugation steps. The final pellet was suspended in tissuebuffer, and protein concentrations were determined using thebicinchoninic acid (BCA) assay (Pierce, Rockford, IL) with bovineserum albumin as standard. The membrane proteins preparedfrom human CRF1 HEK293e cells (5– 8 �g), rat cortex, pituitary,and human CRF2� HEK293e cells, respectively (40 – 60 �g), wereused in receptor binding experiments.
Equilibrium competition binding experiments were performedusing a modification of the methods described previously to deter-mine binding potencies (IC50 values) of compounds at CRF1 andCRF2� receptors (Rominger et al., 1998). All small molecule li-gands were initially prepared in 100% dimethyl sulfoxide at aconcentration of 10�2 M and diluted in assay buffer that wasidentical to the tissue buffer except for inclusion of 0.15 mMbacitracin and 0.1% (w/v) ovalbumin. Competition assays wereconducted in disposable polypropylene 96-well plates (Costar,Cambridge, MA) and initiated by the addition of 150 �l of mem-brane homogenate to 150 �l of assay buffer containing the radio-ligand (100 �l) in a final concentration of 150 pM ([125I]oCRF forthe CRF1 or [125I]sauvagine for CRF2� receptors) and competingligands (50 �l). The reaction mixtures were incubated to equilib-rium for 2 h at 23°C. Specific binding was defined in the presence
of 1 �M DMP696 for CRF1 receptors and 1 �M �-helical CRF forCRF2� receptors, respectively. Binding assays were terminated byrapid filtration over GF/C glass-fibers [presoaked in 0.3% (v/v)polyethyleneimine for more than 2 h] using a 96-well PerkinElmerUnifilter harvester followed by three washes with 0.3 ml of coldwash buffer (PBS, pH 7.0, containing 0.01% Triton X-100). Afterthe filter was dried, scintillation cocktail was added and the platewas counted in a 96-well PerkinElmer Top Counter. The CRF1
competition binding to membranes from rat frontal cortex andpituitary was performed similarly except for the radioligand con-centration of [125I]oCRF (200 pM) used in the binding.
To determine the nature of inhibition (competitive and noncom-petitive) of SN003, homologous isotopic displacement of [125I]oCRF(150 pM) binding by ovine CRF was conducted in membranes pre-pared from HEK293e cells expressing human CRF1 receptors in theabsence and presence of SN003. The KD and Bmax values from ho-mologous competition curves were calculated using the nonlinearregression analysis in Prism (1999; GraphPad Software, Inc., SanDiego, CA). A Scatchard plot of the homologous binding with[125I]oCRF was generated for visualization of any KD or Bmax
changes. Specific bound (femtomoles per milligram of protein) andBound/Free ligand data were transformed. These transformationswere performed according to similar procedures described in Prism(1999; GraphPad Software, Inc.).
[3H]SN003 Binding to Membranes. Equilibrium binding exper-iments in cell or tissue homogenates were performed under condi-tions similar to those described for the [125I]oCRF binding with a fewexceptions. GF/B filters were used in the filtration assay. The satu-ration experiments using [3H]SN003 as a radioligand and rat cortexand HEK293e cell homogenates as CRF1 receptor sources were con-ducted in 12 concentrations of [3H]SN003 (0.60–40 nM) in triplicateat 23°C for 2 h. The nonspecific binding was defined in the presenceof 5 �M DMP696. Association and dissociation assays were per-formed at the [3H]SN003 KD concentration of 4.8 nM at 23°C. After2 h of incubation, when association equilibrium was reached, disso-ciation reactions were initiated by addition of 5 �M DMP696 andcontinued for an additional 3 h.
cAMP Assays in Recombinant Human CRF1 HEK293eCells. Intracellular cAMP levels were measured using the AdenylCyclase Activation Flash Plate kit purchased from PerkinElmerLife Sciences. This radioimmunoassay-based kit enables directdetection of cAMP generated in live cells in a 96-well format.HEK293e cells expressing CRF1 receptors were grown in theDulbecco’s modified Eagle’s medium supplement with 10% fetalbovine serum, L-glutamine (2 mM), and hygromycin (400 �g/ml) at37°C in a humid environment with 5% CO2. On the assay day,cells were dissociated from flasks and centrifuged down at 1,200rpm for 4 min. Cells were resuspended in 100% stimulation buffer,counted, and diluted to 0.6 � 106 cell/ml. hrCRF (1 nM) in theabsence and presence of SN003 in PBS containing 10% stimula-tion buffer (50 �l) was added to the assay plate. Drug treatmentwas initiated by adding HEK293e cells expressing CRF1 receptors(50,000 cells/50 �l/well) to the Flash Plate and incubated for 15min at 37°C in a final volume of 100 �l. Intracellular cAMP wasreleased from cells through cell lysis resulting from adding detec-tion buffer containing [125I]cAMP (100 �l/well). Assay signal isbased on competition of endogenous cAMP and [125I]cAMP forcAMP antibodies coated on the Flash Plate. Radioactivity frombinding of [125I]cAMP to the plate was assessed 2 h later by a96-well PerkinElmer Top Counter.
ACTH Release Assays in Rat Primary Pituitary Cultures.Primary pituitary culture was established as described previously(Vale et al., 1972). Pituitaries were harvested from 20 to 25 ratsand washed in PBS four to six times. Pituitary cells were disso-ciated in collagenase buffer [1� PBS containing 25 mM HEPES,0.2% glucose, 0.4% (w/v) bovine serum albumin, 80 �g/ml DNaseII, and 0.4% (w/v) collagenase type 2] for 3 h at 37°C. Cells werespun down, decanted, and incubated with 0.25% trypsin solution
A Nonpeptide CRF1 Antagonist Radioligand 59
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
[PBS containing 0.25% (w/v) trypsin, 0.4% (w/v) bovine serumalbumin, 0.2% (w/v) glucose] for 5 to 10 min at 37°C. Cells werebriefly centrifuged and resuspended in M199 culture medium(Invitrogen) containing 10% fetal calf serum, minimal essentialmedium vitamins, streptomycin/penicillin, insulin/transferrin/se-lenium, fibroblast growth factor, and trace elements. Cells(300,000/ml) were added to a 48-well plate (Costar) and grown inculture (37°C, 5% CO2) for 4 days. On the assay day, medium wasremoved and replaced with M199 incubation medium identical tothat used above except for exclusion of serum and addition of 0.1%(w/v) ovalbumin and 1 �g/ml aprotinin. The hrCRF-induced ACTHrelease was conducted at 37°C for 3 h. With pretreatment ofvarious concentrations of SN003 or vehicle (control) to cells for 15min, the antagonist assay of SN003 was performed by addinghrCRF (0.3 nM) and coincubation at 37°C for 3 h. After theincubation period, medium was removed, frozen, and stored at–20°C until assayed for ACTH measurement. ACTH levels weredetermined using a radioimmunoassay kit purchased from Dia-Sorin Inc. (Stillwater, MN).
Data Analyses. The concentrations of compounds to inhibit50% of radioligand binding (IC50) for CRF1 and CRF2� receptorswere calculated by fitting data through a competition equation inthe iterative nonlinear regression curve-fitting program Prism(GraphPad Software, Inc.). Ki values (equilibrium dissociationconstant) for inhibitors in competition experiments were calcu-lated according to the Cheng-Prusoff equation. Saturation datawere fit through hyperbolic equations to estimate apparent equi-librium constant (KD) and the maximal number of binding sites(Bmax) using Prism (GraphPad Software, Inc.). Kinetic studies todetermine kobs (observed association rate constant) and k�1 (dis-sociation rate constant) were generated from a slope of the line vialinear regression of transformed data. The association rate con-stant (k�1) was generated based on the equation k�1 � (kobs �k�1/[L], where L is the ligand concentration. The equilibriumdissociation constant derived from kinetic studies was calculatedas KD � k�1/k�1.
For cell-based functional studies, the potency for agonists (EC50)and for antagonists (IC50) was estimated by fitting raw data throughthe sigmoidal dose-response equation using the iterative nonlinearregression curve-fitting programs in Prism (GraphPad Software,Inc.).
Brain Section Ligand Binding and Storage Phosphorim-aging. Rats were decapitated, and the brain and pituitary wereimmediately collected, embedded in M-1 embedding matrix(Thermo Shandon, Pittsburgh, PA), and frozen in iso-pentanechilled with dry ice. Twenty-micrometer coronal sections were cuton a Cryostat, thaw-mounted on superfrost slides (VWR, WestChester, PA), dried, and stored at –70°C until use. Before in vitrobinding, sections were brought to 23°C and preincubated for 30min in assay solution containing 50 mM HEPES, 10 mM MgCl2, 2mM EGTA, 100 kallikrein-inactivating units/ml aprotinin, 0.1 Mbacitracin, and 0.1% ovalbumin (pH 7.2). Sections were then in-cubated in the same solution containing 4 to 10 nM [3H]SN003 for2 h at 23°C. As a comparison, one set of adjacent sections wasincubated with 200 pM [125I]sauvagine. At this concentration (200pM), [125I]sauvagine binds to both CRF1 and CRF2 receptors(Rominger et al., 1998). Nonspecific binding was defined by inclu-sion of 1 �M DMP696. After incubation, sections were rinsed inPBS with 0.01% Triton X-100 for 10 min and subsequently driedunder a stream of cold air. Slides of the sections were then placedin cassettes against storage phosphorimaging screens(PerkinElmer Life Sciences) for 1 to 4 weeks ([3H]SN003) or for12 h ([125I]sauvagine), respectively. The screens were thenscanned with a Cyclone phosphorimaging scanner, and capturedimages were analyzed with the OptiQuant requisition and analy-sis system (PerkinElmer Life Sciences).
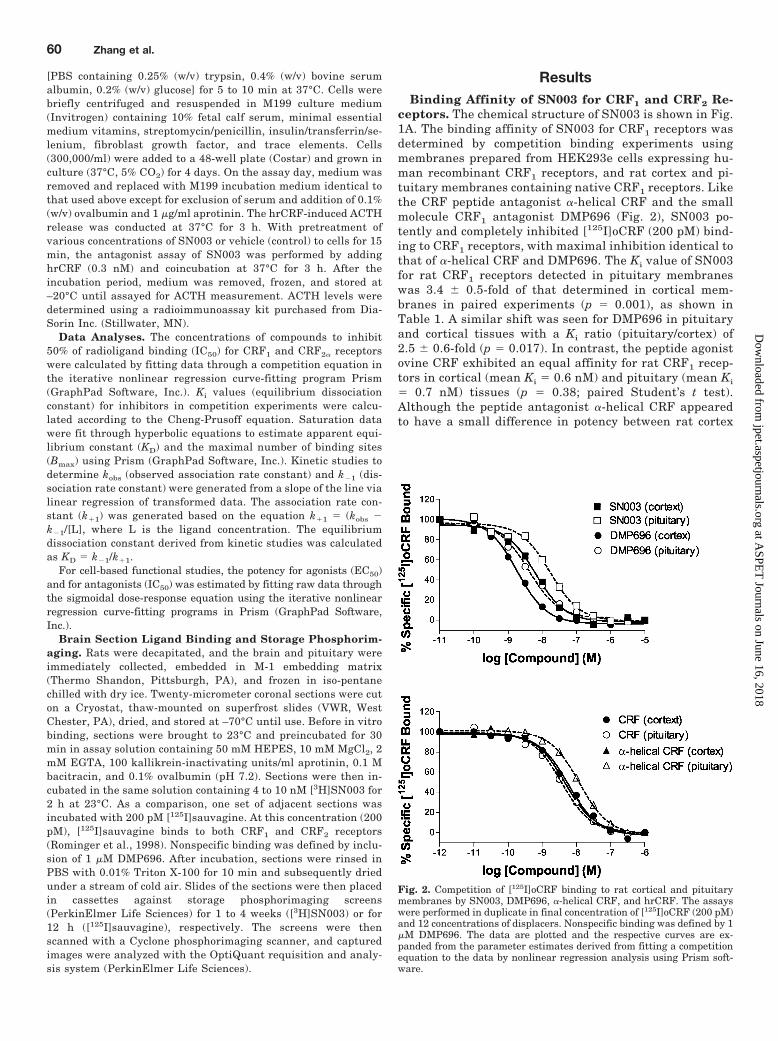
ResultsBinding Affinity of SN003 for CRF1 and CRF2 Re-
ceptors. The chemical structure of SN003 is shown in Fig.1A. The binding affinity of SN003 for CRF1 receptors wasdetermined by competition binding experiments usingmembranes prepared from HEK293e cells expressing hu-man recombinant CRF1 receptors, and rat cortex and pi-tuitary membranes containing native CRF1 receptors. Likethe CRF peptide antagonist �-helical CRF and the smallmolecule CRF1 antagonist DMP696 (Fig. 2), SN003 po-tently and completely inhibited [125I]oCRF (200 pM) bind-ing to CRF1 receptors, with maximal inhibition identical tothat of �-helical CRF and DMP696. The Ki value of SN003for rat CRF1 receptors detected in pituitary membraneswas 3.4 � 0.5-fold of that determined in cortical mem-branes in paired experiments (p � 0.001), as shown inTable 1. A similar shift was seen for DMP696 in pituitaryand cortical tissues with a Ki ratio (pituitary/cortex) of2.5 � 0.6-fold (p � 0.017). In contrast, the peptide agonistovine CRF exhibited an equal affinity for rat CRF1 recep-tors in cortical (mean Ki � 0.6 nM) and pituitary (mean Ki
� 0.7 nM) tissues (p � 0.38; paired Student’s t test).Although the peptide antagonist �-helical CRF appearedto have a small difference in potency between rat cortex
Fig. 2. Competition of [125I]oCRF binding to rat cortical and pituitarymembranes by SN003, DMP696, �-helical CRF, and hrCRF. The assayswere performed in duplicate in final concentration of [125I]oCRF (200 pM)and 12 concentrations of displacers. Nonspecific binding was defined by 1�M DMP696. The data are plotted and the respective curves are ex-panded from the parameter estimates derived from fitting a competitionequation to the data by nonlinear regression analysis using Prism soft-ware.
60 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
(mean Ki � 5.3 nM) and pituitary (mean Ki � 8.1 nM)tissues, this difference was not statistically significant(p � 0.22), as summarized in Table 1.
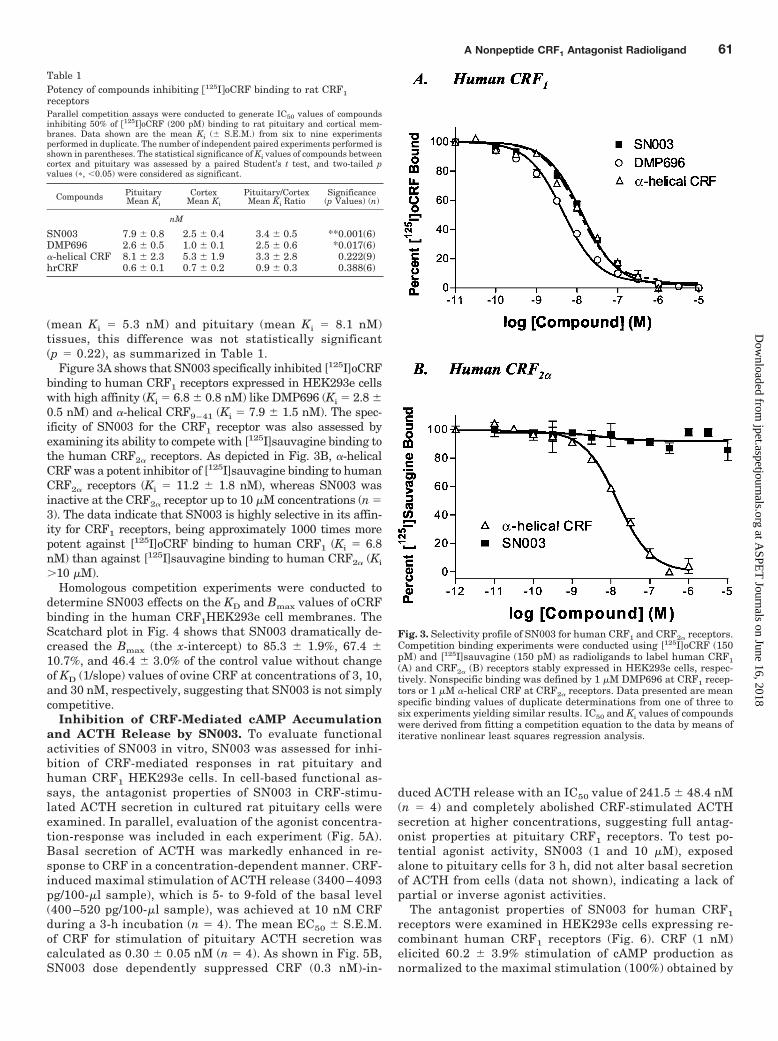
Figure 3A shows that SN003 specifically inhibited [125I]oCRFbinding to human CRF1 receptors expressed in HEK293e cellswith high affinity (Ki � 6.8 � 0.8 nM) like DMP696 (Ki � 2.8 �0.5 nM) and �-helical CRF9–41 (Ki � 7.9 � 1.5 nM). The spec-ificity of SN003 for the CRF1 receptor was also assessed byexamining its ability to compete with [125I]sauvagine binding tothe human CRF2� receptors. As depicted in Fig. 3B, �-helicalCRF was a potent inhibitor of [125I]sauvagine binding to humanCRF2� receptors (Ki � 11.2 � 1.8 nM), whereas SN003 wasinactive at the CRF2� receptor up to 10 �M concentrations (n �3). The data indicate that SN003 is highly selective in its affin-ity for CRF1 receptors, being approximately 1000 times morepotent against [125I]oCRF binding to human CRF1 (Ki � 6.8nM) than against [125I]sauvagine binding to human CRF2� (Ki
�10 �M).Homologous competition experiments were conducted to
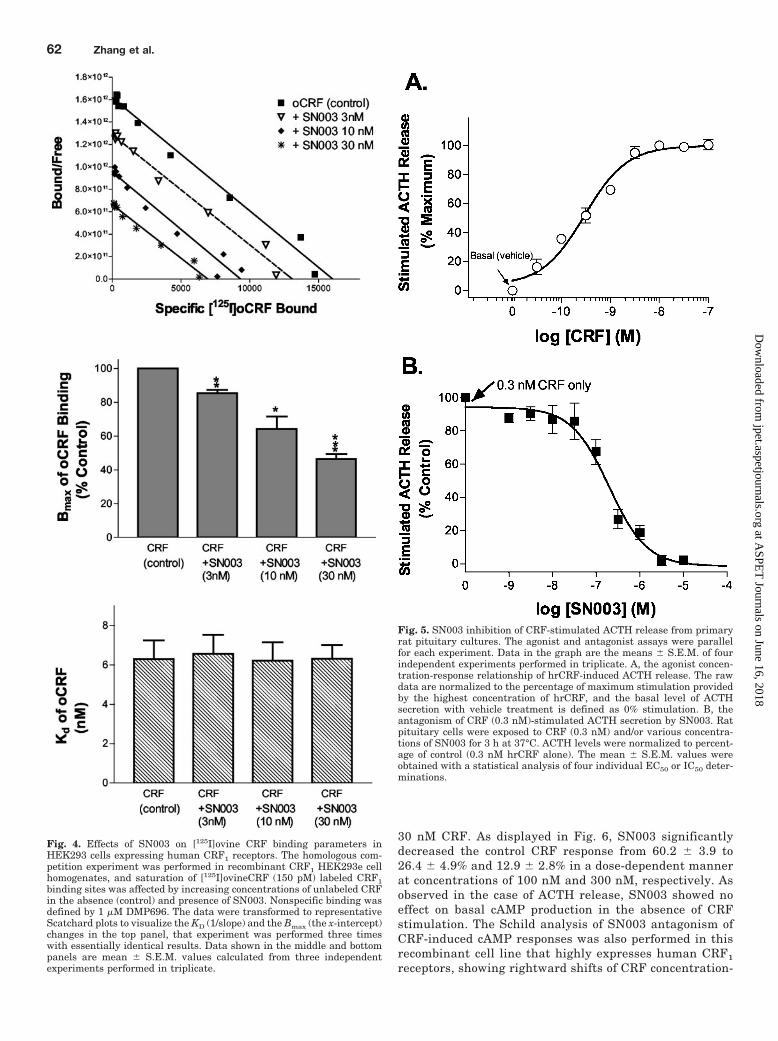
determine SN003 effects on the KD and Bmax values of oCRFbinding in the human CRF1HEK293e cell membranes. TheScatchard plot in Fig. 4 shows that SN003 dramatically de-creased the Bmax (the x-intercept) to 85.3 � 1.9%, 67.4 �10.7%, and 46.4 � 3.0% of the control value without changeof KD (1/slope) values of ovine CRF at concentrations of 3, 10,and 30 nM, respectively, suggesting that SN003 is not simplycompetitive.
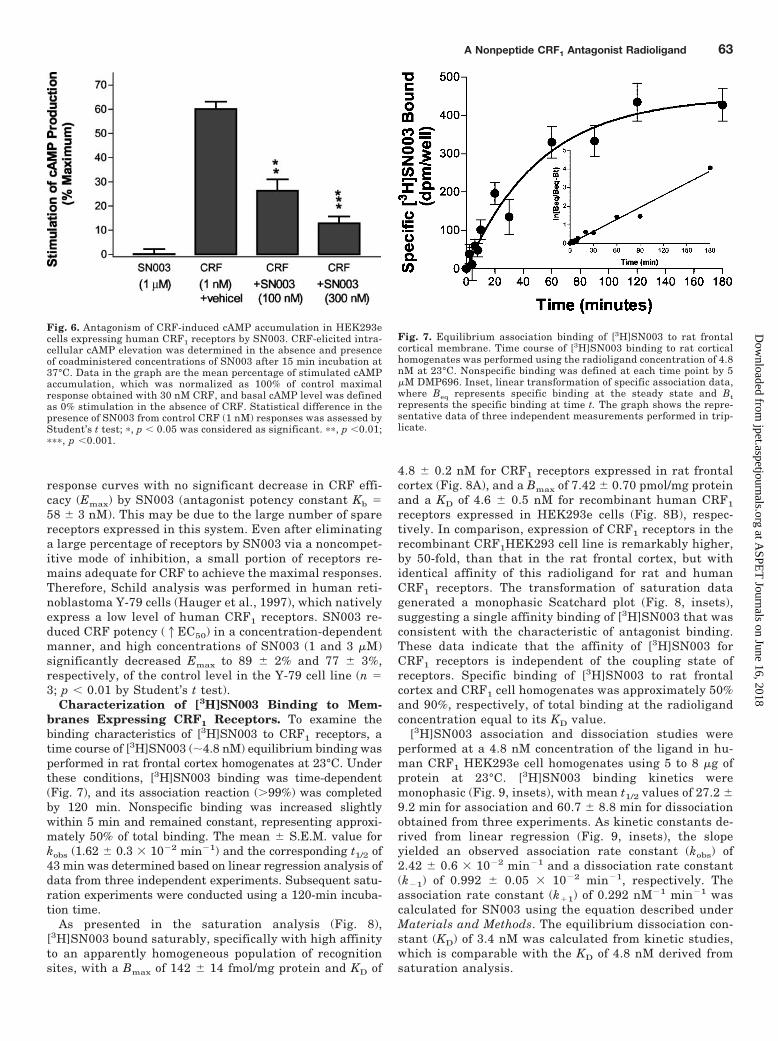
Inhibition of CRF-Mediated cAMP Accumulationand ACTH Release by SN003. To evaluate functionalactivities of SN003 in vitro, SN003 was assessed for inhi-bition of CRF-mediated responses in rat pituitary andhuman CRF1 HEK293e cells. In cell-based functional as-says, the antagonist properties of SN003 in CRF-stimu-lated ACTH secretion in cultured rat pituitary cells wereexamined. In parallel, evaluation of the agonist concentra-tion-response was included in each experiment (Fig. 5A).Basal secretion of ACTH was markedly enhanced in re-sponse to CRF in a concentration-dependent manner. CRF-induced maximal stimulation of ACTH release (3400– 4093pg/100-�l sample), which is 5- to 9-fold of the basal level(400 –520 pg/100-�l sample), was achieved at 10 nM CRFduring a 3-h incubation (n � 4). The mean EC50 � S.E.M.of CRF for stimulation of pituitary ACTH secretion wascalculated as 0.30 � 0.05 nM (n � 4). As shown in Fig. 5B,SN003 dose dependently suppressed CRF (0.3 nM)-in-
duced ACTH release with an IC50 value of 241.5 � 48.4 nM(n � 4) and completely abolished CRF-stimulated ACTHsecretion at higher concentrations, suggesting full antag-onist properties at pituitary CRF1 receptors. To test po-tential agonist activity, SN003 (1 and 10 �M), exposedalone to pituitary cells for 3 h, did not alter basal secretionof ACTH from cells (data not shown), indicating a lack ofpartial or inverse agonist activities.
The antagonist properties of SN003 for human CRF1
receptors were examined in HEK293e cells expressing re-combinant human CRF1 receptors (Fig. 6). CRF (1 nM)elicited 60.2 � 3.9% stimulation of cAMP production asnormalized to the maximal stimulation (100%) obtained by
Table 1Potency of compounds inhibiting �125IoCRF binding to rat CRF1receptorsParallel competition assays were conducted to generate IC50 values of compoundsinhibiting 50% of �125IoCRF (200 pM) binding to rat pituitary and cortical mem-branes. Data shown are the mean Ki (� S.E.M.) from six to nine experimentsperformed in duplicate. The number of independent paired experiments performed isshown in parentheses. The statistical significance of Ki values of compounds betweencortex and pituitary was assessed by a paired Student’s t test, and two-tailed pvalues (�, 0.05) were considered as significant.
Compounds PituitaryMean Ki
CortexMean Ki
Pituitary/CortexMean Ki Ratio
Significance(p Values) (n)
nM
SN003 7.9 � 0.8 2.5 � 0.4 3.4 � 0.5 **0.001(6)DMP696 2.6 � 0.5 1.0 � 0.1 2.5 � 0.6 *0.017(6)�-helical CRF 8.1 � 2.3 5.3 � 1.9 3.3 � 2.8 0.222(9)hrCRF 0.6 � 0.1 0.7 � 0.2 0.9 � 0.3 0.388(6)
Fig. 3. Selectivity profile of SN003 for human CRF1 and CRF2� receptors.Competition binding experiments were conducted using [125I]oCRF (150pM) and [125I]sauvagine (150 pM) as radioligands to label human CRF1(A) and CRF2� (B) receptors stably expressed in HEK293e cells, respec-tively. Nonspecific binding was defined by 1 �M DMP696 at CRF1 recep-tors or 1 �M �-helical CRF at CRF2� receptors. Data presented are meanspecific binding values of duplicate determinations from one of three tosix experiments yielding similar results. IC50 and Ki values of compoundswere derived from fitting a competition equation to the data by means ofiterative nonlinear least squares regression analysis.
A Nonpeptide CRF1 Antagonist Radioligand 61
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
30 nM CRF. As displayed in Fig. 6, SN003 significantlydecreased the control CRF response from 60.2 � 3.9 to26.4 � 4.9% and 12.9 � 2.8% in a dose-dependent mannerat concentrations of 100 nM and 300 nM, respectively. Asobserved in the case of ACTH release, SN003 showed noeffect on basal cAMP production in the absence of CRFstimulation. The Schild analysis of SN003 antagonism ofCRF-induced cAMP responses was also performed in thisrecombinant cell line that highly expresses human CRF1
receptors, showing rightward shifts of CRF concentration-
Fig. 4. Effects of SN003 on [125I]ovine CRF binding parameters inHEK293 cells expressing human CRF1 receptors. The homologous com-petition experiment was performed in recombinant CRF1 HEK293e cellhomogenates, and saturation of [125I]ovineCRF (150 pM) labeled CRF1binding sites was affected by increasing concentrations of unlabeled CRFin the absence (control) and presence of SN003. Nonspecific binding wasdefined by 1 �M DMP696. The data were transformed to representativeScatchard plots to visualize the KD (1/slope) and the Bmax (the x-intercept)changes in the top panel, that experiment was performed three timeswith essentially identical results. Data shown in the middle and bottompanels are mean � S.E.M. values calculated from three independentexperiments performed in triplicate.
Fig. 5. SN003 inhibition of CRF-stimulated ACTH release from primaryrat pituitary cultures. The agonist and antagonist assays were parallelfor each experiment. Data in the graph are the means � S.E.M. of fourindependent experiments performed in triplicate. A, the agonist concen-tration-response relationship of hrCRF-induced ACTH release. The rawdata are normalized to the percentage of maximum stimulation providedby the highest concentration of hrCRF, and the basal level of ACTHsecretion with vehicle treatment is defined as 0% stimulation. B, theantagonism of CRF (0.3 nM)-stimulated ACTH secretion by SN003. Ratpituitary cells were exposed to CRF (0.3 nM) and/or various concentra-tions of SN003 for 3 h at 37°C. ACTH levels were normalized to percent-age of control (0.3 nM hrCRF alone). The mean � S.E.M. values wereobtained with a statistical analysis of four individual EC50 or IC50 deter-minations.
62 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
response curves with no significant decrease in CRF effi-cacy (Emax) by SN003 (antagonist potency constant Kb �58 � 3 nM). This may be due to the large number of sparereceptors expressed in this system. Even after eliminatinga large percentage of receptors by SN003 via a noncompet-itive mode of inhibition, a small portion of receptors re-mains adequate for CRF to achieve the maximal responses.Therefore, Schild analysis was performed in human reti-noblastoma Y-79 cells (Hauger et al., 1997), which nativelyexpress a low level of human CRF1 receptors. SN003 re-duced CRF potency (1EC50) in a concentration-dependentmanner, and high concentrations of SN003 (1 and 3 �M)significantly decreased Emax to 89 � 2% and 77 � 3%,respectively, of the control level in the Y-79 cell line (n �3; p 0.01 by Student’s t test).
Characterization of [3H]SN003 Binding to Mem-branes Expressing CRF1 Receptors. To examine thebinding characteristics of [3H]SN003 to CRF1 receptors, atime course of [3H]SN003 (�4.8 nM) equilibrium binding wasperformed in rat frontal cortex homogenates at 23°C. Underthese conditions, [3H]SN003 binding was time-dependent(Fig. 7), and its association reaction (�99%) was completedby 120 min. Nonspecific binding was increased slightlywithin 5 min and remained constant, representing approxi-mately 50% of total binding. The mean � S.E.M. value forkobs (1.62 � 0.3 � 10�2 min�1) and the corresponding t1/2 of43 min was determined based on linear regression analysis ofdata from three independent experiments. Subsequent satu-ration experiments were conducted using a 120-min incuba-tion time.
As presented in the saturation analysis (Fig. 8),[3H]SN003 bound saturably, specifically with high affinityto an apparently homogeneous population of recognitionsites, with a Bmax of 142 � 14 fmol/mg protein and KD of
4.8 � 0.2 nM for CRF1 receptors expressed in rat frontalcortex (Fig. 8A), and a Bmax of 7.42 � 0.70 pmol/mg proteinand a KD of 4.6 � 0.5 nM for recombinant human CRF1
receptors expressed in HEK293e cells (Fig. 8B), respec-tively. In comparison, expression of CRF1 receptors in therecombinant CRF1HEK293 cell line is remarkably higher,by 50-fold, than that in the rat frontal cortex, but withidentical affinity of this radioligand for rat and humanCRF1 receptors. The transformation of saturation datagenerated a monophasic Scatchard plot (Fig. 8, insets),suggesting a single affinity binding of [3H]SN003 that wasconsistent with the characteristic of antagonist binding.These data indicate that the affinity of [3H]SN003 forCRF1 receptors is independent of the coupling state ofreceptors. Specific binding of [3H]SN003 to rat frontalcortex and CRF1 cell homogenates was approximately 50%and 90%, respectively, of total binding at the radioligandconcentration equal to its KD value.
[3H]SN003 association and dissociation studies wereperformed at a 4.8 nM concentration of the ligand in hu-man CRF1 HEK293e cell homogenates using 5 to 8 �g ofprotein at 23°C. [3H]SN003 binding kinetics weremonophasic (Fig. 9, insets), with mean t1/2 values of 27.2 �9.2 min for association and 60.7 � 8.8 min for dissociationobtained from three experiments. As kinetic constants de-rived from linear regression (Fig. 9, insets), the slopeyielded an observed association rate constant (kobs) of2.42 � 0.6 � 10�2 min�1 and a dissociation rate constant(k�1) of 0.992 � 0.05 � 10�2 min�1, respectively. Theassociation rate constant (k�1) of 0.292 nM�1 min�1 wascalculated for SN003 using the equation described underMaterials and Methods. The equilibrium dissociation con-stant (KD) of 3.4 nM was calculated from kinetic studies,which is comparable with the KD of 4.8 nM derived fromsaturation analysis.
Fig. 6. Antagonism of CRF-induced cAMP accumulation in HEK293ecells expressing human CRF1 receptors by SN003. CRF-elicited intra-cellular cAMP elevation was determined in the absence and presenceof coadministered concentrations of SN003 after 15 min incubation at37°C. Data in the graph are the mean percentage of stimulated cAMPaccumulation, which was normalized as 100% of control maximalresponse obtained with 30 nM CRF, and basal cAMP level was definedas 0% stimulation in the absence of CRF. Statistical difference in thepresence of SN003 from control CRF (1 nM) responses was assessed byStudent’s t test; �, p 0.05 was considered as significant. ��, p 0.01;���, p 0.001.
Fig. 7. Equilibrium association binding of [3H]SN003 to rat frontalcortical membrane. Time course of [3H]SN003 binding to rat corticalhomogenates was performed using the radioligand concentration of 4.8nM at 23°C. Nonspecific binding was defined at each time point by 5�M DMP696. Inset, linear transformation of specific association data,where Beq represents specific binding at the steady state and Btrepresents the specific binding at time t. The graph shows the repre-sentative data of three independent measurements performed in trip-licate.
A Nonpeptide CRF1 Antagonist Radioligand 63
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
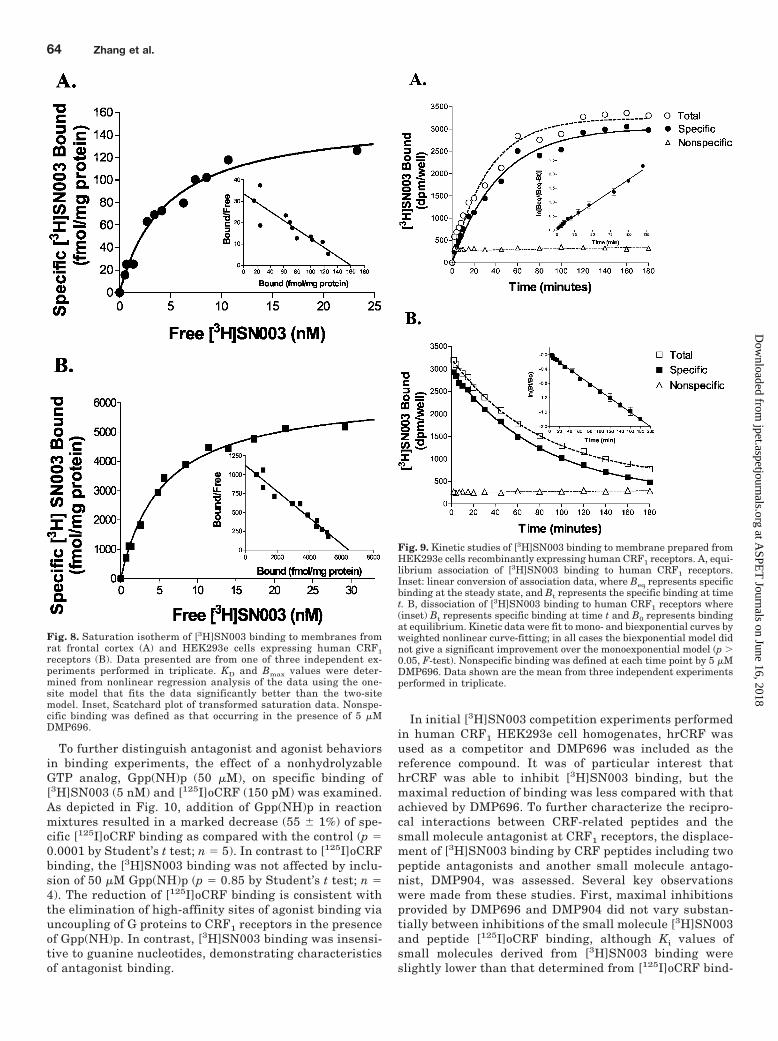
To further distinguish antagonist and agonist behaviorsin binding experiments, the effect of a nonhydrolyzableGTP analog, Gpp(NH)p (50 �M), on specific binding of[3H]SN003 (5 nM) and [125I]oCRF (150 pM) was examined.As depicted in Fig. 10, addition of Gpp(NH)p in reactionmixtures resulted in a marked decrease (55 � 1%) of spe-cific [125I]oCRF binding as compared with the control (p �0.0001 by Student’s t test; n � 5). In contrast to [125I]oCRFbinding, the [3H]SN003 binding was not affected by inclu-sion of 50 �M Gpp(NH)p (p � 0.85 by Student’s t test; n �4). The reduction of [125I]oCRF binding is consistent withthe elimination of high-affinity sites of agonist binding viauncoupling of G proteins to CRF1 receptors in the presenceof Gpp(NH)p. In contrast, [3H]SN003 binding was insensi-tive to guanine nucleotides, demonstrating characteristicsof antagonist binding.
In initial [3H]SN003 competition experiments performedin human CRF1 HEK293e cell homogenates, hrCRF wasused as a competitor and DMP696 was included as thereference compound. It was of particular interest thathrCRF was able to inhibit [3H]SN003 binding, but themaximal reduction of binding was less compared with thatachieved by DMP696. To further characterize the recipro-cal interactions between CRF-related peptides and thesmall molecule antagonist at CRF1 receptors, the displace-ment of [3H]SN003 binding by CRF peptides including twopeptide antagonists and another small molecule antago-nist, DMP904, was assessed. Several key observationswere made from these studies. First, maximal inhibitionsprovided by DMP696 and DMP904 did not vary substan-tially between inhibitions of the small molecule [3H]SN003and peptide [125I]oCRF binding, although Ki values ofsmall molecules derived from [3H]SN003 binding wereslightly lower than that determined from [125I]oCRF bind-
Fig. 8. Saturation isotherm of [3H]SN003 binding to membranes fromrat frontal cortex (A) and HEK293e cells expressing human CRF1receptors (B). Data presented are from one of three independent ex-periments performed in triplicate. KD and Bmax values were deter-mined from nonlinear regression analysis of the data using the one-site model that fits the data significantly better than the two-sitemodel. Inset, Scatchard plot of transformed saturation data. Nonspe-cific binding was defined as that occurring in the presence of 5 �MDMP696.
Fig. 9. Kinetic studies of [3H]SN003 binding to membrane prepared fromHEK293e cells recombinantly expressing human CRF1 receptors. A, equi-librium association of [3H]SN003 binding to human CRF1 receptors.Inset: linear conversion of association data, where Beq represents specificbinding at the steady state, and Bt represents the specific binding at timet. B, dissociation of [3H]SN003 binding to human CRF1 receptors where(inset) Bt represents specific binding at time t and B0 represents bindingat equilibrium. Kinetic data were fit to mono- and biexponential curves byweighted nonlinear curve-fitting; in all cases the biexponential model didnot give a significant improvement over the monoexponential model (p �0.05, F-test). Nonspecific binding was defined at each time point by 5 �MDMP696. Data shown are the mean from three independent experimentsperformed in triplicate.
64 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
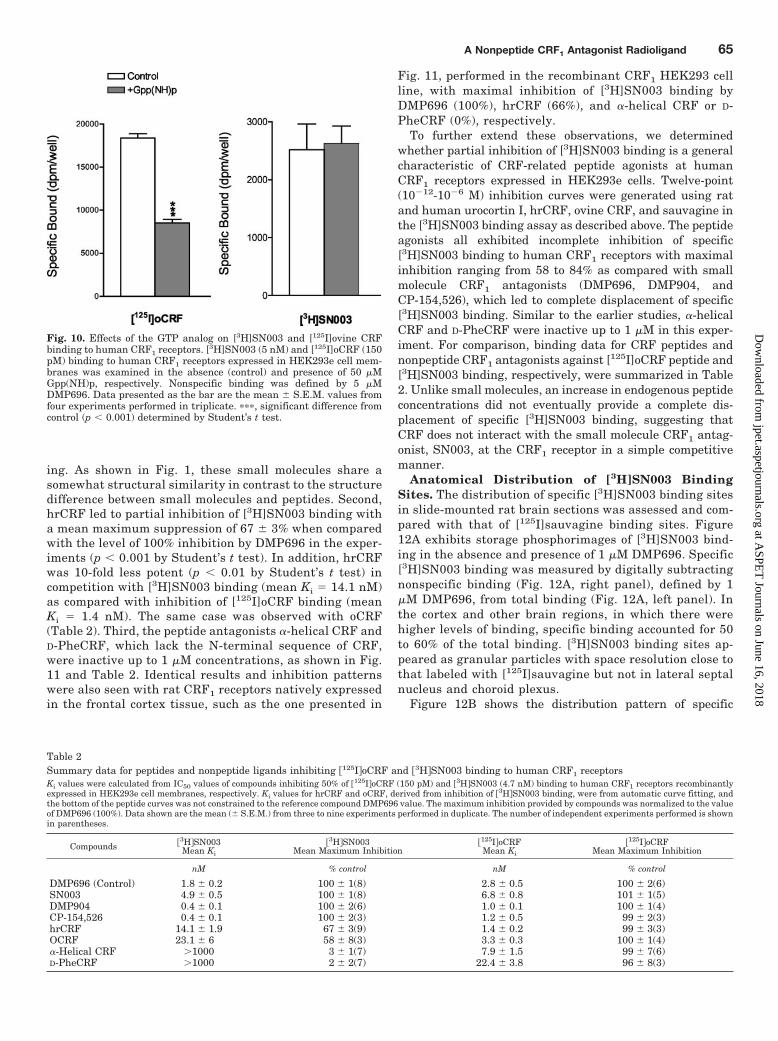
ing. As shown in Fig. 1, these small molecules share asomewhat structural similarity in contrast to the structuredifference between small molecules and peptides. Second,hrCRF led to partial inhibition of [3H]SN003 binding witha mean maximum suppression of 67 � 3% when comparedwith the level of 100% inhibition by DMP696 in the exper-iments (p 0.001 by Student’s t test). In addition, hrCRFwas 10-fold less potent (p 0.01 by Student’s t test) incompetition with [3H]SN003 binding (mean Ki � 14.1 nM)as compared with inhibition of [125I]oCRF binding (meanKi � 1.4 nM). The same case was observed with oCRF(Table 2). Third, the peptide antagonists �-helical CRF andD-PheCRF, which lack the N-terminal sequence of CRF,were inactive up to 1 �M concentrations, as shown in Fig.11 and Table 2. Identical results and inhibition patternswere also seen with rat CRF1 receptors natively expressedin the frontal cortex tissue, such as the one presented in
Fig. 11, performed in the recombinant CRF1 HEK293 cellline, with maximal inhibition of [3H]SN003 binding byDMP696 (100%), hrCRF (66%), and �-helical CRF or D-PheCRF (0%), respectively.
To further extend these observations, we determinedwhether partial inhibition of [3H]SN003 binding is a generalcharacteristic of CRF-related peptide agonists at humanCRF1 receptors expressed in HEK293e cells. Twelve-point(10�12-10�6 M) inhibition curves were generated using ratand human urocortin I, hrCRF, ovine CRF, and sauvagine inthe [3H]SN003 binding assay as described above. The peptideagonists all exhibited incomplete inhibition of specific[3H]SN003 binding to human CRF1 receptors with maximalinhibition ranging from 58 to 84% as compared with smallmolecule CRF1 antagonists (DMP696, DMP904, andCP-154,526), which led to complete displacement of specific[3H]SN003 binding. Similar to the earlier studies, �-helicalCRF and D-PheCRF were inactive up to 1 �M in this exper-iment. For comparison, binding data for CRF peptides andnonpeptide CRF1 antagonists against [125I]oCRF peptide and[3H]SN003 binding, respectively, were summarized in Table2. Unlike small molecules, an increase in endogenous peptideconcentrations did not eventually provide a complete dis-placement of specific [3H]SN003 binding, suggesting thatCRF does not interact with the small molecule CRF1 antag-onist, SN003, at the CRF1 receptor in a simple competitivemanner.
Anatomical Distribution of [3H]SN003 BindingSites. The distribution of specific [3H]SN003 binding sitesin slide-mounted rat brain sections was assessed and com-pared with that of [125I]sauvagine binding sites. Figure12A exhibits storage phosphorimages of [3H]SN003 bind-ing in the absence and presence of 1 �M DMP696. Specific[3H]SN003 binding was measured by digitally subtractingnonspecific binding (Fig. 12A, right panel), defined by 1�M DMP696, from total binding (Fig. 12A, left panel). Inthe cortex and other brain regions, in which there werehigher levels of binding, specific binding accounted for 50to 60% of the total binding. [3H]SN003 binding sites ap-peared as granular particles with space resolution close tothat labeled with [125I]sauvagine but not in lateral septalnucleus and choroid plexus.
Figure 12B shows the distribution pattern of specific
Fig. 10. Effects of the GTP analog on [3H]SN003 and [125I]ovine CRFbinding to human CRF1 receptors. [3H]SN003 (5 nM) and [125I]oCRF (150pM) binding to human CRF1 receptors expressed in HEK293e cell mem-branes was examined in the absence (control) and presence of 50 �MGpp(NH)p, respectively. Nonspecific binding was defined by 5 �MDMP696. Data presented as the bar are the mean � S.E.M. values fromfour experiments performed in triplicate. ���, significant difference fromcontrol (p 0.001) determined by Student’s t test.
Table 2Summary data for peptides and nonpeptide ligands inhibiting �125IoCRF and �3HSN003 binding to human CRF1 receptorsKi values were calculated from IC50 values of compounds inhibiting 50% of �125IoCRF (150 pM) and �3HSN003 (4.7 nM) binding to human CRF1 receptors recombinantlyexpressed in HEK293e cell membranes, respectively. Ki values for hrCRF and oCRF, derived from inhibition of �3HSN003 binding, were from automatic curve fitting, andthe bottom of the peptide curves was not constrained to the reference compound DMP696 value. The maximum inhibition provided by compounds was normalized to the valueof DMP696 (100%). Data shown are the mean (� S.E.M.) from three to nine experiments performed in duplicate. The number of independent experiments performed is shownin parentheses.
Compounds �3HSN003Mean Ki
�3HSN003Mean Maximum Inhibition
�125IoCRFMean Ki
�125IoCRFMean Maximum Inhibition
nM % control nM % control
DMP696 (Control) 1.8 � 0.2 100 � 1(8) 2.8 � 0.5 100 � 2(6)SN003 4.9 � 0.5 100 � 1(8) 6.8 � 0.8 101 � 1(5)DMP904 0.4 � 0.1 100 � 2(6) 1.0 � 0.1 100 � 1(4)CP-154,526 0.4 � 0.1 100 � 2(3) 1.2 � 0.5 99 � 2(3)hrCRF 14.1 � 1.9 67 � 3(9) 1.4 � 0.2 99 � 3(3)OCRF 23.1 � 6 58 � 8(3) 3.3 � 0.3 100 � 1(4)�-Helical CRF �1000 3 � 1(7) 7.9 � 1.5 99 � 7(6)D-PheCRF �1000 2 � 2(7) 22.4 � 3.8 96 � 8(3)
A Nonpeptide CRF1 Antagonist Radioligand 65
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from
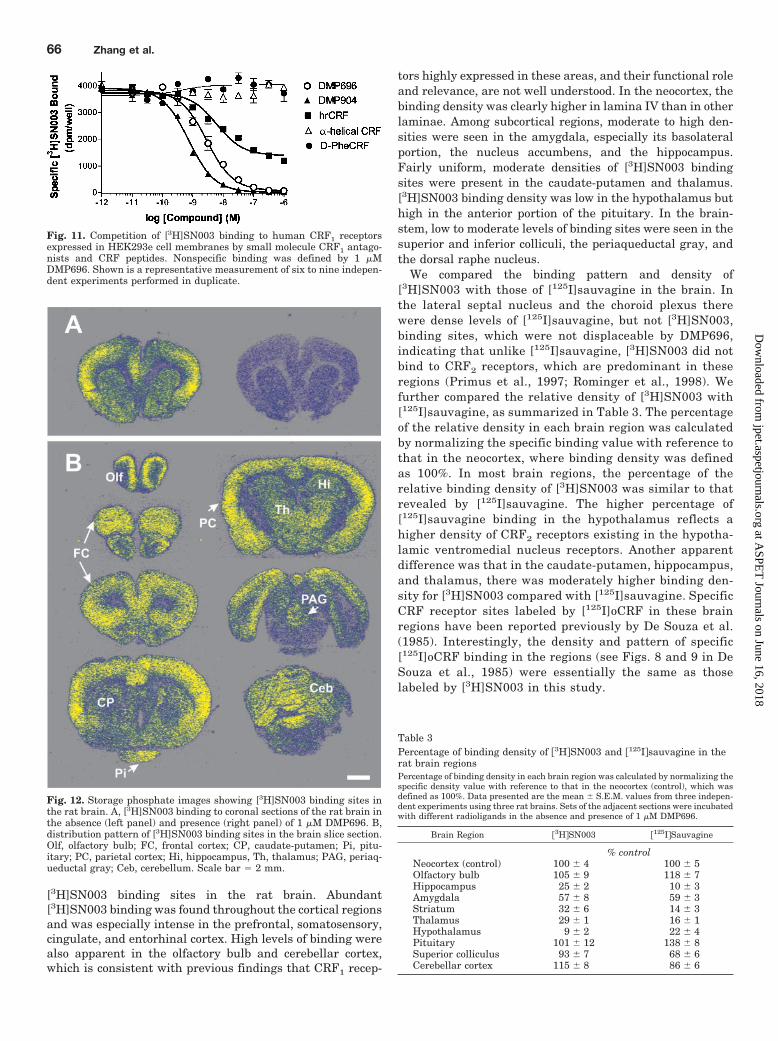
[3H]SN003 binding sites in the rat brain. Abundant[3H]SN003 binding was found throughout the cortical regionsand was especially intense in the prefrontal, somatosensory,cingulate, and entorhinal cortex. High levels of binding werealso apparent in the olfactory bulb and cerebellar cortex,which is consistent with previous findings that CRF1 recep-
tors highly expressed in these areas, and their functional roleand relevance, are not well understood. In the neocortex, thebinding density was clearly higher in lamina IV than in otherlaminae. Among subcortical regions, moderate to high den-sities were seen in the amygdala, especially its basolateralportion, the nucleus accumbens, and the hippocampus.Fairly uniform, moderate densities of [3H]SN003 bindingsites were present in the caudate-putamen and thalamus.[3H]SN003 binding density was low in the hypothalamus buthigh in the anterior portion of the pituitary. In the brain-stem, low to moderate levels of binding sites were seen in thesuperior and inferior colliculi, the periaqueductal gray, andthe dorsal raphe nucleus.
We compared the binding pattern and density of[3H]SN003 with those of [125I]sauvagine in the brain. Inthe lateral septal nucleus and the choroid plexus therewere dense levels of [125I]sauvagine, but not [3H]SN003,binding sites, which were not displaceable by DMP696,indicating that unlike [125I]sauvagine, [3H]SN003 did notbind to CRF2 receptors, which are predominant in theseregions (Primus et al., 1997; Rominger et al., 1998). Wefurther compared the relative density of [3H]SN003 with[125I]sauvagine, as summarized in Table 3. The percentageof the relative density in each brain region was calculatedby normalizing the specific binding value with reference tothat in the neocortex, where binding density was definedas 100%. In most brain regions, the percentage of therelative binding density of [3H]SN003 was similar to thatrevealed by [125I]sauvagine. The higher percentage of[125I]sauvagine binding in the hypothalamus reflects ahigher density of CRF2 receptors existing in the hypotha-lamic ventromedial nucleus receptors. Another apparentdifference was that in the caudate-putamen, hippocampus,and thalamus, there was moderately higher binding den-sity for [3H]SN003 compared with [125I]sauvagine. SpecificCRF receptor sites labeled by [125I]oCRF in these brainregions have been reported previously by De Souza et al.(1985). Interestingly, the density and pattern of specific[125I]oCRF binding in the regions (see Figs. 8 and 9 in DeSouza et al., 1985) were essentially the same as thoselabeled by [3H]SN003 in this study.
Fig. 11. Competition of [3H]SN003 binding to human CRF1 receptorsexpressed in HEK293e cell membranes by small molecule CRF1 antago-nists and CRF peptides. Nonspecific binding was defined by 1 �MDMP696. Shown is a representative measurement of six to nine indepen-dent experiments performed in duplicate.
Fig. 12. Storage phosphate images showing [3H]SN003 binding sites inthe rat brain. A, [3H]SN003 binding to coronal sections of the rat brain inthe absence (left panel) and presence (right panel) of 1 �M DMP696. B,distribution pattern of [3H]SN003 binding sites in the brain slice section.Olf, olfactory bulb; FC, frontal cortex; CP, caudate-putamen; Pi, pitu-itary; PC, parietal cortex; Hi, hippocampus, Th, thalamus; PAG, periaq-ueductal gray; Ceb, cerebellum. Scale bar � 2 mm.
Table 3Percentage of binding density of �3HSN003 and �125Isauvagine in therat brain regionsPercentage of binding density in each brain region was calculated by normalizing thespecific density value with reference to that in the neocortex (control), which wasdefined as 100%. Data presented are the mean � S.E.M. values from three indepen-dent experiments using three rat brains. Sets of the adjacent sections were incubatedwith different radioligands in the absence and presence of 1 �M DMP696.
Brain Region �3HSN003 �125ISauvagine
% controlNeocortex (control) 100 � 4 100 � 5Olfactory bulb 105 � 9 118 � 7Hippocampus 25 � 2 10 � 3Amygdala 57 � 8 59 � 3Striatum 32 � 6 14 � 3Thalamus 29 � 1 16 � 1Hypothalamus 9 � 2 22 � 4Pituitary 101 � 12 138 � 8Superior colliculus 93 � 7 68 � 6Cerebellar cortex 115 � 8 86 � 6
66 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from

DiscussionThe present study describes the in vitro pharmacological
characteristics of a novel nonpeptide radioligand,[3H]SN003, for the rat and human CRF1 receptors. SN003is a high-affinity and selective ligand for rat and humanCRF1 receptors having more than 1000-fold selectivityover human CRF2� receptors. SN003 in vitro functions asa CRF1 receptor antagonist against CRF-mediated re-sponses. Equilibrium binding studies revealed that spe-cific binding of tritium-labeled SN003 to CRF1 receptorswas saturable, reversible, and of high affinity. Antagonistcharacteristics of [3H]SN003 were also suggested by asingle class of [3H]SN003 binding sites and insensitivity toguanine nucleotides. The distribution pattern of[3H]SN003 binding sites was consistent with CRF1 recep-tor mapping in the brain, further supporting the specificlabeling of CRF1 receptors by [3H]SN003.
Both SN003 and DMP696 had 2- to 3-fold higher affini-ties (lower Ki values) for rat CRF1 receptors in the frontalcortex than in the pituitary and in recombinant CRF1
receptors expressed in HEK293e cells. The lower potencyof small molecules against CRF binding in the pituitaryand the recombinant cell line may be in part attributableto the higher density of CRF1 receptors in the membranesfrom these receptor sources. On the other hand, the dis-parity in inhibition of CRF binding observed for smallmolecules in these two rat tissues may reflect a differentialnature of CRF1 receptors expressed in various cell types.The differential effects of the small molecule antagonistson CRF binding in rat pituitary and cortical tissues couldbe the result of differing post-translational modification oraccessory proteins of CRF1 receptors from various celltypes (Grigoriadis and De Souza, 1988). Differential glyco-sylation of CRF1 receptor proteins in anterior pituitaryand brain tissues during post-translational modification(Grigoriadis and De Souza, 1989) has been demonstrated.Additional experiments are required to determine whetherdifferential post-translational modifications of CRF1 re-ceptors have functional consequences.
In functional assays of CRF-mediated ACTH release andcAMP accumulation, SN003 completely antagonized CRF ef-fects without partial or inverse agonist properties. We notedthat the functional potency of SN003 as determined in theACTH assay was approximately 10-fold lower than its bind-ing affinity at rat pituitary CRF1 receptors. This weakerpotency in functional assays is also seen with other GPCRantagonists and, more specifically, CRF1 antagonists (By-master and Falcone, 2000; Heinrichs et al., 2002). The dis-crepancy between binding and functional potencies of SN003is not well understood. It could result, in part, from differentassay conditions in the functional and binding assays, inparticular, the use of intact cells versus membrane prepara-tions and physiological buffer versus hypotonic buffer (By-master and Falcone, 2000). Besides assay conditions, differ-ential properties of small molecule CRF antagonists such astheir binding affinity and coupling, as well as competitiveversus noncompetitive nature, may also have an impact onthe potency of the difference between binding and functionalactivity of CRF ligands in the hypothalamic-pituitary-adre-nal axis.
The present work suggests that SN003 is not simply
competitive with CRF at the CRF1 receptor. The maximalnumber of binding sites for CRF is significantly reduced bySN003 in a concentration-dependent manner, indicative ofa noncompetitive interaction. Incomplete reciprocal bind-ing inhibition of peptide agonists and the small moleculeantagonist also suggests that the interaction is not simplycompetitive at CRF1 receptors; at least SN003 may providemixed competitive and/or noncompetitive inhibition. Par-tial inhibition could result from allosteric modulation andnoncompetitive inhibition by small molecules or accessibil-ity of peptides to the small molecule binding domains. Theunderlying mechanisms need to be explored in a futureinvestigation.
These data indicate that the recognition sites for smallmolecule antagonists and peptide agonists are not mutuallyexclusive on CRF1 receptors. In general, the peptide agonistbinding domain of class B receptors of the GPCR family, suchas CRF1 receptors, is predominantly formed from the largeextracellular N-terminal domain and portions of the extra-cellular loops of the receptors. Small molecule CRF1 antago-nists appear to bind to transmembrane sites in a pocketformed by helices III to VII (Liaw et al., 1997). It is possiblethat, depending on the affinity state of the receptor, limitedoverlap of binding valencies may result in a low-affinityinteraction of the N-terminal sequence of CRF with the smallmolecule binding site. The fact that CRF peptide antagonistswithout N-terminal amino acids fail to inhibit [3H]SN003binding suggests that it is the N-terminus of CRF that inter-acts with the small molecule recognition sites of the receptor,possibly sharing binding valences in the transmembrane re-gion. The findings of previous studies demonstrating that theN-terminus of CRF is important for receptor activation (Valeet al., 1981; De Souza, 1987; Beyermann et al., 1996) may berelevant to the present findings in which the N-terminus ofCRF was also crucial for CRF inhibition of small moleculebinding.
Further studies employing radiolabeled CRF peptidesand small molecule antagonists will greatly aid under-standing of molecular aspects of CRF1 receptor signalingand small molecule antagonism. Additionally, this infor-mation may have implications for other G protein-coupledreceptors with endogenous peptide agonists. This does ap-pear to be the case for the neurokinin-1 and -2 receptors,since partial inhibition and low potencies of endogenouspeptides, substance P and neurokinin A, in displacement ofsmall molecule antagonist radioligand binding (Rosenkildeet al., 1994) have been observed with those receptors.Considered together, the data suggest that the interac-tions between small molecules are more competitive thanthat between the small molecule and peptide, and bindingdomains for peptides and small molecule antagonists arenot mutually exclusive.
Two nonpeptide CRF1 antagonists, CP-154,526 and anantalarmin analog, have recently been radiolabeled (Tianet al., 2001; Keller et al., 2002). The tritium-labeled CP-154,526 was used for assessing pharmacokinetics andblood-brain barrier penetration, and the antalarmin ana-log was developed as a positron emission tomography li-gand. However, in these studies there are no descriptionsof radioligand binding profiles in either tissues or cells ex-pressing CRF1 receptors. Tritiated SN003 is the first nonpep-tidic CRF1 antagonist radioligand shown to be capable of de-
A Nonpeptide CRF1 Antagonist Radioligand 67
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from

tecting specific binding in rat brain tissues. The bindingpattern of [3H]SN003 in the brain was consistent with thedistribution of CRF1 receptors revealed previously by bind-ing autoradiography using the radioligand [125I]oCRF (DeSouza et al., 1985; Aguilera et al., 1987) or by in situhybridization of CRF1 mRNA (Potter et al., 1994). Thesensitivity of [3H]SN003 binding was moderate consider-ing that it was 50 to 60% specific over total binding in boththe homogenized membrane and slide-mounted brain sec-tion binding assays. The moderate sensitivity was, per-haps, caused by several factors, including the ”sticky“ li-pophilic property of SN003 and the low energy status of thelabeled isotope tritium. In addition, storage phosphorim-aging used in the autoradiography study may have com-promised the sensitivity in terms of space resolution. How-ever, this technique proved to be very effective foracquiring images marked with tritium-labeled ligandssuch as [3H]SN003 in as little as 7 days, compared withlonger exposure times required for autoradiographic films.These studies may provide a foundation for studying invivo binding of CRF1 antagonists and developing high-energy isotope-labeled, small molecule ligands with appli-cations in clinical positron emission tomography/singlephoton emission computed tomography studies of CRF1
antagonists.In summary, we describe the in vitro characterization of a
high-affinity, selective nonpeptidic antagonist radioligand,[3H]SN003, for CRF1 receptors. Since this radioligand pos-sesses an excellent signal/noise ratio in a recombinant hu-man CRF1 cell line and distinct features compared withpeptide ligands, it provides a useful tool to understand smallmolecule antagonism of CRF at CRF1 receptors. This infor-mation may prove helpful in drug design and development ofsmall molecule CRF1 antagonists for treatment of affectivedisorders and stress-related diseases.
Acknowledgments
We would like to thank Dr. Rebecca Taub for review and helpfulsuggestions on the manuscript.
ReferencesAguilera G, Millan MA, Hauger RL, and Catt KJ (1987) Corticotropin-releasing
factor receptors: distribution and regulation in brain, pituitary, and peripheraltissues. Ann NY Acad Sci 512:48–66.
Bakshi VP, Smith-Roe S, Newman SM, Grigoriadis DE, and Kalin NH (2002)Reduction of stress-induced behavior by antagonism of corticotropin-releasinghormone 2 (CRH2) receptors in lateral septum or CRH1 receptors in amygdala.J Neurosci 22:2926–2935.
Bakthavatchalam R, Arvanitis AG, Beck JP, Cain GA, Chorvat RJ, Gilligan PJ, andOlson RE (1997) Arglamino fused pyridines and pyrimidines. International Pub-lication Number WO 97/35539.
Beyermann M, Fechner K, Furkert J, Krause E, and Bienert M (1996) A single-pointslight alteration set as a tool for structure-activity relationship studies of ovinecorticotropin releasing factor. J Med Chem 39:3324–3330.
Bymaster FP and Falcone JF (2000) Decreased binding affinity of olanzapine andclozapine for human muscarinic receptors in intact clonal cells in physiologicalmedium. Eur J Pharmacol 390:245–248.
Dautzenberg FM and Hauger RL (2002) The CRF peptide family and their receptors:yet more partners discovered. Trends Pharmacol Sci 23:71–77.
Davis M (1992) The role of the amygdala in fear-potentiated startle: implications foranimal models of anxiety. Trends Pharmacol Sci 13:35–41.
De Souza EB (1987) Corticotropin-releasing factor receptors in the rat central ner-vous system: Characterization and regional distribution. J Neurosci 7:88–100.
De Souza EB and Grigoriadis DE (1998) Corticotropin-releasing factor. Physiology,pharmacology and role in central nervous system and immune disorders, in Psy-chopharmacology: The Fourth Generation of Progress (Bloom FE and Kupfer DJeds) pp 505–517, Raven Press, New York.
De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, and Kuhar MJ (1985)Corticotropin-releasing factor receptors are widely distributed within the rat cen-tral nervous system: an autoradiographic study. J Neurosci 5:3189–3203.
Gilligan PJ, Robertson DW, and Zaczek R (2000) Corticotropin releasing factor (CRF)
receptor modulators: progress and opportunities for new therapeutic agents.J Med Chem 43:1641–1660.
Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P, Geslin M, Scatton B,Maffrand JP, and Soubrie P (2002) 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotro-phin-releasing factor1 receptor antagonist. II. Characterization in rodent modelsof stress-related disorders. J Pharmacol Exp Ther 301:333–345.
Grigoriadis DE and De Souza EB (1988) The brain corticotropin-releasing factor(CRF) receptor is of lower apparent molecular weight than the CRF receptor inanterior pituitary: evidence from chemical cross-linking studies. J Biol Chem263:10927–10931.
Grigoriadis DE and De Souza EB (1989) Heterogeneity between brain and pituitarycorticotropin-releasing factor receptors is due to differential glycosylation. Endo-crinology 125:1877–1888.
Gully D, Geslin M, Serva L, Fontaine E, Roger P, Lair C, Darre V, Marcy C, RoubyPE, Simiand J, et al. (2002) 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A): a potent and selective corticotrophin-releasing factor1 receptor antagonist. I. Biochemical and pharmacologicalcharacterization. J Pharmacol Exp Ther 301: 322–332.
Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL,Atkinson AJ, Schulkin J, Contoreggi C, et al. (2000) Oral administration of acorticotropin-releasing hormone receptor antagonist significantly attenuates be-havioral, neuroendocrine and autonomic responses to stress in primates. Proc NatlAcad Sci USA 97:6079–6084.
Hauger RL, Dautzenberg FM, Flaccus A, Liepold T, and Spiess J (1997) Regulationof corticotropin-releasing factor receptor function in human Y-79 retinoblastomacells: rapid and reversible homologous desensitization but prolonged recovery.J Neurochem 68:2308–2316.
He L, Gilligan PJ, Zaczek R, Fitzgerald LW, McElroy J, Shen HS, Saye JA, KalinNH, Shelton S, Christ D, et al. (2000) 4-(1,3-Dimethylthoxyprop-2-ylamino)-2,7-dimethyl-8-(2,4-dichlorophenyl)-pyrazolo[1,5-a]-1,3,5-triazine: a potent, orally bio-available CRF1 receptor antagonist. J Med Chem 43:449–456.
Heinrichs SC, De Souza EB, Schulteis G, Lapsansky JL, and Grigoriadis DE (2002)Brain penetrance, receptor occupancy and antistress in vivo efficacy of a smallmolecule corticotropin releasing factor type I receptor selective antagonist. Neu-ropsychopharmacology 27:194–202.
Horlick RA, Sperle K, Breth LA, Reid CC, Shen ES, Robbins AK, Cooke GM, andLargent BL (1997) Rapid generation of stable cell lines expressing corticotropin-releasing hormone receptor for drug discovery. Protein Expr Purif 9:301–308.
Keck ME and Holsboer F (2001) Hyperactivity of CRH neuronal circuits as a targetfor therapeutic interventions in affective disorders. Peptides 22:835–844.
Keller C, Bruelisauer A, Lemaire M, and Enz A (2002) Brain pharmacokinetics of anon-peptidic corticotropin-releasing factor antagonist. Drug MetabDispos 30:173–176.
Kostich WA, Chen A, Sperle K, and Largent BL (1998) Molecular identification andanalysis of a novel human corticotropin-releasing factor (CRF) receptor: theCRF2� receptor. Mol Endocrinol 12:1077–1085.
Lewis K, Li C, Perrin MH, Blount A, Kunitake KS, Donaldson C, Vaughan J, ReyesTM, Gulyas J, Fischer W, et al. (2001) Identification of urocortin III, an additionalmember of the corticotropin-releasing factor (CRF) with high affinity for the CRF2receptors. Proc Natl Acad Sci USA 98:7570–7575.
Li YW, Yan XX, Hill G, Gilligan PJ, Bakthavatchalam R, Prakash SR, and RobertZaczek (2001) Autoradiographic of [3H]-SN003, a small molecule corticotropinreleasing factor receptor type-1 antagonist, in the rat brain. Soc Neurosci Abstr27:414.1.
Liaw CW, Grigoriadis DE, Lorang MT, De Souza EB, and Maki RA (1997) Localiza-tion of agonist- and antagonist-binding domains of human corticotropin-releasingfactor receptors. Mol Endocrinol 11:2048–2053.
Maciag CM, Gersham D, Gilligan PJ, He L, Dowling K, Ko T, Levine S, and SmithMA (2002) Effects of a non-peptide CRF antagonist (DMP696) on the behavioraland endocrine sequelae of maternal separation. Neuropsychopharmacology 26:574–582.
McCarthy JR, Heinrichs SC, and Grigoriadis DE (1999) Recent advances with theCRF1 receptor: design of small molecule inhibitors, receptor subtypes and clinicalindications. Curr Pharm Des 5:289–315.
McElroy JF, Ward KA, Zeller KL, Jones KW, Gilligan PJ, He L, and Lelas S (2002)The CRF1 receptor antagonist DMP696 produces anxiolytic effects and inhibits thestress-induced hypothalamic-pituitary-adrenal axis activation without sedation orataxia in rats. Psychopharmacology 165:86–92.
Owens MJ and Nemeroff (1991) Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev 43:425–473.
Potter E, Sutton S, Donaldson C, Chen R, Perrin M, Lewis K, Sawchenko PE, andVale W (1994) Distribution of corticotropin-releasing factor receptor mRNA ex-pression in the rat brain and pituitary. Proc Natl Acad Sci USA 9:8777–8781.
Primus RJ, Ywvich E, Baltazar C, and Gallager W (1997) Autoradiographic local-ization of CRF1 and CRF2 binding sites in adult rat brain. Neuropyschopharma-cology 17:308–316.
Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB,Gulyas J, Rivier W, Vale W, et al. (2001) Urocortin II: a member of the cortico-tropin-releasing factor (CRF) neuropeptide family that is selectively bound by type2 CRF receptors. Proc Natl Acad Sci USA 98:2843–2848.
Rominger DH, Rominger CM, Fitzgerald LW, Grzanna R, Largent BL, and ZaczekR (1998) Characterization of [125I]sauvagine binding to CRH2 receptors: mem-brane homogenate and autoradiographic studies. J Pharmacol Exp Ther 286:459 – 468.
Rosenkilde MM, Cahir M, Gether U, Hjorth SA, and Schwartz TW (1994) Mutationsalong transmembrane segment II of the NK-1 receptor affect substance P compe-
68 Zhang et al.
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from

tition with non-peptide antagonists but not substance P binding. J Biol Chem269:28160–28164.
Schulz DW, Mansbach RS, Sprouse J, Braselton JP, Collins J, Corman M, DunaiskisA, Faraci S, Schmidt AW, Seeger T, et al. (1996) CP-154, 526: a potent andselective nonpeptide antagonist of corticotropin releasing factor receptors. ProcNatl Acad Sci USA 93:10477–10482.
Tian X, Hsin LW, Webster EL, Contoreggi C, Chrousos GP, Gold PW, Habib K, AyalaA, Eckelman WC, Jacobson AE, et al. (2001) The development of a potential singlephoton emission computed tomography (SPECT) imaging agent for the corticotrop-in-releasing hormone receptor type1. Bioorg Med Chem Lett 11:331–333.
Vale W, Grant G, Amoss M, Blackwell R, and Guillemin R (1972) Culture of enzy-matically dispersed anterior pituitary cells: functional validation of a method.Endocrinology 91:562–572.
Vale W, Spiess J, Rivier C, and Rivier J (1981) Characterization of a 41 residue ovinehypothalamic peptide that stimulates secretion of corticotropin and �-endorphin.Science (Wash DC) 213:1394–1397.
Zhang G, Huang N, Bakthavatchalam R, Prakash SR, Gilligan PJ, and Zaczek R(2001) Characterization of a non-peptide antagonist radioligand [3H]SN003at corticotropin releasing factor 1 (CRF1) receptors. Soc Neurosci Abstr 27:179.17.
Address correspondence to: Dr. Ge Zhang, The Bristol-Myers Squibb Com-pany, Building 21/Room 2344A, 311 Pennington-Rocky Hill Rd., Hopewell, NJ08534. E-mail: [email protected]
A Nonpeptide CRF1 Antagonist Radioligand 69
at ASPE
T Journals on June 16, 2018
jpet.aspetjournals.orgD
ownloaded from





























