Embed Size (px)
Citation preview

THERAPY
Pharmacokinetic and Pharmacodynamic Effects of Diltiazem
MARK S. SMITH, MD, CHACKO P. VERGHESE, MS,
DAVID G. SHAND, MB, PhD, and EDWARD L. C. PRITCHEIT, MD
The pharmacokinetic and pharmacodynamic effects of diltiazem were studied in 8 patients after a short intravenous infusion (20 mg over 10 minutes), a single oral dose (80 or 90 mg), and repeated oral administration (80 or 90 mg every 8 hours for 18 doses). Dlltiaxem levels decreased in a triexpo- nential manner after intravenous infusion. Terminal hatf-lives after intravenous, single oral, and repeated oral administration were not significantly different (4.5 f 1.3,3.7 f 0.8, and 4.9 f 0.4 hours, respec- tively). The kinetic effects of oral diltlazem were nonlinear. With repeated oral administration, there was accumulation of both diltiazem and its metab- olite, deacetyldlltiazem. The diltiazem area under the time versus concentration curve increased by
a factor of 2.39 f 0.42 (p = 0.00002). Most patients showed a double peaked time versus concentration curve after oral administration, indicating possible enterohepatic recirculation.
After intravenous administration, there was a substantial increase in the P-R interval (14.3 f 5.4% ). Although only small changes in P-R interval were seen with a single oral dose, with chronic ad- ministration there was persistent P-R interval pro- longation, peaking at 17.3 f 5.8% over control. Counterclockwise hysteresis was present in the P-R interval versus plasma diltiazem concentration curve afler intravenous administration. Only small changes were seen in heart rate and blood pres- sure.
Diltiazem hydrochloride (d-cis-3-acetoxy-2,3-dihy- dro - 5 - [2 - (dimethyl - amino) -ethyl] -2- (p-methoxy- phenyl)-l,Sbenzothiazepin-4(5H)-one hydrochloride) is a new benzothiazepine derivative with cardiac and smooth muscle calcium-channel blocking activity. It is a potent vasodilator useful in the treatment of angina pectoris (due to coronary artery spasm or atheroscle- rosis), systemic hypertension, pulmonary hypertension, and hypertrophic cardiomyopathies.1-5 Limited infor- mation is available about the clinical pharmacologic effects of diltiazem because the assay procedure for plasma drug levels has been cumbersome and not widely available. In this report we describe the detailed phar- macokinetic effects of oral and intravenous diltiazem measured using a new sensitive and specific high per- formance liquid cbromatographic assay. In addition, we
From the Divisions of Cardiology and Clinical Pharmacology, Depart- ments of Medicine and Pharmacology. Duke University Medical Center, Durham, North Carolina. This study was supported in part by &ant RR-30 from the The General Clinical Research Centers Program, Di- vision of Research Resources, Grants HL24920, LM03373, HL07101, and HL28899 from the Naticnal institutes of Health, Bethesda. Maryland; and by (fant 500-08Y from Mari Laboratories, Kansas Cii, Missoui. Manuscript received December 3, 1982; revised manuscript received January 31, 1983, accepted February 2, 1983.
Address for reprints: Edward L. C. Pritchett, MD. Box 3477, Duke University Medical Center, Durham, North Carolina 27710.
describe the pharmacodynamic aspects of P-R interval prolongation with diltiazem, which is a measure of its negative dromotropic effect.
Methods
Eight patients with a history of paroxysmal atria1 tachy- cardia (3 women and 5 men, aged 22 to 58 years) were admit- ted to the Duke University Clinical Research Unit. One pa- tient had a delta wave on the electrocardiogram recorded during sinus rhythm (Wolff-Parkinson-White syndrome). Three patients had coexisting diseases: 1 each with chemical diabetes, chronic obstructive pulmonary disease, and type IV hyperlipidemia. All medications were discontinued at least 5 half-lives before admission, except in 1 patient who required continuous theophylline therapy.
All studies were performed while patients were in normal sinus rhythm. Patients were given nothing by mouth and were kept at complete bed rest from midnight forward on each study day. Between 7 and 8 A.M. a Teflon@ catheter (Quick Cath 2N114, Travenol Laboratories) was inserted into a su- perficial forearm vein and capped with a sterile obturator (Quick Cath 2N1152, Travenol Laboratories). The catheters were kept open by the sterile obturators and heparin was not used. On days when intravenous studies were performed, a second Teflon catheter was inserted into the opposite arm and connected to a continuous infusion of 5% dextrose solution. An electrocardiographic telemetry transmitter (Hewlett- Packard model 78100A and 78101A) was used to record a

1370 DILTIAZEM
modified electrocardiographic lead II (right shoulder negative, cardiac apex positive). The electrocardiogram was displayed continuously on an oscilloscope and recorded on an Elema Mingograph model 1600 ink-jet recorder. Between 8 and 9 A.M., electrocardiogram rhythm strips that included at least 10 consecutive sinus beats were recorded at a paper speed of 100 mm/s, and blood pressure was measured by a sphygmo- manometer at 15-minute intervals. At exactly 9 A.M. the medication was given. For intravenous administration, 20 mg of diltiazem (Cardizem, Marion Laboratories) in a solution of 1 mg/ml of sterile water was given over 10 minutes by con- stant infusion pump (Harvard Apparatus infusion withdrawal pump). Blood samples were obtained before the infusion began and at t = 0,2,3,5,7,10,15,30,45, and 60 minutes and 2,3,4,5,6,8,10,12,24, and 48 hours after completion of the infusion. Blood pressure was measured and electrocardiogram rhythm strips were recorded at the time each blood sample was drawn.
Two days after the intravenous infusion study, a single oral dose of diltiazem was given (60 or 90 mg as two or three 30 mg tablets of the Tanabe formulation). Blood samples were ob- tained, electrocardiographic rhythm strips were recorded, and blood pressure was measured immediately before the tablets were given and at t = 0,0.5,1,1.5,2,2.5,3,3.5,4,4.5,5,5.5,6, 6.5,7,8,9,10,11, and 12 hours after the dose was given. After the 12-hour sample was obtained, patients were given oral diltiazem every 6 hours, continuing the dose they received for the initial oral study (60 mg for the first 4 patients and 90 mg for the last 4 patients). Blood samples were obtained each morning at 9 A.M. to determine trough plasma diltiazem levels. Four days after the single-dose study, a repeat pharmacoki- netic study was done over the same 12-hour period to assess steady-state pharmacokinetic effects after the 16th dose.
Blood samples were drawn into plastic syringes and im- mediately transferred to silanized glass tubes with 10 U/ml of heparin. Plasma was separated and frozen at -2O’C and later analyzed for diltiazem and deacetyldiltiazem using a high performance liquid chromatographic method described by Verghese et al.‘j The mean P-R interval and cardiac cycle on 10 consecutive heart beats recorded at the specified sampling times were measured using an interactive computer program, except for the patient with the Wolff-Parkinson-Wh% syn- drome who was excluded from analysis of electrocardiographic data. Electrocardiograms were measured and blood samples assayed blindly by personnel in separate laboratories.
Initial estimates of the intravenous parameters were ob- tained using linear regression of time versus log of concen- tration plots. These estimates were refined by nonlinear re- gression analysis for both a 2 and a 3 exponential model. Half-lives (t I/‘) for the oral studies were determined using linear regression of the terminal portion of the time versus concentration curves. The area under the time versus con- centration curve (AUC) was estimated by the trapezoid rule. The accumulation factor (A) was calculated using the fol- lowing equation: A = AUCiaO - G/AUCiO - ~0 where AUCi60 - 6 is the AUC after the 16th oral dose from 0 to 6 hours, and AUCiO - m is the AUC after the first oral dose extrapolated to infinity. The apparent bioavailability (F), was calculated using the equation: F = (AUC&AUCi,) X (DiJDorai), where D,, is the intravenous dose and Dora1 the oral dose given. Volume of distribution at steady state (Vd,,) was calculated using the equation: Vd,, = [(A/a2 + B/p2 + Gl~2)/(AUCi~0 - -)I x clearance. The half-lives of the intravenous, the first oral, and the steady-state oral studies were compared using the analysis of variance. Other comparisons were analyzed using the paired t test.
Patient 5 received only 17 mg during the intravenous study because the infusion rate was inappropriately slow. Plasma
May 1. 1983 THE AMERICAN JOURNAL OF CARDIOLOGY Volume 51 1371
levels and all dose-dependent parameters were extrapolated to a dose of 20 mg in this patient.
Results
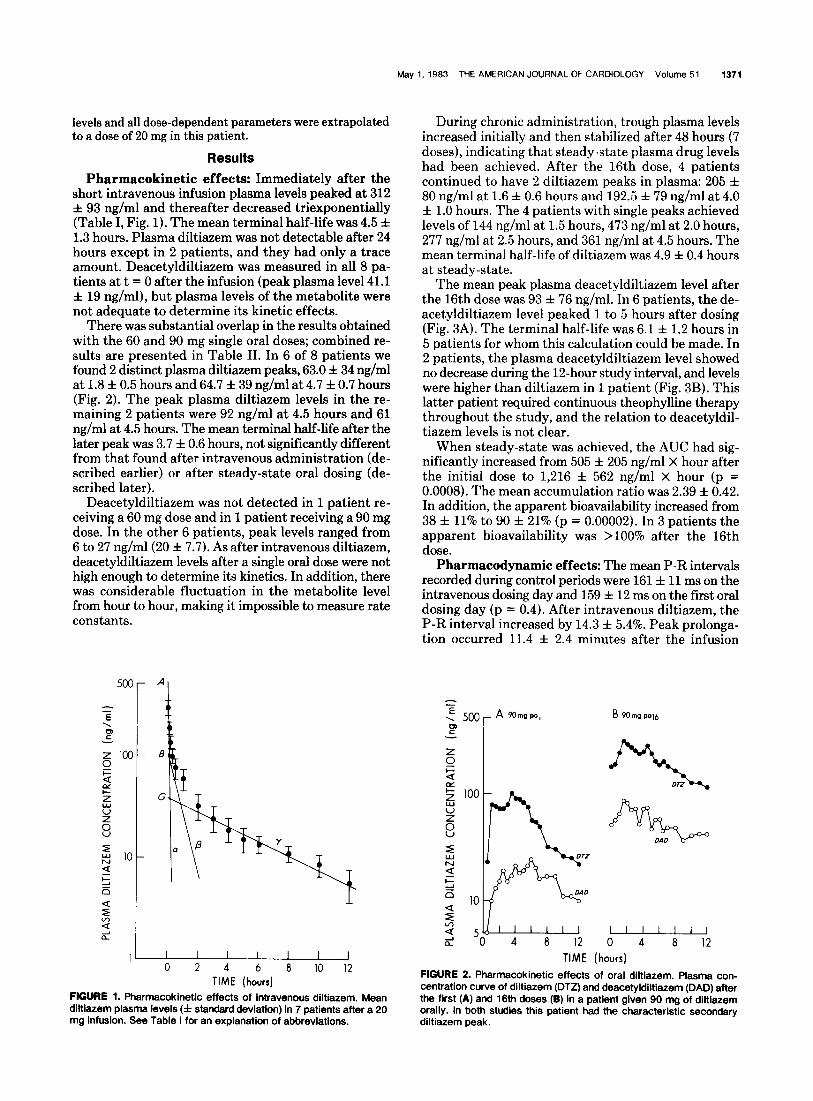
Pharmacokinetic effects: Immediately after the short intravenous infusion plasma levels peaked at 312 f 93 ng/ml and thereafter decreased triexponentially (Table I, Fig. 1). The mean terminal half-life was 4.5 f 1.3 hours. Plasma diltiazem was not detectable after 24 hours except in 2 patients, and they had only a trace amount. Deacetyldiltiazem was measured in all 8 pa- tients at t = 0 after the infusion (peak plasma level 41.1 f 19 ng/ml), but plasma levels of the metabolite were not adequate to determine its kinetic effects.
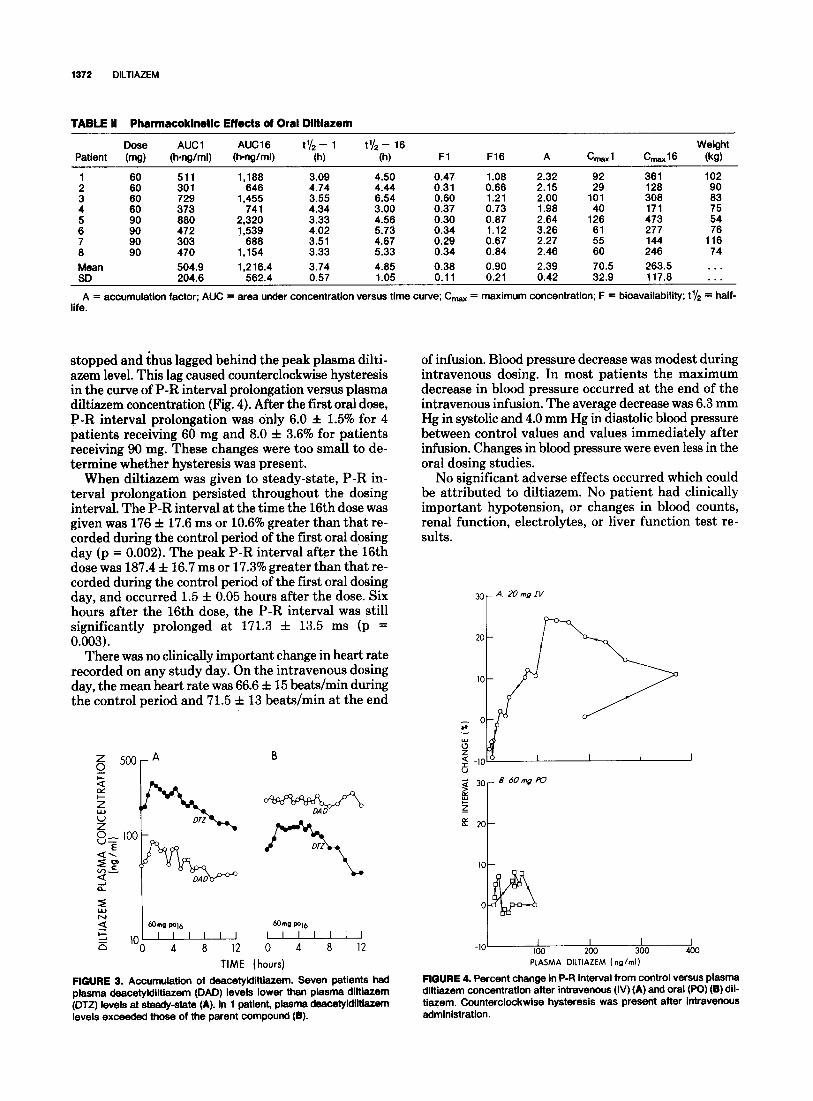
There was substantial overlap in the results obtained with the 60 and 90 mg single oral doses; combined re- sults are presented in Table II. In 6 of 8 patients we found 2 distinct plasma diltiazem peaks, 63.0 f 34 ng/ml at 1.8 f 0.5 hours and 64.7 f 39 ng/ml at 4.7 f 0.7 hours (Fig. 2). The peak plasma diltiazem levels in the re- maining 2 patients were 92 ng/ml at 4.5 hours and 61 ng/ml at 4.5 hours. The mean terminal half-life after the later peak was 3.7 f 0.6 hours, not significantly different from that found after intravenous administration (de- scribed earlier) or after steady-state oral dosing (de- scribed later).
Deacetyldiltiazem was not detected in 1 patient re- ceiving a 60 mg dose and in 1 patient receiving a 90 mg dose. In the other 6 patients, peak levels ranged from 6 to 27 ng/ml(20 f 7.7). As after intravenous diltiazem, deacetyldiltiazem levels after a single oral dose were not high enough to determine its kinetics. In addition, there was considerable fluctuation in the metabolite level from hour to hour, making it impossible to measure rate constants.
500 r
6 8 10 12 TIME (hours]
FIGURE 1. Pharmacokinetic effects of intravenous diltiazem. Mean diltiazem plasma levels (f standard deviation) in 7 patients after a 20 mg infusion. See Table I for an explanation of abbreviations.
During chronic administration, trough plasma levels increased initially and then stabilized after 48 hours (7 doses), indicating that steady-state plasma drug levels had been achieved. After the 16th dose, 4 patients continued to have 2 diltiazem peaks in plasma: 205 f 80 ng/ml at 1.6 f 0.6 hours and 192.5 f 79 ng/ml at 4.0 f 1.0 hours. The 4 patients with single peaks achieved levels of 144 ng/ml at 1.5 hours, 473 ng/ml at 2.0 hours, 277 ng/ml at 2.5 hours, and 361 ng/ml at 4.5 hours. The mean terminal half-life of diltiazem was 4.9 f 0.4 hours at steady-state.
The mean peak plasma deacetyldiltiazem level after the 16th dose was 93 f 76 ng/ml. In 6 patients, the de- acetyldiltiazem level peaked 1 to 5 hours after dosing (Fig. 3A). The terminal half-life was 6.1 f 1.2 hours in 5 patients for whom this calculation could be made. In 2 patients, the plasma deacetyldiltiazem level showed no decrease during the 12-hour study interval, and levels were higher than diltiazem in 1 patient (Fig. 3B). This latter patient required continuous theophylline therapy throughout the study, and the relation to deacetyldil- tiazem levels is not clear.
When steady-state was achieved, the AUC had sig- nificantly increased from 505 f 205 ng/ml X hour after the initial dose to 1,216 f 562 ng/ml X hour (p = 0.0008). The mean accumulation ratio was 2.39 f 0.42. In addition, the apparent bioavailability increased from 38 f 11% to 90 f 21% (p = 0.00002). In 3 patients the apparent bioavailability was >lOO% after the 16th dose.
Pharmacodynamic effects: The mean P-R intervals recorded during control periods were 161 f 11 ms on the intravenous dosing day and 159 f 12 ms on the first oral dosing day (p = 0.4). After intravenous diltiazem, the P-R interval increased by 14.3 f 5.4%. Peak prolonga- tion occurred 11.4 f 2.4 minutes after the infusion
TIME (hours)
FIGURE 2. Pharmacokinetic effects of oral diltiazem. Plasma con- centration curve of diltiazem (DTZ) and deacetyldiltiazem (DAD) after the first (A) and 16th doses (B) in a patient given 90 mg of diltiazem orally. In both studies this patient had the characteristic secondary diltiazem peak.
1272 DILTIAZEM
TABLE II Pharmacokinetlc Effects of Oral Dlltlazem
Dose Patient (mg)
- AUCl AUC16 t’/* - 1 t’/s - 16 Weight
(hang/ml) (h-ng/ml) (h) (h) Fl F16 A Cmaxl C-16 (kg)
1 60 511
: :8 301 729 4
: t:
373
660 472
3 ix 90 303 470
1,188 3.09 4.50 0.47 1.08 2.32 92 646 4.74 4.44 0.31 0.66 2.15 29
1,455 3.55 6.54 0.80 1.21 2.00 101 741 4.34 3.00 0.37 0.73 1.98 40
2.320 3.33 4.56 0.30 0.87 2.64 126 1,539 4.02 5.73 0.34 1.12 3.26
688 3.51 4.67 0.29 0.67 2.27 :: 1,154 3.33 5.33 0.34 0.84 2.48 60
- 361 102 128 308 :: 171 473 5: 277 144 1;: 246 74
Mean 504.9 1,216.4 3.74 4.85 0.38 0.90 2.39 70.5 263.5 . . . SD 204.8 562.4 0.57 1.05 0.11 0.21 0.42 32.9 117.8 . .
A = accumulation factor: AlJC = area under concentration versus time curve; C, = maximum concentration; F = bioavailability; t’/s = half- life.
stopped and thus lagged behind the peak plasma dilti- of infusion. Blood pressure decrease was modest during azem level. This lag caused counterclockwise hysteresis intravenous dosing. In most patients the maximum in the curve of P-R interval prolongation versus plasma decrease in blood pressure occurred at the end of the diltiazem concentration (Fig. 4). After the first oral dose, intravenous infusion. The average decrease was 6.3 mm P-R interval prolongation was only 6.0 f 1.5% for 4 Hg in systolic and 4.0 mm Hg in diastolic blood pressure patients receiving 60 mg and 8.0 f 3.6% for patients between control values and values immediately after receiving 90 mg. These changes were too small to de- infusion. Changes in blood pressure were even less in the termine whether hysteresis was present. oral dosing studies.
When diltiazem was given to steady-state, P-R in- terval prolongation persisted throughout the dosing interval. The P-R interval at the time the 16th dose was given was 176 f 17.6 ms or 10.6% greater than that re- corded during the control period of the first oral dosing day (p = 0.002). The peak P-R interval after the 16th dose was 187.4 f 16.7 ms or 17.3% greater than that re- corded during the control period of the first oral dosing day, and occurred 1.5 f 0.05 hours after the dose. Six hours after the 16th dose, the P-R interval was still significantly prolonged at 171.3 f 13.5 ms (p = 0.003).
No significant adverse effects occurred which could be attributed to diltiazem. No patient had clinically important hypotension, or changes in blood counts, renal function, electrolytes, or liver function test re- sults.
There was no clinically important change in heart rate recorded on any study day. On the intravenous dosing day, the mean heart rate was 66.6 f 15 beats/min during the control period and 71.5 f 13 beats/min at the end
i ,,1”;.:“, , , 12 ] 0 4 a
0 ,“f’?, 4 a , , 12 ,
TIME (hours)
FIGURE 3. Accumulation of deacetyldittiazem. Seven patients had plasma deacetyldilttazem (DAD) levels lower than plasma diltiazem (DTZ) levels at steady-state (A). In 1 patient, plasma deacetyldittazem levels exceeded those of the parent compound (B).
-IO/’ loo 200 300
PLASMA DILTIAZEM ( nglml)
FIGURE 4. Percent change in P-R interval from control versus plasma dittfazem concentration after intravenous (IV) (A) and oral (PO)(B) dil- tiazem. Counterclockwise hysteresis was present after intravenous administration.

May 1. 1983 THE AMERICAN JOURNAL OF CARDIOLOGY Volume 51 1373
Discussion
Diltiazem is a new calcium-channel blocking drug that probably will soon be widely used for the treatment of angina. Important new findings in our study are (1) nonlinear kinetic effects leading to a significant accu- mulation with chronic oral dosing, (2) substantial levels of deacetyldiltiazem (exceeding those of the parent compound in some patients) that might contribute to therapeutic or toxic effects, (3) double peak in plasma drug concentration versus time curve consistent with enterohepatic recirculation, (4) sustained atrioven- tricular nodal slowing measured as P-R interval pro- longation with chronic dosing, (5) terminal half-life 4.5 hours and triexponential elimination after intravenous administration, and (6) counterclockwise hysteresis in P-R interval versus plasma drug concentration curves after rapid infusion.
Oyama et al7 previously reported a l&‘-life of 2 hours for intravenous diltiazem, which is substantially shorter than our value of 4.5 hours. This discrepancy may be explained because Oyama et al used a 2-compartment model to describe their data. In the present study, dil- tiazem elimination was clearly triexponential in all patients. Our values for the half-life after first oral and steady-state oral dosing (3.7 and 4.9 hours, respectively) are more consistent with those previously reported for single oral doses.s1° In contrast to verapamil, which has a half-life that increases with chronic oral dosing, we found that diltiazem’s half-life was not affected by route of administration or length of treatment.ll Like those of verapamil, our pharmacokinetic data are consistent with high “first-pass elimination” in the liver with nonlinear kinetic effects that cause unexpected drug accumulation during chronic administration. Therefore, bioavailability was low with the first oral dose (38%) but high (90%) with steady-state oral dosing, This finding differs from the findings of Rovei et a1,8 who reported only slight accumulation, and Morselli et al9 who re- ported plasma drug level actually decreasing over time in some patients. The reason for this discrepancy is not obvious since we saw 1100% increase in the AUC (or 250% reduction in clearance) in each patient studied. The importance of this finding is clear: like verapamil and propranolol, diltiazem given on a long-term basis leads to unexpected accumulation.li~iz This now ap- pears to be the rule rather than the exception for drugs with high first-pass metabolism; this is presumably due to saturation of the elimination pathway for diltiazem. This mechanism is also suggested by previous work showing a nonlinear increase in AUC with increasing dose and higher bioavailability with larger doses.lc,la The increase we found in apparent bioavailability with time also suggests saturation.
The double peak in plasma concentration-time curves after oral administration is a new and unusual finding, but not entirely unexpected. While diltiazem is rapidly and almost completely absorbed, its main route of ex- cretion is in the feces.14 Using radiolabeled diltiazem, Sakuma et all5 showed excretion into the stomach from fundic glands and into the small intestines from the bile ducts. Meshi et all4 also showed enterohepatic recir-
culation using a cross-cannulation experiment in rats. Piepho et a113 reported that 3 of 6 dogs given oral dilti- azem also had secondary peaks. While this could be explained by conversion of deacetyldiltiazem back to diltiazem or the presence of an immediate and delayed release of diltiazem from the tablets, recirculation seems more likely. The finding of >lOO% bioavailability in 3 patients is probably due to a combination of a small amount of enterohepatic recirculation amplified by a reduction in clearance with long-term therapy.
The metabolite deacetyldiltiazem also accumulated with time in all patients, and had an elimination half-life considerably longer than its parent compound. We also found that the plasma level of the metabolite exceeded the level of diltiazem during long-term therapy in some patients. A considerably longer period of time may be required to reach steady-state levels of deacetyldiltia- zem. Potential therapeutic benefits or toxic effects of deacetyldiltiazem are not known.
Although diltiazem is being introduced as an anti- angina1 agent, it also slows atrioventricular nodal con- duction (negative dromotropic effect) like verapamil. The 14% P-R interval prolongation that we found with our short infusion (20 mg over 10 minutes) is less than the 25% prolongation reported with intravenous vera- pamil(10 mg over 2 minutes) by Reiter et a1.16 As with verapamil, peak P-R interval prolongation occurred 11 minutes after peak plasma drug levels, causing coun- terclockwise hysteresis (Fig. 4), which has been reported previously only for verapamil among the calcium- channel blocking drugs. 17-lg. This phenomenon is probably due to a delay in the transfer of diltiazem from the plasma to the tissue compartment, estimated by Kohno et a120 to take 11 minutes in dogs. Counter- clockwise hysteresis was also found after single oral dosing with verapamil, but with diltiazem, P-R interval prolongation and plasma levels were insufficient to as- sess hysteresis after a single oral dose. However, with long-term administration, as diltiazem accumulated, the P-R interval was significantly prolonged. This suggests that diltiazem will be useful in the chronic management of supraventricular tachycardias.
Diltiazem users should be aware of its unique phar- macokinetic properties. Significant accumulation occurs when the drug is given orally, which may enhance its therapeutic benefits or may possibly increase toxicity. The role of deacetyldiltiazem in the efficacy of diltiazem is uncertain, but it is present in substantial quantities. In addition, the negative dromotropic effect of both intravenous and oral diltiazem suggests that it may be useful in the treatment of supraventricular tachycar- dias.
Acknowledgment: We wish to acknowledge the technical assistance of Betty McCarthy, RN, Linda Aanonsen, Robert Wagoner, and Khalil Jaber, and the secretarial assistance of Ann Clayton.
References
1. Peplne a, Feldman RL, Whittle J, Curry C, Contl CR. Effect of diltiazem in patients with Variant angina: a randomized double-blind trial. Am Heart J 1981;101:719-725.
2. Howck KF, Bruce RA. Improved exercise performance in persons with stable angina peCtoriS receiving diltiazem. Am J Cardiol 1981;47:95- 101

1374 DILTIAZEM
3. Crevey RI, Dmtzkr DR. Sewer JS, Popat KD, Watker SD. Hemodynamic 12. Evans GE, Shand DG. Disposition of propranolol. V. Drug accumulation and gei exchange effects of lnlIavenous dimazem in patients wnh pulmonary hvoertension. Am J Cardlol 1982:49:578-583.
4. &da K. Takaa@ T, Taukane ‘i, Tanaka Y, Shbta K. Clinical study on the hypotensive effecf of diltiaxem hyckochlorfde. Int J Clln Pharmacol Ther Toxlcol 1981;19:47-55.
5. Naaao M. ¬e S, Hyon H, Horde M, Tanaka S. Diltiazeminduced de-
6.
7.
a.
9.
10.
11.
cm&se of exercise-elevated pulmonary arterial diastolic pressure in hy- pertrophic cardiomyopathy patients. Am Heart J 1961;102:789-790. Verghese C, SmRh MS, Aanoneen L, Prftchett ELC, Shand DG. High per- formance liquid chromatographic analysis of diltiazem and Rs metabollte in plasma. J Chromatcgr 1983;272:149-155. Gyama Y. Hemodynamic and electrophysiological evaluations of diltiazem hydrochloride: a clinical study. In: Bing RI. ed. New Drug Therapy with a Calcium Antagonist. Diltiiem Hakone Symposium. 1978. Amsterdam: Exerpta Medical, 1978:169-189. Ravel v, Gemenl R. Yfkrhard M, LanfbaM J, Statrfx C. lhebaiit JJ, Morselli PL. Pharmacokinetics and metabolism of diltiazem in man. Acta Cardiol 1980;35:35-45. Morsetll Pt., Rove1 V, M&hard M. Durand A, Gemenf R, Larribaud J. Pharmacokinetics and metabolism of diltiaxem in man (observations on healthy volunteers and angina pectoris patients). In Ref 7:152-167. Zelfs RF, Klmey EL Ths pham%scokinetics of diltiiem in healthy American men. Am J Cardiol 1982; 49529-532. Shand Do, Hammlll SC, Aanomen L, Prftchett ELC. Reduced verapamil clearance during long-term oral administration. Clin Pharmacol Ther 1981;30:701-703.
and steady-state concentrations during chronic oral administration in man. Clin Pharmacol Ther 1973;14:487-493.
13. Piephe RW, Sloedow DC, Lacy JP, Runser GJ, Dimmlt DC, Srowne RK. Phsrmacoldnetics of diltlazem in selected animal species and human beings. Am J Cardiol 1982;49:525-528.
14. Mesh1 T, Suglhara J, Sato Y. Metabolic fate of d-cis+acetoxy-5- f2-fdimethvlamino)_ ethvll-2.3dihvdro-2-fo-methoxvohenvll-1.5-ben- iothffepi&t (BH)&e hyd;&hl&de (CRD491). Chem %armOBull’(Tokyo) 1971:19:1546-1556.
15. Sakuma M, Yoshfkawa M, Sato Y. Ths whole body autoradiiaphic stud& on the distribution of 14C-labeled new 1.5benzothiazepine derivative (14C-CRD401) in mice. Chem Pharm Bull (Tokyo) 1971;19:995-1005.
16. Refter MJ, Shand 00, Aanonsen L, Wagoner R, MoCatthy E, Prftchett ELC. Pharmacokinetics of verapamil. Experience with a sustained intravenous infusion regimen. Am J Cardiol 1982;50:716-721.
17. Refter MJ, Shand DG, Prltchett ELC. Comparison of intravenous and oral administration. Clin Pharmacol Ther 1982;32:71 l-720.
18. Elchelbaum M, Blrkel P, Grube E, Gutffemann U, Somogyl A. Effects of verapamil on P&intervals in relation to verapamil plasma-ievels following single i.v. and oral administration and during chronic treatment. Klin WC+ chenschr 1980;58:919-925.
19. McAllister RJ Jr, Klrsten EB. The pharmacology of verapamil. IV. Kinetic and dynamic effects after single intravenous and oral doses. Clin Pharmacol Ther 1962;31:418-426.
20. Kohno K, Takeuchi Y, Etoh A, Noda K. Pharmacddnetiis and biosvailabilii of diltiazem (CRD-401) in dog. Arzneimittelforsch 1977;27:1424-1428.





























