Embed Size (px)
Citation preview

Abstract
Active transport processes are now recognized as important determinants of drugabsorption and elimination. The role of the MDR1 gene product P-glycoproteinfor drug disposition in humans is now well established. It is an ATP-dependentefflux transporter which translocates its substrates out of cells. Multiple struc-turally unrelated drugs including HIV protease inhibitors, immunosuppressants,cardiac drugs and �-adrenoceptor antagonists are substrates of P-glycoprotein. In-hibition and induction of P-glycoprotein have been identified as mechanisms ofdrug interactions in humans. Recently, multiple mutations in the MDR1 genehave been identified. The C3435T mutation was associated with reduced intestinalP-glycoprotein levels and higher plasma concentrations of the P-glycoprotein sub-strate digoxin. The implications of genetically determined differences in P-glyco-protein function for drug disposition, therapeutic outcome and risk for develop-ment of certain diseases will be summarized in this chapter.
8.1Introduction: Importance of Active Transport Mechanisms for Uptake,Tissue Distribution and Elimination of Xenobiotics
The MDR1 gene product P-glycoprotein (ABCB1) is a membrane protein whichfunctions as an ATP-dependent exporter of xenobiotics from cells. It was first de-scribed in tumor cells where it contributed to the occurrence of multidrug resis-tance (MDR) against anti-cancer agents [1]. In addition, it is expressed in normaltissues with excretory function such as intestine, liver and kidneys, in capillary en-dothelial cells of brain, placenta and testis and in peripheral blood cells [2–4]. Ex-pression of P-glycoprotein in these normal tissues is believed to be a protectivemechanism against xenobiotics. Intestinal P-glycoprotein has been shown to limitabsorption of the immunosuppressant cyclosporine, the cardiac glycoside digoxinand the �-adrenoceptor antagonist talinolol in humans [5–7]. Moreover, inductionand inhibition of P-glycoprotein are underlying mechanisms of drug interactions[6, 8].
159
8
The Pharmacogenomics of Human P-GlycoproteinMartin F. Fromm and Michel Eichelbaum
Pharmacogenomics: The Search for Individualized Therapies.Edited by J. Licinio and M.-L. Wong
Copyright © 2002 Wiley-VCH Verlag GmbH & Co. KGaAISBNs: 3-527-30380-4 (Paper); 3-527-60075-2 (Electronic)

It is well established that mutations of genes encoding xenobiotic metabolizingenzymes (e.g., members of the cytochromeP450 family such as CYP2D6) and drugtargets (e.g., receptors) determine the efficacy or toxicity of a broad variety of drugsand carcinogens. Recently, multiple mutations were also found in the human MDR1gene [9, 10]. One of those mutations (C3435T), which is located at a wobble positionin exon 26 and has a frequency of 28.6% for the homozygous genotype in Cauca-sians [10, 11], was associated with a lower P-glycoprotein expression in the smallintestine in comparison to subjects homozygous for the wild-type allele [10]. Accord-ingly, after oral administration of the P-glycoprotein substrate digoxin subjectshomozygous for this mutation had significantly higher plasma concentrations thanthe remainder of the population. Moreover, a reduced P-glycoprotein function wasobserved in peripheral blood cells of subjects with the TT genotype.
8.2Structure of the Human MDR1 Gene
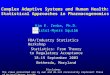
P-glycoprotein is a member of the ATP-binding cassette (ABC) superfamily ofmembrane proteins. After its first description in cultured cells selected for MDR[1], it was cloned from mouse and human cells [12, 13]. The MDR1 gene is lo-cated on chromosome 7q21. P-glycoprotein is a 170kDa phosphorylated and glyco-sylated protein and consists of 1,280 amino acids. It has two homologous halves,which contain six hydrophobic transmembrane domains and an ATP-binding site.Both halves are separated by a flexible linker polypeptide. A hypothetical two-di-mensional model of human P-glycoprotein is shown in Figure 8.1. In mice two
8 The Pharmacogenomics of Human P-Glycoprotein160
Fig. 8.1 Hypothetical two-dimensional model of human P-glycoprotein. Small circles: amino acidresidues; large circles: ATP sites; squiggly lines: N-linked glycosylation sites (modified from [15]).

drug-transporting mdr1 genes have been described, mdr1a and mdr1b. Structuralanalysis by electron microscopy and image analysis revealed that P-glycoproteinapproximates a cylinder of about 10 nm in diameter with a maximum height of 8nm. Since the depth of the lipid bilayer is about 4nm, about one half of the mole-cule is within the membrane. P-glycoprotein has a large central pore of about5 nm. It forms a large aqueous chamber within the membrane, which is open atthe extracellular face and closed at the cytoplasmic face of the membrane [14]. Ex-tensive mutational analysis revealed that two halves of the human P-glycoproteininteract to form a single transporter and that the major drug binding domains re-side in or near transmembrane domains 5, 6 and 11, 12 [15]. Since it appears thatP-glycoprotein detects and ejects its substrates before they reach the cytoplasm, itwas suggested that P-glycoprotein acts as a “hydrophobic vacuum cleaner”, whichdetects and removes its substrate from the lipid bilayer [16]. Another model sug-gests that P-glycoprotein acts as a flippase, carrying its substrate from the innerleaflet of the lipid bilayer to the outer leaflet [17].
8.3P-glycoprotein Expression in Healthy Tissues
P-glycoprotein is not only expressed in tumor cells, but also in cells of severalhealthy tissues. In liver it was detected in the biliary canalicular surface of hepato-cytes and the apical surface of small biliary ductules. In the small intestine andcolon, it is localized in the apical surface of columnar epithelial cells, and in kid-neys it is found in the brush border membrane of proximal tubules. Moreover, itis detectable on the apical surface of small ductules in the pancreas and on thesurface of cells in the medulla and cortex of adrenals [2].
P-glycoprotein is also expressed in endothelial cells of capillaries of the centralnervous system [3] and the sub-apical surface of the choroid plexus epithelium[18], which forms the blood-cerebrospinal fluid barrier. In addition to the blood-brain barrier it is expressed in capillary endothelial cells forming the blood-testisbarrier [3]. The microvillus border of syncytiotrophoblasts of human placenta alsoexpressed P-glycoprotein [19]. Finally, MDR1 mRNA was found in several leuko-cyte lineages with highest expression in CD56+ cells followed by lower levels inCD8+, CD4+, CD15+, CD19+ and CD14+ cells [4].
8.4Function of P-Glycoprotein
The tissue distribution of P-glycoprotein yields important clues to its function. Inmost tissues it is localized to the apical (luminal) membrane of polarized epithe-lial cell layers. This location suggests that P-glycoprotein extrudes its substratesfrom the epithelial cells into the adjacent lumen. It is anticipated that P-glycopro-tein plays an important role as a protective mechanism against naturally occur-
8.4 Function of P-Glycoprotein 161

ring toxins ingested with food. Intestinal P-glycoprotein will limit absorption oftoxins, whereas renal (via direct secretion and possibly prevention of re-absorptionfrom the lumen of tubules) and hepatic P-glycoprotein promotes elimination ofthese toxins into urine and bile. Moreover, P-glycoprotein expression in the blood-brain and blood-testis barrier will limit entry of these substances into brain andtestis. P-glycoprotein also appears to be associated with hormone transport and re-production. It is expressed in adrenal gland, uterus and placenta. Since P-glyco-protein is capable of transporting the corticosteroid hormones cortisol, corticoster-one and aldosterone [20], it was suggested that P-glycoprotein contributes to secre-tion of steroids from these tissues. Possible roles of P-glycoprotein function havebeen described for the hematological compartment, for cell volume regulation, lip-id transport and cell death and differentiation [21, 22].
Studies on P-glycoprotein expressing cell lines (Caco-2, L-MDR1) and P-glyco-protein knock-out mice yielded important insights into the role of this protein indrug transport [23, 24]. P-glycoprotein transports a wide range of structurally un-related, hydrophobic and amphiphatic drugs such as anticancer agents, cardiacdrugs, HIV protease inhibitors, �-adrenoceptor antagonists and antihistamines.A list of P-glycoprotein substrates is provided in Table 8.1. For example, the cardi-ac glycoside digoxin is a substrate of P-glycoprotein [25–28]. P-glycoprotein(mdr1a) knock-out mice had 35-fold and 2-fold higher concentrations than P-glyco-protein-expressing control animals in brain and plasma, respectively [27]. More-over, control animals excreted about 16% of an intravenously administered dosewithin 90 min directly into the gut lumen [29]. In contrast, only 2% of a givendose is found in the gut lumen of P-glycoprotein knock-out mice. Intestinal secre-tion of digoxin in P-glycoprotein-expressing animals could completely be blockedby oral administration of the P-glycoprotein inhibitor PSC-833 [30].
Digoxin is a substrate of P-glycoprotein and it is not metabolized to a major ex-tent in humans. Nevertheless, multiple drug interactions have been observed lead-ing to increased or decreased digoxin plasma concentrations. A common and ser-ious drug interaction occurs between digoxin and quinidine resulting in 2- to 3-fold increased digoxin plasma concentrations and digoxin toxicity [31, 32]. Phar-macokinetic studies reported enhanced absorption, reduced biliary and renal ex-cretion of digoxin during administration of quinidine [33–36]. The following linesof evidence indicate that inhibition of P-glycoprotein-mediated digoxin transport isa major mechanism underlying the digoxin-quinidine interaction. First, polarized,P-glycoprotein-mediated digoxin transport in Caco-2 cell monolayers could be in-hibited by low concentrations of quinidine [8]. Second, identical quinidine serumconcentrations caused a significant increase in digoxin plasma concentrations inP-glycoprotein-expressing animals, whereas this interaction was not observed in P-glycoprotein knock-out mice [8]. Increased digoxin plasma concentrations havenot only been reported during concomitant quinidine therapy, but also with pa-tients simultaneously treated with verapamil, propafenone, cyclosporine, itracona-zole, and amiodarone. Many of these drugs are also known to interact with P-gly-coprotein; thus, inhibition of P-glycoprotein-mediated digoxin elimination may bea common mechanism leading to increased digoxin plasma concentrations.
8 The Pharmacogenomics of Human P-Glycoprotein162

8.4 Function of P-Glycoprotein 163
Tab. 8.1 Summary of drugs, which are substrates of P-glycoprotein
Drug Reference Drug Reference
Anticancer agents– actinomycin D– etoposide– docetaxel– doxorubicin– daunorubicin– irinotecan– mitomycin C– mitoxantrone– paclitaxel– teniposide– topotecan– vinblastine– vincristine
Cardiac drugs– �-acetyldigoxin– �-methyldigoxin– digitoxin– digoxin– quinidine
HIV protease inhibitors– amprenavir– indinavir– nelfinavir– saquinavir– ritonavir
Immunosuppressants– cyclosporine A– tacrolimus
Antiemetic drugs– domperidon– ondansetron
Antidiarrheal agents– loperamide
[73][41][74][75][75][76][77][77][78][77][77][79][24]
[80][80][80][27][8]
[56][38][38][38][40]
[81][81]
[41][41]
[41]
Antibiotics– erythromycin– levofloxacin– sparfloxacin
Steroids– dexamethasone
Lipid-lowering agents– atorvastatin– lovastatin
Cacium channel blockerand metabolites– diltiazem– mibefradil– verapamil– D-617– D-620
�-Adrenoceptor antagonists– bunitrolol– celiprolol– talinolol
H1 antihistamines– fexofenadine– terfenadine
H2 antihistamines– cimetidine– ranitidine
Others– debrisoquine– losartan– morphine– phenytoin– rifampin
[82][83][84]
[27]
[85][86]
[87][88][89][89][89]
[90][91][92]
[47][86]
[93][93]
[86][94][27][41][95]

8.4.1Intestinal P-Glycoprotein
In addition to the observations of increased digoxin plasma concentrations due tocomedications, there have been reports of reduced digoxin plasma concentrationsduring treatment with the antibiotic rifampin, which has been shown to induceP-glycoprotein in human colon carcinoma cell lines [37]. Since intestinal P-glyco-protein appears to have a major impact on drug absorption, the hypothesis wastested, whether induction of intestinal P-glycoprotein could contribute to reduceddigoxin plasma concentrations during treatment with rifampin in humans [6].Indeed, rifampin significantly reduced AUC (area under the plasma concentra-tion-time curve) of orally administered digoxin and caused a 3.5-fold increase inintestinal P-glycoprotein obtained from duodenal biopsies. Moreover, AUC of oraldigoxin was negatively correlated with intestinal P-glycoprotein content, indicatingthat P-glycoprotein in the duodenum is a determinant of digoxin plasma concen-trations [6].
The introduction of HIV protease inhibitors was a considerable step forward in thetreatment of HIV infection. However, bioavailability of HIV protease inhibitors islow or variable. Recently indinavir, nelfinavir, saquinavir and ritonavir were identi-fied as substrates of P-glycoprotein [38–40]. Transepithelial translocation of thesedrugs across a polarized, P-glycoprotein-expressing monolayer of Caco-2 cells,which is a well established system for studying intestinal drug transport, was consid-erably higher in the basal to apical direction (corresponding to drug secretion intothe gut lumen) than to the opposite direction. The influence of intestinal P-glycopro-tein on bioavailability of HIV protease inhibitors was further highlighted by intrave-nous and oral administration of these drugs to P-glycoprotein knock-out mice andcontrol animals [38]. After intravenous administration of indinavir, nelfinavir andsaquinavir, plasma concentrations were not different between both groups of ani-mals indicating that mechanisms other than P-glycoprotein (such as CYP3A-mediated drug metabolism) are more important for elimination after intravenousdrug administration. However, after oral administration of these drugs, plasma con-centrations were 2- to 5-fold higher in P-glycoprotein knock-out mice as compared tocontrol animals. Taken together these data point to a major role of intestinal P-gly-coprotein for limiting bioavailability of HIV protease inhibitors.
8.4.2P-Glycoprotein and the Blood-Brain Barrier
The blood-brain barrier is formed by capillary endothelial cells in the brain. Simi-lar to gut wall mucosa, the blood-brain barrier was considered a passive anatomi-cal structure determining brain entry of molecules, e.g., by lipophilicity and pro-tein binding. It is now well established that active efflux by P-glycoprotein contrib-utes to brain permeability of drugs in addition to the factors mentioned above. In-sights into the role of P-glycoprotein in the blood-brain barrier were again ob-tained by P-glycoprotein knock-out mice. The particular importance of P-glycopro-
8 The Pharmacogenomics of Human P-Glycoprotein164

tein to the brain in comparison to most other tissues is shown by the consider-ably higher accumulation of several drugs in the brain of P-glycoprotein knock-out mice than in the plasma. Brain entry of the HIV protease inhibitors indinavir,nelfinavir and saquinavir is reduced due to P-glycoprotein expression in the endo-thelial cells of the blood-brain barrier. Thus, it was speculated that the ability ofthese drugs to achieve therapeutic concentrations in the brain is limited, therebycreating a potential sanctuary for viral replication [38].
Therapeutic effects of centrally active drugs also depend on adequate brain con-centrations. Accordingly, centrally acting drugs such as haloperidol, clozapine,midazolam and flunitrazepam easily enter the CNS and are not substrates ofP-glycoprotein [41]. The peripherally acting opioid loperamide, which penetratesthe CNS poorly and is used as an antidiarrheal agent, is a substrate of P-glycopro-tein. P-glycoprotein knock-out animals have 14-fold higher brain concentrationsafter oral administration of loperamide in comparison to control animals. More-over, P-glycoprotein-deficient mice show typical, morphine-like effects after admin-istration of loperamide [41]. The relevance of these data was confirmed by a studyin healthy volunteers. Administration of loperamide alone did not cause any respi-ratory depression, but respiratory depression occurred during co-administration ofloperamide with the P-glycoprotein inhibitor quinidine [42]. Taken together, thesedata indicate that P-glycoprotein in the blood-brain barrier is the major cause ofthe selective peripheral effects of loperamide in humans.
8.4.3P-Glycoprotein in Other Tissues
The importance of P-glycoprotein expression in the blood-testis barrier, peripheralleukocytes and the kidneys for drug disposition and disease risk is discussed inSections 8.7.2 and 8.7.3.
8.5Identification of MDR1 Mutations and Their Consequences for Function
In addition to the environmental factors described above, which modify expres-sion and function of human P-glycoprotein, there is also increasing knowledge onnaturally occurring mutations in the MDR1 gene and their potential relevance todrug disposition. A list of MDR1 gene mutations and their allelic frequencies isprovided in Table 8.2. The first mutations in normal cells were described by Mick-ley et al. (G2677T, G2995A, [9]). A first systematic screen of the MDR1 gene wasconducted by Hoffmeyer et al. [10]. They analyzed all 28 exons of the MDR1 geneincluding the core promoter region and the intron-exon boundaries from healthyCaucasian individuals and detected 15 SNPs (single nucleotide polymorphisms)with 6 SNPs located in the coding region. Subsequently, Cascorbi et al. [11] ana-lyzed 461 German volunteers for the frequencies of the previously described mu-tations and the presence of additional mutations. Taken together, 20 SNPs have
8.5 Identification of MDR1 Mutations and Their Consequences for Function 165

8 The Pharmacogenomics of Human P-Glycoprotein166
Tab. 8.2 MDR1 genetic variants in Caucasian individuals
# Location Position Allele Effect Allelicfrequency[%]
Genotype Genotypefrequency[%]
Reference
1 exon 1b exon 1b/12 TC
noncoding(?)
T/TT/CC/C
88.211.80.0
[10]
2 intron 1 exon 2 –1 GA
initiationoftranslation
91.0 G9.0 A
G/GG/AA/A
82.018.00.0
[10]
3 exon 2 cDNA 61 AG
21 Asn21 Asp
88.811.2
A/AA/GG/G
78.520.60.9
[10, 11]
4 intron 4 exon 5 –35 GC
G/GG/CC/C
98.81.20.0
[10]
5 intron 4 exon 5 –25 GT
G/GG/TT/T
70.526.03.5
[10]
6 intron 6 exon 6 +139 CT
62.837.2
C/CC/TT/T
39.047.513.4
[10]
7 intron 6 exon 6 +145 CT
C/CC/TT/T
97.62.40.0
[10]
8 exon 11 cDNA 1199 GA
400 Ser400 Asn
94.55.5
G/GG/AA/A
88.911.10.0
[10]
9 exon 12 cDNA 1236 CT
wobble 59.041.0
C/CC/TT/T
34.449.216.4
[10, 11]
10 intron 12 exon 12+ 44 CT
95.14.9
C/CC/TT/T
90.29.80.0
[10]
11 intron 16 exon 17–76 TA
53.846.2
T/TT/AA/A
28.450.820.8
[10]
12 intron 17 exon 17+137 AG
A/AA/GG/G
98.81.20.0
[10]

been described so far in Caucasian individuals. These mutations including allelicand genotype frequencies are summarized in Table 8.2. Seven of those mutationsalter the amino acid sequence of P-glycoprotein. The A61G SNP, which is locatedclose to the N-terminus of P-glycoprotein, leads to an amino acid exchange fromAsn to Asp. The G1199A mutation (Ser400Asn) is located in the cytoplasmic loopclose to the first ATP-binding domain. The most frequent SNP (G2677T/A) lead-ing to amino acid exchanges from Ala to Ser or Thr is located in the second trans-membrane domain. The G2995A mutation is also located in the second trans-membrane domain, but closer to the second ATP-binding domain. Finally, tworare SNPs located in exon 26 (A3320C and T3421A) leading to amino acidchanges were reported. The T307C (Phe-Leu) mutation described in the initial pa-per by Hoffmeyer [10] was not detectable in the larger population investigated byCascorbi et al. [11]. The location of these mutations in the MDR1 gene and withinthe structure of P-glycoprotein are shown in Figure 8.2.
8.5 Identification of MDR1 Mutations and Their Consequences for Function 167
Tab. 8.2 (continued)
# Location Position Allele Effect Allelicfrequency[%]
Genotype Genotypefrequency[%]
Reference
13 exon 21 cDNA 2677 GTA
893 Ala893 Ser893 Thr
56.541.61.9
G/GG/TT/TG/AT/AA/A
30.949.216.12.01.80.0
[11]
15 exon 24 cDNA 2995 GA
999 Ala999 Thr
G/GG/AA/A
89.011.00.0
[9]
16 exon 26 cDNA 3320 AC
1107 Gln1107 Pro
99.80.2
A/AA/CC/C
99.60.40.0
[11]
17 exon 26 cDNA 3396 CT
wobble C/CC/TT/T
99.50.50.0
[10]
18 exon 26 cDNA 3421 TA
1141 Ser1141 Thr
T/TT/AA/A
99.50.50.0
[49]
19 exon 26 cDNA 3435 CT
wobble 46.153.9
C/CC/TT/T
20.850.528.6
[10]
The positions of the polymorphisms correspond to positions of MDR1 cDNA with the first base ofthe ATG start codon set to 1 (GenBank accession number M14758). Mutations located in introns aregiven as position downstream (–) or upstream (+) of the respective exon according to the genomicorganization of MDR1 as described by Chen et al. [96].

The only polymorphism described so far that is associated with altered P-glyco-protein function is the silent mutation in exon 26 at position 3435 (C3435T). Sev-eral lines of evidence indicate an association of this polymorphism with alteredtransporter function. First, individuals homozygous for the mutation at position3435 (TT) had significantly lower P-glycoprotein levels in the small intestine incomparison to the remainder of the population (Figure 8.3, [10]). Second, thesame genotype-dependent differences were found in human kidneys (H. Brauchand U. Brinkmann, personal communication). Third, in accordance with low in-testinal P-glycoprotein content, subjects with the TT genotype had higher plasmaconcentrations of the P-glycoprotein substrate digoxin as compared to subjectswith the wild-type genotype [10]. Finally, P-glycoprotein function determined by ef-flux of the P-glycoprotein substrate rhodamine123 from CD56+ natural killer cells
8 The Pharmacogenomics of Human P-Glycoprotein168
Fig. 8.2 Single nucleotide polymorphisms ofthe human MDR1 gene (compiled from [10, 11,97]). The locations of the identified polymorph-isms (white arrow: non-coding polymorphisms;black arrow: polymorphisms leading to aminoacid exchanges) in relation to the exon-intron
structure of the MDR1 gene and the corre-sponding P-glycoprotein structure. * Mutationwith described functional consequences(modified from [97]; with permission fromDr. U. Brinkmann).
Fig. 8.3 Expression of P-glycopro-tein in human small intestine ac-cording to genotype at position3435 of the MDR1 gene (modifiedfrom [10]; with permission fromDr. U. Brinkmann).

was impaired in healthy volunteers with the TT genotype, unlike in individualswith the CC genotype (Figure 8.4, [43]). Moreover, MDR1 mRNA showed a trendtowards lower values in the TT as compared to subjects with the CT or CC geno-type [43]. The C3435T polymorphism has a high frequency in the Caucasian popu-lation with genotype frequencies of 20.8, 50.5 and 28.6% for CC, CT and TT, re-spectively [11]. The underlying mechanism for the association of the silentC3435T mutation with altered P-glycoprotein expression and function has not yetbeen established. It could be speculated that the functional consequences of thispolymorphism arise from its linkage to the G2677T mutation (Ala893Ser) or toother unidentified changes in the promoter/enhancer region or in sequences thatare important for mRNA processing [10].
Additional studies on the impact of MDR1 polymorphisms on the disposition ofthe H1 antagonist fexofenadine, which is a P-glycoprotein substrate and that isnot metabolized in humans, revealed conflicting results. It was reported in an ab-stract that genetic variations in the MDR1 gene (wild-type 1236C and 2677G and3435C vs. mutations 1236T and 2677T and 3435T) impact on fexofenadine disposi-
8.5 Identification of MDR1 Mutations and Their Consequences for Function 169
Fig. 8.4 P-glycoprotein function in CD56+ nat-ural killer cells of healthy volunteers accordingto MDR1 genotype at position 3435. Panel A:Rhodamine fluorescence of CD56+ natural killercells in two healthy individuals with differentMDR1 genotypes in exon 26 (position 3435)after an efflux period of 10min (gray line) com-pared to control cells (i.e., fluorescence of cellsfrom the same individuals incubated for 10min
in medium containing 2.5�M of the P-glyco-protein inhibitor PSC-833; black line). Panel B:Individual results (and mean± SD) of rhoda-mine fluorescence of CD56+ natural killer cellsin 31 healthy individuals with different MDR1genotypes in exon 26 (position 3435) after anefflux period of 10min as % of control values(** CC vs. TT: p< 0.01, # CC vs. CT: p < 0.05;modified from [43]).

tion in U.S. Caucasians. Those individuals who carry wild-type alleles at therespective positions have higher AUC [44]. These data stand in contrast to theresults of a fexofenadine study in Caucasians living in Germany (no differences infexofenadine disposition in subjects with 3435 TT vs. CC or 2677GG vs. TT; owndata) and to the increased digoxin plasma concentrations in subjects carrying mu-tations at position 3435 [10]. The reasons for those discrepancies are unknown.One could speculate that differences in European and North American diet (e.g.,salt content) account for differential regulation and expression of drug trans-porters determining fexofenadine disposition [45, 46]. Moreover, fexofenadine dis-position might not only depend on P-glycoprotein function, since OATP-A hasbeen shown to mediate cellular uptake of this antihistaminic drug [47].
8.6Racial Differences in Frequency of MDR1 Mutations
Recent studies indicate that the 3435C high-expression allele is considerably morefrequent in African populations in comparison to Caucasian and Asian popula-tions [48, 49]. The interethnic differences observed for the C3435T polymorphismare summarized in Table 8.3. One could speculate that the higher frequency ofthe CC genotype observed in Africans as compared to the Caucasian and Asianpopulations results from a selective advantage offered by this genotype againstgastrointestinal tract infections, which are endemic in tropical countries. This hy-
8 The Pharmacogenomics of Human P-Glycoprotein170
Tab. 8.3 Interethnic differences in allele and genotype frequencies of the MDR1 exon 26 C3435Tpolymorphism
Population Allele frequency Genotype frequency Reference
n C T CC CT TT
Caucasian, Germany 188 0.52 0.48 0.28 0.48 0.24 [10]Caucasian, Germany 461 0.46 0.54 0.21 0.51 0.29 [11]Caucasian, UK 190 0.48 0.52 0.24 0.48 0.28 [48]Portuguese 100 0.43 0.57 0.22 0.42 0.36 [48]Japanese 50 0.57 0.43 0.34 0.46 0.20 [49]South-west Asians 89 0.34 0.66 0.15 0.38 0.47 [48]Chinese 132 0.53 0.47 0.32 0.42 0.26 [48]Filipino 60 0.59 0.41 0.38 0.42 0.20 [48]Saudi 96 0.55 0.45 0.37 0.38 0.26 [48]Ghanaian 206 0.83 0.17 0.67 0.34 0.00 [48]Ghanaian 172 0.90 0.10 0.83 0.16 0.02 [49]Kenyan 80 0.83 0.17 0.70 0.26 0.04 [48]Sudanese 51 0.73 0.27 0.52 0.43 0.06 [48]African American 88 0.84 0.16 0.68 0.31 0.01 [48]African American 41 0.78 0.22 0.61 0.34 0.05 [49]

pothesis is supported by findings that P-glycoprotein plays a role in the defenseagainst both bacterial and viral infections (see below). It was recently reportedthat clearance of the P-glycoprotein substrates cyclosporine and tacrolimus isreduced in Caucasians compared to African Americans [50, 51]. Since the MDR1genotype was not reported in these studies, it remains to be determined whetherthe lower frequency of the low expression T-allele in the African American thanin the Caucasian population contributed to interethnic differences in dispositionof these P-glycoprotein substrates.
In accordance with the allele frequencies mentioned above, it was reported thatthe 2677T mutation was present in 42% of Caucasians and only 13% of AfricanAmericans with the 2677T mutation being frequently linked to the 3435T muta-tion [44]. In addition, a new mutation in exon 26 (T3421A) leading to an aminoacid exchange was identified in Ghanaians and African Americans (AA genotypeGhanaians vs. African Americans vs. Caucasians: 1.2% vs. 2.4% vs. 0%; [49]). An-other report indicates that the CC genotype at position 1236 is considerably morefrequent among Caucasians than in Japanese subjects (34.4% vs. 14.6%, [52]).Whether this difference is of any functional relevance remains to be determined.
8.7MDR1 Mutations and the Potential Risk for Idiopathic or Spontaneous Diseases
8.7.1P-Glycoprotein and Ulcerative Colitis
The etiology of inflammatory bowel diseases such as ulcerative colitis is not com-pletely understood. One of the current hypotheses suggests that either an en-hanced or aberrant immunologic responsiveness to constituents of the GI lumenis involved in chronic inflammation. There is evidence indicating that the intest-inal microflora is one factor in the pathogenesis of intestinal inflammation andthat disruption of the protective epithelial cell barrier promotes development of in-flammatory bowel disease [53, 54]. The physiological role of intestinal P-glycopro-tein might be to prevent entry of bacterial toxins into the gut wall mucosa. Thishypothesis is supported by the work of Panwala et al. [55], who showed thatmdr1a P-glycoprotein knock-out mice are susceptible to developing a severe, spon-taneous intestinal inflammation when maintained under specific pathogen-freeconditions. Moreover, treatment of the P-glycoprotein knock-out mice with oralantibiotics prevented development of disease and resolved active intestinal inflam-mation. Studies are currently underway to determine whether individuals carryingthe C3435T low-expression mutation in the human MDR1 gene are more suscep-tible to developing ulcerative colitis.
8.7 MDR1 Mutations and the Potential Risk for Idiopathic or Spontaneous Diseases 171

8.7.2P-Glycoprotein and HIV Infection
There is increasing evidence that disease risk and therapeutic outcome of the HIVinfection are linked to P-glycoprotein function in several ways. Highly active anti-retroviral therapy (HAART), which includes treatment with HIV protease inhibi-tors, has considerably improved the clinical management of HIV infection. TheHIV protease inhibitors indinavir, nelfinavir, saquinavir, ritonavir and amprenavirare transported by P-glycoprotein [38, 40, 56]. Some of them have a low bioavail-ability (e.g., saquinavir) and highly variable plasma concentrations, which mightlead to subtherapeutic plasma concentrations in some patients. Although intest-inal CYP3A4 certainly contributes to that variability, experiments with P-glycopro-tein knock-out mice indicate that P-glycoprotein localized in the brush bordermembrane of enterocytes limits absorption by pumping the drug back into thegut lumen [38]. Whether genetically determined differences in P-glycoproteinfunction affect plasma concentrations of HIV protease inhibitors in humans hasto be determined. Therapeutic efficacy of HIV protease inhibitors might furtherbe affected in humans by P-glycoprotein expression at the blood-brain barrier andthe blood-testis barrier, thereby limiting drug entry in these compartments andcreating sanctuary sites for the virus. Animal experiments have shown consider-ably reduced brain and testis concentrations of HIV protease inhibitors comparedto plasma [38, 56, 57]. Pharmacological inhibition of P-glycoprotein resulted inincreased tissue concentrations of the HIV protease inhibitors in these tissues.Studies are currently underway in order to determine the impact of mutations inthe MDR1 gene (e.g., C3435T) for P-glycoprotein expression in the blood-brainbarrier.
P-glycoprotein is also expressed in peripheral blood subpopulations, with thehighest expression in CD56+ natural killer cells followed by lower expression inCD8+, CD4+, CD15+, CD19+ and CD14+ cells [4]. P-glycoprotein expressing CD56+
natural killer cells from individuals homozygous for the C3435T mutation of thehuman MDR1 gene have reduced P-glycoprotein function (determined by inhibi-tion of rhodamine 123 efflux by PSC-833) as compared to the rest of the popula-tion [43]. Lee et al. [39] found a reduced inhibition of HIV replication by ritonavir,saquinavir and indinavir in cells with a high P-glycoprotein content, which couldbe restored by inhibitors of P-glycoprotein function. Although P-glycoprotein func-tion is highly heterogeneous within CD4+ cells [58], it will be important to deter-mine possible genetically determined differences in P-glycoprotein function forthis major cellular target of HIV protease inhibitors. The data described above in-dicate that individuals with a low P-glycoprotein expression might benefit morefrom HIV protease inhibitor therapy. However, using a human CD4+ T-leukemiccell line, Lee et al. [59] showed that HIV virus production was greatly reducedwhen P-glycoprotein was overexpressed. This could indicate that cells with high P-glycoprotein expression may be relatively resistant to HIV infection, but once theyare infected, it might be more difficult to eradicate the virus using HIV proteaseinhibitors [59].
8 The Pharmacogenomics of Human P-Glycoprotein172

8.7.3P-Glycoprotein and Renal Cell Carcinoma
P-glycoprotein is expressed in the brush border membrane of proximal tubularcells [2, 3]. It mediates active secretion of its substrates (e.g., digoxin) into urine.Similar to its role as a protective barrier in the gut wall mucosa and at the blood-brain barrier, renal P-glycoprotein is likely to function as a protective mechanismagainst toxic substances in the glomerulum filtrate by prevention of re-absorption.Thus, individuals with a low renal P-glycoprotein expression would potentially beexposed to higher concentrations of toxic agents and should be more susceptibleto their damaging effects. Indeed, there is data that indicates that the C3435Tpolymorphism is associated with reduced renal P-glycoprotein expression (H.Brauch, personal communication). In normal renal tissues quantitative immuno-histochemistry revealed a lower P-glycoprotein expression in individuals homozy-gous for the mutation (TT) than in patients homozygous for the wild-type allele.Moreover, the TT genotype appears to be a risk factor for the development ofrenal cell carcinoma. In patients with non-clear cell renal cell carcinoma, the fre-quency of homozygous carriers of the T allele was significantly increased in com-parison to healthy controls. Thus, the results of this study are in accordance withthe hypothesis that subjects with low renal P-glycoprotein expression are less pro-tected from intracellular accumulation of carcinogenic compounds. However, itmust be emphasized that the quantity of P-glycoprotein expression is not the onlyfactor determining the risk of renal cell carcinoma. For example, Longuemaux etal. [60] provided evidence that the presence of mutations in several xenobiotic-me-tabolizing enzymes (CYP1A1, GSTs, NAT2) was also associated with an increasedrisk for the development of renal cell carcinoma.
8.8Aspects of P-Glycoprotein Regulation
The factors controlling basal expression of P-glycoprotein in human tissues arepoorly understood. The transcriptional control of mouse mdr1a and mdr1b and ofhuman MDR1 has recently been summarized by M. Müller [61]. In brief, the hu-man MDR1 promoter contains an inverted CCAAT box, which is known to be acore sequence of the Y-box, a GC element and several putative recognition sitesfor transcription factors including AP1, NF-Y, Y-box binding protein (YB) andpregnane X receptor (PXR). Recently, a role for both NF-Y and Sp1 in the tran-scriptional activation of the MDR1 gene by genotoxic stress has been reported[62]. Moreover, p53 inactivation that is seen in cancers most likely leads to selec-tive resistance to chemotherapeutic agents because of up-regulation of P-glycopro-tein expression [63]. Its expression is also increased by reactive oxygen species dueto a process involving NF-�B activation [64]. Finally, it is now well established thatinduction of intestinal P-glycoprotein is the mechanism involved in the reductionof plasma digoxin concentrations during concomitant treatment with rifampin
8.8 Aspects of P-Glycoprotein Regulation 173

(see above). Similarly, the induction of intestinal P-glycoprotein by rifampin wasthe underlying mechanism for reduced plasma concentrations of the P-glycopro-tein substrate talinolol [7]. Due to the fact that the nuclear receptor PXR is in-volved in the induction of CYP3A4 by xenobiotics [65–67] and that CYP3A4 andP-glycoprotein are often co-induced, it was speculated whether a similar mecha-nism is involved in the induction of P-glycoprotein. Indeed, using the human co-lon carcinoma cell line LS174T it could be shown that induction of P-glycoproteinby rifampin is mediated by a DR4 motif in the upstream enhancer at about 8kb,to which PXR binds [68].
Similar to rifampin, the herbal medicine St. John’s wort frequently used as atreatment for mild depression, induces CYP3A4 via activation of PXR [69, 70],which can cause severe drug interactions with cyclosporine and HIV protease in-hibitors [71, 72]. It has recently been shown that during treatment with St. John’swort reduced plasma concentrations of the P-glycoprotein substrate digoxin aredue to the induction of intestinal P-glycoprotein by constituents of St. John’s wort[70]. It should be noted that induction of P-glycoprotein (e.g., by rifampin) showsconsiderable interindividual variability, with some subjects having almost no in-crease in P-glycoprotein expression. Whether this variability is genetically deter-mined or not remains to be clarified.
8.9Conclusions
P-glycoprotein is expressed in organs with excretory function such as small andlarge intestine, liver and kidneys. Moreover, it limits tissue penetration of drugsdue to its expression in the blood-brain and blood-testis barriers and in the pla-centa. It determines absorption, tissue distribution and elimination of a widerange of structurally unrelated drugs such as anticancer agents, HIV protease in-hibitors, cardiac drugs and �-adrenoceptor antagonists. Inhibition of P-glycopro-tein function results in increased drug concentrations and is now a well recog-nized mechanism of drug interactions in humans. On the other hand, P-glycopro-tein is induced by rifampin resulting in reduced drug concentrations of concomi-tantly administered P-glycoprotein substrates. In addition to these exogenous fac-tors affecting P-glycoprotein function, we are now beginning to understand genet-ic factors determining inter-individual variability in P-glycoprotein expression andfunction. It appears that mutations in the MDR1 gene have an impact on drugdisposition within and among ethnic populations. Moreover, an individual’s P-gly-coprotein function might determine the risk for certain diseases and the therapeu-tic outcome from treatment with P-glycoprotein substrates. However, the relativeimportance of variability in P-glycoprotein function due to exogenous and geneticfactors for drug disposition, therapeutic outcome and disease risk needs to beclarified in future studies.
8 The Pharmacogenomics of Human P-Glycoprotein174

AcknowledgementsOur work cited is supported by grants of Deutsche Forschungsgemeinschaft(FR 1298/2-1, Bonn, Germany), Bundesministerium für Bildung und Forschung(01 GG 9846 and 31 0311782, Bonn, Germany), and Robert Bosch Foundation(Stuttgart, Germany).
8.10 References 175
8.10References
1 ������� � ��� �. Biochim BiophysActa 1976; 455:152–162.
2 ������� � ����� ������ � ���������� �� ������ � ������������. Proc Natl Acad Sci USA 1987; 84,7735–7738.
3 ������������ � � !���� �� ������" �������������� !������ �������� �� !������ ��. Proc NatlAcad Sci USA 1989; 86:695–698.
4 #����$%� � ����$��� !� ������ � "����� �&. Blood 1994; 83:2451–2458.
5 �'� #& ��(� �� ��$����� )!����� � ������ "# &$��������� !��'� �! ��� � ����� &� !����* ���%��� �!. Clin Pharmacol Ther1997; 62:248–260.
6 ������� ! +�$������� � ����, �#���$������ ��� -�� ��$���� �*������ � #������ �#. J Clin Invest1999; 104:147–153.
7 ����.��� # ����������� )*�$����$�� � ����%� � #��%� ������� � ����, � -�� ��$���� ����,�% � ��$������� #��//������ &$����% " ������ ! #�������# &������� �. Clin Pharmacol Ther2000, 68:345–355.
8 ����� �� #�� �! &���� �� ���%������ �� ����� "�. Circulation 1999;99:552–557.
9 ��$%��( ) �� �& ���� * *��� *)�-���, � ������ � !���� &+ ��0� . Blood 1998; 91:1749–1756.
10 ��//��(�� & !��% � -�� ��$���� �)����� �� !��$%�1���� � ����� )���$���� � �����// ����� � +�$�������� � !���%���� 2. Proc Natl AcadSci USA 2000; 97:3473–3478.
11 ���$���� � �����// ����� ) ������� ��//��(�� & &$�'�� � &$�3//����� + +�$������� � !���%���� 2����� �. Clin Pharmacol Ther 2001;69:169–174.
12 ���� �� ���� �+ 2��� # ����% "������� � ��������� �� ���������!. Cell 1986; 47:381–389.
13 ���� � !�� 45 ����. �� �������"+. Nature 1986; 323:728–731.
14 ��������� �� ��������� � ������ ������� ��. J Biol Chem 1997;272:10685–10694.
15 )����%�� &� "�( & ��($(�� �)����$������ � ������ � �����������. Annu Rev Pharmacol Toxicol 1999;39:361–398.
16 ��������� �� ������ �. Annu RevBiochem 1993; 62:385–427.
17 ������� �� ��������� ��. TrendsBiochem Sci 1992; 17:18–21.
18 ��� �� "��������� � !������� �+&�(��� )* ���$� �) &��������� )���'��$������� ". Proc Natl Acad SciUSA 1999; 96:3900–3905.
19 ��$������� ) )�����-�$� "� +'��&� ������� �#. Histochem J 1994;26:417–423.
20 2��� # �%����� 4 ����� � ������'��� 5 &��%� #��%� 4 #����� ���� �. J Biol Chem 1992; 267:24248–24252.
21 &$���%�� )�. Sem Cancer Biol 1997;8:161–170.
22 ��������� �� ���/�� )) &�(�� ��.Trends Biochem Sci 2000; 25:1–6.
23 &$���%�� )� ��(�� 2 �������� +��� �) -�� "������ &��� �� -����� ���% �) �������' )� &.��� �-�� �������� � *�0����� �� �����

8 The Pharmacogenomics of Human P-Glycoprotein176
�+ !���� �. Proc Natl Acad Sci USA1997; 94:4028–4033.
24 &$���%�� )� &��� �� -�� �������� �!��0��� �� �������� + -�� "������ ��� �) -�� ��� ���% �) ��������������� +� �� ����� �� !���� )��!���� �. Cell 1994; 77:491–502.
25 �����'��� 5 �%����� 4 ����� �5������� � 2��� # #��%� 4 #������ ���� �. J Pharmacol Exp Ther1992; 263:840–845.
26 �� ����( �) &��-����� �. BiochemBiophys Res Commun 1992; 189:551–557.
27 &$���%�� )� �������� + -�� "������� ��� �) !���� �6 J Clin Invest1995; 96:1698–1705.
28 ��-�� �+ ���� � &������ 4. Br JPharmacol 1996; 118:1389–1396.
29 ��(�� 2 �������� + !��0��� ��&��� �� ���0�� "# -�� )�.���� �!���� � &$���%�� )�. Br J Pharmacol1996; 119:1038–1044.
30 ��(�� 2 �������� + "�����% !!��0��� �� !���� � &$���%�� )�.J Clin Invest 1997; 100:2430–2436.
31 ����( +! Jr., ���//�� �) "����� �+������������� ��6 �-�0�( ��!����� � , Jr. JAMA 1978; 240:533–534.
32 "������ � #1��� +. Med Klinik 1978;73:1085–1088.
33 ����� �" ������� � ��(������ �������� " ���-�� � ���$�� �� �������� &. N Engl J Med 1979; 300:1238–1241.
34 �������� #+ ������������ !" #��������� 4) 4�������#���% �. Eur J ClinPharmacol 1983; 24:41–47.
35 )������ ! )�-������ ) "���7-��� ������� ) &$���$%������/���� #.Eur J Clin Invest 1987; 17:262–265.
36 �� ����( �) #���� � #���� ������% � &��-����� �. Am J Physiol1992; 263:F613–622.
37 &$����, +� !�$% � &$����, �".Mol Pharmacol 1996; 49:311–318.
38 #�� �! ����� �� ������ � ��%�! ���� )�� ����� "� ���%�������. J Clin Invest 1998; 101:289–294.
39 �� �� ��������� �� ������������ ����$������ � ����� # )�����%�� &� ������ � "�( &. Biochemis-try 1998; 37:3594–3601.
40 )����, � &��//�� � )��8 �. PharmRes 1998; 15:423–428.
41 &$���%�� )� �������� + ��� �)-�� "������ . J Clin Invest 1996;97:2517–2524.
42 &���7�� )� ������ � �� � &��� &���� )�. Clin Pharmacol Ther 2000;68:231–237.
43 ���,� � "���$��� & -�� ��� #��. �&$�3//���� + ���$��� � &$�'�� �+�$������� � ����� ��. Pharmaco-genetics 2001; 11:1–6.
44 #�� �! ��%� ! ���� + "������ �##���� &� &$�'��, 2� �(��� ) 9����� &���� �� ���� )�� �$#����( �&$����, +� &$����, �" ���%�������. Drug Metab Rev 2000; 32:199 (Ab-stract).
45 ���%����� ��. Adv Drug Devel Rev1997; 27:129–159.
46 "����� " ����� �� "��� ���� &#�� �! #������ �# +�$������� ������ "�. Circulation 1998; 98:2702–2708.
47 �-��%�-�$ � ��%� ! ����� �����%����� �� #�� �!. Drug MetabDispos 1999; 27:866–871.
48 )��(�' ��� ��������� � � �� 9 ���7 � ������% ) ������� 4����(�� �� ������� � � ������ )�/����)�0�� " ���$��+-��� " �$����. Pharmacogenetics 2001; 11:217–221.
49 &$���//���� + +�$������� � !���%����� 2 ������ ) )��������%� &*����� 2� &$�'�� �. Lancet 2001;358:383–384.
50 ��� "� �� � #� 5� �������� �.Clin Pharmacol Ther 2000; 68:478–486.
51 ���$������ � ��������� ������� "������� " ������� & !�%���%( �!���� * ���������� 2. Clin Pharma-col Ther 2001; 69:24–31.
52 ��� & ����� � ����� � &�,�%� )����$�� & ������ #. Pharmaco-genetics 2001; 11:175–184.
53 ��������� � ������ ��. Science1995; 270:1203–1207.
54 "����� ����( )� ������ 4)&�������( ) ��(��( )� �'�� ��.Am J Pathol 1997; 150:91–97.
55 ���'��� �� ����� �� ����( �. J Im-munol 1998; 161:5733–5744.

8.10 References 177
56 ����� �� ������� � &��������� &"���.���(� �+ "����� &� !���'��#� ������( �. Pharm Res 1999;16:1206–1212.
57 ���� +� ��%� ! ������ � �������� ���� )� ���%����� �� #�� �!.Drug Metab Dispos 2000; 28:655–660.
58 ������� � &��� �� &$���%�� )�.AIDS 2000; 14:237–242.
59 �� �� ����$������ � ����� # ������ �) ������ � ��������� ��.FASEB J 2000; 14:516–522.
60 ��������8 & "�������� � ������� ��0��� ) ���$�������( � !��-���� "��, " #������������( � �������� �� ������ � !����� � "�.�����. Cancer Res 1999; 59:2903–2908.
61 �:���� �. Semin Liver Dis 2000;20:323–337.
62 �� * ��� & &$���� #�. J Biol Chem2000; 275:2979–2985.
63 ���������( �� *������� �� )������# &$����, +� &$����, �". Proc NatlAcad Sci USA 1997; 94:11037–11042.
64 ��-���� � ��������� �� #�����)" ������ �). J Biol Chem 2000;275:1887–1896.
65 !��������� � ������$� � &-������ #)���� � ��������� &(��'�!�$%���� � ������� � �������� � !����7���� � !��%������ ). Proc Natl AcadSci USA 1998; 95:12208–12213.
66 !������� ! +-��� ��. Genes Dev1998; 12:3149–3155.
67 ������ �� �$#�� "" ������ �)������� � ����� � #���'�� &).J Clin Invest 1998; 102:1016–1023.
68 ���$% ) +�$������� � !��% �. J BiolChem 2001; 276:14581–14587.
69 ����� ! ����'�� ! ����� &)�����( �! &����0���&���� �� �������� � ������� � #���'�� &). ProcNatl Acad Sci USA 2000; 97:7500–7502.
70 ":�� " &������ ! #����%�2���$% �)�����$� #� &������� �� ����� ����������� #. Clin Pharmacol Ther 2000;68:598–604.
71 �����!����� ). Lancet 2000; 355:134–138.
72 ���$������ &� !������� )� ������ ")�/��� �� ������� �. Lancet 2000;355:547–548.
73 ����� ���.�( �� �$���$ �� !����-��� �. Biochem Pharmacol 1995;50:1701–1709.
74 ���� � ������!� � ������( )�$������� " ������� & ������� ��&$������ ". Biochem Pharmacol 1994;48:1528–1530.
75 !��� � ����� �� ������%�� 4� -����� ����/ � �������� � �� �����+�. Cancer Treat Rev 2000; 26:449–462.
76 &���(��� 5 #��� 5 ��� 9. CancerChemother Pharmacol 1998; 42(Suppl):S44–S49
77 ������� ��. Ther Drug Monit 1996;18:350–356.
78 &.�������� ) -�� )�.���� � ��(��2 &$���%�� )� &��� �� ���0�� "#!���� � 4���0�� �� !��0��� �� -�� �������� �. Proc Natl Acad Sci USA1997; 94:2031–2035.
79 ��%� 5 ��0����� ) ������� 5. CancerChemother Pharmacol 1997; 40:433–438.
80 ������������ � �:����� ����� )������� +�$������� � #���, 2����� ��. Naunyn SchmiedebergsArch Pharmacol 2001; 363:337–343.
81 &��%� 2��� # �����'��� 5 ����� #����� . J Biol Chem 1993;268:6077–6080.
82 &$����, +� 5����� # )������ #&$����, �". Arch Biochem Biophys1998; 350:340–347.
83 ��� 5��� � ���%� # ���� #�. JPharmacol Exp Ther 1997; 282:955–960.
84 �� ���� + ���$���� & -�� ���!��� " -�� ��� &���� �� �� !���)� "���� ) !��7��� & ����� �.Eur J Pharm Sci 2000; 12:85–93.
85 �� 9 ����/���� � &��'��� !�.Pharm Res 2000; 17:209–215.
86 #�� �! ������ � ��%� ! �-��%�-�$� ����� �� "��.��( �� ������� !���� � ��������( )# �����"� ���� )�� ���%����� ��. PharmRes 1999; 16:408–414.
87 &��%� 2��� # �����'��� 5 ����� #����� . FEBS Lett 1993; 324:99–102.
88 ������ � #�� �! ��������$� ������ )�. Drug Metab Dispos 2000;28:895–898.
89 ������������ � -�� ��$���� � !��%� *������ ) ������� +�$�������

8 The Pharmacogenomics of Human P-Glycoprotein178
� ����� ��. J Pharmacol Exp Ther2000; 293:376–382.
90 �����,�%� � 5������� � ��(��� �%����� � ������ � ����,�%� �#�'����� 5 #�'��� � 4���� � ����� &�'��� 5. Biopharm DrugDispos 1999; 20:85–90.
91 #������� � #�� &� *������% �)�������� �. Br J Pharmacol 1993;110:1009–1016.
92 �������$� 2 &.����������� �����$���� + ������ ! ���$� �������� �. Pharm Res 1996; 13:514–522.
93 ������� ) ����� 4! &��� + ��'����� �������� �. J Pharmacol Exp Ther1999; 288:171–178.
94 &������ ) !���� * ����$���� +���������� 2. Br J Pharmacol 2000;129:1235–1243.
95 &$����, +� &$���%�� )� ��������� &$����, �". Proc Natl Acad SciUSA 1996; 93:4001–4005.
96 ���� �� ����% " 2��� # ������ ���������� �� �������� �!. J BiolChem 1990; 265:506–514.
97 #��� � ��//��(�� & !���%���� 2.Pharmacogenomics 2001; 2:51–64.