Embed Size (px)
Citation preview

PHARMACOGENETIC ANALYSIS OF GLUTAMATE SYSTEM GENE VARIANTS AND CLINICAL RESPONSE
TO CLOZAPINE
by
Danielle L. Taylor
A thesis submitted in conformity with the requirements for the degree of Master of Science
Institute of Medical Science University of Toronto
© Copyright by Danielle L. Taylor (2015)

Taylor DL
ii"""
Pharmacogenetic Analysis of Glutamate System Gene Variants
and Clinical Response to Clozapine
Danielle L. Taylor
Master of Science
Institute of Medical Science University of Toronto
2015
ABSTRACT
Altered glutamatergic neurotransmission is implicated in schizophrenia etiology and
response to the atypical antipsychotic clozapine. Response to clozapine is highly variable and
partly depends on genetic factors as indicated by twin studies. The association of 17
glutamate system gene variants and clinical response to clozapine was investigated in a
sample of European schizophrenia/schizoaffective patients deemed resistant/intolerant to
previous pharmacotherapy. Categorical and continuous response measures were assessed
following six months of clozapine treatment using change in Brief Psychiatric Rating Scale
Scores, and were analyzed using Pearson’s chi-squared test or analysis of covariance,
respectively. No significant associations were observed for all analyses after correction for
multiple testing. Prior to correction, several nominally significant associations were
observed, the most notable being between the SLC6A9 rs16831558 variant and change in
positive symptoms (puncorrected=0.008, pcorrected=0.08). This finding warrants further
investigation in larger well-characterized samples to clarify the role of SLC6A9 in clinical
response to clozapine.

Taylor DL
iii""
ACKNOWLEDGEMENTS
First and foremost, I would like to thank my thesis supervisor Dr. James Kennedy for
providing me with the opportunity to undertake a Master’s degree in his lab. His continued
support, guidance and witty sense of humor have made these last few years in graduate
school an altogether enriching experience. I would also like to thank the members of my
Program Advisory Committee, Dr. Joanne Knight, Dr. Daniel Müller and Dr. Gary
Remington for their patience and constructive feedback. I extend many thanks to my fellow
colleagues in the Neurogenetics Department for providing guidance on protocols and
statistical analyses, and for helping to troubleshoot obstacles that popped up along the way.
To the Pharmacogenetics crew, thank you all for the many laughs, good food and good times.
Much recognition goes out to my friends on the volleyball team for providing a carefree
environment where I could forget about the worries of the work week. Thank you to my
family for continuing to support me on my long scholarly journey. Finally, I would like to
acknowledge the lab technicians, trainees, collaborators, faculty mentors and funding sources
for their contribution to this project.

Taylor DL
iv"""
CONTRIBUTIONS
Dr. James Kennedy, Dr. Daniel Müller, Dr. Joanne Knight and Dr. Gary Remington helped
design this thesis project. Patient blood samples were provided by Dr. Herbert Meltzer, Dr.
Jeffrey Lieberman and Dr. Steven Potkin. Dr. Arun Tiwari provided guidance regarding
statistical analyses of genotype data and Dr. Vanessa Gonçalves provided guidance regarding
use of the Applied Biosystems OpenArray® genotyping platform. A very special thank you
is extended to the patients for their willingness to participate.

Taylor DL
v"""
TABLE OF CONTENTS
CHAPTER SECTION PAGE
ABSTRACT ii
ACKNOWLEDGEMENTS iii
CONTRIBUTIONS iv
TABLE OF CONTENTS v
LIST OF ABBREVIATIONS viii
LIST OF FIGURES xix
LIST OF TABLES xx
1 INTRODUCTION 1 1.1 Schizophrenia Overview 1
1.1.1 Epidemiology and Time Course 2 1.1.2 Deficits in Quality of Life 3 1.1.3 Burden to Society, Patients and Caregivers 4
1.2 Etiology and Pathophysiology 5 1.2.1 Environmental Risk Factors 5 1.2.2 Genetic Predisposition 5
1.2.2.1 Chromosome 6: 6p24-p22 7 1.2.2.2 Chromosome 8: 8p22-p21 8 1.2.2.3 Chromosome 11: 11q22-q21 8 1.2.2.4 Chromosome 13: 13q14-q32 8 1.2.2.5 Polygenic Inheritance 9
1.2.3 Brain Structural Abnormalities 11 1.2.4 Neurochemical Abnormalities 11
1.2.4.1 The Dopamine Hypothesis 11 1.2.4.2 The Glutamate Hypothesis 13
1.3 Glutamatergic Dysfunction in Schizophrenia 14 1.3.1 The Glutamate System 14
1.3.1.1 Ionotropic Glutamate Receptors 16 1.3.1.2 Metabotropic Glutamate Receptors 20 1.3.1.3 Amino Acid Transporters 21 1.3.1.4 Glutamate System Enzymes 23
1.3.2 Glutamate System Abnormalities in Schizophrenia 24 1.4 Schizophrenia Pharmacotherapy 26
1.4.1 Antipsychotic Medication Overview 26

Taylor DL
vi"""
1.4.2 The Mechanism of Action of Antipsychotic Drugs 27 1.4.3 Treatment Response Measures 28 1.4.4 Clozapine for Treatment-Resistant Schizophrenia 29 1.4.5 Variability in Clozapine Response 31
1.5 Pharmacogenetics of Clozapine Response 33 1.5.1 Pharmacogenetics Overview 33 1.5.2 Clozapine and Glutamatergic Neurotransmission 34 1.5.3 Polymorphisms in Glutamate System and Related Genes 38 1.5.4 Identifying Functional Gene Variants Using ENCODE 40
1.6 Study Rationale 44 1.6.1 Summary and Hypothesis 44
2 GENETIC ASSOCIATION ANALYSIS OF THE NMDA 45
RECEPTOR SUBUNIT GENE GRIN2B WITH RESPONSE TO CLOZAPINE (Submitted to Human Psychopharmacology: Clinical & Experimental) 2.1 Abstract 46 2.2 Introduction 47 2.3 Methods 50 2.4 Results 55 2.5 Discussion 56
3 PHARMACOGENETIC ANALYSIS OF GLUTAMATE 67
SYSTEM GENE VARIANTS AND CLINICAL RESPONSE TO CLOZAPINE (Submitted to Molecular Neuropsychiatry) 3.1 Abstract 68 3.2 Introduction 69 3.3 Methods 72 3.4 Results 76 3.5 Discussion 79
4 DISCUSSION 95
4.0 Overview 95 4.1 Summary of Findings 95
4.1.1 Association of GRIN2B with Clozapine Response 95 4.1.2 Association of Glutamate System Variants with Clozapine 96
Response 4.2 Implications of Findings 97
4.2.1 Glycine in Schizophrenia Etiology and Treatment 98 4.2.2 Interaction of Clozapine with the Glycine System 99

Taylor DL
vii""
4.2.3 Promoter Variant Confers Differential Response to 100 Clozapine
4.3 Limitations & Considerations 102 4.3.1 Study Heterogeneity 102 4.3.2 Inter-Study Variability 104 4.3.3 Statistical Power 106 4.3.4 Multiple Testing 108 4.3.5 Deviation from Hardy-Weinberg Equilibrium 110 4.3.6 Incomplete Outcome Data 111
4.4 Future Directions 112 4.4.1 Characterization of Glycine Transporter 1 Promoter 112
Variant 4.4.2 Gene-Environment Interactions 115 4.4.3 Gene-Gene Interaction s 117 4.4.4 Next Generation Sequencing 120 4.4.5 Personalized Medicine in Psychiatry 123
4.5 Concluding Remarks 126
5 REFERENCES 128

Taylor DL
viii""
LIST OF ABBREVIATIONS
Δ change
α alpha; probability associated with a type I error, statistical significance criteria
α-keto acid α-ketoglutaric acid
α-KG alpha-ketoglutarate
β probability associated with making a type II error
γ gamma
oC" degrees Celsius
µL microlitre
χ2 Pearson’s chi-squared goodness of fit test
5-HT1A 5-hydroxytryptamine (serotonin) type 1A receptor
5-HT2A 5-hydroxytryptamine (serotonin) type 2A receptor
5-HTTLPR serotonin-transporter-linked polymorphic region
A adenine nucleotide base
ABI Applied Biosystems Incorporated
Ac-CoA acetyl coenzyme A
AD antidepressant
ADHD attention deficit hyperactivity disorder
ADRA1A adrenergic receptor α-1°
ADRA2A adrenergic receptor α-2°
AFR African American sample
AIWG antipsychotic-induced weight gain
AMPA α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor
AMR ad mixed American sample

Taylor DL
ix""
ANCOVA analysis of covariance
ANOVA analysis of variance
AP antipsychotic
APA American Psychiatric Association
ASD autism spectrum disorder
ASN Asian sample
BDNF brain derived neurotrophic factor gene
BDNF brain derived neurotrophic factor protein
Bill S-201 Genetic Non-discrimination Act (Canada)
BNEG Brief Psychiatric Rating Scale negative symptom subscale
bp nucleotide basepairs
BP bipolar disorder
BPOS Brief Psychiatric Rating Scale positive symptom subscale
BPRS Brief Psychiatric Rating Scale
C cytosine nucleotide base
Ca2+ calcium ion
CAMH Centre for Addiction and Mental Health
CaMKII calcium/calmodulin dependent kinase II
cAMP cyclic adenosine monophosphate
CATIE Clinical Antipsychotic Trials of Intervention Effectiveness Project
CDN Canadian dollars
cDNA complimentary deoxyribonucleic acid
CGI Clinical Global Impressions Scale
ChIP-seq chromatin immunoprecipitation sequencing
CHOR Clozapine, Haloperidol, Olanzapine, Risperidone Study

Taylor DL
x"""
CHRM1 cholinergic receptor, muscarinic 1
chrN chromosome number
CI confidence interval
CIA clozapine-induced agranulocytosis
CI-OCD clozapine-induced obsessive compulsive disorder
CLZ clozapine
CNS central nervous system
Cons evolutionarily conserved variant
COS childhood onset schizophrenia
CPZ chlorpromazine
CREB cyclic AMP response element-binding protein
CRESTAR clozapine pharmacogenetics study
CSF cerebrospinal fluid
CVD cardiovascular disease
CYP1A2 cytochrome-P450 isoenzyme family 1, subfamily A, polypeptide 2 gene
CYP2D6 cytochrome-P450 isoenzyme family 2, subfamily D, polypeptide 6 gene
D’ Normalized linkage disequilibrium constant between two loci
DAAO d-amino acid oxidase gene
DAAO d-amino acid oxidase enzyme; DAO
DAOA d-amino acid oxidase activator gene
DAOA d-amino acid oxidase activator protein; G72
dbSNP National Centre for Biotechnology Information short genetic information database
DISC1 disrupted in schizophrenia 1 gene

Taylor DL
xi""
DLPFC dorsolateral prefrontal cortex
DNA deoxyribonucleic acid
DNAse-seq DNAse I hypersensitivity site sequencing
DRD1 dopamine D1 receptor gene
DRD2/D2 dopamine D2 receptor
DRD2 dopamine D2 receptor gene
DRD3 dopamine D3 receptor gene
D-ser D-serine
DSM-III-R Diagnostic and Statistical Manual, 3rd Edition Revised
DSM-IV Diagnostic and Statistical Manual, 4th Edition
DSM-5 Diagnostic and Statistical Manual, 5th Edition
DTNBP1 dystrobrevin binding protein 1 (dysbindin) gene
DTNBP1 dystrobrevin binding protein 1
DZ dizygotic
EAAT1 excitatory amino acid transporter 1
EAAT2/GLT1 excitatory amino acid transporter 2; glutamate transporter 1
EHM enhancer associated histone mark
emPCR emulsion polymerase chain reaction
ENCODE Encyclopedia of DNA Elements Project
EPS extrapyramidal symptoms
EPSCs excitatory postsynaptic currents
EPSPs excitatory postsynaptic potentials
eQTL expression quantitative trait loci
ESE/ESS exonic splicing enhancer/exonic splicing silencer
EUR European sample

Taylor DL
xii""
FDR false discovery rate
FuncPred National Institute for Environmental Health Sciences SNP Functional Prediction database
FWER family-wise error rate
G guanine nucleotide base
GABA γ-aminobutyric acid
GAD1 glutamate decarboxylase 1 gene
GAD1 glutamate decarboxylase 1 protein
GAIN Genetics Association Information Network
GATA1 GATA-binding factor 1; globin transcription factor 1; erythroid transcription factor 1
GATA4 GATA-binding factor 4; transcription factor GATA-4
GCS glycine cleavage system
GDH glutamate dehydrogenase
GERP genomic evolutionary rate profiling
GINA Genetic Information Non-discrimination Act (US)
Gln glutamine
GLS glutaminase; L-glutamine aminohydrolase
Glu glutamate
GLUR1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor subunit 1 protein; GluA1
Gly glycine
GlyT1 glycine transporter 1
GRIA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor subunit 1 gene
GRIN1 N-methyl-D-aspartate receptor subunit 1 gene
GRIN2A N-methyl-D-aspartate receptor subunit 2A gene

Taylor DL
xiii""
GRIN2B N-methyl-D-aspartate receptor subunit 2B gene
GRM2 metabotropic glutamate receptor 2 gene
GRM3 metabotropic glutamate receptor 3 gene
GS glutamine synthetase
GWAS genome wide association study
H2O2 hydrogen peroxide
Haploreg Broad Institute Genomic Functional Annotation database
HRH1 histamine receptor H1 protein
HRH2 histamine receptor H2 gene
HTR1A 5-hydroxytryptamine (serotonin) receptor type 1A gene
HTR2A 5-hydroxytryptamine (serotonin) receptor type 2A gene
HTR2C 5-hydroxytryptamine (serotonin) receptor type 2C gene
HWE Hardy-Weinberg equilibrium
IP3 inositol 1,4,5-triphosphate
k independent number of tests conducted in a statistical analyses
K+ potassium ion
kb nucleotide kilobases
kDa kilodalton
kg kilogram
LD linkage disequilibrium
LGC Laboratory of the Government Chemist
LOD log of the likelihood odds ratio; confidence in D’
L-ser L-serine
LTP long term potentiation

Taylor DL
xiv""
LY2140023 pomaglumetad methionil; metabotropic glutamate receptor 2/3 agonist
MAF minor allele frequency
MAPK mitogen activated protein kinase
MC motif change
MC4R melanocortin receptor 4 gene
MDD major depressive disorder
MDR multifactor dimensionality reduction
METH-IP methamphetamine-induced psychosis
mg milligrams
Mg+ magnesium ion
mg/d milligrams per day
mGluR2 metabotropic glutamate receptor 2
mGluR3 metabotropic glutamate receptor 3
mGluR5 metabotropic glutamate receptor 5
MIM Mendelian inheritance in man gene code
min(s) minute(s)
miRNA micro ribonucleic acid
MK-801 (+)-D-aspartate 5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-iminedizocilpine (dizocilpine)
mL millilitres
Mn/Mj major/minor allele
mPFC medial prefrontal cortex
mRNA messenger ribonucleic acid
MZ monozygotic
n number; study sample size

Taylor DL
xv""
NA not available
Na+ sodium ion
Nacc nucleus accumbens
NB neuroblastoma cell line
NCBI US National Centre for Biotechnology Information
ng nanograms
ng/mL nanograms per millilitre
NGS next generation sequencing
NH3 ammonia
NHGRI US National Human Genome Research Institute
NIEHS National Institute of Environmental Health Sciences
NMDA N-methyl-D-aspartate
NMDAR N-methyl-D-aspartate receptor
NR1 N-methyl-D-aspartate receptor subunit 1
NR2A N-methyl-D-aspartate receptor subunit 2A
NR2B N-methyl-D-aspartate receptor subunit 2B
NRG1 neuregulin 1 gene
NRG1 neuregulin 1 protein
NXRN1 neurexin 1 gene
NXRN1 neurexin 1 protein
OCD obsessive compulsive disorder
OLZ olanzapine
OR odds ratio
PANSS Positive and Negative Syndrome Scale
PB protein bound

Taylor DL
xvi""
PCA principle components analysis
PCP phencyclidine
PCR polymerase chain reaction
PFC prefrontal cortex
PGC Psychiatric Genomics Consortium
PGM personal genome machine
PGx pharmacogenetics
PHM promoter-associated histone mark
PKA protein kinase A
PKC protein kinase C
pos position
PPI pre-pulse inhibition
PSD postsynaptic density
PSD-95 postsynaptic density protein 95
PubMed United States National Library of Medicine database
p-value probability value
QC quality control
QLS quality of life score
q-value false discovery rate adjusted p-value
r2 correlation coefficient between loci
RegulomeDB Stanford University Centre for Genomics and Personalized Medicine SNP annotation database
RG1678 bitopertin; glycine transporter 1 inhibitor
RNA ribonucleic acid
R/NR responder/non-responder

Taylor DL
xvii""
RR relative risk; risk ratio
rs# reference single nucleotide polymorphism identification number
s seconds
SA schizoaffective
SCZ schizophrenia
SD standard deviation
SLC1A2 solute carrier family 1, member 2 gene; excitatory amino acid transporter 2 gene; glutamate transporter 1 gene
SLC6A4 solute carrier family 6 (neurotransmitter transporter, serotonin), member 4 gene; serotonin transporter 1 gene
SLC6A9 solute carrier family 6, member 9 gene; glycine transporter 1 gene
SMR standardized mortality ratio
SNARE soluble N-ethylmaleimide sensitive factor (NSF) attachment protein receptor
SNP single nucleotide polymorphism
SPSS Statistical Package for the Social Sciences
SRR serine racemase
Suc succinate
T thymine nucleotide base
TBX18 T-box transcription factor 18 protein
TCA tricarboxylic acid cycle ; Krebs cycle ; citric acid cycle
TD tardive dyskinesia
TF transcription factor
TFBS transcription factor binding site
TRD treatment-resistant depression
TRS treatment-resistant schizophrenia

Taylor DL
xviii""
UCSC University of California, Santa Cruz
URS ultra treatment-resistant schizophrenia
UTR untranslated region
VGLUT/vGluT vesicular glutamate transporter
VKORC1 vitamin K epoxide reductase complex subunit 1 gene
VTA ventral tegmental area

Taylor DL
xix""
LIST OF FIGURES
Figure 1.1 Schematic Representation of a Glutamatergic Synapse p.15
Figure 1.2 The Ionotropic N-methyl-D-aspartate Glutamate p.19 Receptor
Figure 1.3 Clozapine is Superior to Chlorpromazine for p.30 Treatment-Resistant Schizophrenia
Figure 1.4 Identifying Functional Promoter Variants in SLC6A9 p.41 Using ENCODE Data
Figure 2.1 Human GRIN2B Gene Diagram p.60
Figure 2.2 GRIN2B Linkage Disequilibrium Map p.62
Figure 2.3 Genotype Analysis of GRIN2B Variant rs1072388 and p.65 Clozapine Response
Figure 3.1 Glutamate System Gene Diagrams p.83
Figure 3.2 Haploreg Results for Functional Glutamate System p.85 Gene Variants
Figure 3.3 GRM2, SLC6A9 and SLC1A2 Linkage Disequilibrium p.86 Maps
Figure 3.4 Genotype Analysis of SLC6A9 rs16831558 with p.92 Clozapine Response

Taylor DL
xx"""
LIST OF TABLES
Table 1.1 Select Candidate Genes and Chromosomal Regions p.10 Associated with Schizophrenia
Table 1.2 Evidence Supporting Clozapine Interacts with the p.35 Glutamate System
Table 1.3 Genetic Associations of Glutamate System Genes and p.39 Clozapine Response
Table 2.1 GRIN2B Gene Variants Genotyped in the Sample p.61
Table 2.2 Study Sample Demographics and Clinical Information p.63
Table 2.3 Association Analysis of GRIN2B Variants and p.64 Response to Clozapine
Table 2.4 Haplotype Analysis of GRIN2B Variants and Response p.66 to Clozapine
Table 3.1 Glutamate System Gene Variants Genotyped in the p.87 Study Sample
Table 3.2 Study Sample Demographics and Clinical Information p.89
Table 3.3 Associations Among Glutamate System Gene Variants p.90 and Clozapine Response
Table 3.4 Haplotype Analysis of GRM2 Variants and Response p.93 to Clozapine
Table 3.5 Haplotype Analysis of SLC6A9 Variants and Response p.94 to Clozapine
"
"
"
"
"
"

Taylor DL I. INTRODUCTION !
1!
!
Chapter 1 !
INTRODUCTION
1.1 Schizophrenia Overview
The disease concept of schizophrenia (SCZ) first emerged in the middle of the 19th century as
psychiatrists within Europe began noting disorders of mental dysfunction that arose in early
adulthood and often progressed to chronic deterioration. Several early terms were used to
refer to patients presenting with these symptoms, including “démence précoce” meaning
precocious onset of cognitive decline in France (Morel, 1860), and “adolescent insanity” in
Scotland (Clouston, 1904). Borrowing from Morel’s term, German neurologist Emil
Kraepelin (1856-1926) termed this condition ‘dementia praecox’ in 1887 and characterized
the disorder as nine different “clinical forms” (Kraepelin, 1899). Approximately two decades
later, the Swiss psychiatrist Eugen Bleuer (1857-1939) coined the term ‘schizophrenia’ on
the 24th of April, 1908 (Bleuer, 1911). The term was derived from two Greek words, ‘schizo’
meaning to tear or to split, and ‘phrenia’ meaning the mind.
In the present day, SCZ is characterized as a chronic, lifelong neuropsychiatric disorder
accompanied by positive, negative and cognitive symptoms that are diagnosed according to
the Diagnostic and Statistical Manual of Mental Health Disorders 5th Edition (DSM-5)
(American Psychiatric Association, 2013). According to this recent version of the DSM, a
patient presenting with positive symptoms, or psychosis, exhibits psychotic behaviours that
are not present in healthy individuals. The hallmark positive symptoms of SCZ include
auditory hallucinations (hearing voices), delusions (fixed false beliefs), disorganized speech,
and grossly disorganized or catatonic behaviour. In contrast, the negative and cognitive

2!
!
!
!
symptoms of SCZ manifest as deficits in normal functioning and include characteristics such
as affective flattening, poverty of speech (alogia), lack of motivation (avolition) and the
inability to experience pleasure (anhedonia). Cognitive symptoms include deficits in
attention, working memory, reasoning and processing speed. Affective symptoms and social
interaction deficits are also considered appropriate symptom categories of SCZ. Together,
these symptoms collectively contribute to the debilitating social and occupational
dysfunction commonly observed in patients suffering from this disorder.
1.1.1 Epidemiology and Time Course
SCZ affects approximately 15 out of every 100,000 people each year (median incidence,
central 80% range: 7.7-43) and almost 1% of the world’s population will suffer from SCZ
sometime in their lifetime (median lifetime morbid risk: 7.2 per 1000 persons) (Jablensky et
al., 1992; McGrath et al., 2008). The onset of SCZ usually occurs during adolescence
between the ages of 18 and 35 years old (Hafner et al., 1994). Interestingly, the disease
manifests two to three years later in females than in males (Reicher et al., 1990) and may be
caused, in part, by the anti-dopaminergic effects of estrogen (Seeman & Lang, 1990).
The time course of SCZ typically follows three distinct phases: the prodrome, the acute or
active phase and the residual phase. SCZ prodrome is the period leading up to psychosis and
is marked by abnormal behaviours and psychological states relating to cognition, emotion,
perception, communication, motivation and sleep (Klosterkotter et al., 2001). Nonspecific
clinical symptoms such as depression, anxiety, social isolation and school and/or
occupational dysfunction may also present during this time (Larson et al., 2010). The acute
phase of SCZ, otherwise known as the ‘first episode’ of psychosis, occurs once the classic
positive symptoms of SCZ manifest. This period is often accompanied by cognitive and

3!
!
!
!
social decline (Hafner et al., 1999). Following the first episode, patients are commonly
hospitalized, given a psychological assessment and diagnosed according to their symptoms.
Patients often begin a trial with antipsychotic (AP) medication, which is the mainstay in SCZ
treatment. Once positive symptoms are brought under control through the use of APs, the
residual phase begins. Unfortunately, this phase is often associated with a number of adverse
drug side effects and the presence of persistent negative and cognitive symptoms.
1.1.2 Deficits in Quality of Life
Individuals suffering from SCZ live shorter, lower quality lives in comparison to members of
the general population (standardized mortality ratio (SMR): 2.6, central 80% range: 1.2-5.8)
(McGrath et al., 2008). In addition, SCZ patients are often at higher risk of suicide (SMR:
12.9) (Saha et al., 2007; Zai et al., 2012). Indeed, approximately 5% of patients commit
suicide in their lifetime, usually near disease onset (Palmer et al., 2005). Cardiovascular
disease (CVD) is also more common in patients with SCZ and is primarily attributable to
unhealthy diet, lack of exercise, excessive smoking and co-morbid substance abuse (Laursen,
Munk-Olsen & Vestergaard, 2012). Compared to the general population, SCZ patients are
35% more likely to smoke cigarettes and 20% more likely to abuse alcohol and other
substances. A subset of patients who are diagnosed with treatment-resistant schizophrenia
(TRS) exhibit even higher rates of these health risk behaviours. In addition, patients often
suffer from motor side effects collectively called extrapyramidal symptoms (EPS), which
include tardive dyskinesia (TD) (30%), akathisia (36%) and parkinsonism (37%) (reviewed
by Kennedy et al., 2014).
Health utility scales are useful for assessing the effect of disease status on a patient’s quality
of life. On a scale from 0 to 1, zero representing death and one representing perfect health,

4!
!
!
!
severe, moderate and mild forms of SCZ have been assigned health utility scores of 0.56
(standard deviation (SD): 0.11), 0.62 (SD: 0.22) and 0.79 (SD: 0.09), respectively (Centre for
the Evaluation of Value and Risk in Health, 2011). TRS is also associated with poorer
quality of life that is approximately 20% lower than patients with SCZ who achieve
remission (0.61 vs. 0.75) (Kennedy et al., 2014). Alleviation of SCZ symptomatology
through successful therapeutic intervention usually increases a patient’s quality of life and
stresses the need for optimizing treatment outcomes (Bobes et al., 2007).
1.1.3 Burden to Society, Patients and Caregivers
The total financial burden of SCZ in Canada in 2004 was estimated to be $6.85 billion CDN,
accounting for direct healthcare and non-healthcare costs, as well as productivity lost due to
unemployment (Goeree et al., 2005; Kennedy et al., 2014). Furthermore, TRS patients report
annual healthcare costs ranging 3-to-11 times higher than schizophrenia patients who
respond to treatment ($66,360-$163,795 vs. $15,500-$22,300 per year). In addition, SCZ is
ranked as one of the top 10 causes of disability among people in developed countries
(Murray & Lopez, 1996; Barbato, 1998). Areas that are most prone to disability include
social functioning in relation to self-care, occupational performance, family relationships and
relationships in society as a whole (Janca et al., 1996). Stigma against individuals with
mental health disorders is lower than in the past, however, individuals with SCZ still
experience high degrees of rejection, discrimination and social isolation that ultimately
affects their sense of self and well-being (Goffman, 1963). Schizophrenia also represents a
substantial burden to caregivers (Schene et al., 1998; Gutierrez-Maldonado et al., 2005;
Vasudeva et al., 2013), with approximately 25% of patients living with a relative after
diagnosis (Torrey, 2013).

5!
!
!
!
1.2 Etiology and Pathophysiology
The cause of SCZ is not known, however, various hypotheses have been posited to explain
why this disorder manifests in particular individuals. These hypotheses propose SCZ
emerges due to a combination of risk factors that include environmental exposures and
genetic predisposition that ultimately lead to abnormalities in brain structure and function. A
brief overview of the etiology and pathophysiology of SCZ is presented below.
1.2.1 Environmental Risk Factors
Several environmental risk factors are thought to increase the likelihood of developing SCZ.
These factors include: male sex (relative risk (RR): 1.42, 95% Confidence Interval (CI):
1.30-1.56) (Aleman et al., 2003), first- and second-generation migrant status (RR: 2.7, 95%
CI: 2.3-3.2 and 4.5, 95% CI: 1.5-13.1, respectively) (McGrath et al., 2004; Cantor-Graae &
Selten, 2005), urban residence (RR: 2.4, 95% CI: 2.13-2.70) (Marcelis et al., 1998;
Mortensen et al., 1999), advanced paternal age (Malaspina et al., 2001; El-Saadi et al., 2004;
Sipos et al., 2004) and cannabis use (Boydell et al., 2007; De Sousa et al., 2013). Pre- and
postnatal exposure to stress and infection (Buka et al., 2001; Brown et al., 2004; Mortensen
et al., 2007), as well as prenatal nutritional deprivation (Susser & Lin, 1992; St Clair et al.,
2005) may also increase one’s risk for developing SCZ.
1.2.2 Genetic Predisposition
SCZ is also caused in part by genetic factors as evidenced by twin, family and adoption
studies. For instance, studies analyzing the incidence of SCZ within families indicate that
children are at an increased risk for developing SCZ if other family members are affected.
This risk increases as the degree of relation to the affected family member increases. As was

6!
!
!
!
mentioned earlier in Section 1.1, the risk of developing SCZ in the general population is
approximately 1%, however this risk jumps to 6% for a child born to an afflicted parent and
even higher to 9% for a person who has an afflicted sibling (Gottesman, 1991).
Twin studies have been particularly useful in understanding the contribution of genes to SCZ
etiology because twins share varying degrees of genetic similarity (reviewed by Cardno &
Gottesman, 2000). More specifically, dizygotic twins share exactly half their genes while
monozygotic twins, or identical twins, have genotypes that are exact duplicates. Twin studies
in SCZ research have yielded proband-wise concordance rates of 41-65% in monozygotic
twins and 0-28% in dizygotic pairs (reviewed by Gottesman, 1991). The amount of
concordance observed between twins as well as observed and expected resemblance between
relatives is useful for estimating trait heritability (Visscher et al., 2008). Researchers
involved in twin and family studies estimate the heritability of SCZ to be approximately 80-
85% (reviewed by Kendler, 1983). Thus, genetic factors are the largest known risk factor for
SCZ.
Adoption studies are another important tool for distinguishing the influence of heredity and
environment on human traits. Generally, traits shared between biological parents and a child
that is given up for adoption is explained by genetic factors, while traits shared
between adoptive or ‘environmental’ parents and an adopted child is attributed to
environmental influence (Plomen et al., 1997). The first adoption study in SCZ was proposed
in 1959 (Kety, 1959) and investigated whether adopted away children of parents (usually
mothers) with SCZ were at increased risk for developing the disorder. Adoption studies that
followed found that children who are raised in foster homes or institutions and whose
biological parent had SCZ developed SCZ more often than children of controls (Heston,

7!
!
!
!
1966). Similar studies have replicated these findings and provide sufficient evidence to
support a role of genetics in SCZ (Higgins, 1966; Rosenthal et al., 1968; Higgins, 1976;
Tienari & Wynne, 1994).
Molecular genetic studies aim to identify genes involved in disease etiology. Genetic linkage
studies follow the segregation of marker alleles from an affected lineage to offspring in
families (Riley & McGuffin, 2000), while genetic association studies assess the contribution
of loci to disease presentation in populations. Results from linkage studies in SCZ have been
mixed, with limited replication of findings. Positive results are generally accepted as
significant if they replicate in several additional studies. To date, SCZ has been linked to
numerous chromosomal regions including chromosome 1, 2, 4, 5-10, 13, 15, 18, 22 and the
X chromosome. The breadth of these findings exceeds the scope of this paper and have been
reviewed elsewhere (Riley & McGuffin, 2000; Riley & Kendler, 2006). Select candidate
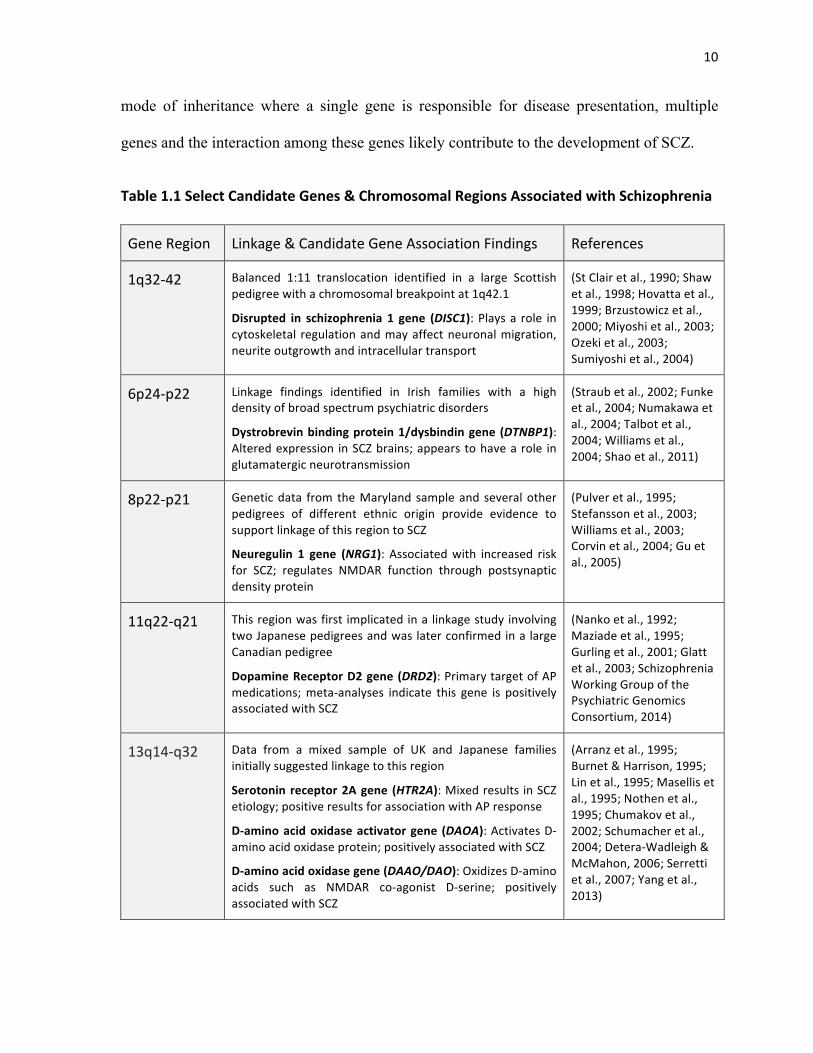
genes and chromosomal regions are summarized in Table 1.1.
1.2.2.1 Chromosome 6: 6p24-p22
Linkage findings for chromosome 6 first emerged from studies involving Irish families with
a high density of broad-spectrum psychiatric disorders (Straub et al., 2002). One particular
gene within this region of chromosome 6, the dystrobrevin binding protein 1/dysbindin gene
(DTNBP1) has been, in general, positively associated with SCZ (Funke et al., 2004;
Numakawa et al., 2004; Williams et al., 2004). Altered expression of DTNBP1 in certain
brain regions has also been reported in patients with SCZ (Weickert et al., 2004). Although
the exact function of DTNBP1 in the brain is unknown, this protein appears to have a role in
glutamatergic neurotransmission (Talbot et al., 2004; Shao et al., 2011).

8!
!
!
!
1.2.2.2 Chromosome 8: 8p22-p21
Findings from the Maryland family sample, as well as data from numerous pedigrees of
individuals with differing ethnic background have provided linkage support for chromosome
8 (Pulver et al., 1995). Association findings have confirmed that a candidate gene located
within this region called neuregulin 1 (NRG1) is associated with increased risk for SCZ
(Stefansson et al., 2003; Williams et al., 2003; Corvin et al., 2004). The neuregulin 1 protein
(NRG1) is a CNS-expressed molecule involved in the expression and activation of
neurotransmitter receptors (reviewed by Harrison & Law, 2006). Of interest to the
glutamatergic neurotransmitter system, NRG1 regulates N-methyl-D-aspartate glutamate
receptor (NMDAR) function via a postsynaptic density protein (PSD-95) (Gu et al., 2005).
1.2.2.3 Chromosome 11: 11q22-q21
The chromosome 11 region was first implicated in SCZ etiology in a linkage study involving
two Japanese pedigrees (Nanko et al., 1992), and was later confirmed in a separate study
involving a large Canadian pedigree (Maziade et al., 1995; Gurling et al., 2001). One
particular gene of interest lying within chromosome region 11q22 is the dopamine receptor
D2 gene (DRD2). This gene is of particular interest because all antipsychotic medications on
the market for the treatment of SCZ symptoms have blockade of the dopamine D2 receptor
(DRD2) as a common mechanism of action (Seeman & Lee, 1975). A large meta-analysis of
variants within DRD2 indicates this gene is associated with SCZ (Glatt et al., 2003).
1.2.2.4 Chromosome 13: 13q14-q32
Data from a mixed sample of UK and Japanese families initially suggested linkage to
chromosome 13 (Lin et al., 1995). This region is of particular interest because approximately
120 known genes of interest are located here including the serotonin receptor type 2A gene

9!
!
!
!
(HTR2A), D-amino acid oxidase activator gene (DAOA) and the d-amino acid oxidase gene
(DAAO). Association studies involving HTR2A variants in SCZ risk have produced mixed
results (reviewed by Serretti et al., 2007), while studies assessing association with response
to atypical APs have yielded more consistently positive findings (Arranz et al., 1995; Burnet
& Harrison, 1995; Masellis et al., 1995; Nothen et al., 1995). Based on meta-analysis, DAOA
has been implicated in SCZ risk (Detera-Wadleigh & McMahon, 2006), as well as DAAO
(Chumakov et al., 2002; Schumacher et al., 2004; Yang et al., 2013). Functionally, the d-
amino acid oxidase activator protein (DAOA) activates d-amino acid oxidase enzyme
(DAAO/DAO) that is responsible for the oxidation of D-amino acids such as D-serine in the
brain. Because D-serine acts as a co-agonist at the NMDAR, variable activity of DAOA
and/or DAAO may therefore contribute to SCZ pathophysiology by affecting glutamatergic
neurotransmission.
1.2.2.5 Polygenic Inheritance
Recently, a very large genome wide association study (GWAS) consisting of up to 36,989
cases and 113,075 controls from the psychiatric genomics consortium (PGC) identified 128
independent associations spanning 108 conservatively defined loci with SCZ (Schizophrenia
Working Group of the Psychiatric Genomics Consortium, 2014). The top 128 loci included
DRD2 and many other genes related to glutamatergic function such as the metabotropic
glutamate receptor 3 gene (GRM3), the NMDAR subunit 2A gene (GRIN2A) and the α-
amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR) subunit 1 gene
(GRIA1). As implicated by this recent GWAS, SCZ is considered a complex trait that almost
certainly follows a polygenic mode of inheritance (Gottesman & Shields, 1967; McClellan et
al., 2007; International Schizophrenia Consortium et al., 2009). As opposed to a Mendelian
10!
!
!
!
mode of inheritance where a single gene is responsible for disease presentation, multiple
genes and the interaction among these genes likely contribute to the development of SCZ.
Table&1.1&Select&Candidate&Genes&&&Chromosomal&Regions&Associated&with&Schizophrenia&
Gene!Region! Linkage!&!Candidate!Gene!Association!Findings! References!
1q32A42! Balanced! 1:11! translocation! identified! in! a! large! Scottish!
pedigree!with!a!chromosomal!breakpoint!at!1q42.1!
Disrupted& in& schizophrenia& 1& gene& (DISC1):!Plays!a! role! in!cytoskeletal! regulation!and!may!affect!neuronal!migration,!
neurite!outgrowth!and!intracellular!transport!
(St!Clair!et!al.,!1990;!Shaw!
et!al.,!1998;!Hovatta!et!al.,!
1999;!Brzustowicz!et!al.,!
2000;!Miyoshi!et!al.,!2003;!
Ozeki!et!al.,!2003;!
Sumiyoshi!et!al.,!2004)!
6p24Ap22! Linkage! findings! identified! in! Irish! families! with! a! high!
density!of!broad!spectrum!psychiatric!disorders!!
Dystrobrevin&binding&protein&1/dysbindin&gene&(DTNBP1):!Altered!expression! in!SCZ!brains;!appears!to!have!a!role! in!
glutamatergic!neurotransmission!!
(Straub!et!al.,!2002;!Funke!
et!al.,!2004;!Numakawa!et!
al.,!2004;!Talbot!et!al.,!
2004;!Williams!et!al.,!
2004;!Shao!et!al.,!2011)!
8p22Ap21! Genetic!data! from!the!Maryland! sample!and! several!other!
pedigrees! of! different! ethnic! origin! provide! evidence! to!
support!linkage!of!this!region!to!SCZ!
Neuregulin& 1& gene& (NRG1):! Associated!with! increased! risk!for! SCZ;! regulates! NMDAR! function! through! postsynaptic!
density!protein!
(Pulver!et!al.,!1995;!
Stefansson!et!al.,!2003;!
Williams!et!al.,!2003;!
Corvin!et!al.,!2004;!Gu!et!
al.,!2005)!
11q22Aq21! This!region!was!first! implicated!in!a! linkage!study!involving!
two!Japanese!pedigrees!and!was!later!confirmed!in!a!large!
Canadian!pedigree!
Dopamine&Receptor&D2&gene&(DRD2):!Primary!target!of!AP!
medications;!metaAanalyses! indicate! this! gene! is! positively!
associated!with!SCZ!
(Nanko!et!al.,!1992;!
Maziade!et!al.,!1995;!
Gurling!et!al.,!2001;!Glatt!
et!al.,!2003;!Schizophrenia!
Working!Group!of!the!
Psychiatric!Genomics!
Consortium,!2014)!
13q14Aq32! Data! from! a! mixed! sample! of! UK! and! Japanese! families!
initially!suggested!linkage!to!this!region!
Serotonin& receptor&2A&gene& (HTR2A):!Mixed!results! in!SCZ!
etiology;!positive!results!for!association!with!AP!response!
DFamino&acid&oxidase&activator&gene& (DAOA):!Activates!DAamino!acid!oxidase!protein;!positively!associated!with!SCZ!!
DFamino&acid&oxidase&gene&(DAAO/DAO):!Oxidizes!DAamino!
acids! such! as! NMDAR! coAagonist! DAserine;! positively!
associated!with!SCZ!
(Arranz!et!al.,!1995;!
Burnet!&!Harrison,!1995;!
Lin!et!al.,!1995;!Masellis!et!
al.,!1995;!Nothen!et!al.,!
1995;!Chumakov!et!al.,!
2002;!Schumacher!et!al.,!
2004;!DeteraAWadleigh!&!
McMahon,!2006;!Serretti!
et!al.,!2007;!Yang!et!al.,!
2013)!

11!
!
!
!
1.2.3 Brain Structural Abnormalities
Several brain structural abnormalities have been observed in patients with SCZ (reviewed by
Buckley, 2005). In brief, these structural abnormalities include: reduced cortical thickness
(Voineskos et al., 2013), gradual lateral and third ventricle enlargement (Johnstone et al.,
1976; Mathalon et al., 2001; Kasai et al., 2003), subtle decreases in thalamic volume
(Andreasen et al., 1994; Kemether et al., 2003), an enlarged caudate nucleus (Jernigan et al.,
1991), smaller temporal lobes (Wright et al., 2000; Csernansky et al., 2002) and reduced left-
right asymmetry (anisotropy) (Nakamura et al., 2012; Lee et al., 2013). Brain structural
abnormalities in patients with SCZ have been difficult to reproduce across studies because
they are relatively subtle and are also influenced by patient characteristics such as age,
gender, chronicity of illness, medication use and co-morbid substance abuse (McCarley et
al., 1999; Arango et al., 2003).
1.2.4 Neurochemical Abnormalities
In addition to brain structural aberrations, several neurochemical abnormalities have been
observed in patients with SCZ. The majority of these abnormalities relate primarily to the
dopamine, glutamate, serotonin and the inhibitory neurotransmitter γ-aminobutyric acid
(GABA) neurotransmitter systems. The roles of serotonin and GABA in SCZ etiology are
beyond the scope of this text and have been reviewed elsewhere (Meltzer, 1995a; Aghajanian
& Marek, 2000; Wassaf & Kochan, 2003; Nakazawa et al., 2012).
1.2.4.1 The Dopamine Hypothesis
Much of SCZ research has focused on perturbations in the dopamine neurotransmitter
system. The groundwork for the ‘dopamine hypothesis of SCZ’ was conducted by Arvid
Carlsson (1923-) in the late 1950s, who won the Nobel prize in 2000 for his contribution, and

12!
!
!
!
by Jacques van Rosum in the late 1960s (reviewed by Seeman, 1987). Furthermore, Seeman
& colleagues showed in 1975 that the efficacy of AP drugs was tightly correlated with
affinity of the drugs for DRD2 receptors (Seeman & Lee, 1975). Hence, the dopamine
hypothesis was based largely on observations that dopamine-enhancing drugs, such as
amphetamines, were capable of reproducing positive symptoms observed in patients with
SCZ (Lieberman et al., 1987), and that AP drugs which treat psychosis do so primarily by
blocking DRD2 receptors (Carlsson & Lindqvist, 1963; Seeman & Lee, 1975).
Biological data has also provided evidence to support the dopamine hypothesis of SCZ. A
large proportion of the dopaminergic neurons in the human brain originate in the ventral
tegmental area (VTA), and project to the prefrontal cortex (mesocortical pathway) and to
subcortical limbic brain regions (mesolimbic pathway). Dopamine activity in prefrontal
regions appears to play an inhibitory role on dopamine release in subcortical areas (Pycock,
Kerwin & Carter, 1980; Kolachana, Saunders & Weinberger, 1995; Karreman &
Moghaddam, 1996). From these findings, SCZ has been characterized as a disorder caused
by deficits in dopamine transmission (hypodopaminergia) in the prefrontal cortex,
consequently leading to over-activity of dopamine circuits (hyperdopaminergia) in the
striatum (reviewed by Davis et al., 1991). Hypofrontality in SCZ patients is evidenced by
reduced cerebral blood flow in the frontal cortex and may be related to low dopaminergic
activity in that region (Davis et al., 1991; Rubin et al., 1991; Andreasen et al., 1992). Positive
symptoms are thought to arise due to striatal hyperdopaminergia, while dopamine
hypoactivity in the prefrontal cortex is proposed to account for negative and cognitive
symptoms.

13!
!
!
!
1.2.4.2 The Glutamate Hypothesis
Despite the usefulness of the dopamine hypothesis, cortical abnormalities observed in SCZ
appear to be more complex than the originally proposed hypofrontality. Some posit that the
dopamine system of SCZ patients may in fact be normal, yet suffer from abnormal regulation
(Grace, 2012), and hints towards the involvement of several neurotransmitter systems in
SCZ’s etiology. For instance, a large amount of evidence has characterized SCZ as a neuro-
developmental disorder accompanied by perturbations in glutamatergic neurotransmission
(Weinberger, 1987; Olney & Farber, 1995; Mohn et al., 1999; Olney et al., 1999). Much of
the rationale supporting this ‘glutamate hypothesis of SCZ’ stems from observations that
administration of NMDAR antagonists, ketamine and phencyclidine (PCP), induce SCZ-like
symptoms in healthy controls and exacerbate psychotic symptoms in SCZ patients (Luby et
al., 1959; Luisada, 1978; Lahti et al., 2001). Interestingly, glutamate action, specifically at
the NMDAR, has been shown to regulate dopaminergic neurotransmission (Zweifel et al.,
2008; Parker et al., 2010).
Several theories regarding glutamate’s involvement in SCZ etiology have been proposed.
One overarching theory suggests that NMDAR glutamatergic hypofunction is responsible for
creating the positive and negative symptoms of SCZ through dysregulation of mesolimbic
and mesocortical dopamine neurons, respectively. More specifically, hypofunction of the
inhibitory glutamatergic pathway that projects from cortical pyramidal neurons to dopamine
neurons in the VTA may cause tonically uninhibited dopamine release from the mesolimbic
pathways and result in positive symptoms, while hypofunction of the stimulatory
glutamatergic pathway that acts on mesocortical dopamine neurons could cause the

14!
!
!
!
hypodopaminergia that is responsible for negative, cognitive and affective symptoms (Stahl,
2007). The glutamate hypothesis of SCZ is discussed in more detail in the following section.!!
1.3 Glutamatergic Dysfunction in Schizophrenia
A great deal of evidence has characterized SCZ as a disorder related to aberrant glutamate
signalling. The following section provides a general overview of the glutamate system with
particular emphasis placed on receptors, transporters and enzymes involved in glutamatergic
neurotransmission. This section concludes with a thorough summary of literature on
glutamate abnormalities in SCZ.
1.3.1 The Glutamate System
Glutamate is the primary excitatory neurotransmitter in the mammalian central nervous
system (CNS) (Curtis et al., 1960). Glutamate neurotransmission plays an essential role in
physiological processes such as synaptic plasticity and induction of long term potentiation
(LTP), processes that are both implicated in memory and learning (Harris et al., 1984; Morris
et al., 1986). Various receptors, transporter and enzymes are involved in the proper
functioning of glutamatergic neurotransmission. There are two main groups of excitatory
glutamate receptors, the ligand gated ion channels which include the NMDAR, AMPAR and
kainate types of receptors, and the metabotropic G-protein coupled receptors (mGluRs)
which are subdivided into group I, group II and group III (Foster & Roberts, 1981; Watkins
& Evans, 1981; Watkins & Collingridge, 1994; Pin & Duvoisin, 1995). Extracellular levels
of glutamate and other relevant neurotransmitters such as glycine are carefully regulated by
the action of transporters. Once taken up into neurons and/or surrounding microglia,
glutamate and glycine are either converted to other chemical compounds or metabolized
15!
!
!
!
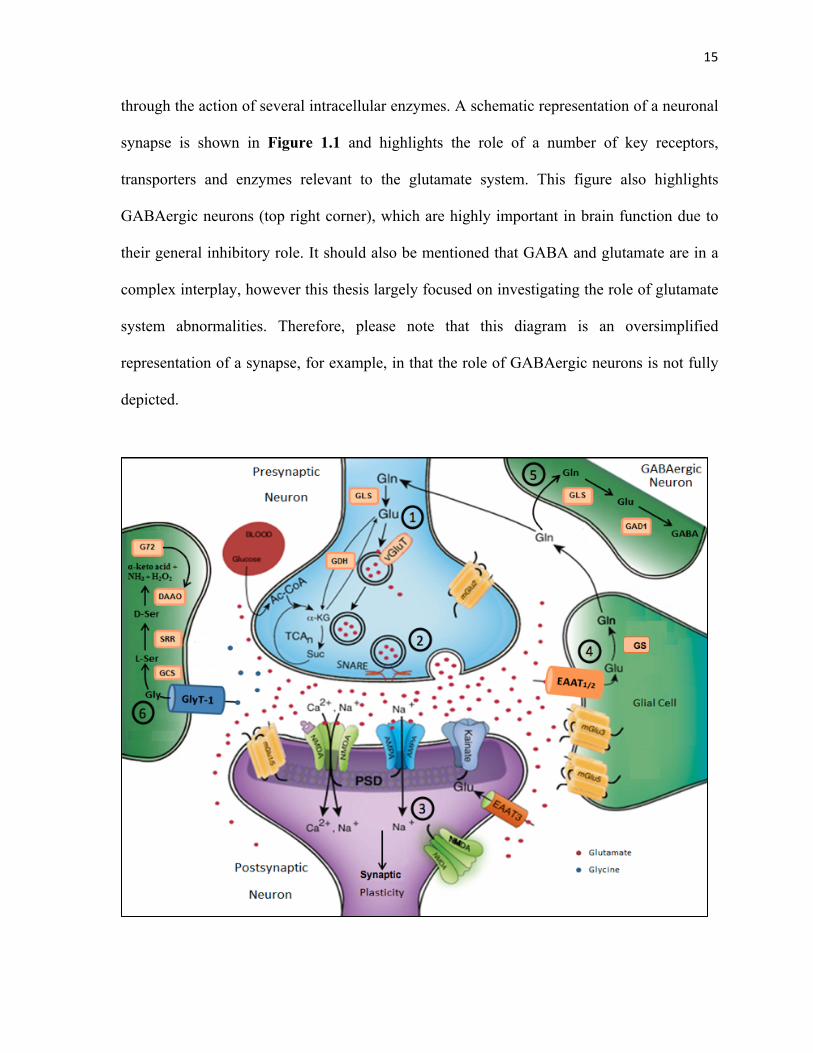
through the action of several intracellular enzymes. A schematic representation of a neuronal
synapse is shown in Figure 1.1 and highlights the role of a number of key receptors,
transporters and enzymes relevant to the glutamate system. This figure also highlights
GABAergic neurons (top right corner), which are highly important in brain function due to
their general inhibitory role. It should also be mentioned that GABA and glutamate are in a
complex interplay, however this thesis largely focused on investigating the role of glutamate
system abnormalities. Therefore, please note that this diagram is an oversimplified
representation of a synapse, for example, in that the role of GABAergic neurons is not fully
depicted.
&

16!
!
!
!
Figure&1.1&Schematic&Representation&of&a&Glutamatergic&Synapse&
Glutamate!(Glu)!is!the!major!excitatory!neurotransmitter!in!the!mammalian!central!nervous!
system! with! roles! in! cognition,! memory! and! learning.! Precise! regulation! of! glutamate!
signaling! must! be! maintained! as! excitotoxicity! can! result! from! excessive! glutamatergic!
stimulation.! (1)! Glutamate! is! derived! from! glutamine! (Gln)! through! the! action! of! the!
enzyme!glutaminase! (GLS)!or! from!the!tricarboxylic!acid! (TCA)!cycle,!and! is!packaged! into!
presynaptic!vesicles!by!vesicular!glutamate!transporters!(vGluTs).!(2)!Vesicles!release!their!
contents! into! the! synaptic! cleft! through! vesicleAmembrane! fusion,! which! is! facilitated! by!
SNARE!proteins.!(3)!Presynaptic!and!postsynaptic!glutamate!binding!activates!metabotropic!
(mGlu)! and! ionotropic! (NMDA,! AMPA,! Kainate)! glutamate! receptors,! downstream! signal!
transduction!cascades!and!synaptic!plasticity.!(4)!Glutamate!is!recycled!from!the!synapse!by!
glutamate! transporters! (EAAT1/2),! which! are! expressed! predominantly! on! astroglia! (glial!
cells).! Once! inside! astroglia,! glutamate! is! converted! to! glutamine! through! glutamine!
synthetase!(GS)!and!is!then!exported!for!reAuptake!by!glutamatergic!neurons.!(5)!Glutamine!
may! also! be! taken! up! by!GABAergic! interneurons,! converted! first! into! glutamate! by!GLS,!
and!then!to!GABA!by!glutamate!decarboxylase!1!(GAD1).!Glycine!(Gly)!acts! independently!
as!an!inhibitory!neurotransmitter,!or!in!concert!with!glutamate!neurotransmission!as!a!coA
agonist! at! the! NMDAR.! (6)! Glycine! is! recycled! from! the! synapse! by! glycine! transporters!
(GlyT1)!which!are!located!primarily!on!astrocytes.!Once!taken!up!by!astrocytes,!glycine!can!
be!metabolized!to!αAketoglutaric!acid!(αAketo!acid),!ammonia!(NH3)!and!hydrogen!peroxide!
(H2O2)! through! a! series! of! intermediate! steps:! glycine! to! LAserine! (LAser)! catalyzed!by! the!
glycine! cleavage! system! (GCS),! LAserine! to! DAserine! (DAser)! catalyzed! by! serine! racemase!
(SRR),!DAserine!to!the!final!products!via!the!DAamino!acid!oxidase!enzyme,!that!is!activated!
by!DAamino!acid!oxidase!activator!protein!(G72).!Adapted!from!Pharmacology+Biochemistry+
and+ Behavior,! 100(4).!Mark! J.!Niciu,! Benjamin! Kelmendi!&!Gerard! Sanacora.!Overview!of!
Glutamatergic! Neurotransmission! in! the! Nervous! System,! 656A64.! Copyright! (2012)! with!
permission!from!Elsevier.!
!

17!
!
!
!
1.3.1.1 Ionotropic Glutamate Receptors
NMDAR and AMPAR are principally expressed on dendritic spines of postsynaptic neurons
(Kharazia et al., 1996) where they function in signaling during induction of synaptic
plasticity and long-term modulation of synaptic strength (Asztely & Gustafsson, 1996;
Shapiro, 2001). Kainate receptors mostly localize to postsynaptic membranes where they
function to modulate synaptic response to glutamate (Van den Pol, 1994; Haak, 1999).
Kainate receptors also localize to surrounding glia and presynaptic neurons where they
modulate a variety of cellular functions including glutamate reuptake and release, and
presynaptic excitability (Cartmell & Schoepp, 2000; Huettner, 2003; Grueter & Winder,
2005; Rodriguez-Moreno, 2006).
The NMDAR is a hetero-tetramer composed of two obligatory GluN1 (NR1) subunits and
either two GluN2 (NR2A-D) or two GluN3 (NR3A-B) subunits (Monyer et al., 1992)
(Figure 1.2). Subunit composition varies during development and results in various receptor
isoforms with different physiological and pharmacological properties (McBain & Mayer,
1994). The NMDAR is implicated in additional processes such as neuronal migration
(Komuro & Rakic, 1993) and differentiation (Pearce et al., 1987), response to trophic factors
(Black, 1999), dendritic spine development (Tian et al., 2007), LTP, and memory and
learning (Massey et al., 2004). Given the importance of the NMDAR in neuronal processes,
abnormal function and/or development of this receptor system could lead to the deficits in
learning and memory that are observed in patients with SCZ.
NMDAR channel activation utilizes a coincidence detection type mechanism that requires
simultaneous binding of glutamate to the NR2 subunit (Laube et al., 1997; Anson et al.,
1998), binding of an endogenous co-agonist, either glycine or D-serine, to the NR1 subunit

18!
!
!
!
(Kuryatov et al., 1994; Hirai et al., 1996) and removal of a voltage-dependent block by
external magnesium (Mg2+) (Mayer et al., 1984; Nowak et al., 1984). Channel activation
produces excitatory postsynaptic potentials (EPSPs) and increases calcium (Ca2+) and
sodium (Na+) concentration inside the cell (Schiller et al., 1998). Intracellularly, calcium acts
as a secondary messenger with downstream effects on protein kinases such as mitogen
activated protein kinase (MAPK) (Xia et al., 1996) and transcription factors such as cyclic
adenosine monophosphate (cAMP) response element binding protein (CREB) (Sala et al.,
2000). NMDAR action is also associated with effector kinases such as protein kinase C
(PKC) (Zheng et al., 1997), protein kinase A (PKA) (Tingley et al., 1997) and
calcium/calmodulin-dependent kinase II (CaMKII) (Omkumar et al., 1996).
The remaining two ionotropic glutamate receptors, AMPA and kainate, both form tetrameric
complexes consisting of GluR1-4 subunits (AMPA) or GluR5-7 and KA1-2 subunits
(kainate). Both receptors are widely expressed in the mammalian CNS and mediate fast
excitatory neurotransmission in response to the binding of glutamate (Khakh & Henderson,
2000). The function of AMPA and kainate ionotropic glutamate receptors is controlled in
part by RNA editing of subunits (Higuchi et al., 1993), as well as alternative splicing of
messenger RNA (mRNA) transcripts (Sommer et al., 1990). Ionotropic glutamate receptor
function, the AMPAR in particular, is also regulated by receptor number, localization and
facilitation at synapses ultimately controlling such processes as LTP-like strengthening of
neocortical synapses after sensory stimulation (Takahashi et al., 2003), learning in the
hippocampus (Whitlock et al., 2006) and response to stress hormones (Groc et al., 2008;
Martin et al., 2009; Yuen et al., 2011).

19!
!
!
!
Figure&1.2!The&Ionotropic&NFmethylFDFaspartate&Glutamate&Receptor!
Functional!NMDARs!are!constructed!from!assemblies!of!four!subunits!(two!NR1!and!either!
two! NR2AAD! or! NR3AAB).! Subunit! composition! varies! and! results! in! various! receptor!
isoforms! that!have!different!physiological! and!pharmacological!properties.!NR1! subunit! is!
encoded!by!eight!alternatively!spliced!variants!of!GRIN1,!and!contains!the!binding!site!for!
endogenous! coAagonists! glycine! and! DAserine.! NR2! subunits! contain! the! glutamate!
recognition! site! and! binding! sites! for! various! other! antagonists.! The! NMDAR! channel! is!
normally! blocked! at! resting! membrane! potential! by! magnesium! (Mg+)! in! a! voltageA
dependent! manner! and! is! activated! following! depolarization! of! the! postsynaptic!
membrane.! Receptor! activation! occurs! through! a! coincidence! detection! type!mechanism!
and!requires!both!glutamate!and!glycine!binding.!Receptor!activation!leads!to!calcium!(Ca2+)!
and!sodium! (Na+)! influx,!and!potassium! (K
+)!efflux,! facilitating! longAterm!potentiation!and!
synaptic! plasticity.! Antagonists! of! the! NMDAR! include:! Memantine! (treats! Alzheimer’s!
disease),! Selfotel! (discontinued! anxiolytic/anticonvulsant),!MRZ! 2/576! (research! use)! and!
Ifenprodil! (discriminates!NMDAR!subpopulations).!Adapted!from!Pharmacological+Reviews!
50.!Wojciech! Danysz! and! Chris! G.! Parsons.! Glycine! and! NAMethylADAAspartate! Receptors:!
Physiological!Significance!and!Possible!Therapeutic!Applications,!597A664.!Copyright!(1998)!
with!permission!from!American!Society!for!Pharmacology!and!Experimental!Therapeutics.!!!

20!
!
!
!
1.3.1.2 Metabotropic Glutamate Receptors
Like the kainate receptors, mGluRs modulate synaptic responses to glutamate at postsynaptic
membranes (Van den Pol, 1994; Haak, 1999) and modulate a variety of cellular functions at
presynaptic neurons (Cartmell & Schoepp, 2000; Huettner, 2003; Grueter & Winder, 2005;
Rodriguez-Moreno, 2006). The metabotropic glutamate receptors mediate signaling through
7-transmembrane region G-protein coupled receptors (Pin et al., 2003). The three subclasses
are groups I, II and III and they signal through different mechanisms. Group I (mGluR1, 5)
signals through G-protein activation that is coupled to protein kinase activation (ie PLC), and
subsequently leads to the formation of inositol 1,4,5-triphosphate (IP3) and intracellular
release of calcium (Hermans & Challiss, 2001). In contrast, group II (mGluR2, 3) and group
III (mGluR4, 6, 7, 8) receptors couple to inhibitory G-protein signaling whereby inhibition of
adenylyl cyclase activity subsequently leads to reduction in cAMP production (Conn & Pin,
1997). The mGluR receptors are tightly regulated by formation of various alternatively
spliced isoforms which control several aspects of receptor function, including surface
receptor expression, G-protein coupling, and interaction with scaffolding proteins in the
postsynaptic density (PSD) (Stowell & Craig, 1999; Tu et al., 1999; Pin et al., 2003).
Drugs targeting metabotropic glutamate receptors have been investigated for the treatment of
SCZ (reviewed by Li et al., 2015). LY404039, a specific and potent mGluR2/3 agonist
developed by Eli Lilly & Co, is one example of a drug from this class (Monn et al., 2005).
Experiments with mGluR2/3 knockout mice suggest that the antipsychotic-like behavourial
effects of LY404039 are caused by mGluR2 activation and further implicate the glutamate
system in SCZ etiology (Rorick-Kehn et al., 2007; Fell et al., 2008). Clinical studies
investigating this compound for the treatment of SCZ indicate it was well tolerated and

21!
!
!
!
lacked the adverse side effects typically associated with dopaminergic agents such as weight
gain, EPS and hyperprolactinemia (Adams et al., 2013). Unfortunately, efficacy results for
this compound were inconsistent. More specifically, the initial proof of concept study
showed LY404039 and the active control olanzapine (OLZ) significantly decreased patient
symptoms compared to placebo (Patil et al., 2007), however a Phase 2 (inpatient, dose-
ranging study) reported ‘inconclusive results’ as both LY2140023 and OLZ failed to separate
from placebo (Kinon et al., 2011). Finally, a Phase 2 (acute, fix-dosed study) showed that
LY404039 did not separate from placebo in the primary efficacy endpoint unlike the active
control risperidone (Kinon et al., 2013). Development of this drug was discontinued
following the last study. Further research into LY2140023 by Eli Lilly has indicated that
response to this compound may be more favourable in a subgroup of patients that carry a
select set of 23 single nucleotide polymorphism (SNP) gene variants (Liu et al.,
2012). Interestingly, 16 of these 23 SNPs are located in the HTR2A gene. Studying the
relationship between genetic variation and drug response is called pharmacogenetics and will
be discussed in more detail in Section 1.5.
1.3.1.3 Amino Acid Transporters
Amino acid transporters control the reuptake of amino acids, such as glutamate and glycine,
from the synapse into neurons and/or surrounding neuroglia. Two amino acid transporters are
involved in glutamatergic neurotransmission: the excitatory amino acid transporters
(EAATs) 1-5 and the glycine transporters (GlyTs) 1 and 2. The EAATs are responsible for
the rapid reuptake of synaptic glutamate into astrocytes and control extracellular
concentrations of glutamate (reviewed by O'Shea, 2002).! Strict control of extracellular
glutamate concentrations is necessary to prevent excitotoxicity and cell death (Coyle &

22!
!
!
!
Puttfarcken, 1993; Szatkowski & Attwell, 1994). Of the five EAATs, EAAT2 (otherwise
known as GLT1) is the chief glutamate transporter in the forebrain and is responsible for
more than 90% of glutamate reuptake into surrounding astrocytes (Bar-Peled et al., 1997;
Furuta et al., 1997).
The second type of transporter involved in glutamatergic neurotransmission is responsible
for controlling levels of the amino acid glycine. Within the nervous system, glycine carries
out a dual role as both an inhibitory neurotransmitter at glycine receptors and as a co-agonist
at the NR1 subunit of the NMDAR to mediate excitatory neurotransmission (Johnson &
Ascher, 1987). The primary neuronal glycine transporter, GlyT1, is expressed predominantly
on astrocytes and is responsible for tightly regulating glycine concentrations in the vicinity of
excitatory synapses through reuptake of glycine into neighbouring astroglia (Smith et al.,
1992; Zafra et al., 1995; Cubelos et al., 2005). Interestingly, changes in GlyT1 activity have
been shown to alter NMDAR-mediated neurotransmission as evidenced by increased
NMDAR excitatory postsynaptic currents (EPSCs) following GlyT1 antagonism (Bergeron
et al., 1998; Chen et al., 2003; Kinney et al., 2003) and by increased NMDAR function in
GlyT1 knockout mice (Gabernet et al., 2005).
Drugs targeting the glycine system have also been investigated for SCZ. One such drug
inhibits GlyT1 function in an attempt to ameliorate the NMDAR hypofunction associated
with SCZ etiology (reviewed by Hashimoto, 2010). One such glycine transporter inhibitor
bitopertin (RG1678), developed Roche, has been investigated as an adjunct for persistent
negative symptoms in SCZ. Five clinical trials have been conducted so far, however the
results have been non-significant (ClinicalTrails.gov identifiers: NCT01192867,
NCT01192906, NCT01192880, NCT01235520, and NCT01235559). Various clinical studies

23!
!
!
!
have also investigated the efficacy of adjunct glycine, D-serine or D-cycloserine (NMDAR
co-agonists) for the treatment of SCZ (reviewed by Javitt, 2004). On a more positive note,
the majority of these studies have reported large effect-size improvements in negative and
cognitive symptoms (Potkin et al., 1992; Heresco-Levy et al., 1996; Tsai et al., 1998; Goff et
al., 1999; Javitt et al., 2001).
1.3.1.4 Glutamate System Enzymes
There are several key enzymes involved in regulating glutamatergic neurotransmission.
These enzymes include glutaminase (GLS) and glutamine synthetase (GS), which are
responsible for interconverting glutamate and glutamine, glutamate decarboxylase 1
(GAD1), which converts glutamate to GABA, and various other enzymes involved in glycine
metabolism.
Glutamine synthetase is an astrocytic enzyme responsible for converting glutamate to
glutamine (Meister, 1974; Martinez-Hernandez et al., 1977). Glutamine is released
extracellularly as a neuroprotective form of glutamate where it can be taken up by neurons
and deaminated back into glutamate by the enzyme glutaminase (Goldstein, 1967; Curthoys
& Watford, 1995). This pattern of compartmentalization and interconversion constitutes what
is known as the ‘glutamate-glutamine cycle’ (Shank & Aprison, 1981). This cycle ensures
three events: 1) glutamate is rapidly removed from the synapse to prevent neurotoxicity; 2)
glutamate is converted to the ‘neuroprotective carrier’ glutamine; and 3) glutamine is made
available in neuronal compartments and allows for the regeneration of glutamate for
excitatory signaling (Daikhin & Yudkoff, 2000).

24!
!
!
!
Besides reuptake into glutamatergic neurons, glutamine can also be taken up by GABAergic
interneurons and metabolized to GABA by GAD1 (Erlander et al., 1991). GABA is an
inhibitory neurotransmitter that controls fundamental processes such as neurogenesis
(Fagiolini et al., 2004; Ge et al., 2006), movement, and tissue development (Nakatsu et al.,
1993). A mutation in the glutamate decarboxylase 1 gene (GAD1) in humans results in a
disorder known as spastic cerebral palsy (Lynex et al., 2004), while GAD1 knockout mice
have dramatically reduced GABA levels and die at birth due to a severe cleft palate (Asada et
al., 1997).
Lastly, a series of enzymes are responsible for the metabolism of glycine to α-ketoglutaric
acid (α-keto acid), ammonia (NH3) and hydrogen peroxide (H2O2). Upon entry into glial cells
through the action of GlyT1, glycine is first converted to L-serine (L-ser) by the glycine
cleavage system (GCS) (Kikuchi, 1973). L-serine is then converted to D-serine (D-ser) by
the enzyme serine racemase (SRR) (Wolosker et al., 1999), and then converted to the final
products α-keto acid, NH3 and H2O2 via D-amino acid oxidase (Dixon & Kleppe, 1965). Like
glycine, D-serine is an endogenous agonist at the NR1 subunit of the NMDAR and plays a
role in regulating excitatory neurotransmission (Matsui et al., 1995).
1.3.2 Glutamate System Abnormalities in Schizophrenia
Numerous glutamate system abnormalities have been reported in patients with SCZ giving
further credence to the glutamate hypofunction hypothesis. For instance, postmortem studies
show abnormalities in NMDAR subunit expression in patients with SCZ, including increased
NR2B transcript in the thalamus (Clinton & Meador-Woodruff, 2004)) and temporal lobes
(Grimwood et al., 1999), as well as higher NR1 expression in the dorsolateral prefrontal
cortex (DLPFC) (Dracheva et al., 2001), substantia nigra (Mueller et al., 2004) and superior

25!
!
!
!
temporal cortex (Nudmamud-Thanoi & Reynolds, 2004). In addition, more recent data has
indicated that unmedicated patients with SCZ have increased hippocampal glutamate in
comparison to healthy controls (Kraguljac et al., 2013).
Preclinical models also point to glutamate system abnormalities in modeling SCZ-related
behaviours. Rats administered PCP and ketamine exhibit hyperlocomotion in an open field
taken to emulate positive symptoms (Sturgeon et al., 1979; Nabeshima et al., 1987), deficits
in social behaviour (Sams-Dodd, 1995, 1998) and immobility in the forced swim test (Noda
et al., 1995, 2000) as behavioural models for negative symptoms, and impairments in various
learning paradigms to imitate the cognitive deficits seen in patients with SCZ (reviewed by
Mouri et al., 2007). Another behavioural model for SCZ involves a phenomenon called pre-
pulse inhibition (PPI) wherein, in healthy individuals, the application of a small stimulus
(typically auditory) before a strong stimulus reduces or inhibits the response to the stronger
stimulus. This PPI function is impaired in most patients with SCZ and is linked to
dysfunctions in sensorimotor gating and protection from sensory overload that can be
modeled in animals (Braff et al., 1992; Lipska et al., 1995). Administration of PCP or (+)-D-
aspartate 5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-iminedizocilpine (MK-
801) to postnatal rats during key neurodevelopmental periods induces PPI deficits in
adulthood, linking glutamate to this important model of SCZ (Harris et al., 2003; Takahashi
et al., 2006).

26!
!
!
!
1.4 Schizophrenia Pharmacotherapy
Antipsychotic medications are the mainstay in the pharmacological treatment of SCZ
(reviewed by Tandon, 2011). This section begins with an overview on the history and
mechanism of action of antipsychotic drugs in psychiatry, with a particular focus on
clozapine. Next, a brief review on the use of symptom rating scales in measuring response
will be presented. This section concludes with a discussion on CLZ’s unique efficacy for the
treatment of TRS and factors that contribute to variability in CLZ response.
1.4.1 Antipsychotic Medication Overview
Prior to the advent of antipsychotic therapy, patients suffering from SCZ were chronically ill,
confined to mental health hospitals and subjected to alternative treatment options such as
electroconvulsive shock therapy (ECT) and lobotomies, in which a portion of the patient’s
brain was removed (Moniz, 1937). The pioneer typical (first-generation) antipsychotic drug,
chlorpromazine (CLZ), was serendipitously discovered in the early 1950s following attempts
to develop a synthetic anti-malaria drug during WWII (Gilman et al., 1944). Rather than use
as an anti-malaria agent, CPZ was found to have anaesthetic-like properties with added
benefits: patients administered CPZ required lower doses of aneasthetic and were able to
better withstand the stress of surgical trauma (Laborit & Huguenard, 1951). CPZ was
initially supplied to two hospitals in Paris – the Central Military Hospital called Val-de-
Grâce and the psychiatric clinic of Sainte-Anne Hospital, to treat psychiatric disorders
(Delay et al., 1952). The first patient administered CPZ was a 57-year-old laborer admitted to
hospital due to erratic and uncontrollable behaviour. Following 3-weeks of CPZ treatment,
the patient appeared normal and was discharged (Hamon et al., 1952). In the decades

27!
!
!
!
following, other typical APs were developed including: haloperidol, fluphenazine,
thioridazine, loxapine, perphenazine, trifluoperazine and thiothixene.
Very shortly after the introduction of CPZ and its derivatives, extrapyramidal symptoms
including parkinsonism and akathisia became recognized as side effects associated with the
use of APs (Steck, 1954; Sigwald et al., 1959). From efforts to develop an AP that was free
of EPS came the birth of the second generation (atypical) APs, the first of which was
clozapine (CLZ). First manufactured by G. Stille at Wander Pharmaceuticals in Bern,
Switzerland in the early 1960s, CLZ was associated with little to no EPS (Hippius, 1996).
After two open studies in 1966 (Gross & Langner, 1966; Bente et al., 1966) and one double-
blind study in 1971 (Angst et al., 1971), CLZ was briefly marketed yet quickly withdrawn
due to reports of a life-threatening side effect in Finland populations (reviewed by Idanpaan-
Heikkila et al., 1997). This side effect is now recognized as clozapine-induced
agranulocytosis (CIA), which occurs in ~1% of CLZ-treated patients. CLZ was reintroduced
into the US market in 1990 following a landmark clinical trial that proved CLZ’s superior
efficacy for TRS patients (Kane et al., 1988). CLZ is currently a 3rd line treatment for
patients with treatment-resistant or intolerant forms of SCZ and is prescribed in combination
with mandatory blood monitoring (Moore et al., 2007). CLZ also has the propensity to cause
weight gain and metabolic syndrome. CLZ’s success quickly led to the development of
additional drugs in the atypical class including: risperidone, olanzapine, quetiapine,
ziprasidone, aripiprazole, paliperidone, iloperidone, asenapine and lurasidone.
1.4.2 The Mechanism of Action of Antipsychotic Drugs
All AP medications share a common primary mechanism of action, which involves blockade
of DRD2 (Carlsson & Lindqvist, 1963; Seeman & Lee, 1975; Creese et al., 1976). For this

28!
!
!
!
reason, DRD2 has been an important access point to the circuitry underlying the symptoms
of SCZ, however, may not be the primary mechanism through which CLZ achieves
therapeutic effect. All AP medications have variable binding profiles and affinities for
different combinations of receptors. CLZ’s multi-receptor profile likely accounts for its
ability to improve some domains of cognition and social function for patients with TRS
(Hagger et al., 1993; McGurk, 1999). Besides having a weaker affinity for DRD2, CLZ has a
high affinity for 5-HT2A, histamine H1 (HRH1/H1), muscarinic M1 (CHRM1/M1) and
adrenergic α-1A receptor (ADRA1A) (Meltzer et al., 1989a), and a moderate affinity for the
serotonin type 1A receptor (5-HT1A) and the adrenergic receptor α-2A receptor (ADRA2A)
(Schotte et al., 1996). This binding profile, more specifically the weak affinity for 5-HT2A
compared to DRD2, is thought to account for CLZ’s lower propensity to cause EPS (Meltzer
et al., 1989b).
1.4.3 Treatment Response Measures
Response to AP medication is commonly assessed using symptom rating scales (reviewed by
Mortimer, 2007). Several scales assessing symptom severity in SCZ have been used for this
purpose, the most common of which are the Brief Psychiatric Rating Scale (BPRS) (Overall
& Gorham, 1962), the Positive and Negative Syndrome Scale (PANSS) (Kay et al., 1987)
and the Clinical Global Impression Scale (CGI) (Guy, 1976). Symptom severity is assessed
at baseline prior to treatment and then again following a trial of AP medication. Change in
symptom scores from baseline can be used as either a categorical measure of response in
which ‘responders’ are those who exhibit a decrease in scores beyond a certain threshold, or
a continuous response measure whereby percent score reduction is calculated using the
following formula: % score change = [(6 month score - baseline score)/(baseline score)] x

29!
!
!
!
100%. The threshold used to define categorical response in this thesis was a 20-point or more
decrease in BPRS scores. This measure of response is commonly used, however, is
somewhat arbitrary and fails to reflect a patient’s personal, social and cognitive functioning –
factors that hold great weight as to how a patient will function in society (Mortimer, 2007).
Despite this limitation, the use of rating scales to assess response has proven useful in
research studies.
1.4.4 Clozapine for Treatment-Resistant Schizophrenia
As mentioned in Section 1.1.3, TRS is particularly burdensome to the healthcare system and
drastically decreases the likelihood of optimal disease outcome. The most commonly used
definition of TRS involves: 1) failure to respond to at least two periods of treatment in the
preceding five years with neuroleptic agents from at least two different chemical classes
(doses equivalent ≥1000 mg/day of CPZ for six weeks) without significant symptomatic
relief; and 2) no period of good functioning within the preceding five years (Kane et al.,
1988). This definition of TRS first emerged from the landmark clinical trial, which proved
CLZ was superior to CPZ for treatment resistant forms of SCZ (Figure 1.3). More
specifically, 268 patients who met criteria for TRS were entered into a double-blind
comparison study that lasted 6 weeks. Patients were randomly assigned to either one of two
groups: CLZ (up to 900mg/day) or CPZ (up to 1,800mg/day). Response was defined as: 1) a
20% decrease or more in BPRS total score; 2) a CGI score less than or equal to 3; and 3) a
BPRS total score equal to or less than 35, using 0-6 ratings. Results showed that a
significantly greater number of treatment-resistant patients responded to CLZ than to the
prototypical AP CPZ (30% vs 4%, p=0.001). The results of this trial were responsible for
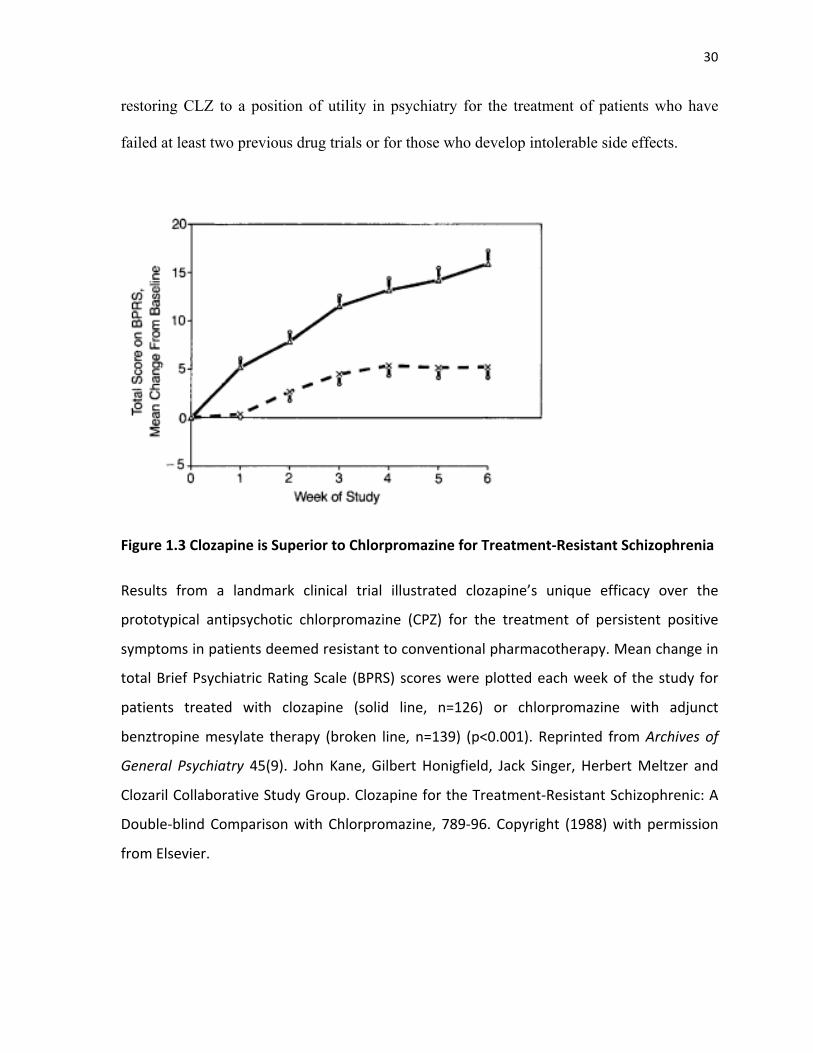
30!
!
!
!
restoring CLZ to a position of utility in psychiatry for the treatment of patients who have
failed at least two previous drug trials or for those who develop intolerable side effects.
&
Figure&1.3!Clozapine&is&Superior&to&Chlorpromazine&for&TreatmentFResistant&Schizophrenia&
Results! from! a! landmark! clinical! trial! illustrated! clozapine’s! unique! efficacy! over! the!
prototypical! antipsychotic! chlorpromazine! (CPZ)! for! the! treatment! of! persistent! positive!
symptoms!in!patients!deemed!resistant!to!conventional!pharmacotherapy.!Mean!change!in!
total!Brief!Psychiatric!Rating!Scale! (BPRS)!scores!were!plotted!each!week!of! the!study! for!
patients! treated! with! clozapine! (solid! line,! n=126)! or! chlorpromazine! with! adjunct!
benztropine!mesylate! therapy! (broken! line,! n=139)! (p<0.001).! Reprinted! from!Archives+ of+
General+ Psychiatry! 45(9).! John! Kane,! Gilbert! Honigfield,! Jack! Singer,! Herbert!Meltzer! and!
Clozaril!Collaborative!Study!Group.!Clozapine!for!the!TreatmentAResistant!Schizophrenic:!A!
DoubleAblind!Comparison!with!Chlorpromazine,! 789A96.! Copyright! (1988)!with!permission!
from!Elsevier.

31!
!
!
!
Several additional trials have been conducted assessing CLZ’s unique efficacy for TRS. For
instance, one such study comparing clozapine, haloperidol, olanzapine and risperidone
(CHOR) in TRS found that CLZ and OLZ had similar antipsychotic efficacy, however, CLZ
was the most effective for treating negative symptoms (Volavka et al., 2002). Additional
studies indicate CLZ shows greater benefits for TRS as compared to OLZ (Conley et al.,
1999; Meltzer et al., 2008). Results from the Clinical Antipsychotic Trials of Intervention
Effectiveness Project (CATIE) also suggest that CLZ is superior to other AP drugs for
treatment resistance (McEvoy et al., 2006). Briefly, CATIE was a double-blind randomized
clinical trial funded by the National Institute of Mental Health (NIMH) that compared the
effectiveness of first- and second-generation APs used to treat SCZ. Patients who
discontinued treatment in the first phase with one of four newer atypical APs (olanzapine,
quetiapine, risperidone or ziprasidone) because of inadequate efficacy (n=99) were randomly
assigned to either open-label CLZ (n=49) or blinded treatment to an alternative atypical in
the second phase. Discontinuation due to lack of efficacy was significantly less in the CLZ
group than in the other four groups (11% vs ~40%). It should be noted that this study
disallows for firm conclusions regarding the advantages of CLZ because of the unblinded
nature with which CLZ was used. However, given the results from other studies, CLZ does
appear to be superior for TRS.
1.4.5 Variability in Clozapine Response
Response to CLZ is highly variable and likely influenced by a combination of clinical,
demographic, environmental and genetic factors (reviewed by Arranz & Munro, 2011). Non-
genetic factors that alter clinical response include: treatment adherence (Dunbar-Jacob &
Mortimer-Stephens, 2001), previous drug exposure and duration of illness (Perkins et al.,

32!
!
!
!
2005), diet, smoking and concomitant medications (Meyer, 2001; de Leon, 2004), as well as
gender, ethnicity and age (Palego et al., 2002). Response to APs is also overshadowed by the
occurrence of adverse side effects that result in less than optimal treatment outcomes.
Regarding genetic factors, twin and family studies of CLZ response indicate concordance for
response (Vojvoda et al., 1996). Like SCZ, response to CLZ is thought to be a complex trait
likely depending on the multiplicative effect of several genes that are involved in drug action
(International Schizophrenia Consortium et al., 2009; Sadee, 2013). However, drug response
phenotypes likely depend on fewer genes than those involved in disease etiology due to the
relatively narrow pathway a drug follows through the body to mediate therapeutic effect
(Johnson et al., 2011; Ma & Lu, 2011). Identifying genetic variation with the ability to
improve treatment outcome remains an active area of research and is discussed in the
following section.

33!
!
!
!
1.5 Pharmacogenetics of Clozapine Response
Pharmacogenetics (PGx) is the study of gene variation as it relates to drug response and the
development of adverse side effects. This section will touch on the history of PGx,
summarize findings from PGx studies on CLZ response with a particular focus on glutamate
system related genes, and will conclude by exhibiting how ENCODE data can be used to
select functional candidate SNPs for PGx studies.
1.5.1 Pharmacogenetics Overview
The roots of pharmacogenetics travel as far back as 1866 with the work of Gregor Johann
Mendel (1822-1884) on the garden pea (Pisum sativum L.) and the establishment of the rules
of heredity (Mendel, 1866). Since Mendel’s time, there have been several noteworthy
individuals who have contributed to our present day understanding of the relationship
between genetics and drug response. Noteworthy contributions include: 1) twin studies
indicating the influence of multiple genes on the pharmacokinetics of various drugs between
the years of 1957-1970 (Vesell, 1978); 2) development of the term ‘pharmacogenetics’ by
Friedrich Vogel in 1959 (Vogel, 1959); and 3) revolutionary works by a man named Werner
Kalow (1917-2008), deemed the ‘grandfather of pharmacogenetics’, who published the first
monograph on PGx entitled ‘Phamacogenetics – Heredity and the Response to Drugs’ in
1962 (Kalow, 1962). The advent of polymerase chain reaction (PCR) in 1985 was also a
significant contribution to PGx as it paved the way for gene cloning experiments and allowed
for further characterization of genes involved in drug response (Mullis et al., 1986). Shortly
thereafter in 1987, a very important family of liver enzymes called the cytochrome P450
enzymes (CYPs), such as CYP2D6 and CYP1A2, were cloned and polymorphisms within
these genes were linked to drug response (Distlerath et al., 1985). CYP enzymes are

34!
!
!
!
particularly relevant to the field of psychiatry because they are responsible for metabolizing a
large proportion of the currently prescribed antipsychotics and antidepressants (reviewed by
Guengerich, 2008).
Present day pharmacogenetics studies in psychiatry hope to elucidate gene variants that alter
response to psychotropic medications. The current prescribing method for AP medications
utilizes a ‘one size fits all’ approach and a suitable drug is identified through trial and error.
During this trial and error period, a subset of patients is plagued by lack of efficacy and side
effects due to toxicity. Generalized treatment plans are designed to satisfy the needs of the
majority of patients and do not account for patients that lie at the extremes of the distribution.
Treatment plans also fail to consider the biological patient-specific differences that cause
certain individuals sharing similar symptoms to react differently to the same medication.
Pharmacogenetics studies seek to identify the portion of patient-specific differences that lie
within the genome. Once predictive genetic markers are identified, they can be used to guide
therapeutic dosing and to warn against increase risk for developing adverse drug reactions
(Phillips et al., 2001). To illustrate current advancements in the field of psychiatry thus far,
positive results involving the use of genetic testing in depression have been reported (Hall-
Flavin et al., 2013; Winner et al., 2013; Altar et al., 2015). In addition, our Neurogenetics
section at the Centre for Addiction and Mental Health (CAMH) in Toronto is currently
leading a randomized control trial of an enhanced version of the GeneSight Psychotropic
Test for treatment response in SCZ.
1.5.2 Clozapine and Glutamatergic Neurotransmission
An important aspect of PGx studies is variant selection. Variants are often selected from
genes involved in known pharmacodynamic and pharmacokinetic pathways that relate to
35!
!
!
!
drug action, and often include variants that are located within drug metabolizing enzymes,
membrane transporters, receptors and target proteins (Sadee, 2013). A thorough
understanding of drug action can help guide selection of genetic variants that may play a role
in response. As mentioned above, CLZ has a unique binding profile and may achieve
therapeutic effect in part by modulating glutamatergic neurotransmission. A great deal of
evidence supports this theory and is summarized in Table 1.2. The information presented in
this table was used to guide gene selection for the work presented herein.
&
Table&1.2&Evidence&Supporting&Clozapine&Interacts&with&the&Glutamate&System&
Main!Findings! References!
CLZ!Attenuates!NMDAR!AntagonistAinduced!Behaviours!in!Preclinical!Models!
PPI!Impairments!
Clozapine!(5.0mg/kg)!antagonized!the!ability!of!PCP!(1.0mg/kg)!as!well!as!MKA801!
(0.5mg/kg)!to!repeatedly!and!robustly!decrease!PPI!in!a!rat!model,!without!
interfering!with!PPI!itself!
(Bakshi!et!al.,!1994)!!
Clozapine!(7.5kg/mg),!but!not!haloperidol!(0.1mg/kg),!significantly!restored!PPI!in!
ketamine!(1,!3,!or!6mg/kg)!treated!rats!
(Swerdlow!et!al.,!1998)!
Clozapine!(2.5mg/kg)!but!not!haloperidol!(0.035mg/kg)!successfully!reversed!PCPA
induced!PPI!deficits!in!a!primate!model!(Cebus!apella)!!
(Linn!et!al.,!2003)!
Clozapine!(5!or!10!mg/kg),!but!not!risperidone!(0.1!and!1mg/kg)!restored!deficits!
in!PPI!induced!by!MKA801!(0.3mg/kg)!in!a!rat!model!
(Bubenikova!et!al.,!2005)!!
Clozapine!(1.25!or!2.5mg/kg)!significantly!attenuated!PPIAimpairments!induced!by!
MKA801!(0.05mg/kg)!in!a!rat!model!
(Levin!et!al.,!2007)!
Hyperlocomotion!!!
Clozapine!and!haloperidol!(variable!doses)!were!moderately!effective!in!blocking!
PCPAinduced!(5.0mg/kg)!locomotor!stimulation!in!rats!
(Freed!et!al.,!1984)!
Clozapine!(2.5!or!5mg/day)!and!olanzapine!(0.25!or!0.5mg/kg)!potently!
antagonized!MKA801Ainduced!(0.05!or!1.1mg/kg)!locomotion!and!falling!assay!
(MKA801ALF)!in!rats!!
(Corbett!et!al.,!1995)!
Clozapine!(5A20mg/kg)!and!haloperidol!(0.01A0.1mg/kg)!progressively!potentiated!
hyperlocomotion!induced!by!PCP!administration!(3.2mg/kg)!in!rats!
!
(Sun,!Hu!&!Li,!2009)!
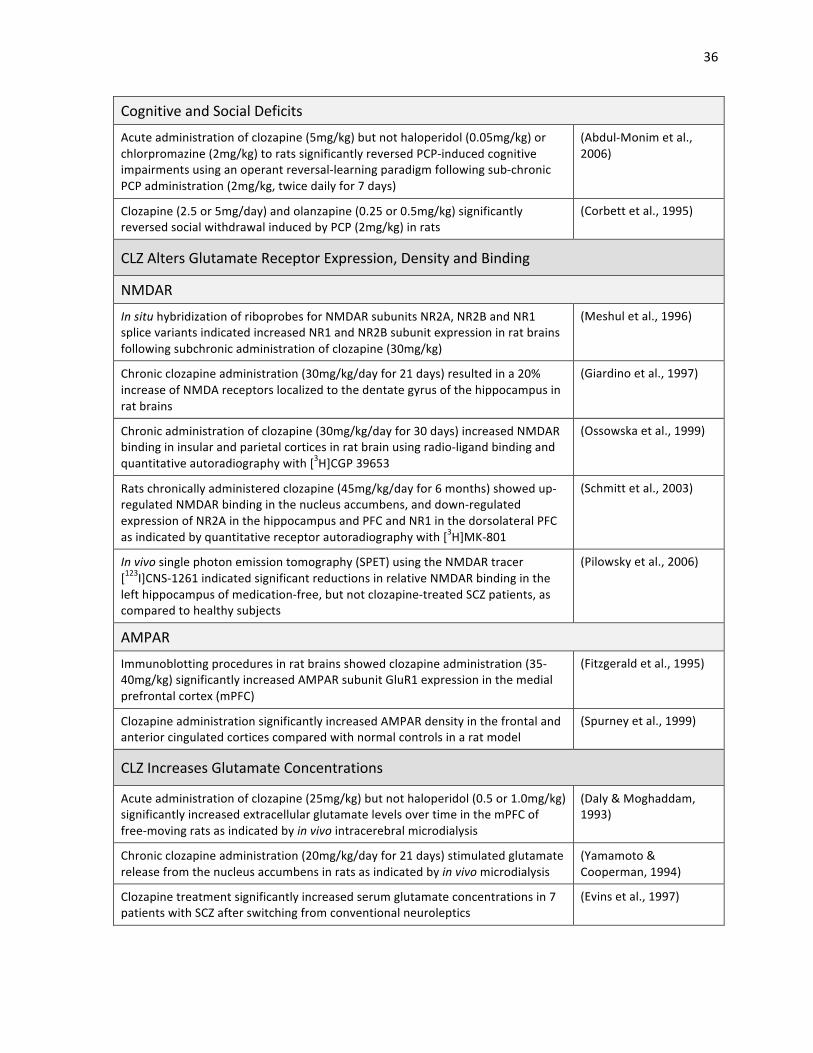
36!
!
!
!
Cognitive!and!Social!Deficits!
Acute!administration!of!clozapine!(5mg/kg)!but!not!haloperidol!(0.05mg/kg)!or!
chlorpromazine!(2mg/kg)!to!rats!significantly!reversed!PCPAinduced!cognitive!
impairments!using!an!operant!reversalAlearning!paradigm!following!subAchronic!
PCP!administration!(2mg/kg,!twice!daily!for!7!days)!
(AbdulAMonim!et!al.,!
2006)!
Clozapine!(2.5!or!5mg/day)!and!olanzapine!(0.25!or!0.5mg/kg)!significantly!
reversed!social!withdrawal!induced!by!PCP!(2mg/kg)!in!rats!!
(Corbett!et!al.,!1995)!
CLZ!Alters!Glutamate!Receptor!Expression,!Density!and!Binding!
NMDAR!
In+situ!hybridization!of!riboprobes!for!NMDAR!subunits!NR2A,!NR2B!and!NR1!
splice!variants!indicated!increased!NR1!and!NR2B!subunit!expression!in!rat!brains!
following!subchronic!administration!of!clozapine!(30mg/kg)!!
(Meshul!et!al.,!1996)!
Chronic!clozapine!administration!(30mg/kg/day!for!21!days)!resulted!in!a!20%!
increase!of!NMDA!receptors!localized!to!the!dentate!gyrus!of!the!hippocampus!in!
rat!brains!
(Giardino!et!al.,!1997)!
Chronic!administration!of!clozapine!(30mg/kg/day!for!30!days)!increased!NMDAR!
binding!in!insular!and!parietal!cortices!in!rat!brain!using!radioAligand!binding!and!
quantitative!autoradiography!with![3H]CGP!39653!
(Ossowska!et!al.,!1999)!
Rats!chronically!administered!clozapine!(45mg/kg/day!for!6!months)!showed!upA
regulated!NMDAR!binding!in!the!nucleus!accumbens,!and!downAregulated!
expression!of!NR2A!in!the!hippocampus!and!PFC!and!NR1!in!the!dorsolateral!PFC!
as!indicated!by!quantitative!receptor!autoradiography!with![3H]MKA801!!!
(Schmitt!et!al.,!2003)!
In+vivo+single!photon!emission!tomography!(SPET)!using!the!NMDAR!tracer!
[123I]CNSA1261!indicated!significant!reductions!in!relative!NMDAR!binding!in!the!
left!hippocampus!of!medicationAfree,!but!not!clozapineAtreated!SCZ!patients,!as!
compared!to!healthy!subjects!
(Pilowsky!et!al.,!2006)!
AMPAR!
Immunoblotting!procedures!in!rat!brains!showed!clozapine!administration!(35A
40mg/kg)!significantly!increased!AMPAR!subunit!GluR1!expression!in!the!medial!
prefrontal!cortex!(mPFC)!
(Fitzgerald!et!al.,!1995)!
Clozapine!administration!significantly!increased!AMPAR!density!in!the!frontal!and!
anterior!cingulated!cortices!compared!with!normal!controls!in!a!rat!model!
(Spurney!et!al.,!1999)!
CLZ!Increases!Glutamate!Concentrations!
Acute!administration!of!clozapine!(25mg/kg)!but!not!haloperidol!(0.5!or!1.0mg/kg)!
significantly!increased!extracellular!glutamate!levels!over!time!in!the!mPFC!of!
freeAmoving!rats!as!indicated!by!in+vivo!intracerebral!microdialysis!!
(Daly!&!Moghaddam,!
1993)!
Chronic!clozapine!administration!(20mg/kg/day!for!21!days)!stimulated!glutamate!
release!from!the!nucleus!accumbens!in!rats!as!indicated!by!in+vivo+microdialysis!
(Yamamoto!&!
Cooperman,!1994)!
Clozapine!treatment!significantly!increased!serum!glutamate!concentrations!in!7!
patients!with!SCZ!after!switching!from!conventional!neuroleptics!
(Evins!et!al.,!1997)!

37!
!
!
!
CLZ!Activates!Excitatory!Glutamatergic!Neurotransmission!!
Clozapine!administration!(20mg/kg)!potentiated!NMDARAmediated!synaptic!
responses!in!the!dentate!gyrus!of!chronically!prepared!rabbits!!
(Kubota!et!al.,!1996,!
2000)!
Clozapine!administration!produced!a!marked!facilitation!(300A400%)!of!NMDARA
evoked!responses!and!produced!bursts!of!EPSPs!caused!by!increased!release!of!
excitatory!amino!acids!in!pyramidal!cells!of!the!mPFC!in!rat!brain!slices!as!
indicated!by!intracellular!recording!and!singleAelectrode!voltage!clamp!
(Arvanov!et!al.,!1997;!
Chen!et!al.,!2003;!Ninan!
et!al.,!2003)!
Administration!of!CLZ!reversed!PCP!effects!on!pyramidal!cell!firing!and!cortical!
synchronization!as!recorded!extracellularly!with!glass!micropipettes,!and!
prevented!PCPAinduced!increases!in!cAfos!expression!in!PFC!pyramidal!neurons!as!
indicated!by!in+situ+hybridization!
(Kargieman!et!al.,!2007)!
To summarize, CLZ is thought to interact with the glutamate system due to its ability to
block NMDAR antagonist-induced behaviours in preclinical models including: PPI
impairments induced by PCP (Bakshi et al., 1994; Linn et al., 2003), MK-801 (Bubenikova
et al., 2005; Levin et al., 2007) and ketamine (Swerdlow et al., 1998), as well as
hyperlocomotion, social deficits and impaired cognitive function induced by PCP and MK-
801 (Freed et al., 1984; Corbett et al., 1995; Abdul-Monim et al., 2006; Sun et al., 2009).
Functional studies in preclinical models also suggest that CLZ alters glutamate receptor
subunit expression, binding and density. More specifically, CLZ administration has been
shown to: increase AMPAR density in the frontal and anterior cingulate cortices; increase
GluR1 expression in the medial prefrontal cortex (mPFC) (Fitzgerald et al., 1995; Spurney et
al., 1999); increase NR1 and NR2B expression in select brain regions (Meshul et al., 1996);
decrease NR2A expression in the PFC and hippocampus (Schmitt et al., 2003); increase
NMDAR expression in the hippocampus (Giardino et al., 1997); increase NMDAR binding
in insular and parietal cortices, as well as nucleus accumbens (NAcc) (Ossowska et al.,
1999); increase mGluR2 signaling (Fribourg et al., 2011); decrease EAAT2 expression in
cerebral cortex (Melone et al., 2001); increase synaptic glycine concentrations (Javitt et al.,

38!
!
!
!
2005); and down-regulate promoter methylation of GAD1 (Dong et al., 2008). Lastly, an in
vivo single photon emission tomography (SPET) study using the NMDAR tracer [123I]CNS-
1261 showed that CLZ significantly reduced relative NMDAR binding in the left
hippocampus of medication-free, but not clozapine treated SCZ patients, as compared to
healthy subjects (Pilowsky et al., 2006).
CLZ also alters glutamate concentrations and activates excitatory glutamate
neurotransmission. More specifically, CLZ administration has been shown to: increase
extracellular glutamate levels in the mPFC and NAcc as evidenced by in vivo micro-dialysis
in free-moving rats (Daly & Moghaddam, 1993; Yamamoto & Cooperman, 1994); increase
amplitude of excitatory postsynaptic potentials (EPSPs) elicited by single electrical
stimulation in the dentate gyrus of chronically prepared rabbits (Kubota et al., 1996, 2000);
increase EPSPs in pyramidal cells of the mPFC in rat brain slices (Arvanov et al., 1997;
Chen &Yang, 2002; Ninan et al., 2003); and increase serum glutamate concentrations in SCZ
subjects switched from conventional neuroleptics to CLZ (Evins et al., 1997). Altogether,
these findings suggest that CLZ’s therapeutic benefits may in part be mediated by re-
establishing a balance in glutamatergic neurotransmission. Therefore, genetic variation
within glutamate system-related receptors, transporters and enzymes may be ideal candidates
for PGx studies in CLZ response.
1.5.3 Polymorphisms in Glutamate System and Related Genes
A comprehensive review on the pharmacogenetics of CLZ response has recently been
conducted (Kohlrausch, 2013). Studies investigating the association among glutamate system
genes and CLZ response are summarized in Table 1.3 below.
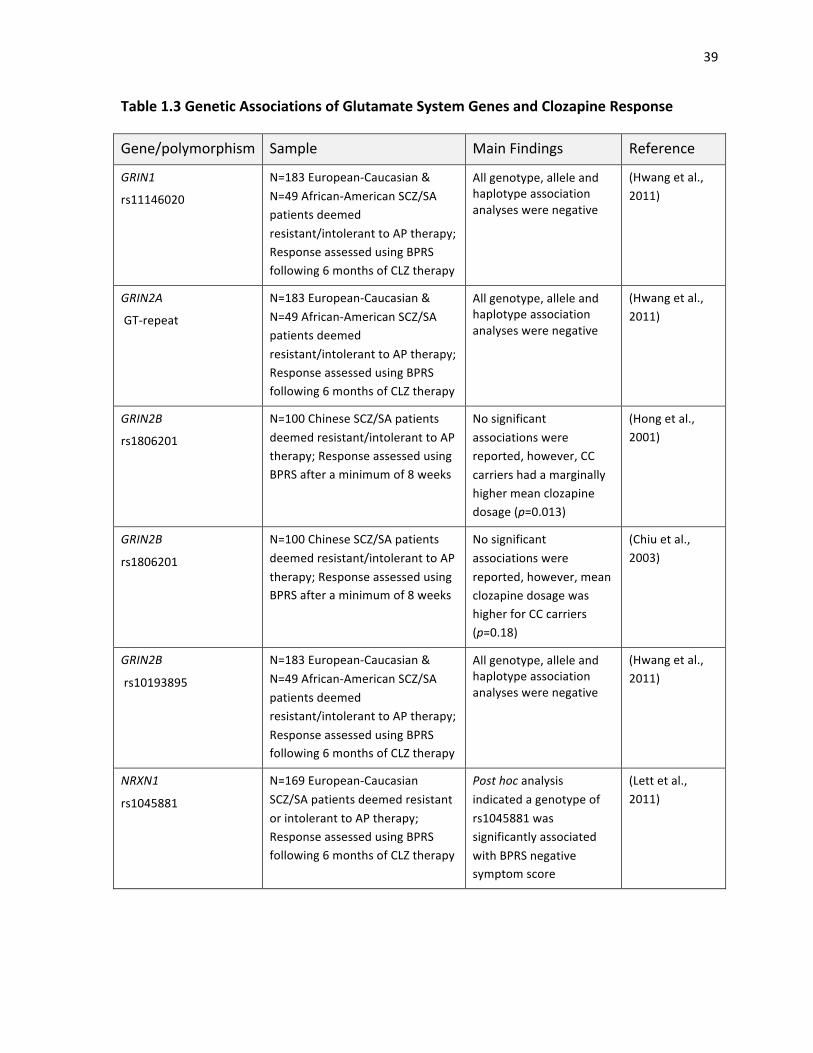
39!
!
!
!
Table&1.3&Genetic&Associations&of&Glutamate&System&Genes&and&Clozapine&Response&
Gene/polymorphism! Sample! Main!Findings! Reference!
GRIN1!!
rs11146020+
N=183!EuropeanACaucasian!&!
N=49!AfricanAAmerican!SCZ/SA!
patients!deemed!
resistant/intolerant!to!AP!therapy;!
Response!assessed!using!BPRS!
following!6!months!of!CLZ!therapy!
All!genotype,!allele!and!
haplotype!association!
analyses!were!negative!
!!
(Hwang!et!al.,!
2011)!
GRIN2A+
!GTArepeat+
N=183!EuropeanACaucasian!&!
N=49!AfricanAAmerican!SCZ/SA!
patients!deemed!
resistant/intolerant!to!AP!therapy;!
Response!assessed!using!BPRS!
following!6!months!of!CLZ!therapy!
All!genotype,!allele!and!
haplotype!association!
analyses!were!negative!
!
(Hwang!et!al.,!
2011)!
GRIN2B+
rs1806201+
N=100!Chinese!SCZ/SA!patients!
deemed!resistant/intolerant!to!AP!
therapy;!Response!assessed!using!
BPRS!after!a!minimum!of!8!weeks!
No!significant!
associations!were!
reported,!however,!CC!
carriers!had!a!marginally!
higher!mean!clozapine!
dosage!(p=0.013)!
(Hong!et!al.,!
2001)!
GRIN2B+
rs1806201+
+
N=100!Chinese!SCZ/SA!patients!
deemed!resistant/intolerant!to!AP!
therapy;!Response!assessed!using!
BPRS!after!a!minimum!of!8!weeks!
No!significant!
associations!were!
reported,!however,!mean!
clozapine!dosage!was!
higher!for!CC!carriers!
(p=0.18)!
(Chiu!et!al.,!
2003)!
GRIN2B+
!rs10193895+
+
N=183!EuropeanACaucasian!&!
N=49!AfricanAAmerican!SCZ/SA!
patients!deemed!
resistant/intolerant!to!AP!therapy;!
Response!assessed!using!BPRS!
following!6!months!of!CLZ!therapy!
All!genotype,!allele!and!
haplotype!association!
analyses!were!negative!
!
(Hwang!et!al.,!
2011)!
NRXN1!
rs1045881+
!
N=169!EuropeanACaucasian!
SCZ/SA!patients!deemed!resistant!
or!intolerant!to!AP!therapy;!
Response!assessed!using!BPRS!
following!6!months!of!CLZ!therapy!
Post+hoc+analysis!indicated!a!genotype!of!
rs1045881!was!
significantly!associated!
with!BPRS!negative!
symptom!score!
(Lett!et!al.,!
2011)!

40!
!
!
!
Briefly, the neurexin-1 gene (NXRN1) was previously associated with CLZ response in our
group (Lett et al., 2011). The rs1045881 variant was significantly associated with change in
BPRS negative symptom score following six months of CLZ monotherapy (p=0.033). The
neurexin-1 protein (NRXN1) is a neuron-specific cell-surface molecule necessary for normal
activity of the NMDAR (Kattenstroth et al., 2004). The remaining glutamate system gene
variants are polymorphisms located in the NMDAR subunit genes GRIN1, GRIN2A and
GRIN2B. Unfortunately, all variants within these genes were reported to be non-significant
with CLZ response (Hong et al., 2001; Chiu et al., 2003; Hwang et al., 2011). Two studies
did, however, report that patients carrying the CC-genotype of GRIN2B variant rs1806201
required marginally higher mean CLZ dosages than the other genotype groups (Hong et al.,
2001; Chiu et al., 2003). Given the fact relatively few pharmacogenetics studies investigating
the contribution of glutamate system gene variation to CLZ response have been conducted
thus far, additional studies are of high priority.
1.5.4 Identifying Functional Gene Variants using ENCODE
Gene variant selection can also be approached through the use of functional annotation
databases such as the University of California Santa Cruz (UCSC) Genome Browser (Kent et
al., 2002), Haploreg (Ward & Kellis, 2012), RegulomeDB (Boyle et al., 2012) and the
National Institute for Environmental Health Sciences (NIEHS) Functional SNP Prediction
Database (Xu & Taylor, 2009). These databases provide access to data generated by the
Encyclopedia of DNA Elements (ENCODE) project. In more detail, the ENCODE project is
a public consortium research effort launched by the US National Human Genome Research
Institute (NHGRI) with the aim to decipher all of the functional elements within the human
genome (ENCODE Project Consortium, 2012). Of interest to candidate SNP selection is

41!
!
!
!
mapping of functional elements such as transcription factor binding sites (TFBS), DNase I
hypersensitivity sites and regions of histone modification from data pooled from 1,640 data
sets and 147 different cell types. Originally, a large portion of non-coding DNA identified
during the Human Genome project was believed to have little importance (International
HapMap Consortium, 2003), however, ENCODE reports the majority of the human genome
(~80%) is found to be functional in some way. In fact, many non-coding variants and SNPs
identified to be associated with disease through GWAS have been found to lie within
ENCODE-annotated functional regions that lie outside of protein-coding genes. To illustrate
the use of ENCODE data in SNP selection, Figure 1.4 shows search results displaying
ENCODE functional data for the glycine transporter 1 gene (SLC6A9) variant rs16831558
genotyped within this study, as presented by the UCSC Genome Browser.
&
Figure&1.4&Identifying&Functional&Promoter&Variants&in&SLC6A94Using&ENCODE&Data&
University! of! California! Santa! Cruz! (UCSC)! Genome! Browser! track! results! for! the! glycine!
transporter!1!gene!(SLC6A9)!approximately!5kb!upstream!from!the!promoter!region.!Search!
results! highlight! the! location! of! the! reference! SNP! rs16831558! under! ‘Single! Nucleotide!
Polymorphisms’! (dbSNP! 142)! (highlighted! in! the! red! box)! relative! to! intron! 1! of! SLC6A9!
(indicated!by!the!green!arrow).!Search!settings!were!customized!using!dropAdown!options!
located! in! the!page!below.!More! specifically,! ‘Integrated!Regulation! from!ENCODE’!under!
H3K27Ac!
!DNAse1!
!Txn!Factor!
!LBNL!Enhancers!
!

42!
!
!
!
‘Regulation’!was!set!to!‘show’!and!‘common!SNPs!(142)’! located!under!‘Variation’!was!set!
to!‘pack’.!Functionally,!the!rs16831558!was!associated!with:!Layered!H3K27Ac!histone!mark!
(lysine!27!of! the!H3!histone!protein!acetylated),!with!colours! indicating!cell! type! in!which!
H3K27Ac! mark! was! identified! [Blue:! K562! (myelogenous! leukocyte),! Purple:! NHEK!
(epidermal! keratinocytes),! Pink:! NHLF! (normal! lung! fibroblast)];! DNAse! I! Hypersensitivity!
Cluster! represents! areas! of! open! chromatin! where! regulatory! regions! likely! reside,! with!
darkness! of! the! boxes! indicating! extent! of! hypersensitivity;! Txn! Factor! ChIP! indicates!
regions! of! transcription! factor! binding! as! determined! by! chromatin! immunoprecipitation!
(ChIPAseq)! DNA! binding! motif! assays;! and! Vista! HMRAConserved! NonACoding! Human!
Enhancers! from! LBNL! that! identify! distantAacting! transcriptional! enhancers! in! the! human!
genome.!This!genome!browser!screen!shot!was!constructed!using!human!genome!assembly!
version! 19! and! can! be! accessed! by! visiting:! http://genome.ucsc.edu/cgiA
bin/hgTracks?db=hg19&position=chr1%3A44494763A44508289!(Kent!et!al.,!2002)!!
!
Several functional elements reported in Figure 1.4 deserve further explanation. For instance,
K27Ac is a histone mark (modified histone protein) in which lysine 27 of the H3 histone
protein has been acetylated as determined by a ChIP-seq assay. This histone mark is thought
to increase transcription possibly by blocking the spread of the repressive histone mark
H3K27Me3 (tri-methylation of lysine 27 on H3) (Bernstein et al., 2005). Another functional
element is the DNAse I Hypersensitivity sites that locate regions of open chromatin where
regulatory elements such as promoters, enhancers and silencers likely reside (Song &
Crawford, 2010). Transcription factor chromatin immunoprecipitation (Txn Factor ChIP) is
also used and identifies regions of DNA where transcription factors (TF) are likely to bind.
Transcription factors are proteins that bind to DNA and regulate gene expression by
interacting with RNA polymerase (Wang et al., 2012). Lastly, the Vista human, mouse, rat
(HMR)-Conserved Non-Coding Human Enhancers from Lawrence Berkeley National

43!
!
!
!
Laboratory (LBNL) identifies distant-acting transcriptional enhancers in the human genome
by coupling evolutionary conserved non-coding sequences with mouse enhancer assays
(Pennacchio et al., 2006).
Pulling from the data presented in the previous sections, a total of 17 gene variants from 6
glutamate-system related genes with evidence of CLZ-interaction were selected for study
inclusion (summarized in Table 2.1 & Table 3.1). Variants were selected based on previous
investigation in neuropsychiatric phenotypes as cited in the literature, and/or potential
functionality from information gathered from functional annotation websites. The first data
chapter of this thesis investigated the association among eight gene variants in GRIN2B with
CLZ response. This gene encodes for the NR2B subunit of the NMDAR, contains 13 exons
and is mapped to region 12p12 (Mandich et al., 1994; Endele et al., 2010). The second data
chapter investigated the association among ten additional glutamate-system gene variants and
response to CLZ. More specifically, the remaining ten variants included two variants in the
mGluR2 gene, GRM2, which maps to 3p21.1 (Joo et al., 2001), one variant in the AMPAR
subunit GluR1 gene, GRIA1, which maps to 5q31.1 (Puckett et al., 1991), three variants in
EAAT2 encoded by SLC1A2 mapped to region 11p13-p12 (Li & Francke, 1995), three
variants in GlyT1 encoded by SLC6A9 mapped to 1p33 (Jones et al., 1995), and lastly, one
variants in the GAD1 enzyme gene, encoded by GAD1 that is mapped to region 2q.31 (Bu et
al., 1992). The 17 glutamate system gene variants investigated in this study include: GRIN2B
(rs7301328, rs1072388, rs12826365, rs2284411, rs1806201, rs1806191, rs3764030, rs890);
GRM2 (rs4067, rs2518461); SLC1A2 (rs4354668, rs4534557, rs2901534); SLC6A9
(rs12037805, rs1978195, rs16831558); GRIA1 (rs2195450); and GAD1 (rs3749034).

44!
!
!
!
1.6 Study Rationale
1.6.1 Summary and Hypothesis
Treatment-resistant schizophrenia is a debilitating, costly and burdensome disorder that
affects approximately 30-40% of patients diagnosed with SCZ. Disease prognosis is highly
dependent upon successful early intervention strategies and stresses the utility of having the
ability to predict likelihood of response before a drug is prescribed. Pharmacogenetics
research aims to unravel the genetic underpinnings of drug response and to develop
personalized treatment plans that are capable of guiding adjusted therapeutic doses,
decreasing adverse drug reactions, improving treatment adherence and providing patients
with a better quality of life.
This study was designed to test for associations among glutamate system gene variants and
clinical response to CLZ in SCZ or schizoaffective (SA) disorder patients deemed resistant
or intolerant to previous AP therapy. We hypothesize that putatively functional variants with
the potential to alter glutamatergic neurotransmission may in some way interact with CLZ’s
ability to achieve therapeutic effect, and may lead to variable response among study
participants. In total, 17 glutamate system gene variants were selected for study inclusion,
many of which were reported to be functional using ENCODE data.
By investing time, effort and resources in studying the relationship between putatively
functional glutamate system gene variants and response to CLZ, we ultimately hope to
contribute to the ever-evolving field of pharmacogenetics, and help make personalized
medicine a better-established approach in psychiatric care.

Taylor DL II. ORIGINAL RESEARCH ARTICLE
45
Chapter 2
GENETIC ASSOCIATION ANALYSIS OF THE NMDA RECEPTOR
SUBUNIT GENE GRIN2B WITH RESPONSE TO CLOZAPINE
Danielle L. Taylor1,2, Arun K. Tiwari1, Jeffrey A. Lieberman3, Steven G. Potkin4, Herbert Y.
Meltzer5, Joanne Knight1,2,6, Gary Remington6,7, Daniel J. Müller1,2,6, James L. Kennedy1,2,6
1Neuroscience Research Department, Campbell Family Research Institute, Centre for Addiction and Mental Health, Toronto, ON, Canada
2Institute of Medical Science, University of Toronto, ON, Canada
3Department of Psychiatry, College of Physicians and Surgeons, Columbia University and the New York State Psychiatric Institute, New York City, NY, USA
4Department of Psychiatry, University of California, Irvine, CA, USA
5Northwestern University Feinberg School of Medicine, Chicago, IL, USA
6Department of Psychiatry, University of Toronto, Toronto, ON, Canada
7Schizophrenia Program, Centre for Addiction and Mental Health, Toronto, ON, Canada
This chapter was modified from the following:
Taylor DL, Tiwari AK, Lieberman JA, Potkin SG, Meltzer HY, Knight J, Remington G, Müller DJ, Kennedy JL. Genetic association analysis of the NMDA receptor subunit gene (GRIN2B) with response to clozapine. Submitted to Hum Psychopharmacol Clin Exp.
Key Words: GRIN2B, pharmacogenetics, clozapine response, glutamate, schizophrenia

46
2.1 ABSTRACT
Approximately 30% of patients with schizophrenia fail to respond to antipsychotic therapy
and are classified as having treatment-resistant schizophrenia. Clozapine is the most
efficacious drug for treatment-resistance and may deliver its superior therapeutic effects
partly by modulating glutamatergic neurotransmission. Response to clozapine is highly
variable and may depend on genetic factors as indicated by twin studies. This study
investigated eight single nucleotide polymorphisms in the N-methyl-D-aspartate glutamate
receptor subunit gene GRIN2B with response to clozapine. Standard TaqMan procedures
were used to type rs7301328, rs1072388, rs12826365, rs2284411, rs1806201, rs1806191,
rs3764030 and rs890 in 175 European schizophrenia/schizoaffective patients deemed
resistant or intolerant to previous antipsychotic therapy. Response was assessed using change
in Brief Psychiatric Rating Scale (BPRS) scores following six months of clozapine therapy.
Categorical and continuous response was assessed using Pearson’s chi-squared test and
analysis of covariance, respectively. No associations were observed between the variants and
response to clozapine. A-allele carriers (AA and AG) of rs1072388 responded marginally
better to clozapine therapy than GG-homozygotes, however the difference was not
statistically significant (puncorrected=0.067, pcorrected=0.440, assuming 6.56 independent tests).
Our findings do not support a role for these GRIN2B variants in altering response to
clozapine in our sample.

47
2.2 INTRODUCTION
Treatment-resistant schizophrenia occurs in approximately 30% (20-40%) of patients treated
with antipsychotic drugs (Borgio et al., 2007; Elkis & Meltzer, 2007; Solanki et al., 2009).
TRS patients experience suboptimal response characterized by persistent positive symptoms
that often result in long periods of hospitalization (McGlashan, 1988) and chronic severe
disability. Treatment resistance remains an immense burden to both patients and their
families and is associated with annual healthcare costs ranging 3-to-11 times higher than
schizophrenia patients who respond to treatment ($66,360-$163,795 vs. $15,500-$22,300 per
year). TRS is also associated with poorer quality of life that is approximately 20% lower than
patients with SCZ who achieve remission (0.61 vs. 0.75) (Kennedy et al., 2014).
The atypical AP drug clozapine is particularly effective for treating the persistent symptoms
experienced by patients with TRS. A landmark clinical trial conducted by Kane et al. showed
that approximately 30% of TRS patients responded to CLZ versus only 4% of patients given
the prototypical AP CPZ (p<0.001) (Kane et al., 1988). As an atypical AP, CLZ differs from
typical APs in particular dimensions, such as decreased DRD2 receptor affinity (Altar et al.,
1986) and lowered risk for adverse effects such as extrapyramidal symptoms and tardive
dyskinesia (Pickar et al., 1992; Tandon and Fleischhacker, 2005). CLZ also has the ability to
alter other neurotransmitter systems such as serotonin and glutamate (reviewed by Miyamoto
et al., 2005).
A great deal of evidence suggests that CLZ augments glutamatergic neurotransmission
(reviewed by Heresco-Levy, 2003). One pillar supporting this theory stems from
observations that CLZ is capable of blunting the psychotomimetic effects of glutamate

48
antagonists in SCZ patients (Malhotra et al., 1997). These antagonists, such as phencyclidine
and ketamine, bind to the NMDAR and elicit schizophrenia-like symptoms in healthy
controls and exacerbate psychotic symptoms in SCZ patients (Luby et al., 1959; Luisada,
1978; Javitt & Zukin, 1991; Lahti et al., 2001) – observations that form the basis of the
‘glutamate hypofunction hypothesis’ of SCZ. Collectively, these findings suggest that
blockade and underactivity of glutamate signaling at the NMDAR contributes to the clinical
presentation of SCZ. In turn, CLZ is theorized to offer therapeutic relief in part by
augmenting glutamatergic signals.
Findings from preclinical models and functional studies support this theory. CLZ
administration reverses PCP-induced psychotic-like behaviours such as hyper-locomotion
(Freed et al., 1984; Sun et al., 2009; Zhao et al., 2012), social deficits (Corbett et al., 1995),
enhanced immobility (Noda et al., 1995) and PPI deficits (Linn et al., 2003). Functional
studies in both preclinical models and SCZ patients also indicate that CLZ alters glutamate
receptor subunit expression (Fitzgerald et al., 1995; Meshul et al., 1996), binding (Giardino
et al., 1997; Pilowsky et al., 2006), and density (Ossowska et al., 1999; Schmitt et al., 2003).
In addition, CLZ appears to alter glutamate concentrations and activity of excitatory
glutamate neurotransmission in different brain regions as exhibited by: micro-dialysis studies
in rodents (Daly & Moghaddam, 1993; Yamamoto & Cooperman, 1994), increased
amplitude of EPSPs in neuronal cell cultures (Banerjee et al., 1995; Arvanov et al., 1997;
Kubota et al., 2000; Ninan et al., 2003; Kargieman et al., 2007), and increased serum
glutamate levels in patients switched to CLZ (Evins et al., 1997). Taken together, these
results provide evidence for CLZ’s ability to restore glutamate signaling – a factor that may
distinguish CLZ as the most efficacious drug for treatment-resistant patients.

49
Clinical response to CLZ is highly variable (Bleehen, 1993; Davis et al., 2003) and an
estimated 30-60% of TRS patients fail to respond to CLZ therapy (Kane et al., 1988;
Lieberman et al., 1994). These patients are diagnosed with ultra treatment-resistant SCZ
(URS) and various augmentation strategies for managing URS have proven inconclusive
(Meltzer et al., 1989b; Lieberman et al., 1994; Remington et al., 2005). Because early
intervention strategies with AP drugs attenuates disease prognosis, the ability to predict
response prior to drug administration would have important clinical utility (Wyatt & Henter,
2001; Gunduz-Bruce et al., 2005; Perkins et al., 2005; Emsley et al., 2007). Given that a
portion of the inter-individual variability in CLZ response is thought to be caused by genetic
variation (Vojvoda et al., 1996; Horacek et al., 2001; Mata et al., 2001; Theisen et al., 2005;
Hoyer et al., 2010), a number of pharmacogenetics studies investigating genetic variability in
CLZ response have been conducted (reviewed by Kohlrausch, 2013). However, no predictive
test for AP response utilizing this genetic information is currently in clinical use. The most
notable gene variants investigated for association with CLZ response include those related to
the dopamine, serotonin and to a lesser degree, glutamate neurotransmitter systems.
Glutamate is becoming increasingly popular in recent years due to novel therapies that target
this neurotransmitter system (Heresco-Levy et al., 1996; Patil et al., 2007; Pinard et al., 2010;
Hopkins, 2011). Additional advancements in SCZ genetics have also yielded promising
genome-wide hits for glutamate genes in risk (GAIN Collaborative Research Group, 2007;
Jia et al., 2012; Schizophrenia Working Group of the Psychiatric Genomics Consortium,
2014). More specifically, genome-wide significant associations with schizophrenia have
been observed in glutamate system genes such as GRM3, GRIN2A, GRIA1 and GRIN2B. The
GRIN2B gene (138252 [MIM]) codes for subunit 2B of the NMDAR and is of particular

50
interest. This gene contains 13 exons and is mapped to 12p12 (Mandich et al., 1994; Endele
et al., 2010). Single nucleotide polymorphisms in GRIN2B have been investigated for
associations with several neuropsychiatric disorders, including drug response phenotypes
(Table 2.1). A small number of GRIN2B variants have also been studied in CLZ response –
while no associations between rs1806201 (2664C/T) (Hong et al., 2001; Chiu et al., 2003) or
rs1019385 (-200T/G) (Hwang et al., 2011) have been observed, the rs1805502 variant
(T5988C) significantly predicted negative symptom change during CLZ therapy (Martucci &
Kennedy, 2010). Associations between additional GRIN2B polymorphisms and CLZ
response have not yet been investigated. This study examined for associations among eight
GRIN2B variants and clinical response to CLZ monotherapy in a sample of treatment-
resistant/intolerant patients.
2.3 METHODS
2.3.1 Subjects
One hundred and seventy-five patients meeting Diagnostic and Statistical Manual of Mental
Disorders III-R or IV (DSM-III-R/IV) criteria for schizophrenia or schizoaffective disorder
were included in this study (American Psychiatric Association, 1987, 1994). Patients were
recruited from three clinical sites: Case Western Reserve University in Cleveland, Ohio (HY
Meltzer, n=74), Hillside Hospital in Glen Oaks, New York (JA Lieberman, n=73), and the
University of California at Irvine (SG Potkin, n=28). All patients were considered European
based on self-reported ancestry. Informed consent was obtained from each study participant
prior to enrollment in accordance with the Ethical Principles for Medical Research Involving

51
Human Subjects at the Centre for Addiction and Mental Health and with the Helsinki
Declaration of 1975, as revised in 1989 (World Medical Association, 2013).
TRS was defined as failure to respond to two or more AP drug trials in the previous five
years (involving drugs from two different chemical classes, with doses ≥1000mg
chlorpromazine equivalents for four to six weeks), accompanied by no period of good
functioning within the preceding five years (Kane et al., 1988). Less than 15% of study
participants met criteria for treatment intolerance, which was defined as the presence of
moderate to severe TD and/or extreme EPS making treatment with therapeutic dosages
intolerable (Lieberman et al., 1994). Before beginning CLZ treatment, patients underwent a
two to four week wash-out period which involved no administration of pharmacotherapy
unless clinically necessary. Following the washout period, all patients were treated with CLZ
monotherapy, with mean dosages of 453 mg/d, for a period of six months or longer.
Benzodiazepines were administered intermittently during the titration period. Throughout
treatment, CLZ serum levels were monitored to ascertain adherence to the medication.
2.3.2 Response Measures
Baseline Brief Psychiatric Rating Scale (Overall & Gorham, 1962) scores were obtained at
time of study enrollment. Following CLZ administration, response was evaluated after six
months using two BPRS scoring methods. The first was a categorical responder/non-
responder response measure that classified responders as individuals who experienced a
≥20% decrease in BPRS total score. The second scoring method was a quantitative response
measure that divided the BPRS items into three subcategories [positive (BPOS), negative
(BNEG), and general (BPRS)]. Percent score reduction for each of these subscales was

52
calculated using the following equation: [(six month score - baseline score)/(baseline score)]
x 100%.
2.3.3 SNP Selection
A literature search was conducted on PubMed for the following terms: “GRIN2B”,
“NMDAR”, “N-methyl-D-aspartate receptor”, “clozapine” and “pharmacogenetics.”
Literature pertaining to genetic association studies in psychiatric disorders and psychotropic
drug response was reviewed to identify GRIN2B SNPs of interest. Variants previously
investigated for an association with CLZ response or other psychiatric phenotypes were
considered for inclusion. Altogether, eight GRIN2B polymorphisms were selected:
rs1072388, rs12826365, rs1806191, rs2284411, rs3764030, rs7301328 (366G>C), rs1806201
(2664C>T), and rs890 (Figure 2.1 & Table 2.1). These eight variants were assessed for
potential functionality using the National Institute of Environmental Health Sciences
Functional SNP Prediction (FuncPred) database (Xu & Taylor, 2009). Variants were deemed
functional if alternate alleles were thought to have differential effects on gene transcription,
translation, or splicing.
Variants rs1072388 and rs12826365 were the most significant p-value SNPs from the
Genetics Association Information Network (GAIN) schizophrenia GWAS from the European
and African samples, respectively (GAIN Collaborative Research Group, 2007; Ayalew et
al., 2012). Variant rs1806191 was previously investigated in SCZ risk and obsessive
compulsive disorder (OCD) (Di Maria et al., 2004; Alonso et al., 2012), while variant
rs2284411 had been investigated in attention deficit hyperactivity disorder (ADHD) (Dorval
et al., 2007; Park et al., 2013). The promoter variant rs3764030 had previously been studied

53
in autism spectrum disorder (ASD) (Yoo et al., 2012) and is proposed to affect GRIN2B
expression by altering transcription factor binding. Both rs7301328 and rs1806201 were
investigated in a number of psychiatric phenotypes, including response to lithium
(Szczepankiewicz et al., 2009) and CLZ (Hong et al., 2001). These SNPs both lie near
intron-exon borders (within two base-pairs, specifically) and are proposed to regulate mRNA
spicing. Lastly, rs890 had been investigated in SCZ risk (Di Maria et al., 2004), treatment-
resistant depression (TRD) (Zhang et al., 2014), CLZ-induced OCD (CI-OCD) (Cai et al.,
2013) and response to lithium (Szczepankiewicz et al., 2009). This variant lies in a micro
RNA (miRNA) binding site at the 3’ untranslated region (UTR) of GRIN2B and is proposed
to alter translation initiation.
2.3.4 DNA Isolation and Genotyping
Venous blood samples were collected from study participants and sent to CAMH in Toronto,
Canada where genomic DNA was isolated using the high salt method (Lahiri & Nurnberger,
1991). GRIN2B genotypes were determined using TaqMan allele specific single tube assays,
the ABI Prism®7500 Sequence Detection System and the ABI allelic discrimination
software according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA,
USA). The reaction mixture consisted of 20ng of genomic DNA, 2X TaqMan® Universal
Master Mix and 40X SNP Genotyping Assay for a final reaction volume of 10 µL. The
polymerase chain reaction (PCR) protocol consisted of a denaturation step for 10 mins at
95°C, followed by 50 cycles of amplification that consisted of denaturation (92°C for 15 s)
and annealing (60°C for 1 min). Genotype calls were confirmed by two independent
researchers and 10% of the total sample was re-genotyped to ensure genotyping accuracy.
Discordant genotypes were set as missing in the statistical analysis.

54
2.3.5 Statistical Analysis
Quality-control (QC) was carried out using PLINK software v1.07 (Purcell et al., 2007). To
pass filtering, SNPs were required to have a ≥90% genotyping rate and a minor allele
frequency (MAF) of ≥0.05. Individuals with a genotype success rate less than 75% across all
markers (samples producing genotypes for five SNPs or less) were also excluded from
analysis. All analyses were carried out using the Statistical Package for the Social Sciences
(SPSS) v20 software (IBM Corp, Armonk NY, USA) and PLINK.
Descriptive statistics were obtained for each clinical site. Mean age and baseline values of
BPRS, BPOS and BNEG subscale scores across sites were compared using Student’s t-test or
analysis of variance (ANOVA), while frequency counts for gender and response rates were
compared using Pearson’s chi-square test (χ2-test). Genotype and allele frequencies in the
responder/non-responder (R/NR) groups were compared using χ2-test or Fisher’s exact test
(for cell counts less than 5). Differences in percent score reduction in BPRS, BPOS and
BNEG subscales between genotype groups were compared using analysis of covariance
(ANCOVA), co-varying for age and baseline scores.
Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) among SNPs were
determined using Haploview v4.2 using the Solid Spine of LD to construct LD blocks
(Figure 2.2) (Barrett et al., 2005). Haplotype analysis was carried out in UNPHASED v3.1.5
(Dudbridge, 2008). Haplotypes with <5% frequency were removed from analysis. Power
calculations were conducted in Quanto v1.2.4 (Gauderman & Morrison, 2006). Correction
for the effective number of independent markers was performed using the method described
by Nyholt and resulted in a statistical significance threshold of p<0.0076 (Nyholt, 2004).

55
2.4 RESULTS
2.4.1 Sample Characteristics
Of the eight SNPs genotyped, seven met QC criteria and were included in subsequent
association analyses. The genotype distribution for variant rs7301328 significantly deviated
from HWE (p=1.93x10-7) and was therefore excluded. The remaining SNPs had a mean
genotyping rate of 99.1%. Regarding study participants, 18 patients were removed due to low
genotyping efficiency (<75% of markers were successfully genotyped). The remaining
sample had a mean genotyping efficiency of 98.9%.
Following QC, 157 European treatment-resistant or intolerant patients were included in the
study (Table 2.2). Our sample had over 80% power to detect an odds ratio (OR) as low as
2.00 (unmatched case-control design: n=157, non-responder frequency=48.4%,
MAF=30.5%, α/2=0.05) and down to 8.5% variance in the quantitative response variable
(continuous design: n=85). The average minor allele frequency across the seven GRIN2B
variants (excluding rs7301328) was used for power calculations. Patients between clinical
sites did not differ in gender, categorical response rates, or BPRS/BPOS/BNEG baseline
scores. The sites did differ, however, in mean age (F(2,157)=7.10, P=0.001). Post hoc
analysis showed that the average age of the HY Meltzer sample was significantly lower than
the SG Potkin sample. In order to combine samples for analysis, age and baseline
BPRS/BPOS/BNEG scores were added as covariates to account for any disparities.
Following QC, the final combined sample (n=157) consisted of 76.4% male patients and had
a total mean age of 35.06 years (SD 8.1). Approximately half (51.6%) of the sample was

56
considered CLZ ‘responders’ (n=81). Quantitative response measures were available for a
subset of patients in the BPRS (n=86), BPOS (n=84), and BNEG (n=85) subscales.
2.4.2 GRIN2B SNP and Haplotype Association Analysis
Genotype and allele frequencies for each of the seven GRIN2B variants were not
significantly different between CLZ responder and non-responder groups or as measured by
the quantitative percent score reduction in BPRS, BPOS and BNEG subscales (Table 2.3).
Variant rs1072388 A-allele carriers (AA and AG) responded marginally better to CLZ
therapy than GG-homozygotes, however this difference was not statistically significant
(puncorrected=0.067) (Figure 2.3). Haplotype association analyses confirmed allele and
genotype findings with no association reported across all haplotypes tested (Table 2.4).
2.5 DISCUSSION
We report no significant associations among the seven GRIN2B variants investigated and
response to CLZ in our sample of patients. Despite A-allele carriers of the intronic variant
rs1072388 responding marginally better to CLZ therapy than GG-homozygotes
(puncorrected=0.067, genotype; puncorrected=0.069, allele), this observation was statistically non-
significant (pcorrected=0.440, assuming 6.56 independent tests). To our knowledge, this is the
first reported study to investigate associations of the remaining seven SNPs (rs1072388,
rs3764030, rs12826365, rs2284411, rs1806191, rs890 and rs7301328) with CLZ response.
The rs1806201 marker has previously been investigated in two other CLZ response samples
(Hong et al., 2001; Chiu et al., 2003). Each aforementioned variant was selected based on
previous investigation in CLZ response or other psychiatric phenotypes. After inclusion, the

57
potential functionality of these variants was assessed using the NIEHS Functional SNP
Prediction Database (Xu & Taylor, 2009).
Our negative finding for rs1806201 (2664C/T) is in accordance with previous reports that
this variant was not associated with CLZ response (Hong et al., 2001; Chiu et al., 2003).
These studies did report a marginally higher mean CLZ dosage for patients carrying the CC
genotype of rs1806201. Due to the inter-individual variability in CLZ metabolism and
absorption, future studies may benefit from measuring serum CLZ levels as opposed to
dosage in order to predict response. An optimal threshold for CLZ serum levels has already
been established (≥450 ng/mL) and may be of clinical utility to identify non-responders
(Lindenmayer & Apergi, 1996).
As a measure of genotyping quality, we tested for HWE to ensure genotype proportions
observed in the study population coincided with those expected under equilibrium conditions
(Hardy, 1908; Weinberg, 1908). The potentially functional variant rs7301328 deviated from
HWE (p<0.05, data not shown). This deviation may have been caused by a number of
reasons including systematic genotyping errors that mistyped heterozygotes as homozygotes
or vice versa (Gomes et al., 1999; Hosking et al., 2004), stochastic variation, or due to the
study population’s biological characteristics. The biological explanation posits that patient
genotypes deviate from HWE because of the effect an allele has on a disease phenotype from
which the sample was non-randomly selected (Wittke-Thompson et al., 2005; Xu & Taylor,
2009). The cause for deviation from HWE in our sample appeared to be genotyping error: no
homozygous recessive genotypes were observed for either rs7301328 responder or non-
responder groups, even though this variant has a MAF of 23% and therefore, was expected to

58
have a homozygous recessive count of approximately eight people out of 157. Because many
genetic tests such as χ2 require HWE to be assumed, rs7301328 was removed from
subsequent analyses (Sasieni, 1997). Retyping of rs7301328 with a newly designed assay
remains a priority for future work.
This study had several limitations. The sample size is relatively small and may not have had
sufficient power to detect the effect sizes of these genes. To increase statistical power in the
future, it will be necessary to collect larger CLZ response samples. One promising sample is
being collected by the CRESTAR consortium in Europe (CRESTAR, 2011). Sample
heterogeneity may also have limited our findings. Patients were collected from three clinical
sites that may have varied in methods and populations. Population ancestry was also based
on self-report and in the future, a principle components analysis (PCA) on the study sample
may provide more accurate ancestry profiles (Paschou et al., 2008; Price et al., 2008; Liu et
al., 2011).
In addition, two environmental factors with the potential to alter CLZ metabolism were not
controlled for in our study: cigarette smoking and caffeine consumption. Cigarette smoking
induces the Cytochrome P450 hepatic enzyme CYP1A2 that is primarily responsible for CLZ
metabolism (Bertilsson et al., 1994; Ghassabian et al., 2010). As a result, smokers are
observed to have lower CLZ serum levels than non-smokers (Seppala et al., 1999; Palego et
al., 2002), and therefore require higher CLZ dosages to achieve similar plasma levels as
patient who do not smoke. In contrast, caffeine inhibits CYP1A2 and increases both CLZ
plasma concentrations and the risk of developing side effects linked to toxicity (Odom-White
& de Leon, 1996; Hagg et al., 2000). Future studies incorporating these environmental

59
factors may help identify more accurate predictors of response and develop dosage
guidelines that are tailored to the individual patient.
Inter-study variability may also have limited comparison of results across studies. We
evaluated treatment response following six months of CLZ therapy, while previous studies
evaluated response after shorter periods, e.g. two months of treatment (Hong et al., 2001;
Chiu et al., 2003). Differences in treatment length are particularly problematic because
response to CLZ is variable and may take up to five months or longer in a subset of patients
(Lieberman et al., 1994; Meltzer & Okayli, 1995b). Thus, patients deemed non-responders
after a two month treatment period have the potential to be reclassified as responders
following a longer trial of CLZ therapy. This disparity makes comparing results across
studies problematic and reaching a consensus regarding treatment duration among
researchers would make meta-analyses more efficient in the future.
To conclude, we report no associations between seven GRIN2B variants and CLZ response in
our sample. Response to CLZ therapy is complex and likely depends on multiple gene
systems and environmental factors. Therefore, investigation of additional glutamate variants
and variants within related neurotransmitter systems is warranted for future studies. Even
though conclusive predictive markers remain elusive thus far, continued research to identify
such markers remains of high interest to improve clinical response and treatment outcomes.

60
Figure'2.1!Human'GRIN2B'Gene'Diagram''
Structure,! location! and! size! of! the! GRIN2B! gene! are! noted! along! with! the! eight! single!
nucleotide! polymorphisms! genotyped! in! this! study.! The! coding! region! of! GRIN2B' is!
represented!by!darkly!shaded!boxes!and!the!5’!and!3’!untranslated!regions!are!represented!
by!lightly!shaded!boxes.!!
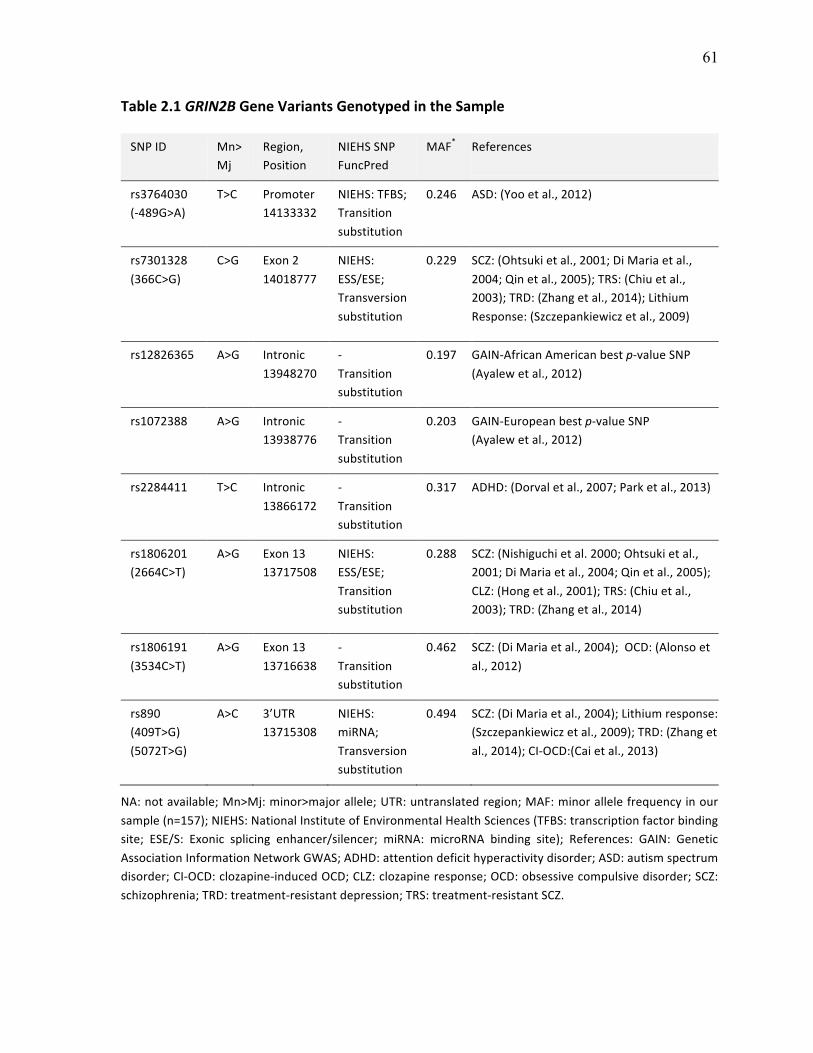
61
Table'2.1!GRIN2B'Gene'Variants'Genotyped'in'the'Sample!
SNP!ID! Mn>Mj!
Region,!Position!!!
NIEHS!SNP!FuncPred!
MAF*! References!
rs3764030!(R489G>A)!
T>C! Promoter!14133332!
NIEHS:!TFBS;!Transition!!substitution!!
0.246! ASD:!(Yoo!et!al.,!2012)!
rs7301328!(366C>G)!!
C>G! Exon!2!14018777!
NIEHS:!ESS/ESE;!Transversion!substitution!
0.229! SCZ:!(Ohtsuki!et!al.,!2001;!Di!Maria!et!al.,!2004;!Qin!et!al.,!2005);!TRS:!(Chiu!et!al.,!2003);!TRD:!(Zhang!et!al.,!2014);!Lithium!Response:!(Szczepankiewicz!et!al.,!2009)!
rs12826365! A>G! Intronic!13948270!
R!Transition!substitution!!
0.197! GAINRAfrican!American!best!pRvalue!SNP!(Ayalew!et!al.,!2012)!
rs1072388! A>G! Intronic!13938776!
R!Transition!substitution!
0.203! GAINREuropean!best!pRvalue!SNP!!(Ayalew!et!al.,!2012)!
rs2284411! T>C! Intronic!13866172!
R!Transition!substitution!
0.317! ADHD:!(Dorval!et!al.,!2007;!Park!et!al.,!2013)!
rs1806201!(2664C>T)!
A>G! Exon!13!13717508!
NIEHS:!ESS/ESE;!Transition!substitution!
0.288! SCZ:!(Nishiguchi!et!al.!2000;!Ohtsuki!et!al.,!2001;!Di!Maria!et!al.,!2004;!Qin!et!al.,!2005);!CLZ:!(Hong!et!al.,!2001);!TRS:!(Chiu!et!al.,!2003);!TRD:!(Zhang!et!al.,!2014)!
rs1806191!(3534C>T)!
A>G! Exon!13!13716638!
R!Transition!substitution!
0.462! SCZ:!(Di!Maria!et!al.,!2004);!!OCD:!(Alonso!et!al.,!2012)!
rs890!(409T>G)!(5072T>G)!
A>C! 3’UTR!13715308!
NIEHS:!miRNA;!!!Transversion!substitution!
0.494! SCZ:!(Di!Maria!et!al.,!2004);!Lithium!response:!(Szczepankiewicz!et!al.,!2009);!TRD:!(Zhang!et!al.,!2014);!CIROCD:(Cai!et!al.,!2013)!
NA:!not!available;!Mn>Mj:!minor>major!allele;!UTR:!untranslated!region;!MAF:!minor!allele!frequency!in!our!sample!(n=157);!NIEHS:!National!Institute!of!Environmental!Health!Sciences!(TFBS:!transcription!factor!binding!site;! ESE/S:! Exonic! splicing! enhancer/silencer;! miRNA:! microRNA! binding! site);! References:! GAIN:! Genetic!Association!Information!Network!GWAS;!ADHD:!attention!deficit!hyperactivity!disorder;!ASD:!autism!spectrum!disorder;!CIROCD:!clozapineRinduced!OCD;!CLZ:!clozapine!response;!OCD:!obsessive!compulsive!disorder;!SCZ:!schizophrenia;!TRD:!treatmentRresistant!depression;!TRS:!treatmentRresistant!SCZ.!
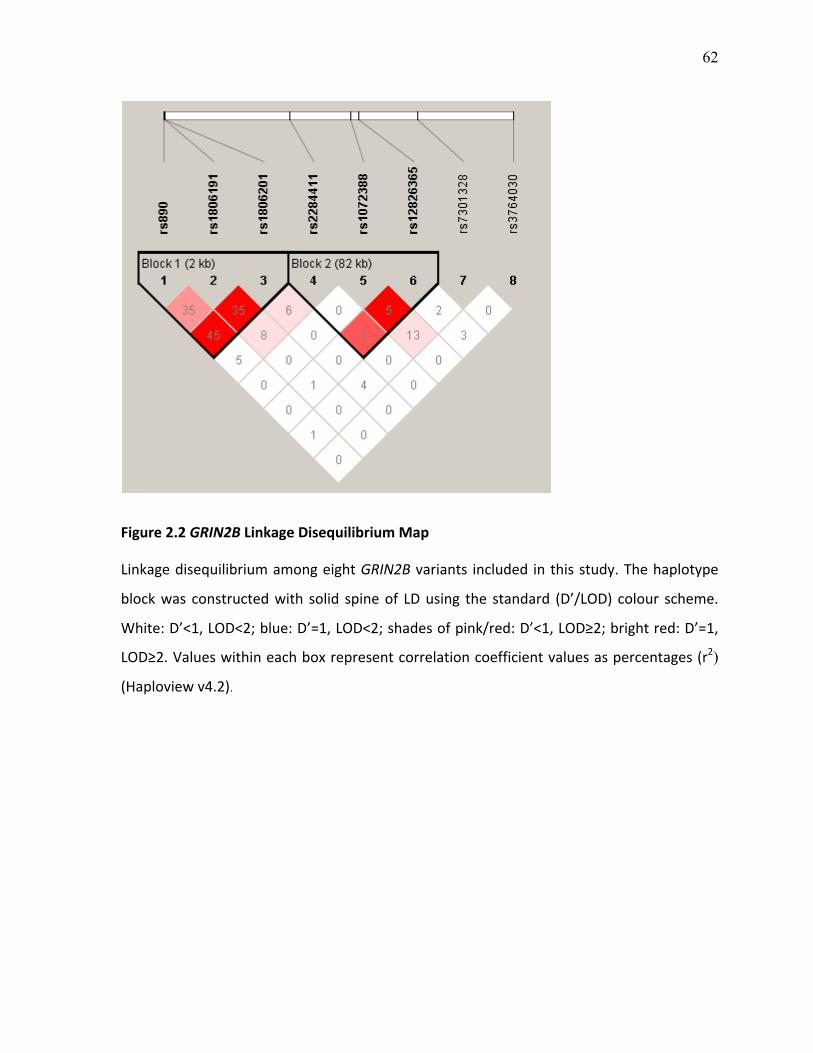
62
Figure'2.2!GRIN2B'Linkage'Disequilibrium'Map''
Linkage!disequilibrium!among!eight!GRIN2B!variants! included! in!this!study.!The!haplotype!
block!was! constructed!with! solid! spine!of! LD!using! the! standard! (D’/LOD)! colour! scheme.!
White:!D’<1,!LOD<2;!blue:!D’=1,!LOD<2;!shades!of!pink/red:!D’<1,!LOD≥2;!bright!red:!D’=1,!
LOD≥2.!Values!within!each!box!represent!correlation!coefficient!values!as!percentages!(r2)
(Haploview!v4.2).!
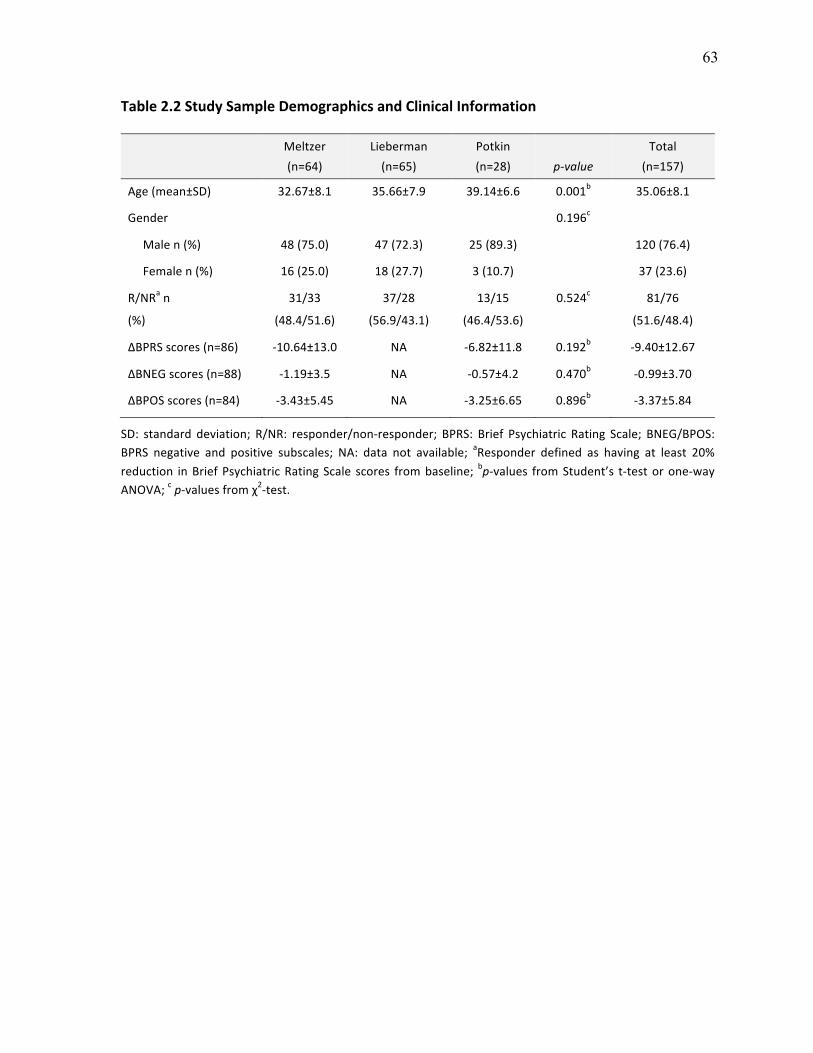
63
Table'2.2!Study'Sample'Demographics'and'Clinical'Information!
! Meltzer!!(n=64)!
Lieberman!!(n=65)!
Potkin!(n=28)!
'p)value'
Total!!(n=157)!
Age!(mean±SD)! 32.67±8.1! 35.66±7.9! 39.14±6.6! 0.001b! 35.06±8.1!
Gender! ! ! ! 0.196c! !
!!!!!Male!n!(%)! 48!(75.0)! 47!(72.3)! 25!(89.3)! ! 120!(76.4)!
!!!!!Female!n!(%)! 16!(25.0)! 18!(27.7)! 3!(10.7)! ! 37!(23.6)!
R/NRa!n!!
(%)!
31/33!!
(48.4/51.6)!
37/28!!
(56.9/43.1)!!
13/15!!
(46.4/53.6)!
0.524c! 81/76!!
(51.6/48.4)!
ΔBPRS!scores!(n=86)! R10.64±13.0! NA! R6.82±11.8! 0.192b! R9.40±12.67!
ΔBNEG!scores!(n=88)! R1.19±3.5! NA! R0.57±4.2! 0.470b! R0.99±3.70!
ΔBPOS!scores!(n=84)! R3.43±5.45! NA! R3.25±6.65! 0.896b! R3.37±5.84!
SD:! standard! deviation;! R/NR:! responder/nonRresponder;! BPRS:! Brief! Psychiatric! Rating! Scale;! BNEG/BPOS:!BPRS! negative! and! positive! subscales;! NA:! data! not! available;! aResponder! defined! as! having! at! least! 20%!reduction! in!Brief!Psychiatric!Rating!Scale! scores! from!baseline;! bpRvalues! from!Student’s! tRtest!or!oneRway!ANOVA;!c!pRvalues!from!χ2Rtest.!
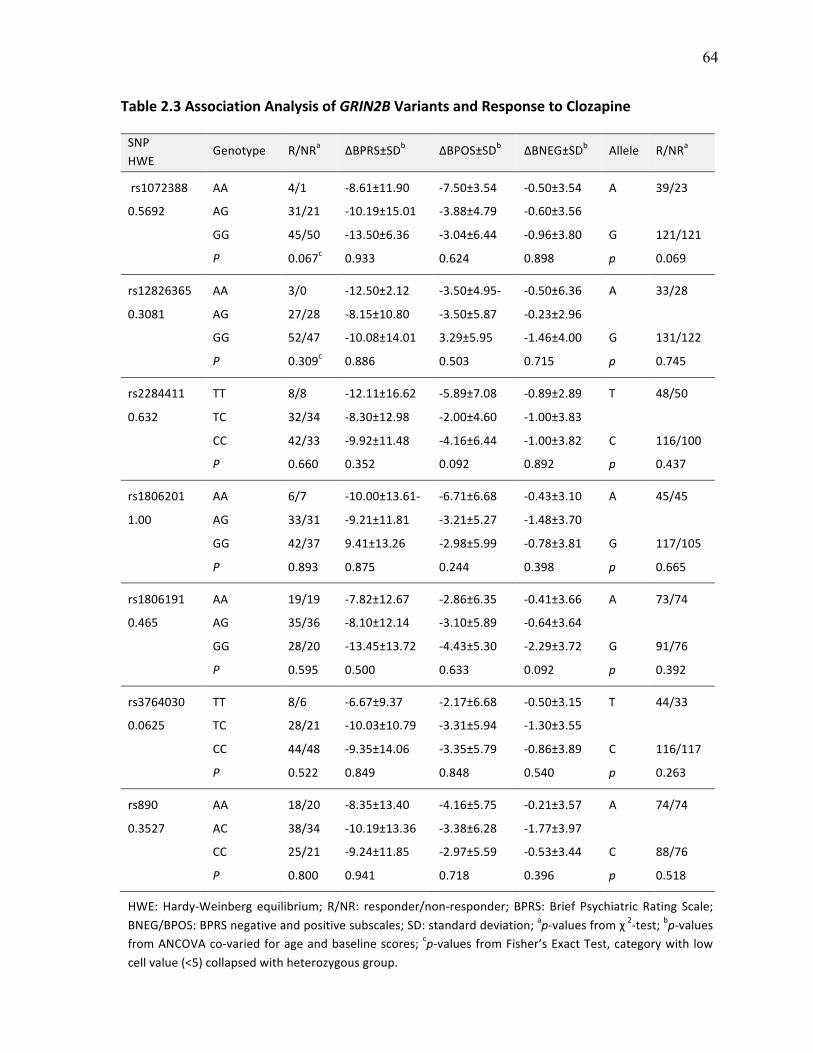
64
Table'2.3!Association'Analysis'of'GRIN2B'Variants'and'Response'to'Clozapine!!
SNP!HWE!
Genotype! R/NRa! ΔBPRS±SDb! ΔBPOS±SDb! ΔBNEG±SDb! Allele! R/NRa!
'rs1072388! AA! 4/1! R8.61±11.90! R7.50±3.54! R0.50±3.54! A! 39/23!
0.5692! AG! 31/21! R10.19±15.01! R3.88±4.79! R0.60±3.56! ! !
! GG! 45/50! R13.50±6.36! R3.04±6.44! R0.96±3.80! G! 121/121!
! P' 0.067c! 0.933! 0.624! 0.898! p' 0.069!
rs12826365! AA! 3/0! R12.50±2.12! R3.50±4.95R! R0.50±6.36! A! 33/28!
0.3081! AG! 27/28! R8.15±10.80! R3.50±5.87! R0.23±2.96! ! !
! GG! 52/47! R10.08±14.01! 3.29±5.95! R1.46±4.00! G! 131/122!
! P' 0.309c! 0.886! 0.503! 0.715! p' 0.745!
rs2284411! TT! 8/8! R12.11±16.62! R5.89±7.08! R0.89±2.89! T! 48/50!
0.632! TC! 32/34! R8.30±12.98! R2.00±4.60! R1.00±3.83! ! !
! CC! 42/33! R9.92±11.48! R4.16±6.44! R1.00±3.82! C! 116/100!
! P' 0.660! 0.352! 0.092! 0.892! p' 0.437!
rs1806201! AA! 6/7! R10.00±13.61R! R6.71±6.68! R0.43±3.10! A! 45/45!
1.00! AG! 33/31! R9.21±11.81! R3.21±5.27! R1.48±3.70! ! !
! GG! 42/37! 9.41±13.26! R2.98±5.99! R0.78±3.81! G! 117/105!
! P' 0.893! 0.875! 0.244! 0.398! p' 0.665!
rs1806191! AA! 19/19! R7.82±12.67! R2.86±6.35! R0.41±3.66! A! 73/74!
0.465! AG! 35/36! R8.10±12.14! R3.10±5.89! R0.64±3.64! ! !
! GG! 28/20! R13.45±13.72! R4.43±5.30! R2.29±3.72! G! 91/76!
! P' 0.595! 0.500! 0.633! 0.092! p' 0.392!
rs3764030! TT! 8/6! R6.67±9.37! R2.17±6.68! R0.50±3.15! T! 44/33!
0.0625! TC! 28/21! R10.03±10.79! R3.31±5.94! R1.30±3.55! ! !
! CC! 44/48! R9.35±14.06! R3.35±5.79! R0.86±3.89! C! 116/117!
! P' 0.522! 0.849! 0.848! 0.540! p' 0.263!
rs890! AA! 18/20! R8.35±13.40! R4.16±5.75! R0.21±3.57! A! 74/74!
0.3527! AC! 38/34! R10.19±13.36! R3.38±6.28! R1.77±3.97! ! !
! CC! 25/21! R9.24±11.85! R2.97±5.59! R0.53±3.44! C! 88/76!
! P' 0.800! 0.941! 0.718! 0.396! p' 0.518!
! HWE:!HardyRWeinberg! equilibrium;! R/NR:! responder/nonRresponder;! BPRS:! Brief! Psychiatric! Rating! Scale;!BNEG/BPOS:!BPRS!negative!and!positive!subscales;!SD:!standard!deviation;!apRvalues!from!χ!2Rtest;!bpRvalues!from!ANCOVA!coRvaried!for!age!and!baseline!scores;! cpRvalues!from!Fisher’s!Exact!Test,!category!with! low!cell!value!(<5)!collapsed!with!heterozygous!group.!
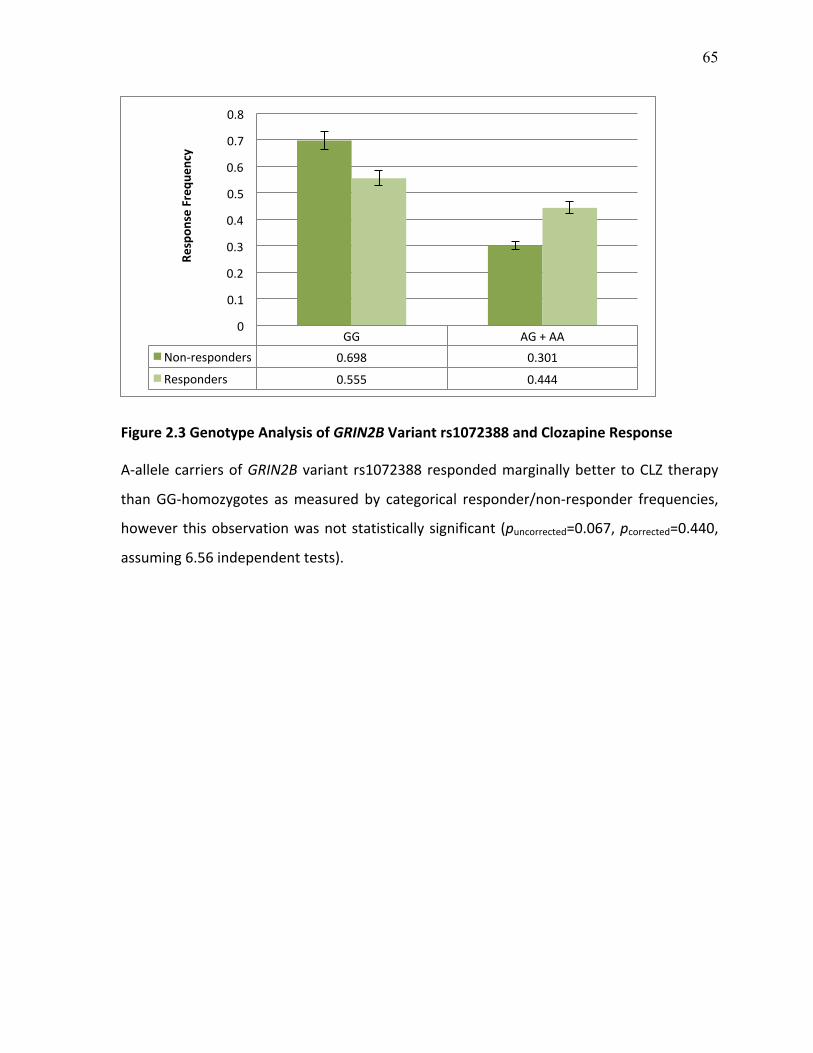
65
Figure'2.3!Genotype'Analysis'of'GRIN2B'Variant'rs1072388'and'Clozapine'Response''
ARallele!carriers!of!GRIN2B! variant! rs1072388!responded!marginally!better! to!CLZ! therapy!
than!GGRhomozygotes! as!measured! by! categorical! responder/nonRresponder! frequencies,!
however!this!observation!was!not!statistically!significant! (puncorrected=0.067,!pcorrected=0.440,!
assuming!6.56!independent!tests).!!
GG! AG!+!AA!NonRresponders! 0.698! 0.301!
Responders! 0.555! 0.444!
0!
0.1!
0.2!
0.3!
0.4!
0.5!
0.6!
0.7!
0.8!
Respon
se'Frequ
ency'
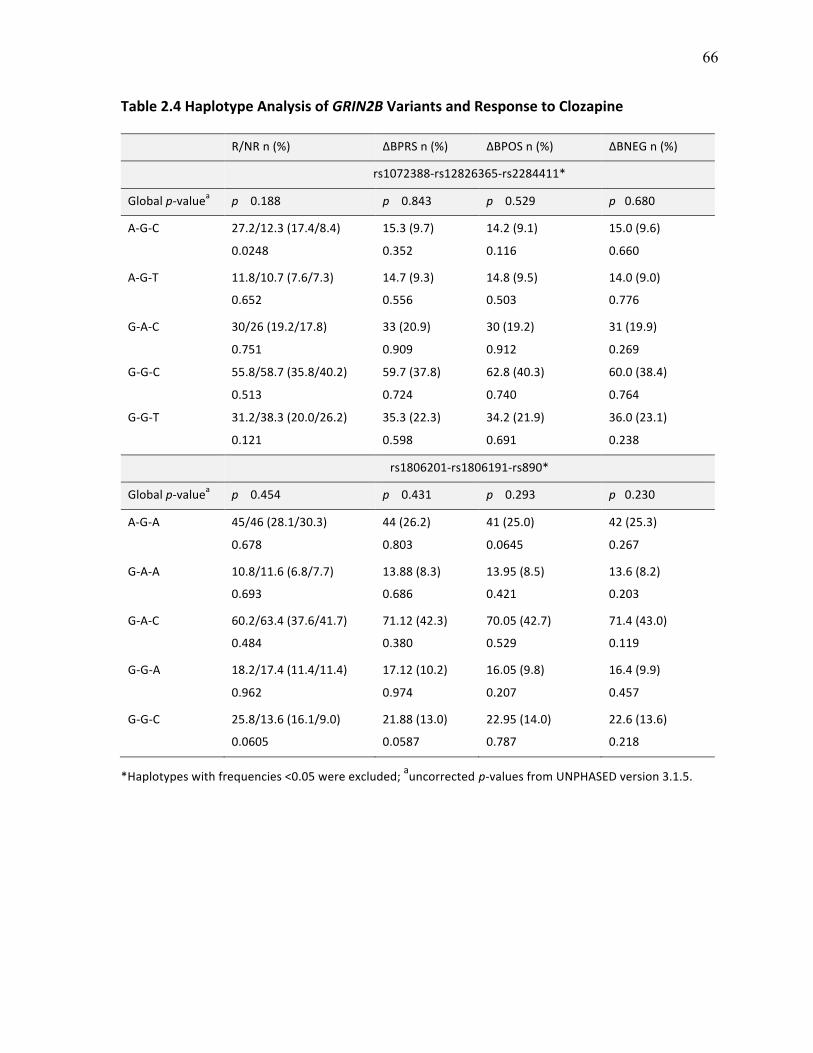
66
Table'2.4!Haplotype'Analysis'of'GRIN2B'Variants'and'Response'to'Clozapine!
! R/NR!n!(%)! ΔBPRS!n!(%)! ΔBPOS!n!(%)! ΔBNEG!n!(%)!
! rs1072388Rrs12826365Rrs2284411*!
Global!p)valuea! p''''0.188! p''''0.843! p''''0.529! p'''0.680!
ARGRC!
!
27.2/12.3!(17.4/8.4)!
0.0248!
15.3!(9.7)!
0.352!
14.2!(9.1)!
0.116!
15.0!(9.6)!
0.660!
ARGRT! 11.8/10.7!(7.6/7.3)!
0.652!
14.7!(9.3)!
0.556!
14.8!(9.5)!
0.503!
14.0!(9.0)!
0.776!
GRARC!
!
GRGRC!
!
GRGRT!
30/26!(19.2/17.8)!
0.751!
55.8/58.7!(35.8/40.2)!
0.513!
31.2/38.3!(20.0/26.2)!
0.121!
33!(20.9)!
0.909!
59.7!(37.8)!
0.724!
35.3!(22.3)!
0.598!
30!(19.2)!
0.912!
62.8!(40.3)!
0.740!
34.2!(21.9)!
0.691!
31!(19.9)!
0.269!
60.0!(38.4)!
0.764!
36.0!(23.1)!
0.238!
! rs1806201Rrs1806191Rrs890*!
Global'pRvaluea! p''''0.454! p''''0.431! p''''0.293! p'''0.230!
ARGRA!
!
45/46!(28.1/30.3)!
0.678!
44!(26.2)!
0.803!
41!(25.0)!
0.0645!
42!(25.3)!
0.267!
GRARA! 10.8/11.6!(6.8/7.7)!
0.693!
13.88!(8.3)!
0.686!
13.95!(8.5)!
0.421!
13.6!(8.2)!
0.203!
GRARC!
!
60.2/63.4!(37.6/41.7)!
0.484!
71.12!(42.3)!
0.380!
70.05!(42.7)!
0.529!
71.4!(43.0)!
0.119!
GRGRA!
!
18.2/17.4!(11.4/11.4)!
0.962!
17.12!(10.2)!
0.974!
16.05!(9.8)!
0.207!
16.4!(9.9)!
0.457!
GRGRC!
!
25.8/13.6!(16.1/9.0)!
0.0605!
21.88!(13.0)!
0.0587!
22.95!(14.0)!
0.787!
22.6!(13.6)!
0.218!
*Haplotypes!with!frequencies!<0.05!were!excluded;!auncorrected!pRvalues!from!UNPHASED!version!3.1.5.!

Taylor DL III. ORIGINAL RESEARCH ARTICLE
67
Chapter 3
PHARMACOGENETIC ANALYSIS OF GLUTAMATE SYSTEM GENE
VARIANTS AND CLINICAL RESPONSE TO CLOZAPINE
Danielle L. Taylor1,2, Arun K. Tiwari1, Jeffrey A. Lieberman3, Steven G. Potkin4, Herbert Y.
Meltzer5, Joanne Knight1,2,6, Gary Remington6,7, Daniel J. Müller1,2,6, James L. Kennedy1,2,6
1Neuroscience Research Department, Campbell Family Research Institute, Centre for Addiction and Mental Health, Toronto, ON, Canada
2Institute of Medical Science, University of Toronto, ON, Canada
3Department of Psychiatry, College of Physicians and Surgeons, Columbia University and the New York State Psychiatric Institute, New York City, NY, USA
4Department of Psychiatry, University of California, Irvine, CA, USA
5Northwestern University Feinberg School of Medicine, Chicago, IL, USA
6Department of Psychiatry, University of Toronto, Toronto, ON, Canada
7Schizophrenia Program, Centre for Addiction and Mental Health, Toronto, ON, Canada
This chapter was modified from the following:
Taylor DL, Tiwari AK, Lieberman JA, Potkin SG, Meltzer HY, Knight J, Remington G, Müller DJ, Kennedy JL. Pharmacogenetic analysis of glutamate system gene variants and clinical response to clozapine. Submitted to Mol Neuropsychiatry.
Keywords: GRM2, SLC1A2, SLC6A9, GRIA1, GAD1, pharmacogenetics, clozapine response,
glutamate

68
3.1 ABSTRACT
Altered glutamatergic neurotransmission is implicated in schizophrenia etiology as well as in
response to clozapine, the drug of choice for treatment-resistant schizophrenia. Response to
antipsychotic therapy is highly variable, although twin studies suggest a genetic component.
We investigated the association among ten glutamate system gene variants with clozapine
response in 175 European schizophrenia or schizoaffective disorder patients deemed resistant
or intolerant to previous pharmacotherapy. GRM2 (rs4067, rs2518461), SLC1A2 (rs4354668,
rs4534557, rs2901534), SLC6A9 (rs12037805, rs1978195, rs16831558), GRIA1 (rs2195450)
and GAD1 (rs3749034) were typed using standard genotyping procedures. Response was
assessed following six months of clozapine treatment using change in Brief Psychiatric
Rating Scale scores. Categorical and continuous response variables were analyzed using
Pearson’s chi-squared test and analysis of covariance, respectively. No significant
associations were observed following correction for multiple testing. Prior to correction,
several nominally significant associations were seen for SLC6A9, SLC1A2, GRM2 and
GRIA1. Most notably, CC-homozygotes of rs16831558 located in the glycine transporter 1
gene exhibited an allele dose-dependent improvement in positive symptoms compared to T-
allele carriers (puncorrected=0.008, pcorrected=0.08). To clarify the role of SLC6A9 in clinical
response to antipsychotic medication and clozapine in particular, this finding warrants further
investigation in larger well-characterized samples.

69
3.2 INTRODUCTION
The glutamate system has emerged as an area of strong interest for both schizophrenia risk
and response to antipsychotic medications (Shan et al., 2012). The glutamate hypothesis of
SCZ first emerged from observations that administration of NMDAR antagonists such as
PCP and ketamine elicit behaviours that mimic the symptoms of SCZ in healthy individuals
and dramatically worsen symptoms in SCZ patients (Luby et al., 1959; Luisada, 1978; Javitt
& Zukin, 1991; Lahti et al., 2001). Glutamate levels also appear to be altered in SCZ brains
and dysregulation of glutamate receptors has been postulated to play an integral role in
hyperdopaminergic states often thought to be the primary cause of psychosis (Moghaddam &
Javitt, 2012).
CLZ is an atypical AP drug that is particularly effective for treating patients with TRS (Kane
et al., 1988). Approximately 30% of patients fail two or more trials with AP drugs, and are
subsequently diagnosed with TRS. CLZ is the one drug that has been approved for treatment
of this particular subpopulation (Borgio et al., 2007; Elkis & Meltzer, 2007; Solanki et al.,
2009). Response to CLZ is complex and thought to depend, at least in part, on genetic factors
as indicated by twin and family studies (Vojvoda et al., 1996; Horacek et al., 2001; Mata et
al., 2001; Hoyer et al., 2010). Like other APs, high inter-individual variability is also
observed with CLZ treatment (Bleehen, 1993; Potkin et al., 1994; Davis et al., 2003), with up
to 50% of eligible patients failing to respond (Kane et al., 1988; Lieberman et al., 1994).
Studies in pharmacogenetics aim to identify gene variants with the potential to predict
dosing, response and side effects prior to starting drug treatment (Müller et al., 2003). The
implications of a genetic test incorporating such variants would have far-reaching clinical
applications; given the fact that early and effective treatment has been associated with

70
favourable outcome (Gunduz-Bruce et al., 2005; Perkins et al., 2005). To date, the majority
of PGx studies investigating CLZ response have focused on gene variants in dopamine and
serotonin neurotransmitter pathways (reviewed by Kohlrausch, 2013).
As noted, there is mounting evidence to suggest that glutamatergic neurotransmission may
also play a role in mediating response to CLZ (Potkin et al., 1999; reviewed by Heresco-
Levy, 2003). The major observation supporting this hypothesis is that if CLZ is co-
administered with NMDAR antagonists, their psychotomimetic effects are blunted (Malhotra
et al., 1997). Glutamate concentrations and excitatory glutamatergic neurotransmission are
also increased by CLZ administration, as shown by micro-dialysis studies in rodents (Daly &
Moghaddam, 1993; Yamamoto & Cooperman, 1994), EPSPs in neuronal cell cultures
(Banerjee et al., 1995; Arvanov et al., 1997; Kubota et al., 2000; Ninan et al., 2003;
Kargieman et al., 2007), and serum glutamate levels in patients switched to CLZ (Evins et
al., 1997).
CLZ is also posited to modulate various receptors, transporters and enzymes that are
involved in glutamate signaling and consequently implicates single nucleotide polymorphism
gene variants within these pathways as strong candidates for assessing susceptibility to CLZ
non-response. In regard to glutamate receptors, CLZ administration has been observed to
increase AMPAR density and GluR1 expression (Fitzgerald et al., 1995; Spurney et al.,
1999). The GluR1 protein is encoded by the GRIA1 gene and has been mapped to 5q31.1
(Puckett et al., 1991). CLZ also increases mGluR2 signaling (Fribourg et al., 2011), which is
encoded by the GRM2 gene, which maps to 3p21.1 (Joo et al., 2001). AMPA and mGluR
glutamate receptors are involved in primary depolarization of glutamate-mediated
neurotransmission and synaptic plasticity (Ichise et al., 2000; Santos et al., 2009).

71
With respect to transporter proteins, CLZ reportedly down-regulates expression of EAAT2 in
rat cerebral cortex (Melone et al., 2001), which is encoded by the SLC1A2 gene that maps to
region 11p13-p12 (Li & Francke, 1995). EAAT2 localizes to astrocytes in the mammalian
CNS and is responsible for the greatest proportion of total glutamate reuptake from the
synapse (Rothstein et al., 1996). Functional inactivation of EAAT2 raises extracellular
glutamate levels – a biological phenomenon implicated in AP non-response (Egerton et al.,
2012; de la Fuente-Sandoval et al., 2013; Demjaha et al., 2014). CLZ also increases synaptic
glycine concentrations (Javitt et al., 2005). In neuronal tissue, the glycine transporter 1
chiefly determines the availability of glycine in the brain by mediating glycine reuptake into
surrounding nerve terminals and glial cells (Kim et al., 1994). The SLC6A9 gene codes for
GlyT1 and is mapped to 1p33 (Jones et al., 1995).
Lastly, CLZ has been shown to down-regulate promoter methylation of the GAD1 gene in
mice (Dong et al., 2008). This gene is mapped to 2q.31 (Bu et al., 1992) and codes for the
GAD1 enzyme, which catalyzes the decarboxylation of glutamic acid to GABA and carbon
dioxide (Erlander et al., 1991). Interestingly, abnormal GABAergic function is also reported
in patients with SCZ (Taylor & Tso, 2014). Based on this aforementioned body of evidence,
the present study set out to investigate the contribution of variants within SLC1A2, SLC6A9,
GRIA1, GRM2 and GAD1 to response to CLZ in a sample of treatment resistant or intolerant
patients.

72
3.3 METHODS
3.3.1 Study Sample
Subjects included in this study were recruited from three clinical sites: Case Western Reserve
University in Cleveland, Ohio (HY Meltzer, n=74); Hillside Hospital in Glen Oaks, New
York (JA Lieberman, n=73); and University of California, Irvine (SG Potkin, n=28) (Total
sample, n=175). All patients had a diagnosis of either SCZ or SA disorder according to the
DSM-III-R/IV (American Psychiatric Association, 1987, 1994) and met criteria for either
TRS or intolerance to standard pharmacotherapy. Treatment-resistant schizophrenia was
defined as failure to respond to two or more antipsychotic trials with drugs from at least two
different chemical classes at doses of ≥1000mg/d chlorpromazine equivalents for four to six
weeks, accompanied by no period of good functioning in the preceding five years (Kane et
al., 1988). A smaller portion of patients (<15%) met criteria for treatment intolerance defined
as the presence of moderate to severe tardive dyskinesia or extreme sensitivity to
extrapyramidal symptoms (Lieberman et al., 1994).
For this genetic study, written informed consent was obtained from all participants in
accordance with the Ethical Principles for Medical Research Involving Human Subjects at
the Centre for Addiction and Mental Health and with the Declaration of Helsinki, as revised
in 1989 (World Medical Association, 2013). Participants underwent a two to four week
washout period during which time no medication was administered unless clinically
necessary. Patients were then placed on CLZ therapy for six months or longer with mean
doses in the range of 450 mg/day. For the duration of the study, patients were seen weekly
for blood draws to monitor white blood cell count as a precaution against clozapine-induced
agranulocytosis.

73
3.3.2 Response Measures
CLZ response was assessed using both categorical and continuous response measures.
Categorical response was assessed using change in Brief Psychiatric Rating Scale scores
(Overall & Gorham, 1962) from baseline and following six months of CLZ therapy.
Participants were considered responders if they experienced a 20% or more decrease in their
BPRS scores. Continuous response was measured using percent score reduction for total
BPRS, as well as for the BPOS and BNEG subscales, using the following calculation: [(six
month score – baseline score)/baseline score] x 100%. Using this formula, scores below zero
indicated symptom improvement.
3.3.3 SNP Selection
In total, ten glutamate system gene variants were included in this study (Figure 3.1 & Table
3.1). Four variants were selected based on previous investigation in other neuropsychiatric
phenotypes as cited in the literature, and the remaining six SNPs were selected from several
functional annotation websites based on their potential to alter gene expression.
3.3.3.1 Identification of SNPs from the Literature
A literature search was conducted on PubMed using the following search terms: “GRM2”,
“Metabotropic Glutamate Receptor 2”, “SLC1A2”, “Glutamate Transporter 1”, “GLT1”,
“Excitatory Amino Acid Transporter 2”, “EAAT2”, “SLC6A9”, “Glycine Transporter 1”,
“GlyT1”, “AMPA”, “GRIA1”, “GluA1”, “GLUR1”, “GAD1” and “Glutamate Decarboxylase
1.” Articles investigating genetic associations (genome-wide and candidate SNP studies)
with neuropsychiatric phenotypes were reviewed. As well, articles investigating the
functional significance (positron emission tomography ligand binding assays, electrophoretic

74
mobility shift assays, luciferase promoter assays, etc.) of variants within SLC1A2, SLC6A9,
GRIA1, GRM2 and GAD1 were reviewed to identify glutamate system SNPs of interest. Four
variants were selected based on previous investigations in other phenotypes: 1) SLC1A2
(rs4354668) in bipolar disorder (BP), response to lithium salts (Dallaspezia et al., 2012),
EAAT2 expression in SCZ subjects (Ohnuma et al., 1998) and plasma glutamate
concentrations (Mallolas et al., 2006); 2) SLC1A2 (rs4534557) in SCZ risk (Deng et al.,
2004; Nagai et al., 2009); 3) GRM2 (rs4067) in major depressive disorder (MDD), BP,
response to fluvoxamine, methamphetamine-induced psychosis and SCZ (Tsunoka et al.,
2009, 2010); and 4) GAD1 (rs3749034) in childhood onset SCZ (COS), cortical gray matter
volume (Addington et al., 2005) and GAD1 mRNA expression (Straub et al., 2007).
3.3.3.2 Identification of SNPs with Potential Functionality
The Broad Institute’s HaploReg database was used to identify glutamate system variants with
potentially functional effects on gene expression (Ward & Kellis, 2012). The gene coding
regions plus an additional 10kb upstream and 2kb downstream were submitted to the ‘Build
Query’ search field on HaploReg. Gene regions (chrN:start-end) were obtained from the
UCSC Genome Browser using Human Genome assembly version 19: GRM2 (chr3:
51,731,081-51,754,625), SLC1A2 (chr11: 35,270,752-35,461,610), SLC6A9 (chr1:
44,455,172-44,507,164) and GRIA1 (chr5: 152,849,175-153,088,732).
Seven criteria to measure functionality were provided for each SNP: 1) sequence
conservation across mammals, 2) promoter histone marks, 3) enhancer histone marks, 4)
DNAse hypersensitivity sites, 5) bound proteins, 6) expression quantitative trait loci (eQTL)
data and 7) effect on regulatory motifs. SNPs with a minor allele frequency of at least 5% in
the European (EUR) population that satisfied at least 3 of these 7 functionality criteria were

75
considered for study inclusion. Six variants satisfying these criteria were selected: SLC1A2
(rs2901534), SLC6A9 (rs12037805, rs1978195, rs16831558), GRIA1 (rs2195450) and GRM2
(rs2518461) (Figure 3.2). RegulomeDB (Boyle et al., 2012) and NIEHS Functional SNP
Prediction (FuncPred) database scores (Xu & Taylor, 2009) were also obtained to further
assess functionality.
3.3.4 DNA Isolation and Genotyping
Venous blood samples were collected from study participants and sent to our Centre
(CAMH, Toronto, Canada) where genomic DNA was extracted using the high salt method
(Lahiri & Nurnberger, 1991). Genotyping was performed using the QuantStudio 12K Flex
Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Final PCR reaction
mixtures consisted of 20ng of genomic DNA and 2µL of 2X TaqMan® OpenArray®
Genotyping Master Mix. Samples were loaded onto a QuantStudio Digital PCR Plate using
QuantStudio 12K Flex AccuFill System and run on the QuantStudio 12K Flex Instrument as
per the manufacturer’s instructions. Genotype calls were visualized using the DigitalSuite
Software and were validated by two independent researchers blind to genotyping conditions.
To ensure genotyping accuracy, 10% of the samples were re-genotyped and conflicting
genotypes were set as missing for analysis.
3.3.5 Statistical Analysis
Quality-control of genotyping data was carried out using PLINK v1.07 (Purcell et al., 2007).
In order to pass QC, SNPs were required to have ≥90% genotyping rate and a MAF of ≥0.05,
and to satisfy HWE (p>0.005). In addition, individuals with genotyping efficiency less than

76
80% across all markers (samples producing genotypes for seven SNPs or less) were excluded
from analysis.
Descriptive statistics were carried out using SPSS v20 (IBM Corp, Armonk NY, USA).
Continuous variables were analyzed using Student’s t-test or ANOVA, and categorical
variables were analyzed using χ2-test. Genotype and allele frequencies in R/NR groups were
compared using χ2-test or Fisher’s Exact for cell counts less than 5. Differences in percent
score reduction in BPRS, BPOS and BNEG subscales among genotypic groups were
compared using ANCOVA, with age and baseline scores added as covariates.
HWE and LD among SNPs were determined using Haploview v4.2 and the Solid spine of
LD to construct LD blocks (Figure 3.3) (Barrett et al., 2005). Haplotype analysis was carried
out using UNPHASED v3.1.5 and p-values were corrected using permutation (10,000 tests)
(Dudbridge, 2008). Haplotypes with a frequency of <0.05 were removed from subsequent
analyses. Power calculations were performed using Quanto v1.2.4 (Gauderman & Morrison,
2006). Multiple testing correction for genotype and allele tests was performed using the
Nyholt method and resulted in a revised statistical significance threshold of p<0.005 (Nyholt,
2004).
3.4 RESULTS
3.4.1 Sample Characteristics
Following QC procedures, twelve individuals were removed due to low genotyping
efficiency (<80% of markers were successfully genotyped). The remaining sample had a
mean genotyping efficiency of 98.9%. All SNPs met QC criteria and had a mean genotyping
rate of 99.3%. In total, 163 European treatment-resistant/intolerant patients were included in

77
the statistical analysis (Table 3.2). Our sample had over 80% power to detect an OR as low
as 2.1 (unmatched case-control design, n=163, non-responder frequency=47.2, MAF=26.5%,
α/2=0.05) and down to 8.5% of the variance in the quantitative response variable (continuous
design, n=91). The average minor allele frequency across all ten glutamate system variants
was used for power calculations.
For the three clinical sites, no statistically significant differences were observed for gender,
response rates, or percent score reduction in BPRS, BPOS and BNEG scales. A statistically
significant difference was observed, however, for age of study participants across clinical
sites (p=0.001). Post hoc analysis revealed that patients originating from the SG Potkin
sample were significantly older than patients from the HY Meltzer sample. Baseline BPRS,
BPOS and BNEG values were also correlated with their respective percent score reductions.
In order to combine the three samples for analysis, both age and baseline scores were
included as covariates in the ANCOVA analysis of continuous response. The combined
sample (n=163) was 74.8% male and had a mean age of 35.1 years (SD 8.1). Following six
months of CLZ therapy, patients experienced a mean percent score reduction of -9.1 points
(SD 12.4), -1.1 (SD 3.6) and -3.2 (SD 5.7) from baseline for the BPRS, BNEG and BPOS
scales, respectively. In addition, 52.8% of patients experienced a ≥20% decrease in BPRS
scores and were subsequently classified as responders.
3.4.2 Genotype and Allele Association Analysis
Following correction for multiple testing, no significant differences between responder and
non-responder groups or change in percent scores using BPRS, BPOS and BNEG scales
were observed for genotype and allele frequencies for each of the ten glutamate system gene

78
variants (Table 3.3). A number of nominally significant associations were observed;
however, none of these findings were significant following correction for multiple testing.
Prior to correction, SLC6A9 rs16831558 C-allele carriers exhibited an allele dose-dependent
reduction (improvement) in BPOS subscale scores following six months of CLZ therapy
(puncorrected=0.008, pcorrected=0.08) (Figure 3.4). CC-homozygotes of rs16831558 also
exhibited greater reduction in BPRS total scores (puncorrected=0.045, pcorrected=0.421). In
addition, greater reduction in BNEG subscale scores were observed for five variants:
SLC1A2 rs4534557 homozygotes (GG and CC) (puncorrected=0.015, pcorrected=0.140), GRIA1
rs2195450 A-allele carriers (puncorrected=0.017, pcorrected=0.162), SLC1A2 rs4354668 TT-
homozygotes (puncorrected=0.026, pcorrected=0.243), GRM2 rs2518461 GG-homozygotes
(puncorrected=0.037, pcorrected=0.346), and SLC1A2 rs2901534 CC-homozygotes
(puncorrected=0.047, pcorrected=0.440).
3.4.3 GRM2 and SLC6A9 Haplotype Analysis
No haplotype blocks within GRM2 rs4067-rs2518461 were significantly different between
responder/non-responder groups, or regarding continuous response measures (Table 3.4).
Initially, the haplotype block rs16831558-rs12037805-rs1978195 T-T-A within SLC6A9
appeared significantly associated with change in BPOS scores following CLZ treatment
(puncorrected=0.001, pcorrected=0.006, permutation corrected, individual haplotype effect).
However, further investigation revealed that the positive association was driven solely by
rs16831558. When the SLC6A9 rs12037805-rs1978195 haplotype was analyzed
independently with the rs16831558 variant added as a conditioning marker, this two-marker
haplotype was no longer significant (Table 3.5).

79
3.5 DISCUSSION
This investigation explored the association between ten glutamate system gene variants and
clinical response to CLZ. No significant associations were observed following correction for
multiple testing in our sample of patients. To our knowledge, this is the first reported study to
investigate the role of SLC1A2 (rs4354668, rs4534557, rs2901534), SLC6A9 (rs12037805,
rs1978195, rs16831558), GRIA1 (rs2195450), GRM2 (rs4067, rs251) and GAD1 (rs3749034)
in clinical response to CLZ. Four of these variants were previously investigated in other
neuropsychiatric phenotypes and the remaining six SNPs were previously unstudied and
were selected using HaploReg.
Several nominally significant associations were observed for SLC6A9, SLC1A2, GRM2 and
GRIA1 with CLZ response before correction for the number of independent tests. Of interest
was the association between C-allele carriers of SLC6A9 rs16831558 and percent score
reduction in the BPOS subscale (puncorrected=0.008). Individuals carrying two copies of the C-
allele showed a 4.25% decrease in the severity of positive symptoms, those carrying one
copy experienced no symptom change, and individuals with two copies of the T-allele
experienced a worsening of their positive symptoms as indicated by BPOS scores that were
approximately 3.00% higher than at baseline. This finding became non-significant following
correction (pcorrected=0.08), but given our relatively small sample size, this nominally
significant finding for SLC6A9 rs16831558 deserves further investigation in larger CLZ
response samples.
The SLC6A9 locus is particularly complex, with 16 different mRNA transcripts and 14
different splice variants (Thierry-Mieg & Thierry-Mieg, 2006). The rs16831558 variant

80
reported herein is located approximately 5kb upstream of the SLC6A9 start-site. This variant
has a RegulomeDB functional score of 2b (‘likely to affect binding’) and is predicted to lie
within an enhancer histone mark, a DNAse I hypersensitivity site, and to affect regulatory
motifs. Therefore, characterization of this SNP’s potential functionality using mobility shift
and luciferase promoter assays remains a priority for future work.
As was mentioned in the introduction, GlyT1 has recently been the focus of several novel
therapies for SCZ (Harvey & Yee, 2013). Five phase III clinical trials investigating
bitopertin, a synthetic GlyT1 inhibitor, as an adjunct to conventional AP therapy for
persistent negative symptoms or partial responders have been conducted, although the results
have been non-significant thus far (ClinicalTrails.gov identifiers: NCT01192867,
NCT01192906, NCT01192880, NCT01235520, and NCT01235559). Because GlyT1
regulates extracellular glycine concentrations in the vicinity of NMDARs (Smith et al.,
1992), GlyT1 is able to regulate NMDAR function (Kuryatov et al., 1994; Laube et al.,
1997). By blocking glycine reuptake from the synapse, GlyT1 inhibitors cause an increase in
NR1 glycine site occupancy and NMDAR activity, ultimately reversing the NMDAR
hypofunction that is thought to contribute to SCZ etiology.
Following the same rationale, various NR1 glycine site agonists including glycine, D-serine
and D-cycloserine have been investigated as adjuncts to conventional AP therapy for the
treatment of persistent negative and cognitive symptoms in SCZ (reviewed by Javitt, 2004).
Even though glycine agonist drugs are not designed to improve the positive symptoms seen
in TRS patients, observations of their use in clinical studies to improve negative symptoms
have shed light on a possible action of CLZ on the glycine system: NR1 glycine site agonists
are generally well received when paired with most APs, however, provide little therapeutic

81
benefit and may in fact worsen symptoms when administered to patients taking CLZ (Potkin
et al., 1999; Singh & Singh, 2011). This incompatibility between NR1 glycine site agonists
and CLZ may be due to CLZ’s pre-existing ability to potentiate NMDAR-mediated
neurotransmission through an as-of-yet-unknown mechanism (Lane et al., 2006). Several
mechanisms have been posited, for instance, CLZ may already increase synaptic glycine
levels (Javitt et al., 2005) or may already act as a partial agonist at the NR1 glycine site of
the NMDAR (Schwieler et al., 2008). Alternatively, CLZ’s ability to alter dopamine activity
and achieve therapeutic effect may depend in part on availability of the NMDAR glycine site
(Schwieler & Erhardt, 2003). Future studies clarifying the role of glycine transporter gene
variants in the mechanism of CLZ’s action are necessary.
There are some limitations inherent in our study that must be mentioned. For instance, our
small sample size may not have had sufficient statistical power to detect the effect sizes of
these gene variants, and going forward the collection of larger samples will help determine
the contribution of gene variants to CLZ response. One such sample is currently being
collected by the CRESTAR consortium in Europe (CRESTAR, 2011) with the goal of
identifying markers involved in CLZ response and side effects. Heterogeneity within our
study sample may also have confounded association findings. Participants were collected
from three clinical sites that may have differed in population substructure. In addition,
patients with a diagnosis of either SCZ or SA disorder were eligible for study inclusion.
Incomplete outcome data may also have limited our findings. Individuals who ceased
treatment prior to study completion invariably have an affect on response rates and may have
confounded our ability to identify genetic variants linked to treatment response.
Unfortunately, rates for treatment discontinuation in AP drug trials are estimated to be as

82
high as 40%, with common reasons for discontinuation being noncompliance, lack of
efficacy and the occurrence of adverse side effects (Lieberman et al., 1994; Meltzer &
Okayli, 1995b; Kinon et al., 2006). In certain patients, response to CLZ may take up to five
months or longer and suggests that a portion of participants who discontinue treatment
because of lack of efficacy most likely do so prematurely. The high rates of discontinuation
during AP drug trials stresses the need for understanding inter-individual variability in drug
response phenotypes and emphasizes the utility associated with identifying clinically relevant
predictive markers.
To conclude, we report no significant associations among ten glutamate system gene variants
and response to CLZ in our sample following correction for multiple testing. A nominally
significant association within the glycine transporter 1 gene variant rs16831558 was
observed prior to correction, and deserves further examination in larger well-characterized
CLZ response samples.
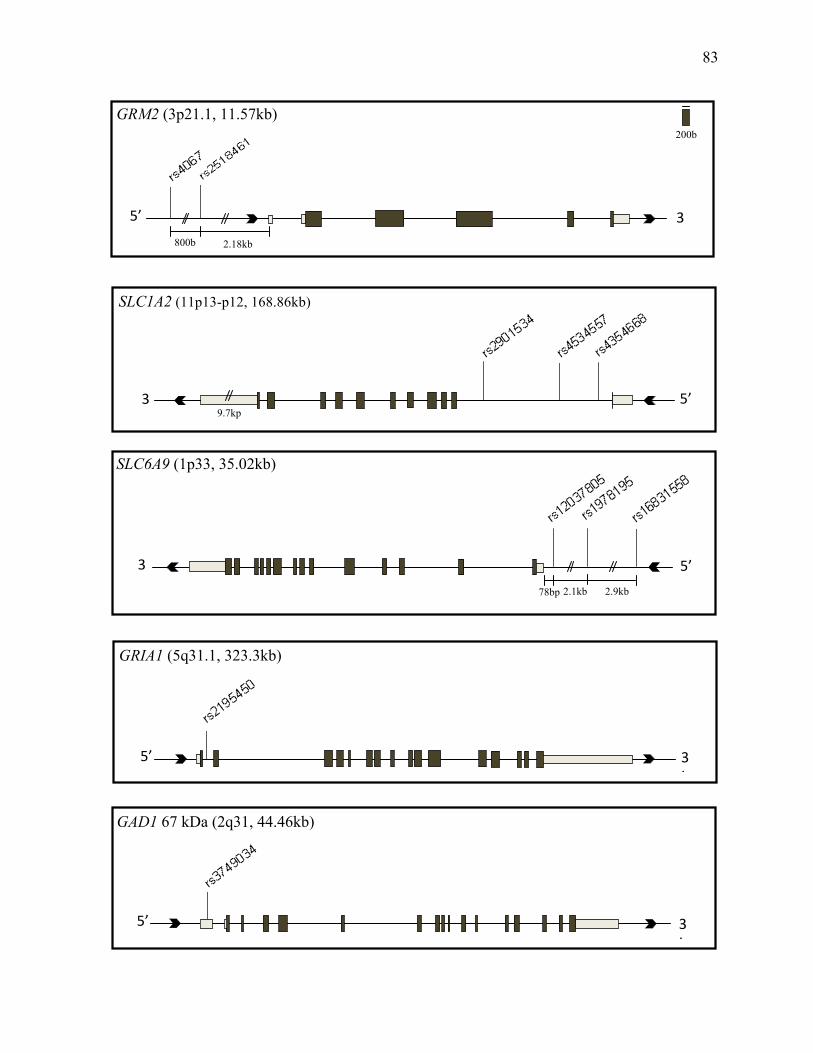
83
!
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
9.7kp
rs 29 01 53 4*
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
Kobil
ka BK,
Frielle
T,
Collin
s S,
Yang-
Feng
T,
Kobil
ka TS,
Franc
ke U,
Lefko
witz
RJ,
Caron
MG, 1
987.
An
intron
less
gene
encod
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
3
’
5’
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
SLC1A2 (11p13-p12, 168.86kb)
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
K
o
b
il
k
a
B
K
,
F
ri
e
ll
e
T
,
C
o
ll
i
n
s
S
,
Y
a
n
g
-
Ko
bil
ka
BK
,
Fri
ell
e
T,
Co
lli
ns
S,
Ya
ng
-
Fe
ng
T,
Ko
bil
ka
TS
,
Fr
an
ck
e
U,
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
3
’
5’
GRM2 (3p21.1, 11.57kb)
800b
p 2.18kb
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
e
T
,
C
o
l
l
i
n
s
S
,
200b
p
78bp
SLC6A9 (1p33, 35.02kb)
5’ 3
’
rs 16 83 15 58*
rs 19 78 19 5*rs 12 03 78 05*
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
Ko
bil
ka
BK
,
Fri
ell
e
T,
Co
lli
ns
S,
Ya
ng
-
Fe
ng
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
2.1kb 2.9kb K
o
b
i
l
k
a
B
K
,
F
r
i
e
l
l
GAD1 67 kDa (2q31, 44.46kb)
rs 37 49 03 4*
3’
5’
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
K
o
b
i
l
Kob
ilka
BK,
Frie
lle
K
o
b
i
l
K
o
b
i
l
GRIA1 (5q31.1, 323.3kb)
*
5’
K
o
b
i
l
k
a
B
K
,
K
o
b
i
l
k
a
B
K
Kobilka BK,
Frielle T,
Collins S,
Yang-Feng
T, Kobilka
TS, Francke
U,
Lefkowitz
RJ, Caron
MG, 1987.
An
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
K
o
b
i
l
k
a
B
K
3’

84
Figure!3.1*Glutamate!System!Gene!Diagrams!!
Schematic* representation* of* GRM2,* SLC1A2,* SLC6A9,* GRIA1* and* GAD1* genes* indicating*
chromosomal* location* and* gene* size.* Single* nucleotide* polymorphisms* genotyped* in* this*
study* are* shown.*Dark*boxes* represent* coding*exons* and* light* boxes* represent* 5’* and*3’*
untranslated* regions* (UTRs).* Gene* diagrams* were* constructed* using* NCBI* Reference*
Sequence*from*Homo1sapiens,*transcript*variant*1.*Alternatively*spliced*variants*exist*for*all*
five*genes*and*can*be*viewed*on*NCBI*AceView*(ThierryVMieg*&*ThierryVMieg,*2006).

85
Figure!3.2*HaploReg!Results!for!Functional!Glutamate!System!Gene!Variants**
To* be* considered* for* study* inclusion,* variants* were* required* to* have* a* minor* allele*
frequency* of* at* least* 5%* and* to* satisfy* at* least* 3* of* 7* functionality* criteria.* Chr:*
chromosome;* pos:* position;* LD:* linkage* disequilibrium;* r2:* correlation* coefficient;* D’:*
Hedrick’s* multiVallelic* D’;* Ref:* reference* allele;* Alt:* alternate* allele;* AFR/AMR/ASN/EUR*
freq:*African/ad*Mixed*American/Asian/European*minor*allele*frequency;*GERP/SiPhy*cons:*
genomic* evolutionary* rate* profiling/SiPhy* mammalian* conservation* algorithms;* DNAse:*
DNAse* I* hypersensitivity* site;* eQTL:* expression* quantitative* trait* loci;* dbSNP* fun* annot:*
dbSNP*functional*annotation*(Ward*&*Kellis,*2012).***
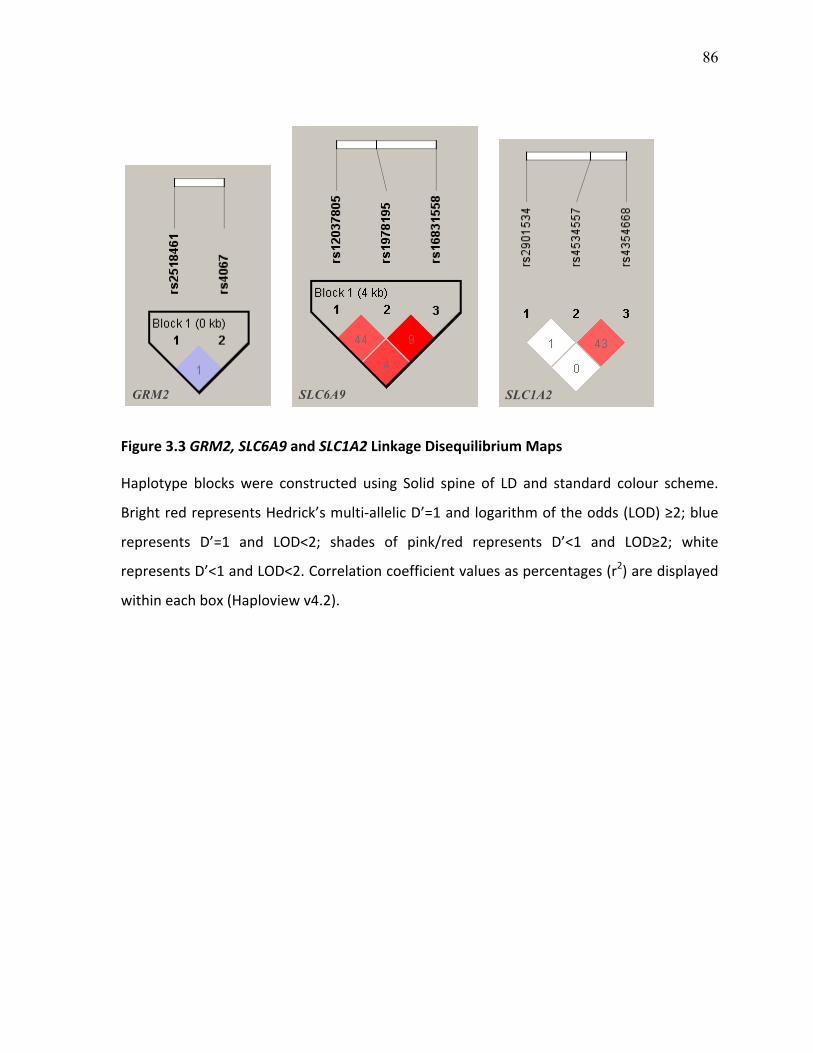
86
Figure!3.3*GRM2,&SLC6A9&and&SLC1A2*Linkage!Disequilibrium!Maps!*
Haplotype* blocks*were* constructed* using* Solid* spine* of* LD* and* standard* colour* scheme.*
Bright*red*represents*Hedrick’s*multiVallelic*D’=1*and*logarithm*of*the*odds*(LOD)*≥2;*blue*
represents* D’=1* and* LOD<2;* shades* of* pink/red* represents* D’<1* and* LOD≥2;* white*
represents*D’<1*and*LOD<2.*Correlation*coefficient*values*as*percentages*(r2)*are*displayed*
within*each*box*(Haploview*v4.2).
SLC6A9 SLC1A2 GRM2
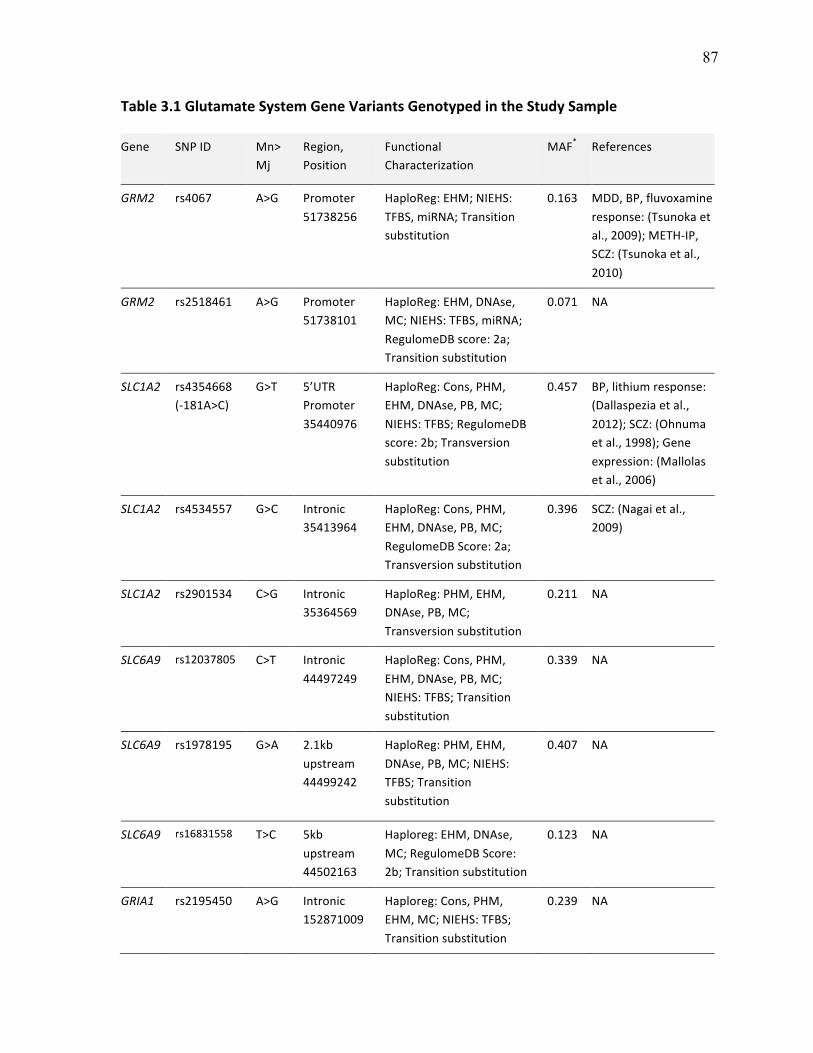
87
Table!3.1*Glutamate!System!Gene!Variants!Genotyped!in!the!Study!Sample*
Gene* SNP*ID* Mn>*Mj*
Region,*Position***
Functional*Characterization**
MAF** References*
GRM21 rs4067* A>G* Promoter*51738256*
HaploReg:*EHM;*NIEHS:*TFBS,*miRNA;*Transition*substitution*
0.163* MDD,*BP,*fluvoxamine*response:*(Tsunoka*et*al.,*2009);*METHVIP,*SCZ:*(Tsunoka*et*al.,*2010)*
GRM2* rs2518461* A>G* Promoter151738101*
HaploReg:*EHM,*DNAse,*MC;*NIEHS:*TFBS,*miRNA;*RegulomeDB*score:*2a;*Transition*substitution**
0.071* NA*
SLC1A21 rs4354668*(V181A>C)*
G>T* 5’UTR*Promoter*35440976*
HaploReg:*Cons,*PHM,*EHM,*DNAse,*PB,*MC;*NIEHS:*TFBS;*RegulomeDB*score:*2b;*Transversion*substitution**
0.457* BP,*lithium*response:*(Dallaspezia*et*al.,*2012);*SCZ:*(Ohnuma*et*al.,*1998);*Gene*expression:*(Mallolas*et*al.,*2006)*
SLC1A2* rs4534557* G>C* Intronic*35413964*
HaploReg:*Cons,*PHM,*EHM,*DNAse,*PB,*MC;*RegulomeDB*Score:*2a;*Transversion*substitution*
0.396* SCZ:*(Nagai*et*al.,*2009)*
SLC1A2* rs2901534* C>G* Intronic*35364569*
HaploReg:*PHM,*EHM,*DNAse,*PB,*MC;*Transversion*substitution**
0.211* NA*
SLC6A9* rs12037805* C>T* Intronic**44497249*
HaploReg:*Cons,*PHM,*EHM,*DNAse,*PB,*MC;*NIEHS:*TFBS;*Transition*substitution*
0.339* NA*
SLC6A91 rs1978195* G>A* 2.1kb*upstream*44499242*
HaploReg:*PHM,*EHM,*DNAse,*PB,*MC;*NIEHS:*TFBS;*Transition*substitution*
0.407* NA*
SLC6A91 rs16831558* T>C* 5kb*upstream*44502163*
Haploreg:*EHM,*DNAse,*MC;*RegulomeDB*Score:*2b;*Transition*substitution**
0.123* NA*
GRIA11 rs2195450* A>G* Intronic*152871009*
Haploreg:*Cons,*PHM,*EHM,*MC;*NIEHS:*TFBS;*Transition*substitution*
0.239* NA**

88
GAD11 rs3749034* A>G* 5’UTR*Exon*1*171673475*
Haploreg:*Cons,*PHM,*DNAse,*PB,*MC;*NIEHS:*TFBS;*Transition*substitution*
0.253* SCZ:*(Addington*et*al.,*2005;*Lundorf*et*al.,*2005;*Straub*et*al.,*2007)*
NA:*not*available;*Mn>Mj:*minor>major*allele;*UTR:*untranslated*region;*MAF:*minor*allele*frequency*in*our*sample* (n=163);* NIEHS:* National* Institute* of* Environmental* Health* Sciences* (TFBS:* transcription* factor*binding* site;*miRNA:*microRNA*binding* site);*Haploreg*Functional*Database* (2a/2b:* likely* to*affect*binding;*Cons:* evolutionarily* conserved* variant;* P/EVHM:* promoter/enhancer* associated* histone* mark;* PB:* protein*bound;*MC:*motifs* changed);* References* (BP:* bipolar* disorder;*MDD:*major* depressive* disorder;*METHVIP:*methamphetamineVinduced*psychosis;*SCZ:*schizophrenia).*
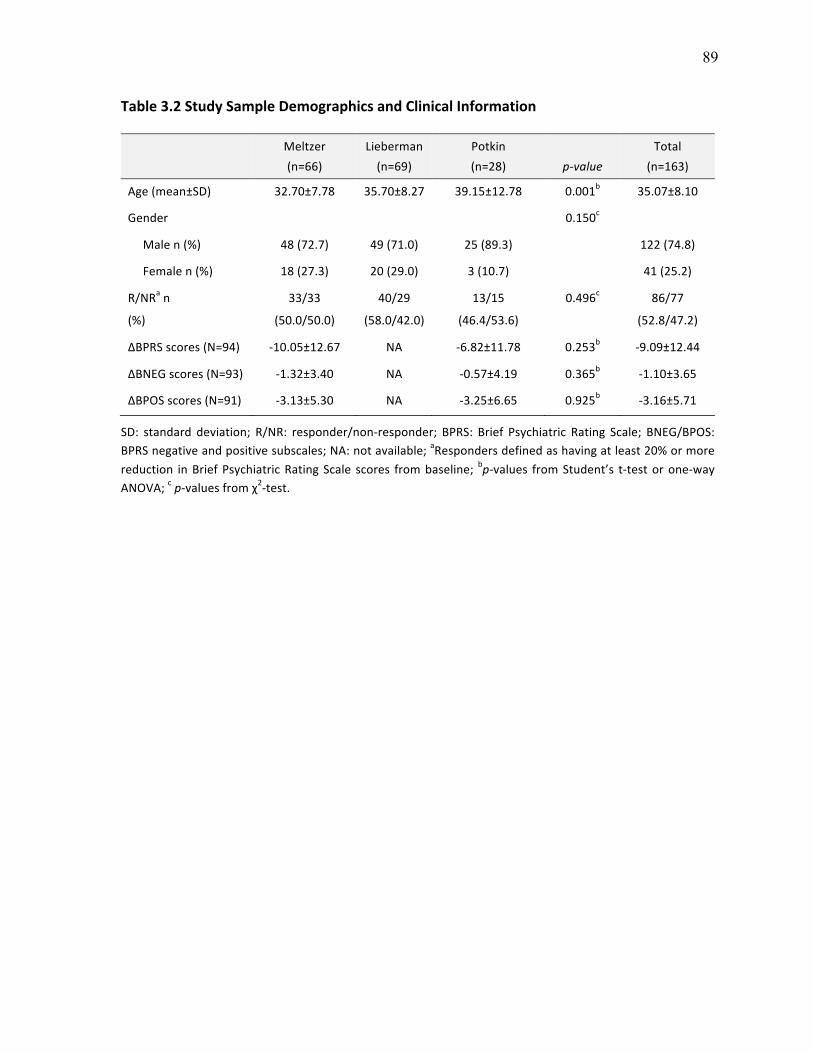
89
Table!3.2*Study!Sample!Demographics!and!Clinical!Information*
* Meltzer**(n=66)*
Lieberman**(n=69)*
Potkin**(n=28)*
1p8value1
Total**(n=163)*
Age*(mean±SD)* 32.70±7.78* 35.70±8.27* 39.15±12.78* 0.001b* 35.07±8.10*
Gender* * * * 0.150c* *
*****Male*n*(%)* 48*(72.7)* 49*(71.0)* 25*(89.3)* * 122*(74.8)*
*****Female*n*(%)* 18*(27.3)* 20*(29.0)* 3*(10.7)* * 41*(25.2)*
R/NRa*n**
(%)*
33/33**
(50.0/50.0)*
40/29**
(58.0/42.0)**
13/15**
(46.4/53.6)*
0.496c* 86/77**
(52.8/47.2)*
ΔBPRS*scores*(N=94)* V10.05±12.67* NA* V6.82±11.78* 0.253b* V9.09±12.44*
ΔBNEG*scores*(N=93)* V1.32±3.40* NA* V0.57±4.19* 0.365b* V1.10±3.65*
ΔBPOS*scores*(N=91)* V3.13±5.30* NA* V3.25±6.65* 0.925b* V3.16±5.71*
SD:* standard* deviation;* R/NR:* responder/nonVresponder;* BPRS:* Brief* Psychiatric* Rating* Scale;* BNEG/BPOS:*BPRS*negative*and*positive*subscales;*NA:*not*available;*aResponders*defined*as*having*at*least*20%*or*more*reduction* in*Brief*Psychiatric*Rating*Scale* scores* from*baseline;* bpVvalues* from*Student’s* tVtest*or*oneVway*ANOVA;*c*pVvalues*from*χ2Vtest.*
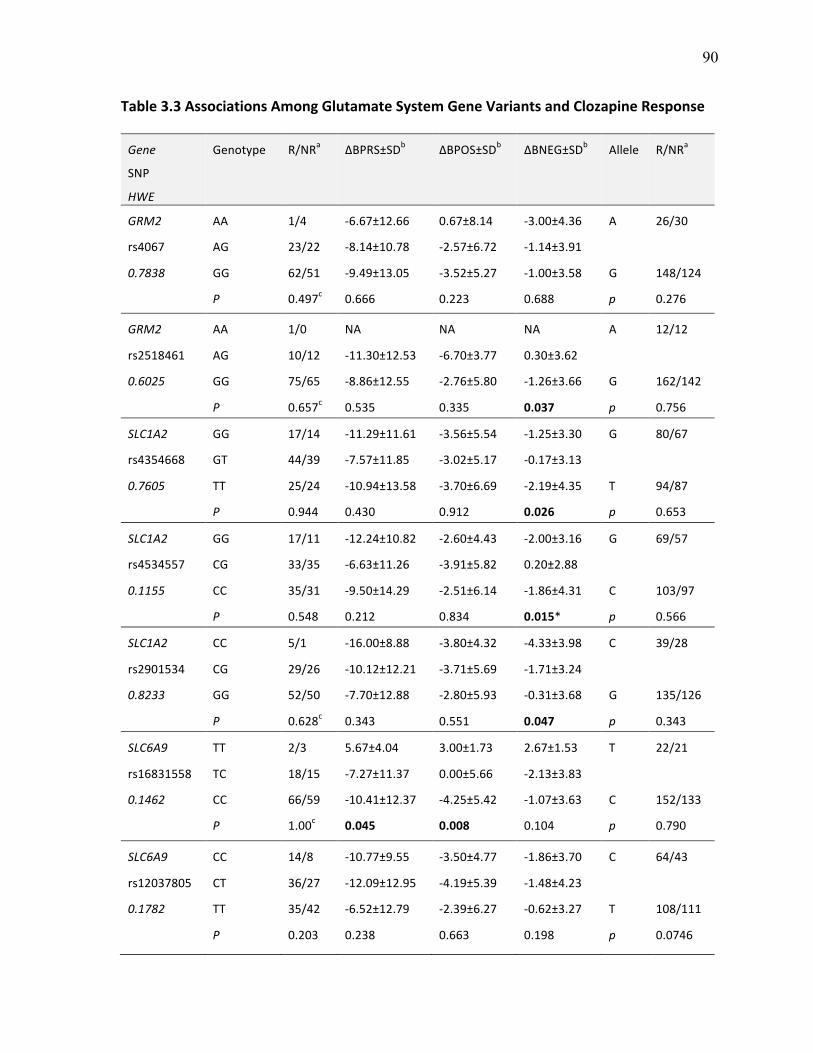
90
Table!3.3*Associations!Among!Glutamate!System!Gene!Variants!and!Clozapine!Response**
Gene1
SNP*
HWE1
Genotype* R/NRa* ΔBPRS±SDb* ΔBPOS±SDb* ΔBNEG±SDb* Allele* R/NRa*
GRM21 AA* 1/4* V6.67±12.66* 0.67±8.14* V3.00±4.36* A* 26/30*
rs4067* AG* 23/22* V8.14±10.78* V2.57±6.72* V1.14±3.91* * *
0.78381 GG* 62/51* V9.49±13.05* V3.52±5.27* V1.00±3.58* G* 148/124*
* P1 0.497c* 0.666* 0.223* 0.688* p1 0.276*
GRM21 AA* 1/0* NA* NA* NA* A* 12/12*
rs2518461* AG* 10/12* V11.30±12.53* V6.70±3.77* 0.30±3.62* * *
0.60251 GG* 75/65* V8.86±12.55* V2.76±5.80* V1.26±3.66* G* 162/142*
* P1 0.657c* 0.535* 0.335* 0.037! p1 0.756*
SLC1A21 GG* 17/14* V11.29±11.61* V3.56±5.54* V1.25±3.30* G* 80/67*
rs4354668* GT* 44/39* V7.57±11.85* V3.02±5.17* V0.17±3.13* * *
0.76051 TT* 25/24* V10.94±13.58* V3.70±6.69* V2.19±4.35* T* 94/87*
* P1 0.944* 0.430* 0.912* 0.026! p1 0.653*
SLC1A2* GG* 17/11* V12.24±10.82* V2.60±4.43* V2.00±3.16* G* 69/57*
rs4534557** CG* 33/35* V6.63±11.26* V3.91±5.82* 0.20±2.88* * *
0.11551 CC* 35/31* V9.50±14.29* V2.51±6.14* V1.86±4.31* C* 103/97*
* P1 0.548* 0.212* 0.834* 0.015** p1 0.566*
SLC1A2* CC* 5/1* V16.00±8.88* V3.80±4.32* V4.33±3.98* C* 39/28*
rs2901534* CG* 29/26* V10.12±12.21* V3.71±5.69* V1.71±3.24* * *
0.82331 GG* 52/50* V7.70±12.88* V2.80±5.93* V0.31±3.68* G* 135/126*
* P1 0.628c* 0.343* 0.551* 0.047! p1 0.343*
SLC6A91 TT* 2/3* 5.67±4.04* 3.00±1.73* 2.67±1.53* T* 22/21*
rs16831558* TC* 18/15* V7.27±11.37* 0.00±5.66* V2.13±3.83* * *
0.14621 CC* 66/59* V10.41±12.37* V4.25±5.42* V1.07±3.63* C* 152/133*
* P1 1.00c* 0.045! 0.008! 0.104* p1 0.790*
SLC6A91 CC* 14/8* V10.77±9.55* V3.50±4.77* V1.86±3.70* C* 64/43*
rs12037805* CT* 36/27* V12.09±12.95* V4.19±5.39* V1.48±4.23* * *
0.17821 TT* 35/42* V6.52±12.79* V2.39±6.27* V0.62±3.27* T* 108/111*
* P1 0.203* 0.238* 0.663* 0.198* p1 0.0746*
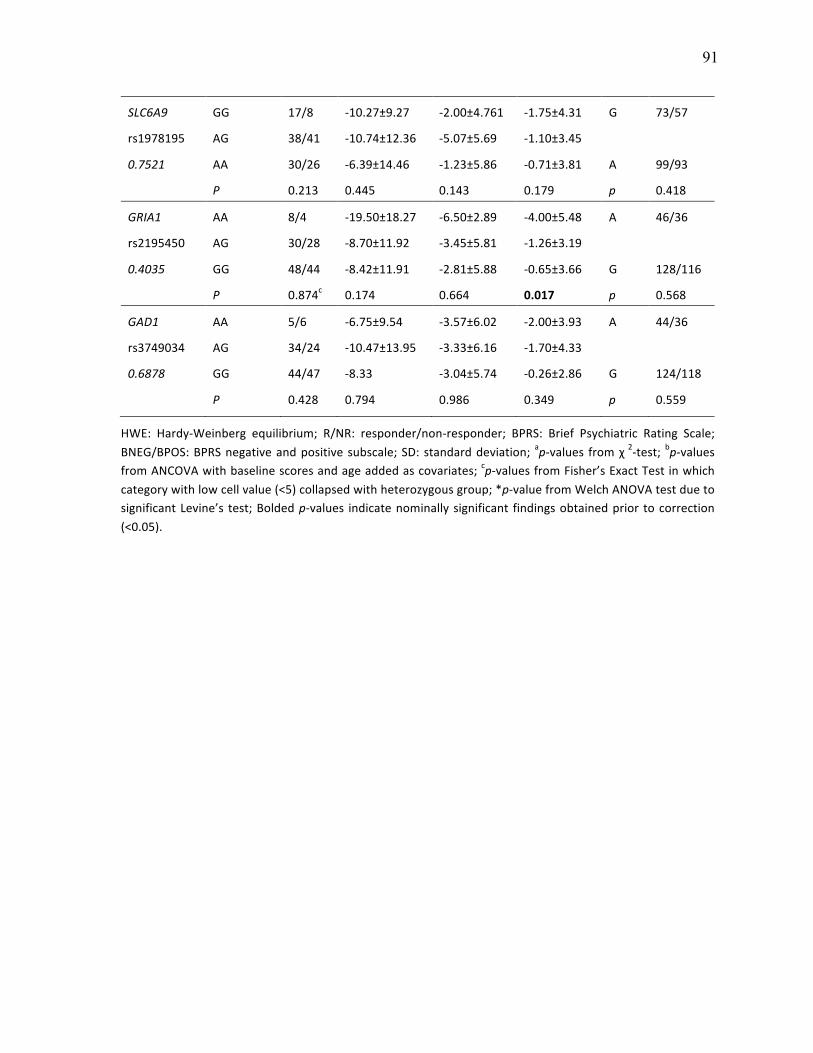
91
SLC6A9* GG* 17/8* V10.27±9.27* V2.00±4.761* V1.75±4.31* G* 73/57*
rs1978195* AG* 38/41* V10.74±12.36* V5.07±5.69* V1.10±3.45* 1 *
0.75211 AA* 30/26* V6.39±14.46* V1.23±5.86* V0.71±3.81* A* 99/93*
* P1 0.213* 0.445* 0.143* 0.179* p1 0.418*
GRIA11 AA* 8/4* V19.50±18.27* V6.50±2.89* V4.00±5.48* A* 46/36*
rs2195450* AG* 30/28* V8.70±11.92* V3.45±5.81* V1.26±3.19* * *
0.40351 GG* 48/44* V8.42±11.91* V2.81±5.88* V0.65±3.66* G* 128/116*
* P1 0.874c* 0.174* 0.664* 0.017! p1 0.568*
GAD11 AA* 5/6* V6.75±9.54* V3.57±6.02* V2.00±3.93* A* 44/36*
rs3749034* AG* 34/24* V10.47±13.95* V3.33±6.16* V1.70±4.33* * *
0.68781 GG* 44/47* V8.33* V3.04±5.74* V0.26±2.86* G* 124/118*
* P1 0.428* 0.794* 0.986* 0.349* p1 0.559*
HWE:* HardyVWeinberg* equilibrium;* R/NR:* responder/nonVresponder;* BPRS:* Brief* Psychiatric* Rating* Scale;*BNEG/BPOS:*BPRS*negative*and*positive*subscale;*SD:* standard*deviation;* apVvalues* from*χ* 2Vtest;* bpVvalues*from*ANCOVA*with*baseline*scores*and*age*added*as*covariates;*cpVvalues*from*Fisher’s*Exact*Test*in*which*category*with*low*cell*value*(<5)*collapsed*with*heterozygous*group;**pVvalue*from*Welch*ANOVA*test*due*to*significant*Levine’s* test;*Bolded*pVvalues* indicate*nominally* significant* findings*obtained*prior* to*correction*(<0.05).*
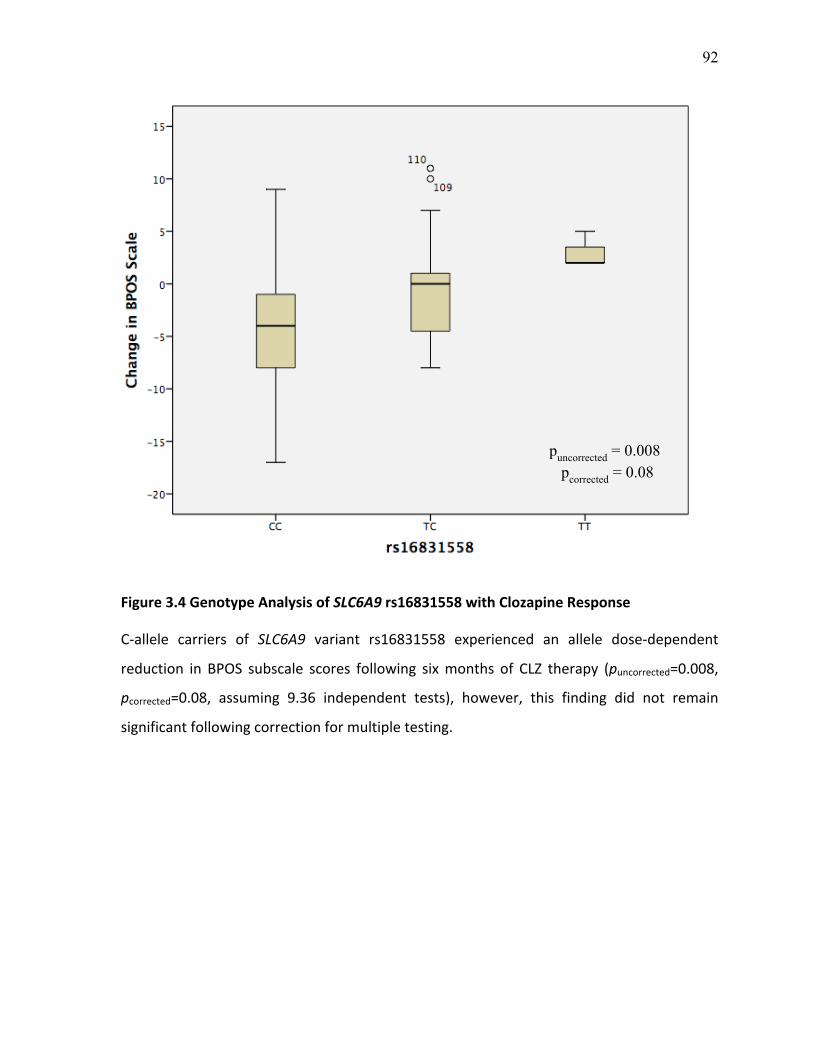
92
Figure!3.4*Genotype!Analysis!of!SLC6A9&rs16831558!with!Clozapine!Response!!
CVallele* carriers* of* SLC6A9* variant* rs16831558* experienced* an* allele* doseVdependent*
reduction* in* BPOS* subscale* scores* following* six*months* of* CLZ* therapy* (puncorrected=0.008,*
pcorrected=0.08,* assuming* 9.36* independent* tests),* however,* this* finding* did* not* remain*
significant*following*correction*for*multiple*testing.*
puncorrected = 0.008 pcorrected = 0.08
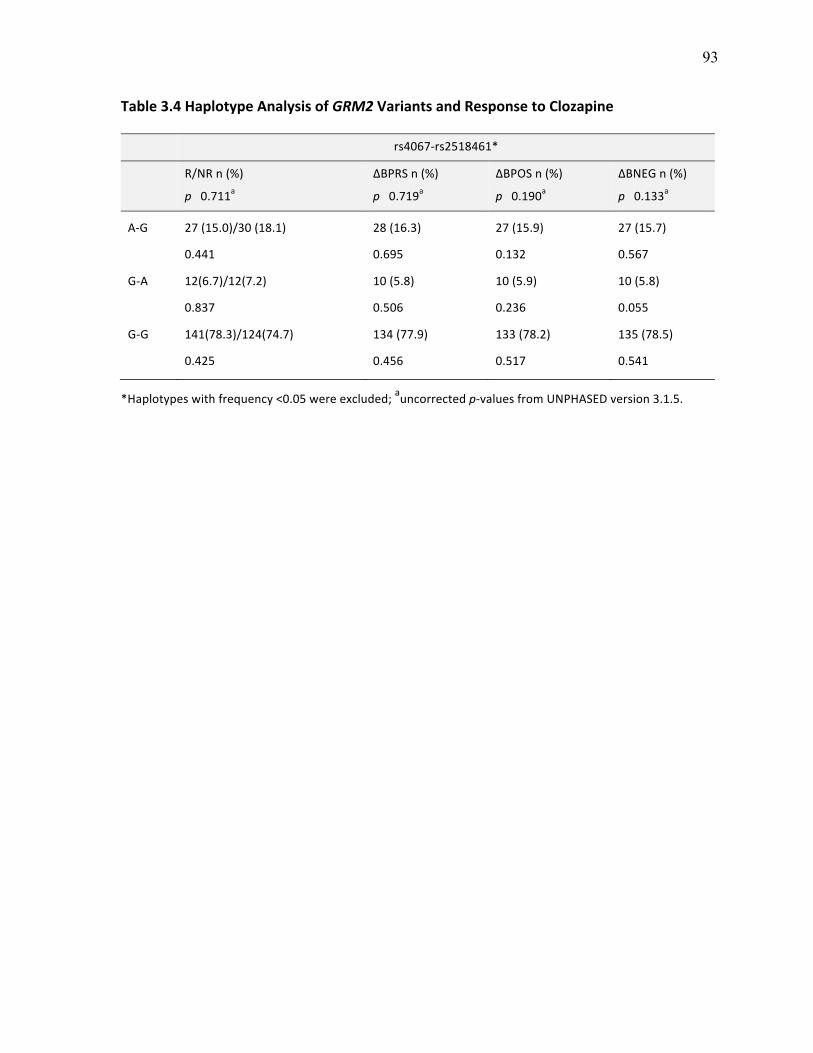
93
Table!3.4*Haplotype!Analysis!of!GRM2!Variants!and!Response!to!Clozapine*
* rs4067Vrs2518461**
* R/NR*n*(%)*
p1110.711a*
ΔBPRS*n*(%)*
p1110.719a*
ΔBPOS*n*(%)*
p1110.190a*
ΔBNEG*n*(%)*
p1110.133a*
AVG*
*
GVA*
*
GVG*
27*(15.0)/30*(18.1)*
0.441*
12(6.7)/12(7.2)*
0.837*
141(78.3)/124(74.7)*
0.425*
28*(16.3)*
0.695*
10*(5.8)*
0.506*
134*(77.9)*
0.456*
27*(15.9)*
0.132*
10*(5.9)*
0.236*
133*(78.2)*
0.517*
27*(15.7)*
0.567*
10*(5.8)*
0.055*
135*(78.5)*
0.541*
*Haplotypes*with*frequency*<0.05*were*excluded;*auncorrected*pVvalues*from*UNPHASED*version*3.1.5.**
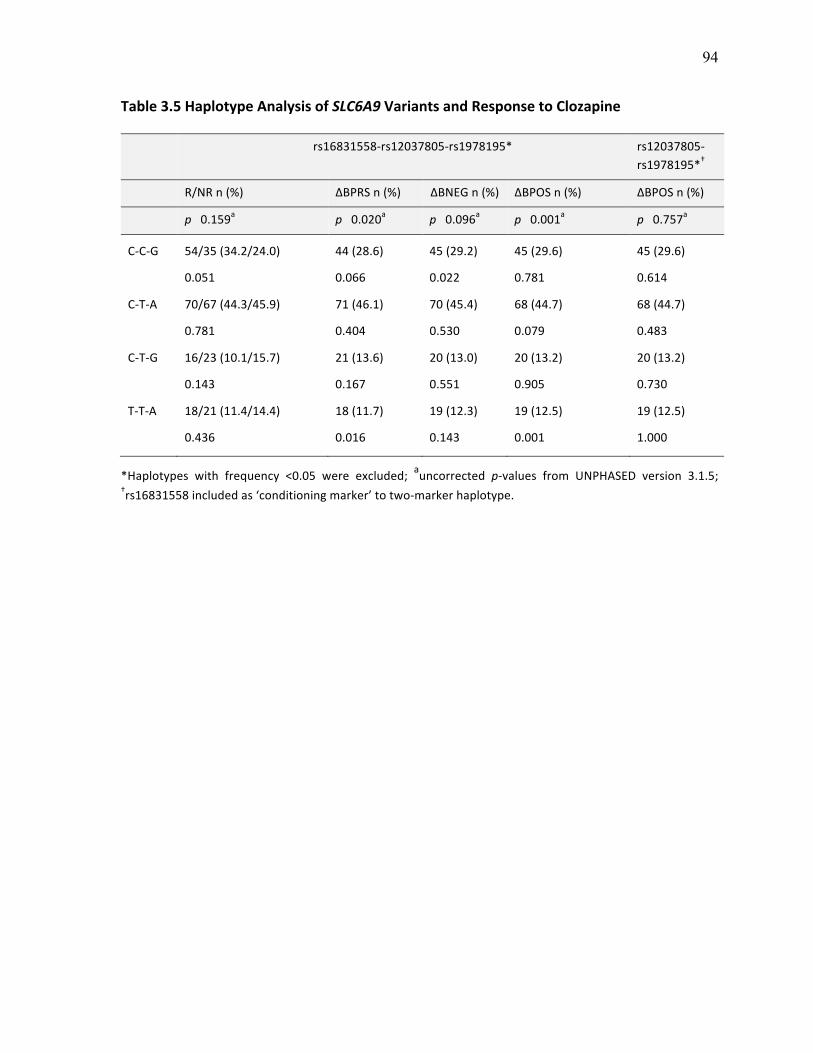
94
Table!3.5*Haplotype!Analysis!of!SLC6A9!Variants!and!Response!to!Clozapine*
* ******rs16831558Vrs12037805Vrs1978195** rs12037805Vrs1978195*†*
* R/NR*n*(%)* ΔBPRS*n*(%)* ΔBNEG*n*(%)* ΔBPOS*n*(%)* ΔBPOS*n*(%)*
* p1110.159a* p1110.020a* p1110.096a* p1110.001a* p1110.757a*
CVCVG*
*
CVTVA*
*
CVTVG*
*
TVTVA*
54/35*(34.2/24.0)*
0.051*
70/67*(44.3/45.9)*
0.781*
16/23*(10.1/15.7)*
0.143*
18/21*(11.4/14.4)*
0.436*
44*(28.6)*
0.066*
71*(46.1)*
0.404*
21*(13.6)*
0.167*
18*(11.7)*
0.016*
45*(29.2)*
0.022*
70*(45.4)*
0.530*
20*(13.0)*
0.551*
19*(12.3)*
0.143*
45*(29.6)*
0.781*
68*(44.7)*
0.079*
20*(13.2)*
0.905*
19*(12.5)*
0.001*
45*(29.6)*
0.614*
68*(44.7)*
0.483*
20*(13.2)*
0.730*
19*(12.5)*
1.000*
*Haplotypes* with* frequency* <0.05* were* excluded;* auncorrected* pVvalues* from* UNPHASED* version* 3.1.5;*†rs16831558*included*as*‘conditioning*marker’*to*twoVmarker*haplotype.*

Taylor DL V. DISCUSSION
95##
Chapter 4 #
DISCUSSION
4.0 OVERVIEW
This thesis work investigated the association of 17 glutamate system gene variants with
clinical response to CLZ. No significant associations for genotype, allele, or haplotype
analyses were observed across all genes following correction for multiple testing. Prior to
correction, several nominally significant associations were observed, the most important of
which was for the glycine transporter 1 gene variant rs16831558 and change in BPRS
positive symptom subscale scores. In this section we discuss main findings, explore their
implications, assess study limitations and conclude with future directions.
4.1 SUMMARY OF FINDINGS
4.1.1 Association of GRIN2B with Clozapine Response
The first chapter of this thesis investigated the association of eight GRIN2B variants with
response to CLZ. To our knowledge, this is the first reported study to investigate associations
among rs7301328, rs1072388, rs3764030, rs12826365, rs2284411, rs1806191 and rs890
with CLZ response, while the rs1806201 variant had previously been investigated in two
other response studies (Hong et al., 2001; Chiu et al., 2003). Variant rs7301328 significantly
deviated from Hardy-Weinberg equilibrium and was therefore excluded from analysis
(pHWE=1.93x10-7). The implications regarding deviation from HWE are discussed in more
detail in Study Limitations Section 4.3.5.

96#
#
For all GRIN2B SNPs investigated, no significant associations were observed for allele,
genotype, or haplotype analyses prior to and after correction for multiple testing. A-allele
carriers of variant rs1072388 responded marginally better to CLZ therapy than GG-
homozygotes, however this finding was not statistically significant (puncorrected=0.067,
pcorrected=0.440) (Chapter 2, Figure 2.3). Our negative association findings for GRIN2B
variant rs1806201 was consistent with previous reports that also found no association
between this variant and CLZ response in two independent samples of Chinese TRS patients
(Hong et al., 2001; Chiu et al., 2003).
4.1.2 Association of Glutamate System Variants with Clozapine Response
The second chapter investigated the association of ten glutamate system gene variants and
response to CLZ. To our knowledge, this is the first reported study to investigate the
association of GRM2 (rs4067, rs2518461), SLC1A2 (rs4354668, rs4534557, rs2905134),
SLC6A9 (rs12037805, rs1978195, rs16831558), GRIA1 (2195450) and GAD1 (rs3709054)
with clinical response to CLZ. No significant genotype, allele, or haplotype associations
were reported across all SNPs following correction for multiple testing.
Prior to correction, several nominally significant associations were observed. Briefly, a
greater reduction in BPRS negative symptom subscale scores was observed for five variants:
SLC1A2 rs4534557 homozygotes (GG and CC) (puncorrected=0.015, pcorrected=0.140); GRIA1
rs2195450 A-allele carriers (puncorrected=0.017, pcorrected=0.162), SLC1A2 rs4354668 TT-
homozygotes (puncorrected=0.026, pcorrected=0.243), GRM2 rs2518461 GG-homozygotes
(puncorrected=0.037, pcorrected=0.346), and SLC1A2 rs2901534 CC-homozygotes
(puncorrected=0.047, pcorrected=0.440).

97#
#
The most interesting association observed before correction was for SLC6A9 rs16831558: C-
allele carriers experienced an allele dose-dependent reduction in BPOS subscale scores
following six months of CLZ therapy (puncorrected=0.008, pcorrected=0.08) (Chapter 3, Figure
3.4). More specifically, individuals carrying two copies of the C-allele showed a 4.25%
decrease in severity of positive symptoms, those carrying one copy experienced no symptom
change, and individuals carrying two copies of the T-allele experienced a 3.00% increase in
positive symptoms compared to baseline. The rs16831558 SNP is a previously unstudied
variant that was identified using HaploReg functional annotation database (Benson et al.,
2013). Located in the promoter region of SLC6A9, this variant lies in an enhancer/promoter-
associated histone mark and is proposed to affect both SLC6A9 transcriptional activity and
protein expression by altering transcription factor binding. This variant’s functional potential
and biological plausibility, in combination with the nominally significant association finding
reported herein makes rs16831558 an ideal candidate to be further investigated in larger CLZ
response samples and for characterization using functional assays.
4.2 IMPLICATIONS OF FINDINGS
The main findings of this work suggest that genetic variation in the glycine transporter 1
gene may confer risk for clozapine non-response. This section begins with a discussion on
the biological significance of the glycine neurotransmitter system in relation to SCZ etiology
and treatment. CLZ’s ability to interact with the glycine system will then be noted. This
section concludes by exploring a possible mechanism through which the SLC6A9 promoter
variant rs16831558 may regulate glycine transporter expression, and lead to differential drug
effects.

98#
#
4.2.1 Glycine in Schizophrenia Etiology and Treatment
Glycine is a necessary co-agonist at the NR1 subunit of the NMDAR. Through a coincidence
detection type mechanism, the NMDAR will not fire without the simultaneous binding of
both glycine and glutamate (Johnson & Ascher, 1987; Mayer et al., 1989; Thomson, 1990).
Therefore, control of glycine levels at glutamatergic synapses is an important regulatory
component for the activation of excitatory neurotransmission. As was mentioned in the
introduction, the glycine transporter 1 protein is localized to glutamatergic neurons and
controls the reuptake of glycine from the synapse (Kleckner & Dingledine, 1988; Bergeron et
al., 1998). Changes in GlyT1 activity alter NMDAR-mediated neurotransmission as
evidenced by increased NMDAR excitatory post-synaptic currents following GlyT1
antagonism (Bergeron et al., 1998) and enhanced NMDAR function in GlyT1 knockout mice
(Gabernet et al., 2005).
Because glycine is necessary for NMDAR activation, abnormalities in the glycine system
may contribute to the hypoglutamatergic states implicated in SCZ etiology. Several
abnormalities related to glycine biology have been reported in patients with SCZ. In
comparison to healthy controls, AP naïve patients have decreased plasma levels of glycine
and low glycine levels have been shown to correlate with more severe negative symptoms
(Sumiyoshi et al., 2004; Neeman et al., 2005). Binding assays with 3H-glycine in
postmortem brain tissue also show patients experience increased NR1 glycine site binding in
sensory and motor cortices (Ishimaru et al., 1992), and may indicate postsynaptic
compensation for glutamatergic hypofunction.
Since glycine abnormalities may contribute to the hypoglutamatergic states that are
implicated in SCZ’s etiology, glycine has been targeted as a plausible mechanism for novel

99#
#
treatments. Glycine was also recognized as having antipsychotic potential upon the discovery
that pretreatment with glycine or glycine-like compounds reverse PCP-induced hyperactivity
in preclinical models (Javitt et al., 1997). The glycine-modulating compounds that exist at
present include the glycine transporter inhibitors, such as bitopertin (RG1678) developed by
Roche, as well as the use of adjunct glycine or glycine-like compounds for the treatment of
persistent negative symptoms. Glycine-modulating compounds share a common mechanism
of action to achieve therapeutic effect: glycine transporter inhibitors antagonize GlyT1
function to restore normal levels of glycine at the synapse, while glycine compounds directly
stimulate NMDAR function.
4.2.2 Interaction of Clozapine with the Glycine System
CLZ is theorized to achieve therapeutic effect, in part, by acting on the glycine system.
Scientific evidence supporting this theory is as follows: 1) CLZ treatment significantly
restores plasma glycine to levels seen in healthy controls (Neeman et al., 2005; Yamamori et
al., 2014); 2) baseline glycine levels have the ability to predict negative symptom response
during CLZ treatment (Sumiyoshi et al., 2004); and 3) glycine/glycine-like compounds have
no effect or worsen patient symptoms when combined specifically with CLZ (Goff et al.,
1999; Tsai et al., 1999; Evins et al., 2000). The mechanism through which CLZ alters glycine
levels is largely unknown, however, one study has suggested that CLZ may do so by
interfering with glycine reuptake from neuronal synapses (Javitt et al., 2005).
Alternative mechanisms have been postulated to explain how glycine compounds interfere
with CLZ’s therapeutic effect. One group of researchers propose that CLZ may directly
interact with the NMDAR by binding to the NR1 glycine site (Schwieler et al., 2004;
Schwieler et al., 2008), however, no direct evidence supporting this theory currently exists,

100#
#
for instance, from co-immunoprecipitation assays. An alternative hypothesis suggests that
CLZ may depend on NR1 glycine site availability to achieve therapeutic effect (Schwieler &
Erhardt, 2003). Despite the mechanism, these findings suggest that response to CLZ depends
in part on plasma glycine levels and/or availability of the NR1 glycine site.
4.2.3 Promoter Variant Confers Differential Response to Clozapine
We observed that individuals carrying two copies of the rs16831558 C-allele showed a
4.25% improvement in positive symptom severity, those carrying one copy experienced no
symptom change, and patients with two copies of the T-allele experienced a 3.00%
worsening of their positive symptoms from baseline. The rs16831558 promoter variant is
proposed to alter GlyT1 expression through altering transcription factor binding. Future work
entails conducting luciferase promoter assays to test if the C-allele has greater transcriptional
activity over the T-allele. Functional characterization of this SLC6A9 promoter variant is
discussed in more detail in Future Directions Section 4.4.1.
If rs16831558 is found to have an affect on GlyT1 expression, we propose that variation in
rs16831558 may lead to differential response to CLZ. This proposition stems from previous
research indicating that CLZ response is contingent upon glycine levels: baseline levels of
glycine inversely correlate with negative symptom response during CLZ treatment
(Sumiyoshi et al., 2004) and increasing levels of NR1 agonists through the addition of
exogenous glycine/glycine-like compounds during CLZ treatment leads to lack of response
or worsening of symptoms (Goff et al., 1999; Tsai et al., 1999; Evins et al., 2000). Therefore,
variants with the ability to alter the quantity or quality of glycine at neuronal synapses may
also affect CLZ efficacy. We now propose a mechanism through which the glycine
transporter 1 gene promoter polymorphism rs16831558 may regulate GlyT1 expression, and

101#
#
subsequently lead to differential response to CLZ. The mechanism is as follows: if the C-
allele is found to increase SLC6A9 transcriptional activity, individuals carrying the CC-
genotype should exhibit increased GlyT1 expression, leading to lower levels of synaptic
glycine and a more favourable response to CLZ. In contrast, T-allele carriers should exhibit
normal-to-low GlyT1 expression, resulting in normal-to-high levels of endogenous synaptic
glycine, which may either have no effect or may interfere with CLZ’s ability to achieve
therapeutic effect. Further characterization of the rs16831558 variant will be needed to give
credence to this proposed mechanism.
The clinical implications of predictive biomarkers that are capable of distinguishing
responders from non-responders prior to treatment are far reaching and would serve as an
invaluable tool for optimizing treatment outcomes for patients with SCZ. Several examples
of promoter variants conferring risk for non-response exist in other areas of medical research
and provide hope for the clinical application of predictive variants in psychiatric care. For
instance, the vitamin K epoxide reductase complex subunit 1 gene (VKORC1) promoter
variant -1639G>A significantly correlates with Warfarin dose (Carlquist et al., 2008). A
luciferase promoter assay was used to show the G-allele of this variant is associated with a
50% increase in VKORC1 transcriptional activity as compared to the A-allele (Yuan et al.,
2005). One additional example demonstrating an interaction between gene promoter
sequence variants and drug treatment response is in asthma (Drazen et al., 1999).
Recent literature has confirmed that polymorphisms in drug targets have the ability to alter
patient sensitivity to treatment and change the pharmacodynamics of treatment response.
Continued research in pharmacogenetics that aims to increase our understanding of the
complex interplay between genetic variants that control the pharmacokinetic and

102#
#
pharmacodynamic factors involved in drug effects is an important step on the road to
personalizing treatment. An overview of the future of personalized medicine in psychiatry is
discussed in more detail in Personalized Medicine in Psychiatry Section 4.4.5.
4.3 LIMITATIONS AND CONSIDERATIONS
Several limitations associated with our study must be considered before interpreting our
findings. This section explores the importance of study heterogeneity, inter-study variability,
statistical power, correction for multiple testing, deviation from Hardy-Weinberg equilibrium
and incomplete outcome data, as they relate to our findings.
4.3.1 Study Heterogeneity
Study heterogeneity refers to the variability inherent in a study and can be caused by
differences in study design (methodological diversity), and variation among participants,
interventions and outcomes (clinical diversity) (Hanson & Levin, 2013). Heterogeneity in
association studies is particularly problematic because it may lead to spurious associations or
the inability to detect true associations between marker loci and phenotypes of interest. Steps
are often taken to limit the heterogeneity in a sample so that results may be interpreted with
the least amount of confounding.
The first source of heterogeneity in our sample was caused by methodological and clinical
diversity. Participants were collected from three clinical sites that may have differed in study
methodology and populations. Combining individuals with different ancestral backgrounds
into a single group for analysis can lead to population stratification where false positive
associations arise because of population substructure rather than true associations (Cardon &
Palmer, 2003). Ad-mixed populations particularly confound drug response phenotypes

103#
#
because response tends to vary according to a patient’s ethnicity (Frackiewicz et al., 1997;
Wilson et al., 2001).
Patients included in our study were considered to be European based on self-reports. Even
though the accuracy between self-reported ancestry and actual ancestry is fairly consistent
(agreement rate of 75.1%) (Choudhry et al., 2007; Lee et al., 2010), the use of a principle
components analysis to experimentally determine ancestry profiles remains a priority for
future work. Briefly, PCA utilizes a panel of SNPs that are known to differ in allele
frequencies across populations of different geographical regions. Ancestry is then determined
by comparing genotyping results to reference panels from well-characterized datasets such as
genetic data from the HapMap Project (Paschou et al., 2008). PCA analysis is also useful for
detecting population stratification (Liu et al., 2011). Ideally, genetic data from admixed
populations should be stratified prior to analysis to maintain as much homogeneity as
possible.
Heterogeneity related to disease diagnosis and differences between SCZ and SA disorder
may also have increased variation in our sample. For instance, these disorders have different
clinical presentations: SA disorder is characterized as periods of psychosis with concurrent
symptoms meeting criteria for a Major Mood Episode, while in comparison, SCZ is
characterized by less frequent/prominent depressed or manic mood states (American
Psychiatric Association, 2013). As well, SA disorder patients tend to experience higher rates
of response and better clinical outcomes than patients with SCZ (Harrow et al., 2000). Taken
together, these differences suggest that SCZ and SA disorder may differ in their underlying
biology (Abrams et al., 2008; American Psychiatric Association, 2013; Cosgrove & Suppes,
2013,). The propensity of SA disorder patients to respond more favourably to treatment may

104#
#
have increased our CLZ response rate, and combining these two disorders into a single
sample for analysis may limit detection of causal variants that contribute to response.
Lastly, our sample consisted of patients who were placed on CLZ for different reasons. CLZ
is a 3rd line treatment for SCZ patients who are diagnosed as having TRS either because they
failed to respond to previous treatments with pharmacotherapy (treatment resistance) or
because they developed intolerable side effects during treatment (treatment intolerance)
(Moore et al., 2007). Patients diagnosed with treatment resistance experience suboptimal
response characterized by persistent psychotic symptoms, while those experiencing
intolerance cease treatment due to intolerable side effects such as TD. Clinical evidence
suggests treatment intolerant and resistant patients respond differently when treated with
CLZ: treatment intolerant patients tend to experience higher rates of response than patients
who are resistant (Claghorn et al., 1987; Owen et al., 1989; Lieberman et al., 1994). In fact,
higher baseline levels of EPS have been shown to correlate with a favourable response to
CLZ in treatment intolerant patients (Pickar et al., 1992; Lieberman et al., 1994). The
implications for combining treatment resistant and intolerant patients into a single group for
association analyses in unknown, and ideally patients should be stratified to maintain as
much homogeneity as possible.
4.3.2 Inter-Study Variability
Inter-study variability limits comparison of results across studies and also complicates the
process of combining data for meta-analysis. Meta-analyses are important because they aim
to reconcile the contribution of common variants to disease risk, when genetic associations
are unclear from individual studies (Lohmueller et al., 2003). To increase comparability of
results across studies, variables such as inclusion criteria, duration of treatment and clinical

105#
#
outcomes should be selected with consideration to readily accepted standards in the
literature.
Many definitions for TRS exist in the literature and these inconsistencies impede
interpretation of results across studies (Suzuki et al., 2011). Studies using less conservative
definitions for TRS are likely to report higher rates of response. This is because the
likelihood of response decreases after each subsequent failed AP drug trial. Herein, we
defined TRS as failure to respond to two or more adequate trials with AP drugs from at least
two different chemical classes (with doses of ≥1000mg chlorpromazine equivalents for 4-6
weeks), and no period of good functioning in the previous five years (Kane et al., 1988). This
definition of TRS is consistent with definitions existing in the literature (Canadian
Psychiatric Association, 2005; Kreyenbuhl et al., 2010; McIlwain et al., 2011; Kennedy et
al., 2014) and accurately reflects CLZ’s current prescribing policy as a 3rd line treatment
(Elkis & Meltzer, 2007; Moore et al., 2007).
The duration of treatment that constitutes an adequate trial with CLZ also varies across
studies (Rosenheck et al., 1999). High inter-individual variability in AP response has been
observed during treatment and response has been reported to occur as soon as one week
(Pickar et al., 1992; Stern et al., 1994) and as late as one year (Meltzer, 1992; Wilson, 1996)
following treatment initiation. In addition, length of CLZ treatment has been found to
correlate with treatment outcomes: approximately 30% of patients respond in the first six
weeks (Kane et al., 1988; Lieberman et al., 2008) and an additional 20% after three months
(Claghorn et al., 1987; Lindstrom, 1988). These findings suggest that studies assessing
response after only one or two months may do so prematurely for a subset of patients. Our
study assessed CLZ response after a six month trial of CLZ to ensure response rates were

106#
#
accurate and accounted for ‘late responders’. Agreement upon the length of time that
constitutes an adequate CLZ trial will help maintain consistency among studies and aid in the
interpretation of results across studies.
The last source of variability in our study stems from differences in the assessment of clinical
outcomes. Various definitions for response exist in the literature; however, most studies
assessing response use change in either the PANSS or the BPRS (reviewed by Suzuki et al.,
2012). The difference between these two scales is negligible and their scores are easily
interchangeable (Overall & Gorham, 1962; Kay et al., 1987). Herein, response was assessed
using change in BPRS scores from baseline following six months of CLZ therapy.
Categorical response was measured using responder/non-responder frequencies and CLZ
responders were classified as those individuals who experienced a ≥20% decrease in BPRS
scores, while continuous response was measured using percent change in scores controlled
for baseline (Kane et al., 1988). This assessment of clinical outcome was in line with similar
studies evaluating CLZ response from the literature (Hong et al., 2001; Chiu et al., 2003).
Despite widespread use, symptom rating scales have been criticized for their inability to
reflect a patient’s personal, social and cognitive functioning – factors that hold greater weight
in regards to how a patient will function in society. To ensure the clinical utility of response
studies, it may be worthwhile to construct a more ‘patient focused’ response assessment
method that takes factors beyond symptom change into consideration (Mortimer, 2007).
4.3.3 Statistical Power
The power of a statistical test is defined as the probability of correctly rejecting the null
hypothesis of ‘no association’ when the null hypothesis is false, or said in another way, the
ability to detect a true effect when it is present. Power is equal to one minus the probability

107#
#
of a type II error (1-β). Type II errors are otherwise known as false negatives and occur when
true effects are not detected even though they are present. The power of an association study
is influenced by a number of factors including sample size, statistical significance criteria (α)
and the magnitude of the desired effect size to be detected (Evans & Purcell, 2012). Power
tends to be highest for studies with large sample sizes, less stringent significance levels and
larger effect sizes.
A common limitation of genetic association studies is insufficient statistical power. Many
pharmacogenetics studies investigating response are underpowered due to limited sample
sizes. Patients placed on CLZ are particularly hard to come by because of the strict
guidelines surrounding its prescribing and the need for mandatory blood monitoring to
protect against the life-threatening adverse effects of CIA (Warnez & Alessi-Severini, 2014).
Averaged between the two manuscripts, our sample of 160 patients with categorical
(responder/non-responder) response data and 88 patients with continuous response data, had
over 80% power to detect an odds ratio (OR) as low as 2.05 and down to 8.5% of the
variance in the quantitative response measure (unmatched case control design: non-responder
frequency=47.8%, MAF=28.5%, α/2=0.05) (Gauderman, 2002; Gauderman & Morrison,
2006).
Similar to the phenotype of SCZ, AP response is a complex trait that follows a polygenic
mode of inheritance. However, unlike risk for SCZ, drugs administered to a patient for
treatment follow a relatively narrow pathway through the body to the brain where they bind
to a limited number of target receptors. AP drugs thereby actively provoke the phenotype of
response by enacting change in the symptoms of the patient. Therefore, drug response
phenotypes should exhibit larger gene effects than seen when studying SCZ diagnosis due to

108#
#
the a priori selection of genes based on the known pharmacologic mechanism of the drug. A
recent example demonstrating this phenomenon is the large effect size of the melanocortin
receptor 4 gene (MC4R) (RR=3.5 or greater, n=139) for predicting antipsychotic-induced
weight gain (Malhotra et al., 2012; Chowdhury et al., 2013). Therefore, our sample of
patients likely had sufficient statistical power to detect the effect sizes of genes contributing
to response.
For other genes with very small effects, the future collection of larger CLZ response samples
will help unravel their contribution to complex phenotypes, such as drug response. A larger
CLZ response sample is currently being collected by the CRESTAR consortium in Europe
with the aim of developing pharmacogenetic biomarkers for treatment response and side
effects (CRESTAR, 2011). To illustrate the effect of sample size on power: a sample size of
373 cases and 392 controls would have sufficient statistical power (80%) to detect an odds
ratio as low as 1.5 (unmatched case-control design, MAF=15%, α/2=0.05) (Gauderman,
2002; Gauderman, Morrison, 2006). Samples of this size, once available, will aid in the
discovery of novel genetic markers with the ability to predict response to CLZ.
4.3.4 Multiple Testing
Genetic studies often test multiple alleles for association with a desired phenotype and
invariably inflate the type I error rate (α), or likelihood of observing a false-positive finding.
To account for multiple comparisons, a more stringent threshold of significance is applied.
The Bonferroni method is a commonly used correction tool that divides the significance
criteria by the number of independent tests that were conducted (k) (Bonferroni, 1936). This
method is considered conservative for correlated tests such as when SNPs are in linkage
disequilibrium, as this method assumes that tests are independent. To account for these

109#
#
correlations, an alternative method was developed by Nyholt that computes a new value for
the number of independent tests that considers linkage among variants (Nyholt, 2004).
For the first data chapter of this thesis, the Nyholt method was used to compute a corrected
significance threshold of 0.008, based on 6.56 independent tests among seven GRIN2B
variants. The Nyholt method was also used for the second data chapter to compute a
corrected significance threshold of 0.005, based on 9.36 independent tests among the ten
glutamate system gene variants. If all tests across the two manuscripts were combined, a
study-wise corrected threshold of 0.003 based on 15.92 tests would be computed. With these
significance threshold adjustments in place, no polymorphism typed in our study would
reach statistical significance.
Our correction criteria considered the number of SNPs to equal to the number of tests being
performed, however, alternative methods for correction do exist. A less stringent method is a
gene-wise correction, in which k is considered to be equal to the number of SNPs present
within a single gene (≥2) (as used in Kalsi et al., 2010). To illustrate, a gene-wise correction
for the three SNPs located in SLC6A9 would result in a corrected significance threshold of
0.019 based on 2.70 tests and would have rendered the association between rs16831558 and
CLZ response statistically significant (puncorrected=0.008). Opinions differ on what level of
stringency should be used for correction and some researchers may consider this gene-wise
correction to be insufficient. On the other hand, a considerably more stringent approach is to
correct for the total number of intragenic SNPs in the human genome. This correction is
analogous to meeting statistical criteria for a genome wide association study, in which p
equal to 10-8 (Risch & Merikangas, 1996). Many believe this correction to be overly stringent
and argue that a genome wide correction would leave the majority of association studies

110#
#
underpowered (Neale & Sham, 2004; Patnala et al., 2013). A final method to consider
corrects for the total number of statistical tests that are performed and in our case, would
include all allele, genotype and haplotype analyses performed for each variant (Lewis &
Knight, 2012).
The Bonferroni and Nyholt methods of correction are considered family-wise error rate
(FWER) procedures that seek to reduce the probability of observing even one false
discovery. FWER correction methods are disadvantageous for studies in which many tests
are performed because the significance threshold becomes increasingly small and often result
in insufficient statistical power to detect true effects (Perneger, 1998). Because the
consequences of false discoveries in complex traits like AP response are less severe than for
single gene disorders (van den Oord & Sullivan, 2003), less conservative methods of
correction may be applied with caution. A more recently developed less conservative method
is the false discovery rate (FDR) approach. Like the Bonferroni and Nyholt methods, FDR
also determines adjusted p-values for each test (q-values), however, does so by controlling
for the number of false positive discoveries in tests for which the null hypothesis was
rejected as opposed to taking all tests into consideration. This method results in greater
statistical power to find true effects while controlling for the type 1 error rate (Benjamini &
Hochberg, 1995).
4.3.5 Deviation from Hardy-Weinberg Equilibrium
Hardy-Weinberg equilibrium is a quality-control measure used to ensure observed
genotyping frequencies in a study population coincide with those expected under equilibrium
conditions where alleles segregate randomly (Hardy, 1908; Weinberg, 1908). HWE is
calculated using χ2-test where observed genotype frequencies in the study population are

111#
#
compared to those expected under HWE (Weir, 1996). One variant in our sample, GRIN2B
rs7301328, significantly deviated from HWE. HWE threshold values were set at the
multiple-testing adjusted significance levels calculated for each study using the Nyholt
method. This SNP was removed from subsequent analyses because chi-square tests require
SNPs to be in HWE (Sasieni, 1997).
Several reasons may have accounted for why this variant deviated from HWE such as:
genotyping errors that mistype heterozygotes as homozygotes or vice versa (Gomes et al.,
1999; Hosking et al., 2004), stochastic variation, or due to the underlying biological
characteristics of the study population (Nielsen et al., 1998). Deviations may also be a
symptom of disease association (Balding, 2006).
4.3.6 Incomplete Outcome Data
Incomplete outcome data is generated when a patient discontinues treatment and is usually as
result of noncompliance, lack of efficacy, the occurrence of adverse effects, or because the
patient was lost to follow-up. Patients who leave a study prematurely contribute to inaccurate
estimations of response rates in response studies and confound the ability of researchers to
identify genetic markers that may have predictive potential. Noncompliance is particularly
common among patients with SCZ and has been linked to relapse, re-hospitalization and
poor outcome (Centorrino et al., 2001). Paranoia, cognitive dysfunction and bad judgment
are potential reasons to explain why patients may not adhere to their medication regimens
(Lacro et al., 2002). Fortunately, routine blood monitoring for patients taking CLZ allows
treating physicians to both ascertain compliance and ensure CLZ serum levels have reached
optimal threshold that is necessary to achieve response (≥450 ng/mL) (Lindenmayer &
Apergi, 1996).

112#
#
Discontinuation due to lack of efficacy and adverse reactions also similarly effect response
rates. Lack of efficacy to a medication may indicate that drug levels are too low, while
toxicity and adverse effects suggest drug levels are too high, and in both cases a dose
adjustment may be necessary. As would be expected for a phenotype that has high inter-
individual variability, optimal doses for achieving maximum therapeutic effect vary greatly
among patients. Doses within the therapeutic window for the majority of patients may be
inadequate or excessive for a subset of patients and may lead to undesirable treatment
outcomes (Ma & Lu, 2011). The high rates of discontinuation in AP drug trails stresses the
importance of understanding this inter-individual variability in drug response phenotypes and
for identifying predictive markers that may have clinical utility.
4.4 FUTURE DIRECTIONS
This section explores future research initiatives assuming time and resources were unlimited.
The first initiative would characterize the potential functionality of the glycine transporter 1
gene variant rs16831558 using a luciferase promoter assay. The second initiative would
investigate the role of gene-gene and gene-environment interactions in drug response
phenotypes. Lastly, the third initiative would investigate the contribution of novel rare
variants in drug response phenotypes using next generation sequencing (NGS) platforms.
This section concludes with a look at the emerging field of personalized medicine in
psychiatry.
4.4.1 Characterization of Glycine Transporter 1 Promoter Variant
The GlyT1 promoter variant rs16831558 emerged as the highest priority SNP from our
study. As mentioned previously, CC-homozygotes exhibited a notable improvement in their

113#
#
positive symptom scores compared to T allele carriers following six months of CLZ therapy
(puncorrected=0.008, pcorrected=0.08, assuming 9.36 independent tests). This novel variant is
located approximately 5kb upstream of the SLC6A9 gene and is proposed to be functional
according to data generated by ENCODE (ENCODE Project Consortium, 2012).
Four functional characteristics have been cited for this variant: 1) rs16831558 is localized to
a region on chromosome 1 where GATA-binding factor 1 (GATA1) binds to the DNA (Hu et
al., 2011). GATA1, otherwise known as erythroid transcription factor, has been shown to
play a role in cell growth and cancer (Ferreira et al., 2005); 2) rs16831558 is localized to a T-
box element DNA-motif where T-box transcription factor 18 (TBX18) binds to the DNA
(Matys et al., 2006; Pique-Regi et al., 2011). TBX18 has been shown to interact with
GATA4 and other transcription factors in gene promoter regions to modulate transcriptional
activity (Farin et al., 2007); 3) rs16831558 is found within a DNAse hypersensitivity site in
13 different cell lines from ENCODE, including a medulloblastoma neuron precursor cell
line (Medullo_D341) and cerebellum tissue from Caucasian donors (Cerebellum_OC)
(ENCODE Project Consortium, 2012). DNAse-sequencing methods map DNAse
hypersensitivity sites to locate regions of open chromatin where gene regulatory elements
such as promoters, enhancers and silencers are likely to reside (Song & Crawford, 2010); and
lastly, 4) rs16831558 is localized to a region of DNA designated as ‘enhancer’ in tissues
from various brain regions (Bernstein et al., 2010; Ernst & Kellis, 2012). Collectively, this
evidence provides sufficient rationale to investigate rs18631558’s potential to regulate
transcription using, for example, a luciferase promoter assay.
Luciferase promoter assays allow researchers to study changes in mammalian gene
expression in vitro by measuring the strength of a luminescent signal that is created when a

114#
#
reporter gene that encodes the luciferase protein is transcribed (Fan & Wood, 2007).
Conducting a luciferase promoter assay for rs16831558 would first involve designing two
promoter DNA fragments: one containing the C-allele and the other containing the T-allele.
These fragments would then be cloned into two different luciferase reporter vectors upstream
from the luciferase complementary DNA (cDNA), which has had its promoter elements
removed. These vectors would then be transfected into a neuronal cell-line to assess the
differential ability of each promoter variant to drive expression of the luciferase protein.
Expression levels are then measured by comparing the luminescent signal generated by each
rs16831558 promoter construct to basal levels of luminescent intensity generated by
promoter-less vector controls (Solberg & Krauss, 2013).
Certain aspects of the study methodology must be considered before designing a luciferase
promoter assay for rs16831558. For instance, the cell-line chosen for the in vitro experiment
is of particular importance: selecting a cell-line that mimics or is closest to the tissue of
interest will yield results that more accurately represent that variant’s function in vivo.
Neuroblastoma (NB) cell lines derived from neural crest cells are often used in neurobiology
studies (Shastry et al., 2001). Because luciferase promoter assays are performed in vitro, they
do not account for certain cellular and environmental factors that are present in vivo.
Furthermore, sites relatively far away from the gene of interest may in reality have the ability
to effect gene expression. This is possible due to looping and folding of the longer stretches
of DNA in its native state, which is a 3D chromatin structure. Therefore, the results obtained
from in vitro studies may only partially reflect the actual ability of the cloned promoter DNA
construct to regulate gene expression in vivo. Consequently, additional studies to further

115#
#
characterize this GlyT1 variant would be necessary. If this variant is found to be functional,
further characterization in transgenic mice, for instance, may be indicated.
4.4.2 Gene-Environment Interactions
Gene-environment interactions occur when the effect of an individual’s genotype on a
particular phenotypic outcome depends on the level of exposure to an environmental factor
(Clayton & McKeigue, 2001). The contribution of gene-environment interactions to risk
phenotypes is often overlooked for reasons such as lack of environmental data or bias in
estimating the effect of exposure (Arranz & Munro, 2011). Gene-environment interactions
also involve additional tests that invariably inflate the type I error rate (Peduzzi et al., 1995;
Peduzzi et al., 1996). Despite these limitations, the combined effect of genetic and
environmental factors such as cigarette smoking, caffeine consumption and concomitant
medication use have been shown to contribute to variation in drug response phenotypes (de
Leon, 2004; Caspi & Moffitt, 2006; Arranz & Munro, 2011). Therefore, future work
characterizing gene-environment interactions in CLZ response may be useful for
personalizing CLZ treatment and developing targeted interventions for patients at high risk
of non-response and adverse side effects.
The first gene-environment interaction worth exploring in CLZ response investigates the
joint effect of cigarette smoking and variation in the GRIN2B gene. The rationale for
investigating this interaction is as follows: 1) genetic variation in GRIN2B is associated with
early onset cigarette smoking and smoking initiation (Vink et al., 2009; Grucza et al., 2010);
2) nicotine administration in rats increases GRIN2B expression in particular brain regions
(Wang et al., 2007); and 3) by-products that are generated from smoking tobacco induce
CLZ metabolism and result in lower CLZ serum levels in patients who smoke versus those

116#
#
who do not (Seppala et al., 1999; Meyer, 2001; Palego et al., 2002). Patients who carry a
particular GRIN2B genotype that predisposes them to smoking behaviours may be more
likely to experience sub-optimal CLZ response due to sub-therapeutic serum levels and/or
alteration of NMDAR subunit expression. Identification of a gene-environment interaction
between cigarette smoking and high-risk genotypes in GRIN2B may have clinical
applications. Should an interaction be identified, for instance, clinicians may recommend that
patients with risk genotypes reduce their cigarette smoking behaviours, or clinicians may
prescribe higher CLZ dosages to patients who smoke.
Caffeine consumption may also interact with glutamate system genes and alter response to
CLZ. The following observations support investigating this interaction: 1) caffeine
consumption enhances glutamate release and excitatory neurotransmission in particular brain
regions by binding to adenosine receptors on glutamatergic neurons (Dunwiddie, 1980;
Dunwiddie et al., 1981; Solinas et al., 2002; Salmi et al., 2005); and 2) during acute
administration, caffeine has been shown to inhibit clozapine metabolism and lead to higher
CLZ serum levels in patients who consume caffeine versus patients who do not (Hagg et al.,
2000; de Leon, 2004). Therefore, higher serum CLZ levels in patients who drink coffee, in
combination with caffeine’s ability to mediate glutamate signaling may have multiplicative
effects on drug response and may increase the risk of developing adverse side effects. Akin
to the first interaction, understanding the relationship between caffeine intake and CLZ
response may have clinical applications. Following Hagg et al., clinicians may initially
prescribe lower CLZ dosages to patients who consume high amounts of caffeinated
beverages, or recommend that patients with risk genotypes either limit caffeine consumption

117#
#
or make attempts to maintain consistency in the amount of caffeine consumed per day (Hagg
et al., 2000).
4.4.3 Gene-Gene Interactions
Gene-gene interaction, or epistasis, is the dynamic interaction among different genetic
variants that occurs when one allele modifies the effect of alleles at other loci, or when two
or more loci interact to create a new phenotype (Phillips, 2008). Epistatic interactions are
thought to account for a portion of the inter-individual variability that is observed in
polygenic traits, such as AP response (Long & Langley, 1999; Sadee, 2012; Zuk et al.,
2012). Treatment outcomes likely depend on the multiplicative effect of several genes that
are involved in drug action, such as enzymes involved in metabolism, membrane
transporters, receptors and target proteins (Sadee, 2013). However, compared to the large
number of genes involved in disease phenotypes such as SCZ, CLZ response likely depends
on a lesser number of genes to determine treatment outcomes as evidenced by
pharmacogenetics studies (Johnson et al., 2011; Ma & Lu, 2011). Therefore, effective
biomarker panels requiring only a limited number of genes may be able to capture much of
the genetic variation contributing to drug response (Sadee, 2013). The clinical utility of such
predictive tests provides sufficient rationale for the investigation of three potential epistatic
interactions involving glutamate genes with response to CLZ.
The first gene-gene interaction worth exploring in CLZ response involves two genes that
were involved in this study: the NMDAR NR2B subunit gene GRIN2B, and the GlyT1 gene
SLC6A9. Three observations provide support for investigating this gene-gene interaction: 1)
Differential expression of either NR2B and GlyT1 are capable of altering NMDAR
activation (Flint et al., 1997); 2) NR2B and GlyT1 co-localize to glutamatergic synapses and

118#
#
may physically interact (Cubelos et al., 2005); and 3) NMDAR activation is thought to
account for a portion of CLZ’s therapeutic effects (Banerjee et al., 1995; Arvanov et al.,
1997; Kubota et al., 2000; Ninan et al., 2003; Kargieman et al., 2007). Collectively, these
findings suggest that differential expression of NR2B and GlyT1 proteins by potentially
functional genetic variants may interact to augment NMDAR neurotransmission, and
subsequently, response to CLZ. A proposed gene-gene interaction analysis would investigate
the putatively functional SLC6A9 variant rs16831558 that was genotyped in this study, and
the GRIN2B variant rs1805502 that was previously associated with negative symptom
change during CLZ therapy (Martucci & Kennedy, 2010).
An epistatic interaction may also exist for the mGluR2 gene GRM2 that was investigated in
this study, and the 5-HT2A gene, HTR2A. The rationale for investigating this interaction is as
follows: 1) In particular brain regions, translation of the 5-HT2A receptor is necessary for
normal mGluR2 expression (Gonzalez-Maeso et al., 2008); 2) mGluR2 has been shown to
form a special hetero-complex with 5-HT2A through specific transmembrane helix domains
(Gonzalez-Maeso et al., 2008); 3) CLZ binds to the 5-HT2A component of this complex and
up-modulates mGluR2-elicited signaling by 40% (Fribourg et al., 2011); and 4) The 5-
HT2A-dependent antipsychotic-like behavioural effects of CLZ require the expression of
mGluR2 (Fribourg et al., 2011). Taken together, these findings suggest that CLZ may act on
the mGluR2-5-HT2A complex to achieve symptom change in SCZ patients and that altered
expression of both HTR2A and GRM2 may have an additive affect on response. Of high
priority is the functional HTR2A variant rs6314 previously associated with CLZ response
(Nothen et al., 1995; Masellis et al., 1995; Burnet & Harrison, 1995; Arranz et al., 1995) and
the potentially functional GRM2 SNP rs2518461 identified in this study.

119#
#
The final interaction worth considering for future work involves GRIN2B and the 5-HT1A
gene, HTR1A. Support for this interaction is as follows: 1) CLZ is a partial agonist at the 5-
HT1A receptor (Newman-Tancredi et al., 1996); 2) CLZ facilitates formation of a synergistic
triad between 5-HT1A, the NMDAR, and CaMKII (Purkayastha et al., 2012); and 3) This
triad is necessary for CLZ-mediated increases in EPSPs in PFC pyramidal neurons
(Purkayastha et al., 2012). Genetic variation in both HTR1A and GRIN2B may have the
potential to alter triad formation and, as a result, alter CLZ’s ability to elicit excitability in
certain brain regions. The functional HTR1A promoter variant rs6295 previously implicated
in depression and antidepressant response (Lemonde et al., 2003; Lemonde et al., 2004;
Parsey et al., 2006) is of high interest and deserves investigation with GRIN2B SNPs of
interest.
There are a number of challenges associated with analysing gene-gene and gene-environment
interactions in drug response phenotypes. One challenge of modelling such relationships is
the increasing dimensionality that results from a large number of possible interactions
(Moore, 2004; Motsinger et al., 2007). As a result, the application of traditional parametric
tests to analyse interaction data has limited use and often increases the risk of type I and type
II errors, while at the same time decreasing power (Moore, 2004). For these reasons, more
advanced statistical approaches have been developed such as the multifactor dimensionality
reduction (MDR) method (Ritchie et al., 2001). MDR reduces dimensionality by dividing
high-risk and low-risk interactions into two separate groups for analysis, and then selects the
model(s) best able to accurately predict response/non-response status in independent testing
sets generated through cross-validation. Models with the highest cross-validation consistency
are used in permutation testing to identify models with statistically significant results not

120#
#
expected by chance alone. Often the most challenging aspect of interaction analysis involves
interpreting the biological plausibility of the results, and follow-up studies using cell-culture
or other in vitro experiments can be useful aids to interpretation (Ritchie & Motsinger, 2005).
4.4.4 Next Generation Sequencing
Next generation sequencing (NGS) is a high-throughput DNA sequencing technology that
has revolutionized the way scientists extract genomic data from biological systems. NGS
utilizes a similar concept to Sanger sequencing whereby bases in a small fragment of DNA
are sequentially identified from fluorescent signals emitted as each fragment is resynthesized
(Sanger et al., 1977). However, NGS carries out this process on a grander scale by
sequencing millions of DNA fragments at once and results in a quicker, less expensive
method for sequencing whole genomes (Shendure & Ji, 2008).
One particular application of NGS that is of interest to disease risk and medication response
is targeted sequencing, wherein a subset of genes is sequenced, as opposed to the whole
genome. Targeted sequencing allows researchers to achieve high sequence coverage, obtain
sequencing results for many individuals, and makes it possible to identify rare variants that
were missed using previous methods. Rare, in this case, refers to variants that occur less than
once per 100 individuals. Rare variants have given rise to what is known as the ‘common
disease-rare variant’ hypothesis, which argues that multiple different rare mutations in
overlapping genomic locations and/or biochemical pathways drive susceptibility to risk
phenotypes, and are likely to contribute to a portion of the missing heritability in complex
traits (Schork et al., 2009; Altmann et al., 2013).

121#
#
NGS technologies have already identified excess rare variants and deletions in glutamate-
related synaptic genes in SCZ and autism (Kelleher et al., 2012; Myles-Worsley et al., 2013;
Kenny et al., 2014). Regarding drug treatment, NGS has mostly been used in cancer research
to identify rare and common variants in response to chemotherapeutic agents (Ross &
Cronin, 2011; Tran et al., 2013; Ong et al., 2014). A current review of the literature does not
reveal any major discoveries using NGS technologies to identify genetic predictors of
response to AP therapy. However, the practical applications of such discoveries cannot be
understated. Discovery of rare variants and predictive markers would allow clinicians to
distinguish between responders and non-responders and identify patients who are at high risk
of failing treatment. The information derived from NGS may also be used to guide
development of novel drugs that target aberrant gene systems in the non-responder group.
Therefore, future research involving targeted sequencing of glutamate system genes is
recommended to help uncover rare variants involved in CLZ response.
To briefly illustrate the methodology behind NGS, a hypothetical workflow for sequencing a
gene of interest using the Ion Torrent Personal Genome Machine (PGM) Sequencer (Life
Technologies) in our CAMH laboratory is proposed. The four steps are as follows: 1) Library
preparation: The first step is to create a library of DNA fragments that are flanked by Ion
Torrent adapters. Adapters can be added to DNA fragments by one of two ways – ligating
the adapter to the PCR product, or by incorporating the adapter sequence automatically
during PCR using specially designed primers; 2) Template preparation: The second step
involves coating special ‘Ion Sphere’ particles with DNA fragments via their adapter
sequences, followed by clonal amplification of the library fragments by emulsion PCR
(emPCR); 3) Sequencing: The Ion Sphere particles coated with library fragments are then

122#
#
deposited onto the sequencing chip, placed into the Ion Torrent machine, and the sequencing
run is begun, 4) Data Analysis: The final step of the work flow analyses sequencing data.
Data is transferred to the appropriate servers, and then run through signal processing and
base-calling algorithms to re-construct DNA sequences. Sequencing data is then imported
into analysis programs such as the Partek® Genomics SuiteTM. One additional point to
consider during study design is the depth of coverage. Depth refers to the number of times
the region of interest is re-sequenced. Rare variants and somatic mutations that are present at
low frequencies require higher depths of coverage to ensure the mutation calls are made with
sufficient statistical confidence (Life Technologies, 2011).
Challenges associated with NGS technology must also be mentioned. Because this
technology produces vast amounts of data in a relatively short period of time, future research
initiatives will need to focus on how to organize and interpret the sheer volume of genomic
data that is produced. In addition, the ability to detect rare variants depends on the
availability of large sample sizes and stresses the need for a collaborative effort in the
research community. Ideally, a common database will be developed where researchers
investigating AP response phenotypes can upload their DNA samples and/or sequencing
results, with the shared goal of advancing medical research within this field. Ethical issues
also abound and stress the need for legislation to protect citizens from discrimination based
on their genome (Mathieu et al., 2013). Several nations have passed legislation barring
insurance companies and employers from discriminating against citizens based on their DNA
sequence. The United States of America passed the Genetic Information Nondiscrimination
Act of 2008 (GINA) (United States of America, 2008), while the United Kingdom passed the
Equality Act of 2010 (United Kingdom of Great Britain, 2010). Canada passed Bill C-445 in

123#
#
2011, which amended the Canadian Human Rights Act to include protection of Canadian
citizens from discrimination based on their ‘genetic characteristics’ (Parliament of Canada,
2011). Shortly afterwards, Bill S-201 was passed by Canadian legislature, otherwise known
as the Genetic Nondiscrimination Act (Parliament of Canada, 2013), to provide Canadian
citizens with further protection against discrimination based on their DNA. Despite these
limitations, NGS has already revolutionized the field of genetics research and is predicted to
lead to many groundbreaking discoveries in the future.
4.4.5 Personalized Medicine in Psychiatry
Personalized medicine in psychiatry is a relatively new field with the goal of tailoring drug
treatment to suit an individual’s personal genetic makeup. The realization that inter-
individual variability in drug response had a genetic underpinning was a pivotal discovery on
the path to personalized medicine (Garrod, 1909; Beutler et al., 1955; Evans et al., 1960).
Furthermore, launch of the Human Genome Project (Lander et al., 2001) and the
development of more cost-effective genome sequencing methods have made genetic
information more easily accessible to the general population. New studies in
pharmacogenetics that investigate how drugs and genetics interact are also of high
importance for the developing field of personalized medicine.
The field of personalized medicine in psychiatry is evolving and seeing the development of
genetic panels and algorithms used as aids in medication prescribing. A number of
pharmacogenetic tests are available on the market and have been reviewed elsewhere (Arranz
& Munro, 2011). One genetic test worth mentioning is the GeneSight® test developed by
AssureRX Health Inc. This test incorporates genotype data for four different CYP enzymes,
as well as the serotonin transporter 1 (SLC6A4) and HTR2A genes. Studies investigating the

124#
#
utility of the test have reported improved outcomes should the test be used in routine clinical
care (Hall-Flavin et al., 2013).
Unfortunately, a scientifically validated test for CLZ response has not yet emerged.
Approximately 15 years ago, researchers Kerwin and Arranz along with their colleagues
published findings on a genetic panel of six polymorphisms that were capable of predicting
CLZ response with a 76.6% success rate in their sample of 200 Caucasian SCZ patients
(p=0.001) (Arranz et al., 2000). The six variants identified were located in neurotransmitter-
receptor related genes including two HTR2A variants (102T/C and His452Tyr), two serotonin
2C receptor gene (HTR2C) variants (-330-GT/-244-CT and Cys23Ser), the serotonin
transporter promoter (5-HTTLPR), and the histamine H2 receptor gene (HRH2) variant (-
1018-G/A). These six polymorphisms, when combined, had a retrospective sensitivity of
95% for identifying patients with ‘satisfactory response’. This report was the first of its kind
attempting to predict CLZ response using genetic markers. Unfortunately, other authors were
unable to replicate these findings (Arranz et al., 2000; Schumacher et al., 2000). A
commercialized test was developed based on Kerwin and Arranz’s findings by a UK
company known as LGC (formerly known as the Laboratory of the Government Chemist that
was privatized in 1996). Unfortunately, no prospective studies assessing the clinical and
economic implications of this test have been conducted.
Despite the lack of success in developing a predictive test for CLZ response, advancements
in other areas of pharmacogenetics such as the ability to predict drug side effects, offer hope
for the future. Relatively recently, our colleagues at CAMH in Toronto developed a gene
panel consisting of 5 gene markers that is able to explain 27% of the variance associated with
developing antipsychotic induced weight gain (AIWG) (Tiwari et al, unpublished). AIWG is

125#
#
a debilitating side effect of second-generation antipsychotics such as CLZ and OLZ that
frequently results in obesity, metabolic syndrome and premature morbidity. The ability to
predict the likelihood of developing such negative side effects will allow physicians to make
more informed decisions regarding medication choice.
The benefits of personalized medicine to psychiatric care cannot be understated: outcomes
from personalized treatment may allow physicians the ability to define diseases more
precisely and to distinguish subtypes/groups of SCZ patients that are based on a drug’s
limited group specificity. In addition, the increased availability of genome sequencing data
may greater our understanding of the underlying pathophysiological basis of SCZ and lead to
the development of novel treatments that are more effective for treating specific disease
subtypes (reviewed by Insel & Scolnick, 2006). For instance, research has suggested that
patients suffering from TRS may present with a different underlying etiology than patients
who respond to conventional dopamine-blocking antipsychotic medications (Egerton et al.,
2012; de la Fuente-Sandoval et al., 2013; Demjaha et al., 2014). Demarcation between
individuals likely to respond and those who are not at first episode would enable physicians
to select a drug, once developed, that is more specific to this subgroup of patients.
There are several issues surrounding the use of genetic testing in personalized medicine that
must be mentioned. For instance, drug response likely results from a complex interaction
between genes, brain and environmental factors such as a patient’s lifestyle, age, gender,
diet, ethnicity and amount of social support. Therefore, it is imperative that researchers
investigating pharmacogenetic processes sufficiently acknowledge the uncertainties
pertaining to their research and report their findings with caution (Evers, 2009). As well, the
contribution of epigenetic interactions must also be considered regarding nonresponse to

126#
#
medications. Additional concerns include increased drug costs associated with targeted drug
development and an increased risk of pharmacological exclusion due to ethnicity or under-
represented genetic groups. As with all novel areas of research, appropriate infrastructure to
handle challenges associated with using pharmacogenomic drugs in psychiatric care will also
need to be implemented. Additional aspects that will need to be addressed are reliability of
genotyping, equal access to testing, as well as clinician and patient education (Costa e Silva,
2013).
Given the advancements in genetic technology and the interest in developing genetic panels
capable of response prediction, the future of personalized medicine in psychiatry looks
promising. As with all new areas of research, educating physicians and patients, developing
the appropriate infrastructure and anticipating ethical issues will need to be considered as the
field of personalized medicine continues to evolve. To highlight the prospective benefits,
personalized medicine in psychiatry has the potential to guide clinician choice regarding
therapeutic dose, decrease the occurrence of adverse drug reactions, and improve overall
disease prognosis. Results from pharmacogenetics studies and personalized treatment may
also guide development of novel treatments and identify more homogeneous disease
subclasses based on the propensity of certain individuals to respond to select medications.
4.5 CONCLUDING REMARKS
Glutamate signaling pathways are emerging as top candidates in SCZ risk and as targets for
novel therapies for the treatment of SCZ. As the field of psychiatric genetics continues to
expand with the advent of more cost effective methods for analysing genomic data,
personalized medicine in psychiatry is becoming more of a reality.

127#
#
Herein, we investigated the association of 17 glutamate system gene variants and clinical
response to CLZ. We reported no significant associations after correction for multiple
testing. Prior to correction, several nominally significant associations were observed. The
most noteworthy preliminary finding was for the glycine transporter 1 gene variant
rs16831558: patients carrying the C-allele exhibited a marked improvement in positive
symptoms compared to T-allele carriers following six months of CLZ therapy. Our findings
provide further credence to the ‘glutamate hypofunction hypothesis’ of SCZ and support
future research initiatives to investigate the potential functionality of rs16831558 and further
explore this variant in larger CLZ response samples.

Taylor DL V. REFERENCES !
128!!
REFERENCES
Abdul*Monim!Z,!Reynolds!GP!&!Neill!JC.!(2006).!The!effect!of!atypical!and!classical!antipsychotics!on!sub*chronic! PCP*induced! cognitive!deficits! in! a! reversal*learning!paradigm.! Behavioural! Brain!Research,!169(2):!263*273.!!
Abrams! DJ,! Rojas! DC! &! Arciniegas! DB.! (2008).! Is! schizoaffective! disorder! a! distinct! categorical!diagnosis?! A! critical! review! of! the! literature.! Neuropsychiatric! Disease! and! Treatment,! 4(6):!1089*1109.!!
Adams!DH,!Kinon!BJ,!Baygani!S,!Millen!BA,!Velona!I,!Kollack*Walker!S!&!Walling!DP.!(2013).!A!long*term,! phase! 2,! multicenter,! randomized,! open*label,! comparative! safety! study! of!pomaglumetad!methionil! (LY2140023!monohydrate)!versus!atypical!antipsychotic!standard!of!care!in!patients!with!schizophrenia.!BMC!Psychiatry,!13:!143*151.!!
Addington!AM,!Gornick!M,!Duckworth! J,!Sporn!A,!Gogtay!N,!Bobb!A,! .! .! .!Straub!RE.! (2005).!GAD1!(2q31.1),!which! encodes! glutamic! acid! decarboxylase! (GAD67),! is! associated!with! childhood*onset!schizophrenia!and!cortical!gray!matter!volume!loss.!Molecular!Psychiatry,!10(6):!581*588.!!
Aghajanian!GK!&!Marek!GJ.! (2000).!Serotonin!model!of!schizophrenia:!Emerging!role!of!glutamate!mechanisms.!Brain!Research!Reviews,!31(2*3):!302*312.!!
Aleman!A,!Kahn!RS!&!Selten!JP.!(2003).!Sex!differences!in!the!risk!of!schizophrenia:!Evidence!from!meta*analysis.!Archives!of!General!Psychiatry,!60(6):!565*571.!!
Alonso!P,!Gratacos!M,!Segalas!C,!Escaramis!G,!Real!E,!Bayes!M,!.!.!.!Menchon!JM.!(2012).!Association!between! the! NMDA! glutamate! receptor! GRIN2B! gene! and! obsessive*compulsive! disorder.!Journal!of!Psychiatry!&!Neuroscience,!37(4):!273*281.!
Altar!CA,!Wasley!AM,!Neale!RF!&!Stone!GA.!(1986).!Typical!and!atypical!antipsychotic!occupancy!of!D2!and!S2!receptors:!An!autoradiographic!analysis! in!rat!brain.!Brain!Research!Bulletin,!16(4):!517*525.!!
Altar! CA,! Carhart! JM,!Allen! JD,!Hall*Flavin!DK,!Dechairo!BM!&!Winner! JG.! (2015).! Clinical! validity:!Combinatorial! pharmacogenomics! predicts! antidepressant! responses! and! healthcare!utilizations! better! than! single! gene! phenotypes.! The! Pharmacogenomics! Journal,! advanced!online!publication.!!
Altmann!A,!Quast!C!&!Weber!P.!(2013).!Detecting!rare!variants!for!psychiatric!disorders!using!next!generation!sequencing:!A!methods!primer.!Current!Psychiatry!Reports,!15(1):!333*141.!!
American! Psychiatric! Association.! (1987).! Diagnostic! and! statistical! manual! of! mental! disorders!(DSMPIIIPR)!(3rd!ed).!Washington!DC:!American!Psychiatric!Association!(revised).!!
American! Psychiatric! Association.! (1994).! Diagnostic! and! statistical! manual! of! mental! disorders!(DSMPV)!(4th!ed).!Washington!DC:!American!Psychiatric!Association.!!

129!!
!!
American! Psychiatric! Association.! (2013).! Diagnostic! and! statistical! manual! of! mental! disorders!(DSMP5)!(5th!ed).!Arlington,!VA:!American!Psychiatric!Publishing.!!
Andreasen! NC,! Rezai! K,! Alliger! R,! Swayze! VW,! Flaum! M,! Kirchner! P,! .! .! .! O'Leary! DS.! (1992).!Hypofrontality! in! neuroleptic*naive! patients! and! in! patients! with! chronic! schizophrenia.!Assessment!with!xenon!133!single*photon!emission!computed!tomography!and!the!Tower!of!London.!Archives!of!General!Psychiatry,!49(12):!943*958.!!
Andreasen!NC,! Arndt! S,! Swayze! V,! Cizadlo! T,! Flaum!M,!O'Leary! D,! .! .! .! Yuh!WT.! (1994).! Thalamic!abnormalities! in! schizophrenia! visualized! through! magnetic! resonance! image! averaging.!Science,!266(5183):!294*298.!!
Angst! J,! Jaenicke!U,!Padrutt!A!&!Scharfetter!C.! (1971).!Ergebnisse!eines!doppelblindversuches!von!HF! 1854! (8*chlor*11*(4*methyl*1*piperazinyl)*5H*dibenzo! (b,e)! (1,4)diazepin)! im! vergleich! zu!levomepromazin.!Pharrnackopsychiatrie,!4:!192*200.!!
Anson! LC,! Chen! PE,! Wyllie! DJ,! Colquhoun! D! &! Schoepfer! R.! (1998).! Identification! of! amino! acid!residues!of!the!NR2A!subunit!that!control!glutamate!potency!in!recombinant!NR1/NR2A!NMDA!receptors.!The!Journal!of!Neuroscience,!18(2):!581*589.!!
Arango! C,! Breier! A,! McMahon! R,! Carpenter! WT! Jr! &! Buchanan! RW.! (2003).! The! relationship! of!clozapine!and!haloperidol! treatment! response! to!prefrontal,!hippocampal,!and!caudate!brain!volumes.!The!American!Journal!of!Psychiatry,!160(8):!1421*1427.!!
Arranz!M,!Collier!D,!Sodhi!M,!Ball!D,!Roberts!G,!Price!J,! .! .! .!Kerwin!R.!(1995).!Association!between!clozapine!response!and!allelic!variation!in!5*HT2A!receptor!gene.!Lancet,!346(8970):!281*282.!!
Arranz! MJ,! Munro! J,! Birkett! J,! Bolonna! A,! Mancama! D,! Sodhi! M,! .! .! .! Kerwin! RW.! (2000).!Pharmacogenetic!prediction!of!clozapine!response.!Lancet,!355(9215):!1615*1616.!!
Arranz!MJ,!Munro!J,!Osborne!S,!Collier!D!&!Kerwin!RW.!(2000).!Difficulties!in!replication!of!results.!Lancet,!356(9238):!1359*1360.!!
Arranz! MJ! &! Munro! JC.! (2011).! Toward! understanding! genetic! risk! for! differential! antipsychotic!response!in! individuals!with!schizophrenia.!Expert!Review!of!Clinical!Pharmacology,!4(3):!389*405.!!
Arvanov! VL,! Liang! X,! Schwartz! J,! Grossman! S! &! Wang! RY.! (1997).! Clozapine! and! haloperidol!modulate! N*methyl*D*aspartate*! and! non*N*methyl*D*aspartate! receptor*mediated!neurotransmission!in!rat!prefrontal!cortical!neurons!in!vitro.!The!Journal!of!Pharmacology!and!Experimental!Therapeutics,!283(1):!226*234.!!
Asada!H,!Kawamura!Y,!Maruyama!K,!Kume!H,!Ding!RG,!Kanbara!N,!.!.!.!Obata!K.!(1997).!Cleft!palate!and!decreased!brain!gamma*aminobutyric!acid!in!mice!lacking!the!67*kDa!isoform!of!glutamic!acid!decarboxylase.! Proceedings!of! the!National!Academy!of! Sciences!of! the!United! States!of!America,!94(12):!6496*6499.!!

130!!
!!
Asztely! F! &! Gustafsson! B.! (1996).! Ionotropic! glutamate! receptors.! Their! possible! role! in! the!expression!of!hippocampal!synaptic!plasticity.!Molecular!Neurobiology,!12(1):!1*11.!!
Ayalew! M,! Le*Niculescu! H,! Levey! DF,! Jain! N,! Changala! B,! Patel! SD,! .! .! .! Niculescu! AB.! (2012).!Convergent! functional! genomics! of! schizophrenia:! From! comprehensive! understanding! to!genetic!risk!prediction.!Molecular!Psychiatry,!17(9):!887*905.!!
Bakshi!VP,!Swerdlow!NR!&!Geyer!MA.!(1994).!Clozapine!antagonizes!phencyclidine*induced!deficits!in!sensorimotor!gating!of!the!startle!response.!The!Journal!of!Pharmacology!and!Experimental!Therapeutics,!271(2):!787*794.!!
Balding! DJ.! (2006).! A! tutorial! on! statistical! methods! for! population! association! studies.! Nature!Reviews,!Genetics,!7(10):!781*791.!!
Banerjee!SP,!Zuck!LG,!Yablonsky*Alter!E!&!Lidsky!TI.!(1995).!Glutamate!agonist!activity:!Implications!for!antipsychotic!drug!action!and!schizophrenia.!Neuroreport,!6(18):!2500*2504.!!
Barbato! A.! (1998).! Schizophrenia! and! public! health,! Chapter! 4:! Consequences! of! schizophrenia.!Geneva:!World!Heath!Organization.!!
Bar*Peled! O,! Ben*Hur! H,! Biegon! A,! Groner! Y,! Dewhurst! S,! Furuta! A! &! Rothstein! JD.! (1997).!Distribution! of! glutamate! transporter! subtypes! during! human! brain! development.! Journal! of!Neurochemistry,!69(6):!2571*2580.!!
Barrett! JC,! Fry! B,! Maller! J! &! Daly! MJ.! (2005).! Haploview:! Analysis! and! visualization! of! LD! and!haplotype!maps.!Bioinformatics,!21(2):!263*265.!!
Benjamini! Y! &! Hochberg! Y.! (1995).! Controlling! the! false! discovery! rate:! A! practical! and! powerful!approach! to! multiple! testing.! Journal! of! the! Royal! Statistical! Society! Series! B,! Statistical!Methodology,!57B(1):!289*300.!!
Benson! DA,! Cavanaugh! M,! Clark! K,! Karsch*Mizrachi! I,! Lipman! DJ,! Ostell! J! &! Sayers! EW.! (2013).!GenBank.!Nucleic!Acids!Research,!Database!Issue,!41:!D36*D42.!!
Bente!D,!Engelmeier!M,!Heinrich!K,!Schmitt!W!&!Hippius!H.!(1966).!Klinische!untersungen!mit!einem!neuroleptisch!wirksamen!dibenzothiazepin*derivat.!Arzneimittelforschung,!16:!314*316.!!
Bergeron! R,! Meyer! TM,! Coyle! JT! &! Greene! RW.! (1998).! Modulation! of! N*methyl*D*aspartate!receptor!function!by!glycine!transport.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!95(26):!15730*15734.!!
Bernstein!BE,!Kamal!M,!Lindblad*Toh!K,!Bekiranov!S,!Bailey!DK,!Huebert!DJ,! .! .! .!Lander!ES.!(2005).!Genomic!maps!and!comparative!analysis!of!histone!modifications! in!human!and!mouse.!Cell,!120(2):!169*181.!!

131!!
!!
Bernstein!BE,!Stamatoyannopoulos!JA,!Costello!JF,!Ren!B,!Milosavljevic!A,!Meissner!A,!.!.!.!Thomson!JA.!(2010).!The!NIH!roadmap!epigenomics!mapping!consortium.!Nature!Biotechnology,!28(10):!1045*1048.!!
Bertilsson!L,!Carrillo! JA,!Dahl!ML,! Llerena!A,!Alm!C,!Bondesson!U,! .! .! .!Benitez! J.! (1994).!Clozapine!disposition! covaries! with! CYP1A2! activity! determined! by! a! caffeine! test.! British! Journal! of!Clinical!Pharmacology,!38(5):!471*473.!!
Beutler!E,!Dern!RJ!&!Alving!AS.! (1955).!The!hemolytic!effect!of!primaquine.!VI.!An! in!vitro! test! for!sensitivity! of! erythrocytes! to! primaquine.! The! Journal! of! Laboratory! and! Clinical! Medicine,!45(1):!40*50.!!
Black!IB.!(1999).!Trophic!regulation!of!synaptic!plasticity.!Journal!of!Neurobiology,!41(1):!108*118.!!
Bleehen!T.!(1993).!Clozapine:!Literature!review.!Basel,!Switzerland:!Sandoz!Pharma.!!
Bleuer!E.!(1911).!Dementia!praecox!oder!gruppe!der!schizophrenien!(dementia!praecox!or!the!group!of!schizophrenias).!Leipzig,!Germany:!Franz!Deuticke.!!
Bobes! J,! Garcia*Portilla! MP,! Bascaran! MT,! Saiz! PA! &! Bousono! M.! (2007).! Quality! of! life! in!schizophrenic!patients.!Dialogues!in!Clinical!Neuroscience,!9(2):!215*226.!!
Bonferroni! C.! (1936).! Teoria! statistica! delle! classi! e! calcolo! delle! probabilità.! Pubblicazioni! Del! R!Istituto!Superiore!Di!Scienze!Economiche!e!Commerciali!Di!Firenze,!8:!3*62.!!
Borgio!JG,!Bressan!RA,!Barbosa!Neto!JB!&!Daltio!CS.! (2007).!Refractory!schizophrenia:!A!neglected!clinical!problem.!Revista!Brasileira!De!Psiquiatria,!29(3):!292*293.!!
Boydell!J,!Dean!K,!Dutta!R,!Giouroukou!E,!Fearon!P!&!Murray!R.!(2007).!A!comparison!of!symptoms!and! family!history! in! schizophrenia!with!and!without!prior! cannabis!use:! Implications! for! the!concept!of!cannabis!psychosis.!Schizophrenia!Research,!93(1*3):!203*210.!!
Boyle! AP,! Hong! EL,! Hariharan! M,! Cheng! Y,! Schaub! MA,! Kasowski! M,! .! .! .! Snyder! M.! (2012).!Annotation!of!functional!variation!in!personal!genomes!using!RegulomeDB.!Genome!Research,!22(9):!1790*1797.!!
Braff!DL,!Grillon!C!&!Geyer!MA.!(1992).!Gating!and!habituation!of!the!startle!reflex!in!schizophrenic!patients.!Archives!of!General!Psychiatry,!49(3):!206*215.!!
Brown!AS,! Begg!MD,! Gravenstein! S,! Schaefer! CA,!Wyatt! RJ,! Bresnahan!M,! .! .! .! Susser! ES.! (2004).!Serologic!evidence!of!prenatal! influenza! in! the!etiology!of! schizophrenia.!Archives!of!General!Psychiatry,!61(8):!774*780.!!
Brzustowicz! LM,!Hodgkinson! KA,! Chow! EW,!Honer!WG!&!Bassett! AS.! (2000).! Location! of! a!major!susceptibility! locus! for! familial! schizophrenia! on! chromosome! 1q21*q22.! Science,! 288(5466):!678*682.!!

132!!
!!
Bu! DF,! Erlander!MG,! Hitz! BC,! Tillakaratne!NJ,! Kaufman!DL,!Wagner*McPherson! CB,! .! .! .! Tobin! AJ.!(1992).!Two!human!glutamate!decarboxylases,!65*kDa!GAD!and!67*kDa!GAD,!are!each!encoded!by! a! single! gene.! Proceedings! of! the! National! Academy! of! Sciences! of! the! United! States! of!America,!89(6):!2115*2119.!!
Bubenikova! V,! Votava! M,! Horacek! J,! Palenicek! T! &! Dockery! C.! (2005).! The! effect! of! zotepine,!risperidone,! clozapine! and! olanzapine! on! MK*801*disrupted! sensorimotor! gating.!Pharmacology,!Biochemistry,!and!Behavior,!80(4):!591*596.!
Buckley!PF.!(2005).!Neuroimaging!of!schizophrenia:!Structural!abnormalities!and!pathophysiological!implications.!Neuropsychiatric!Disease!and!Treatment,!1(3):!193*204.!!
Buka!SL,!Tsuang!MT,!Torrey!EF,!Klebanoff!MA,!Bernstein!D!&!Yolken!RH.!(2001).!Maternal!infections!and!subsequent!psychosis!among!offspring.!Archives!of!General!Psychiatry,!58(11):!1032*1037.!!
Burnet! PW! &! Harrison! PJ.! (1995).! Genetic! variation! of! the! 5*HT2A! receptor! and! response! to!clozapine.!Lancet,!346(8979):!909.!!
Cai!J,!Zhang!W,!Yi!Z,!Lu!W,!Wu!Z,!Chen!J,!.!.!.!Zhang!C.!(2013).!Influence!of!polymorphisms!in!genes!SLC1A1,! GRIN2B,! and! GRIK2! on! clozapine*induced! obsessive*compulsive! symptoms.!Psychopharmacology,!230(1):!49*55.!!
Canadian! Psychiatric! Association.! (2005).! Clinical! practice! guidelines.! Treatment! of! schizophrenia.!Canadian!Journal!of!Psychiatry,!Supplemental,!50(13,!S1):!7S*57S.!!
Cantor*Graae!E!&!Selten!JP.! (2005).!Schizophrenia!and!migration:!A!meta*analysis!and!review.!The!American!Journal!of!Psychiatry,!162(1):!12*24.!!
Cardno! AG! &! Gottesman! II.! (2000).! Twin! studies! of! schizophrenia:! From! bow*and*arrow!concordances!to!star!wars!Mx!and!functional!genomics.!American!Journal!of!Medical!Genetics,!97(1):!12*17.!!
Cardon! LR! &! Palmer! LJ.! (2003).! Population! stratification! and! spurious! allelic! association.! Lancet,!361(9357):!598*604.!
Carlquist!JF,!McKinney!JT,!Nicholas!ZP,!Clark!JL,!Kahn!SF,!Horne!BD,!.! .! .!Anderson!JL.!(2008).!Rapid!melting! curve! analysis! for! genetic! variants! that! underlie! inter*individual! variability! in! stable!warfarin!dosing.!Journal!of!Thrombosis!and!Thrombolysis,!26(1):!1*7.!!
Carlsson! A! &! Lindqvist! M.! (1963).! Effect! of! chlorpromazine! or! haloperidol! on! formation! of! 3*methoxytyramine!and!normetanephrine!in!mouse!brain.!Acta!Pharmacologica!et!Toxicologica,!20:!140*144.!!
Cartmell! J! &! Schoepp! DD.! (2000).! Regulation! of! neurotransmitter! release! by! metabotropic!glutamate!receptors.!Journal!of!Neurochemistry,!75(3):!889*907.!!

133!!
!!
Caspi! A! &! Moffitt! TE.! (2006).! Gene*environment! interactions! in! psychiatry:! Joining! forces! with!neuroscience.!Nature!Reviews,!Neuroscience,!7(7):!583*590.!!
Centorrino! F,! Hernan! MA,! Drago*Ferrante! G,! Rendall! M,! Apicella! A,! Langar! G! &! Baldessarini! RJ.!(2001).! Factors! associated! with! noncompliance! with! psychiatric! outpatient! visits.! Psychiatric!Services,!52(3):!378*380.!!
Centre! for! the! Evaluation! of! Value! and! Risk! in! Health.! (2011).! The! cost*effectiveness! analysis!registry,! Tufts! Medical! Centre.! Retrieved! July! 27th,! 2014,! from! https://research.tufts*nemc.org/cear4/!!
Chen! L,! Muhlhauser! M! &! Yang! CR.! (2003).! Glycine! tranporter*1! blockade! potentiates! NMDA*mediated! responses! in! rat! prefrontal! cortical! neurons! in! vitro! and! in! vivo.! Journal! of!Neurophysiology,!89(2):!691*703.!!
Chen! L! &! Yang! CR.! (2002).! Interaction! of! dopamine! D1! and! NMDA! receptors! mediates! acute!clozapine!potentiation!of!glutamate!EPSPs!in!rat!prefrontal!cortex.!Journal!of!Neurophysiology,!87(5):!2324*2336.!!
Chiu!HJ,!Wang!YC,!Liou!YJ,!Lai!IC!&!Chen!JY.!(2003).!Association!analysis!of!the!genetic!variants!of!the!N*methyl! D*aspartate! receptor! subunit! 2b! (NR2b)! and! treatment*refractory! schizophrenia! in!the!Chinese.!Neuropsychobiology,!47(4):!178*181.!!
Choudhry! S,! Seibold! MA,! Borrell! LN,! Tang! H,! Serebrisky! D,! Chapela! R,! .! .! .! Burchard! EG.! (2007).!Dissecting!complex!diseases!in!complex!populations:!Asthma!in!Latino!Americans.!Proceedings!of!the!American!Thoracic!Society,!4(3):!226*233.!!
Chowdhury! NI,! Tiwari! AK,! Souza! RP,! Zai! CC,! Shaikh! SA,! Chen! S,! .! .! .! Muller! DJ.! (2013).! Genetic!association!study!between!antipsychotic*induced!weight!gain!and!the!melanocortin*4!receptor!gene.!The!Pharmacogenomics!Journal,!13(3):!272*279.!!
Chumakov! I,! Blumenfeld!M,!Guerassimenko!O,!Cavarec! L,! Palicio!M,!Abderrahim!H,! .! .! .! Cohen!D.!(2002).!Genetic!and!physiological!data!implicating!the!new!human!gene!G72!and!the!gene!for!D*amino!acid!oxidase!in!schizophrenia.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!99(21):!13675*13680.!!
Claghorn!J,!Honigfeld!G,!Abuzzahab!FS!Sr,!Wang!R,!Steinbook!R,!Tuason!V!&!Klerman!G.!(1987).!The!risks!and!benefits!of!clozapine!versus!chlorpromazine.!Journal!of!Clinical!Psychopharmacology,!7(6):!377*384.!!
Clayton!D!&!McKeigue!PM.!(2001).!Epidemiological!methods!for!studying!genes!and!environmental!factors!in!complex!diseases.!Lancet,!358(9290):!1356*1360.!!
Clinton! SM!&!Meador*Woodruff! JH.! (2004).! Abnormalities! of! the! NMDA! receptor! and! associated!intracellular! molecules! in! the! thalamus! in! schizophrenia! and! bipolar! disorder.!Neuropsychopharmacology,!29(7):!1353*1362.!!

134!!
!!
Clouston!TS.!(1904).!Clinical!Lectures!on!Mental!Diseases.!6th!ed!London,!UK:!J&A!Churchill.!!
Conley! RR,! Tamminga! CA,! Kelly! DL! &! Richardson! CM.! (1999).! Treatment*resistant! schizophrenic!patients!respond!to!clozapine!after!olanzapine!non*response.!Biological!Psychiatry,!46(1):!73*77.!!
Conn!PJ!&!Pin!JP.!(1997).!Pharmacology!and!functions!of!metabotropic!glutamate!receptors.!Annual!Review!of!Pharmacology!and!Toxicology,!37:!205*237.!!
Corbett!R,!Camacho!F,!Woods!AT,!Kerman!LL,!Fishkin!RJ,!Brooks!K!&!Dunn!RW.!(1995).!Antipsychotic!agents! antagonize! non*competitive! N*methyl*D*aspartate! antagonist*induced! behaviors.!Psychopharmacology,!120(1):!67*74.!!
Corvin!AP,!Morris!DW,!McGhee!K,!Schwaiger!S,!Scully!P,!Quinn! J,! .! .! .!Gill!M.! (2004).!Confirmation!and!refinement!of!an! 'at*risk'!haplotype!for!schizophrenia!suggests!the!EST!cluster,!Hs.97362,!as!a!potential!susceptibility!gene!at!the!neuregulin*1!locus.!Molecular!Psychiatry,!9(2):!208*213.!!
Cosgrove!VE!&!Suppes!T.!(2013).!Informing!DSM*5:!Biological!boundaries!between!bipolar!I!disorder,!schizoaffective!disorder,!and!schizophrenia.!BMC!Medicine,!11(127):!1*7.!!
Costa! e! Silva! JA.! (2013).! Personalized!medicine! in! psychiatry:! New! technologies! and! approaches.!Metabolism:!Clinical!and!Experimental,!Supplemental,!62(S1):!S40*S44.!!
Coyle! JT! &! Puttfarcken! P.! (1993).! Oxidative! stress,! glutamate,! and! neurodegenerative! disorders.!Science,!262(5134):!689*695.!!
Creese! I,! Burt! DR! &! Snyder! SH.! (1976).! Dopamine! receptors! and! average! clinical! doses.! Science,!194(4264):!546.!
CRESTAR.! (2011).! CRESTAR:! Development! of! pharmacogenetic! biomarkers! for! schizophrenia.!Retrieved!March!14th,!2014,!from!http://www.crestar*project.eu/!!
Csernansky!JG,!Wang!L,!Jones!D,!Rastogi*Cruz!D,!Posener!JA,!Heydebrand!G,! .! .! .!Miller!MI.!(2002).!Hippocampal! deformities! in! schizophrenia! characterized! by! high! dimensional! brain!mapping.!The!American!Journal!of!Psychiatry,!159(12):!2000*2006.!!
Cubelos! B,! Gimenez! C! &! Zafra! F.! (2005).! Localization! of! the! GLYT1! glycine! transporter! at!glutamatergic!synapses!in!the!rat!brain.!Cerebral!Cortex,!15(4):!448*459.!!
Curthoys!NP!&!Watford!M.! (1995).! Regulation!of! glutaminase! activity! and! glutamine!metabolism.!Annual!Review!of!Nutrition,!15:!133*159.!
Curtis! DR,! Phillis! JW!&!Watkins! JC.! (1960).! The! chemical! excitation! of! spinal! neurones! by! certain!acidic!amino!acids.!The!Journal!of!Physiology,!150:!656*682.!!
Daikhin!Y!&!Yudkoff!M.! (2000).!Compartmentation!of!brain!glutamate!metabolism! in!neurons!and!glia.!The!Journal!of!Nutrition,!Supplemental,!130(4S):!1026S*31S.!!

135!!
!!
Dallaspezia! S,! Poletti! S,! Lorenzi! C,! Pirovano! A,! Colombo! C!&! Benedetti! F.! (2012).! Influence! of! an!interaction!between! lithium!salts!and!a! functional!polymorphism! in!SLC1A2!on! the!history!of!illness!in!bipolar!disorder.!Molecular!Diagnosis!&!Therapy,!16(5):!303*309.!!
Daly!DA!&!Moghaddam!B.!(1993).!Actions!of!clozapine!and!haloperidol!on!the!extracellular!levels!of!excitatory! amino! acids! in! the! prefrontal! cortex! and! striatum! of! conscious! rats.! Neuroscience!Letters,!152(1*2):!61*64.!!
Danysz! W! &! Parsons! CG.! (1998).! Glycine! and! N*methyl*D*aspartate! receptors:! Physiological!significance!and!possible!therapeutic!applications.!Pharmacological!Reviews,!50(4):!597*664.!!
Davis! JM,! Chen! N! &! Glick! ID.! (2003).! A! meta*analysis! of! the! efficacy! of! second*generation!antipsychotics.!Archives!of!General!Psychiatry,!60(6):!553*564.!!
Davis! KL,! Kahn! RS,! Ko! G! &! Davidson! M.! (1991).! Dopamine! in! schizophrenia:! A! review! and!reconceptualization.!The!American!Journal!of!Psychiatry,!148(11):!1474*1486.!!
de!la!Fuente*Sandoval!C,!Leon*Ortiz!P,!Azcarraga!M,!Favila!R,!Stephano!S!&!Graff*Guerrero!A.!(2013).!Striatal!glutamate!and!the!conversion!to!psychosis:!A!prospective!1H*MRS!imaging!study.!The!International!Journal!of!Neuropsychopharmacology,!16(2):!471*475.!!
de! Leon! J.! (2004).! Atypical! antipsychotic! dosing:! The! effect! of! smoking! and! caffeine.! Psychiatric!Services,!55(5):!491*493.!!
De! Sousa! KR,! Tiwari! AK,! Giuffra! DE,! Mackenzie! B,! Zai! CC! &! Kennedy! JL.! (2013).! Age! at! onset! of!schizophrenia:!Cannabis,!COMT!gene,!and!their!interactions.!Schizophrenia!Research,!151(1*3):!289*290.!!
Delay! J,! Deniker! E! &! Had! JM.! (1952).! Traitement! des! etats! d'excitation! et! d'agitation! par! une!methode!medicamentense!derivee!de!l'hibernotherapie.!Annales!Médico!Psychologiques,!110:!267*273.!!
Demjaha! A,! Egerton! A,! Murray! RM,! Kapur! S,! Howes! OD,! Stone! JM! &! McGuire! PK.! (2014).!Antipsychotic!treatment!resistance!in!schizophrenia!associated!with!elevated!glutamate!levels!but!normal!dopamine!function.!Biological!Psychiatry,!75(5):!11*13.!!
Deng!X,!Shibata!H,!Ninomiya!H,!Tashiro!N,!Iwata!N,!Ozaki!N!&!Fukumaki!Y.!(2004).!Association!study!of!polymorphisms!in!the!excitatory!amino!acid!transporter!2!gene!(SLC1A2)!with!schizophrenia.!BMC!Psychiatry,!4:!21.!!
Detera*Wadleigh!SD!&!McMahon!FJ.!(2006).!G72/G30!in!schizophrenia!and!bipolar!disorder:!Review!and!meta*analysis.!Biological!Psychiatry,!60(2):!106*114.!
!Di!Maria!E,!Gulli!R,!Begni!S,!De!Luca!A,!Bignotti!S,!Pasini!A,!.!.!.!Mandich!P.!(2004).!Variations!in!the!NMDA!receptor!subunit!2B!gene!(GRIN2B)!and!schizophrenia:!A!case*control!study.!American!Journal!of!Medical!Genetics!Part!B,!Neuropsychiatric!Genetics,!128B(1):!27*29.!!

136!!
!!
Distlerath!LM,!Reilly!PE,!Martin!MV,!Davis!GG,!Wilkinson!GR!&!Guengerich!FP.! (1985).!Purification!and! characterization! of! the! human! liver! cytochromes! P*450! involved! in! debrisoquine! 4*hydroxylation! and! phenacetin! O*deethylation,! two! prototypes! for! genetic! polymorphism! in!oxidative!drug!metabolism.!The!Journal!of!Biological!Chemistry,!260(15):!9057*9067.!!
Dixon! M! &! Kleppe! K.! (1965).! D*amino! acid! oxidase.! I.! Dissociation! and! recombination! of! the!holoenzyme.!Biochimica!et!Biophysica!Acta,!96:!357*367.!!
Dong! E,! Nelson! M,! Grayson! DR,! Costa! E! &! Guidotti! A.! (2008).! Clozapine! and! sulpiride! but! not!haloperidol! or! olanzapine! activate! brain! DNA! demethylation.! Proceedings! of! the! National!Academy!of!Sciences!of!the!United!States!of!America,!105(36):!13614*13619.!!
Dorval!KM,!Wigg!KG,!Crosbie!J,!Tannock!R,!Kennedy!JL,!Ickowicz!A,!.!.!.!Barr!CL.!(2007).!Association!of!the! glutamate! receptor! subunit! gene! GRIN2B! with! attention*deficit/hyperactivity! disorder.!Genes,!Brain,!and!Behavior,!6(5):!444*452.!!
Dracheva! S,! Marras! SA,! Elhakem! SL,! Kramer! FR,! Davis! KL! &! Haroutunian! V.! (2001).! N*methyl*D*aspartic!acid!receptor!expression!in!the!dorsolateral!prefrontal!cortex!of!elderly!patients!with!schizophrenia.!The!American!Journal!of!Psychiatry,!158(9):!1400*1410.!!
Drazen! JM,! Yandava! CN,! Dube! L,! Szczerback! N,! Hippensteel! R,! Pillari! A,! .! .! .! Drajesk! J.! (1999).!Pharmacogenetic! association! between!ALOX5! promoter! genotype! and! the! response! to! anti*asthma!treatment.!Nature!Genetics,!22(2):!168*170.!!
Dudbridge! F.! (2008).! Likelihood*based! association! analysis! for! nuclear! families! and! unrelated!subjects!with!missing!genotype!data.!Human!Heredity,!66(2):!87*98.!
Dunbar*Jacob!J!&!Mortimer*Stephens!MK.!(2001).!Treatment!adherence!in!chronic!disease.!Journal!of!Clinical!Epidemiology,!Supplemental,!54(S1):!S57*S60.!!
Dunlop! DS! &! Neidle! A.! (1997).! The! origin! and! turnover! of! D*serine! in! brain.! Biochemical! and!Biophysical!Research!Communications,!235(1):!26*30.!!
Dunwiddie! TV.! (1980).! Endogenously! released! adenosine! regulates! excitability! in! the! in! vitro!hippocampus.!Epilepsia,!21(5):!541*548.!!
Dunwiddie! TV,! Hoffer! BJ! &! Fredholm! BB.! (1981).! Alkylxanthines! elevate! hippocampal! excitability.!evidence! for! a! role! of! endogenous! adenosine.! NaunynPSchmiedeberg's! Archives! of!Pharmacology,!316(4):!326*330.!!
Egerton! A,! Brugger! S,! Raffin!M,! Barker! GJ,! Lythgoe! DJ,!McGuire! PK!&! Stone! JM.! (2012).! Anterior!cingulate! glutamate! levels! related! to! clinical! status! following! treatment! in! first*episode!schizophrenia.!Neuropsychopharmacology,!37(11):!2515*2521.!!
Elkis! H! &! Meltzer! HY.! (2007).! Refractory! schizophrenia.! Revista! Brasileira! De! Psiquiatria,!Supplemental,!29(S2):!S41*S7.!!

137!!
!!
El*Saadi!O,!Pedersen!CB,!McNeil!TF,!Saha!S,!Welham!J,!O'Callaghan!E,!.!.!.!McGrath!J.!(2004).!Paternal!and!maternal!age!as!risk!factors!for!psychosis:!Findings!from!Denmark,!Sweden!and!Australia.!Schizophrenia!Research,!67(2*3):!227*236.!!
Emsley!R,!Rabinowitz!J,!Medori!R!&!Early!Psychosis!Global!Working!Group.!(2007).!Remission!in!early!psychosis:! Rates,! predictors,! and! clinical! and! functional! outcome! correlates.! Schizophrenia!Research,!89(1*3):!129*139.!!
ENCODE! Project! Consortium.! (2012).! An! integrated! encyclopedia! of! DNA! elements! in! the! human!genome.!Nature,!489(7414):!57*74.!!
Endele!S,!Rosenberger!G,!Geider!K,!Popp!B,!Tamer!C,!Stefanova!I,!.!.!.!Kutsche!K.!(2010).!Mutations!in!GRIN2A! and! GRIN2B! encoding! regulatory! subunits! of! NMDA! receptors! cause! variable!neurodevelopmental!phenotypes.!Nature!Genetics,!42(11):!1021*1026.!!
Erlander!MG,! Tillakaratne!NJ,! Feldblum! S,! Patel!N!&! Tobin! AJ.! (1991).! Two! genes! encode! distinct!glutamate!decarboxylases.!Neuron,!7(1):!91*100.!!
Ernst!J!&!Kellis!M.!(2012).!ChromHMM:!Automating!chromatin*state!discovery!and!characterization.!Nature!Methods,!9(3):!215*216.!!
Evans!DA,!Manley!KA!&!Mckusick!VA.!(1960).!Genetic!control!of!isoniazid!metabolism!in!man.!British!Medical!Journal,!2(5197):!485*491.!!
Evans!DM!&!Purcell!S.! (2012).!Power!calculations! in!genetic!studies.!Cold!Spring!Harbor!Protocols,!2012(6):!664*674.!!
Evers!K.!(2009).!Personalized!medicine!in!psychiatry:!Ethical!challenges!and!opportunities.!Dialogues!in!Clinical!Neuroscience,!11(4):!427*434.!!
Evins!AE,!Amico!ET,!Shih!V!&!Goff!DC.!(1997).!Clozapine!treatment!increases!serum!glutamate!and!aspartate! compared! to! conventional! neuroleptics.! Journal! of! Neural! Transmission,! 104(6*7):!761*766.!!
Evins! AE,! Fitzgerald! SM,!Wine! L,! Rosselli! R! &! Goff! DC.! (2000).! Placebo*controlled! trial! of! glycine!added!to!clozapine!in!schizophrenia.!The!American!Journal!of!Psychiatry,!157(5):!826*828.!!
Fagiolini!M,!Fritschy!JM,!Low!K,!Mohler!H,!Rudolph!U!&!Hensch!TK.!(2004).!Specific!GABAA!circuits!for!visual!cortical!plasticity.!Science,!303(5664):!1681*1683.!
Fan! F!&!Wood! KV.! (2007).! Bioluminescent! assays! for! high*throughput! screening.! Assay! and! Drug!Development!Technologies,!5(1):!127*136.!!
Farin!HF,!Bussen!M,!Schmidt!MK,!Singh!MK,!Schuster*Gossler!K!&!Kispert!A.!(2007).!Transcriptional!repression! by! the! T*box! proteins! Tbx18! and! Tbx15! depends! on! groucho! corepressors.! The!Journal!of!Biological!Chemistry,!282(35):!25748*25759.!!

138!!
!!
Fell!MJ,! Svensson! KA,! Johnson! BG!&! Schoepp! DD.! (2008).! Evidence! for! the! role! of!metabotropic!glutamate! (mGlu)2!not!mGlu3! receptors! in! the!preclinical! antipsychotic!pharmacology!of! the!mGlu2/3! receptor! agonist! (*)*(1R,4S,5S,6S)*4*amino*2*sulfonylbicyclo[3.1.0]hexane*4,6*dicarboxylic! acid! (LY404039).! The! Journal! of! Pharmacology! and! Experimental! Therapeutics,!326(1):!209*217.!!
Ferreira! R,! Ohneda! K,! Yamamoto! M! &! Philipsen! S.! (2005).! GATA1! function,! a! paradigm! for!transcription!factors!in!hematopoiesis.!Molecular!and!Cellular!Biology,!25(4):!1215*1227.!!
Fitzgerald! LW,!Deutch!AY,!Gasic!G,!Heinemann!SF!&!Nestler! EJ.! (1995).! Regulation!of! cortical! and!subcortical! glutamate! receptor! subunit! expression! by! antipsychotic! drugs.! The! Journal! of!Neuroscience,!15(3!Pt!2):!2453*2461.!!
Flint! AC,!Maisch! US,!Weishaupt! JH,! Kriegstein! AR! &!Monyer! H.! (1997).! NR2A! subunit! expression!shortens! NMDA! receptor! synaptic! currents! in! developing! neocortex.! The! Journal! of!Neuroscience,!17(7):!2469*2476.!!
Foster!GA!&!Roberts!PJ.!(1981).!Stimulation!of!rat!cerebellar!guanosine!3',5'*cyclic!monophosphate!(cyclic!GMP)! levels:!Effects!of!amino!acid!antagonists.!British! Journal!of!Pharmacology,!74(3):!723*729.!!
Frackiewicz! EJ,! Sramek! JJ,! Herrera! JM,! Kurtz! NM!&! Cutler! NR.! (1997).! Ethnicity! and! antipsychotic!response.!The!Annals!of!Pharmacotherapy,!31(11):!1360*1369.!!
Freed! WJ,! Bing! LA! &! Wyatt! RJ.! (1984).! Effects! of! neuroleptics! on! phencyclidine! (PCP)*induced!locomotor!stimulation!in!mice.!Neuropharmacology,!23(2A):!175*181.!!
Fribourg! M,! Moreno! JL,! Holloway! T,! Provasi! D,! Baki! L,! Mahajan! R,! .! .! .! Logothetis! DE.! (2011).!Decoding!the!signaling!of!a!GPCR!heteromeric!complex!reveals!a!unifying!mechanism!of!action!of!antipsychotic!drugs.!Cell,!147(5):!1011*1023.!!
Funke!B,!Finn!CT,!Plocik!AM,!Lake!S,!DeRosse!P,!Kane!JM,!.!.!.!Malhotra!AK.!(2004).!Association!of!the!DTNBP1! locus!with! schizophrenia! in! a!U.S.! population.! American! Journal! of!Human!Genetics,!75(5):!891*898.!!
Furuta!A,!Rothstein! JD!&!Martin!LJ.! (1997).!Glutamate!transporter!protein!subtypes!are!expressed!differentially!during!rat!CNS!development.!The!Journal!of!Neuroscience,!17(21):!8363*8375.!!
Gabernet! L,! Pauly*Evers!M,! Schwerdel! C,! Lentz!M,! Bluethmann! H,! Vogt! K,! .! .! .! Boison! D.! (2005).!Enhancement!of!the!NMDA!receptor!function!by!reduction!of!glycine!transporter*1!expression.!Neuroscience!Letters,!373(1):!79*84.!!
GAIN!Collaborative!Research!Group,!Manolio!TA,!Rodriguez!LL,!Brooks!L,!Abecasis!G,!Collaborative!Association!Study!of!Psoriasis,!.!.!.!Collins!FS.!(2007).!New!models!of!collaboration!in!genome*wide!association!studies:!The!genetic!association!information!network.!Nature!Genetics,!39(9):!1045*1051.!!

139!!
!!
Garrod!A.!(1909).!Inborn!errors!of!metabolism!(1st!ed).!England:!Oxford!University!Press.!!
Gauderman! WJ.! (2002).! Sample! size! requirements! for! matched! case*control! studies! of! gene*environment!interaction.!Statistics!in!Medicine,!21(1):!35*50.!!
Gauderman!WJ!&!Morrison!JM.! (2006).!QUANTO:!A!computer!program!for!power!and!sample!size!calculations!for!geneticPepidemiology!studies!(Version!1.2.4).!!
Ge!S,!Goh!EL,!Sailor!KA,!Kitabatake!Y,!Ming!GL!&!Song!H.!(2006).!GABA!regulates!synaptic!integration!of!newly!generated!neurons!in!the!adult!brain.!Nature,!439(7076):!589*593.!!
Ghassabian! S,! Chetty!M,! Tattam!BN,!Glen! J,! Rahme! J,! Stankovic! Z,! .! .! .!McLachlan!AJ.! (2010).! The!participation! of! cytochrome! P450! 3A4! in! clozapine! biotransformation! is! detected! in! people!with! schizophrenia! by! high*throughput! in! vivo! phenotyping.! Journal! of! Clinical!Psychopharmacology,!30(5):!629*631.!!
Giardino!L,!Bortolotti!F,!Orazzo!C,!Pozza!M,!Monteleone!P,!Calza!L!&!Maj!M.!(1997).!Effect!of!chronic!clozapine!administration!on![3H]MK801*binding!sites!in!the!rat!brain:!A!side*preference!action!in!cortical!areas.!Brain!Research,!762(1*2):!216*218.!!
Gilman!H,!van!Ess!PR!&!Shirley!DA.!(1944).!The!methalation!of!10*phenylphenothiazine!and!of!10*ethylphenothiazine.!Journal!of!the!American!Chemical!Society,!66:!1214*1216.!!
Glatt! SJ,! Faraone! SV! &! Tsuang! MT.! (2003).! Meta*analysis! identifies! an! association! between! the!dopamine!D2!receptor!gene!and!schizophrenia.!Molecular!Psychiatry,!8(11):!911*915.!!
Goeree!R,!Farahati!F,!Burke!N,!Blackhouse!G,!O'Reilly!D,!Pyne!J!&!Tarride!JE.!(2005).!The!economic!burden! of! schizophrenia! in! Canada! in! 2004.! Current! Medical! Research! and! Opinion,! 21(12):!2017*2028.!!
Goff! DC,! Henderson! DC,! Evins! AE! &! Amico! E.! (1999).! A! placebo*controlled! crossover! trial! of! D*cycloserine!added!to!clozapine!in!patients!with!schizophrenia.!Biological!Psychiatry,!45(4):!512*514.!!
Goff! DC,! Tsai! G,! Levitt! J,! Amico! E,! Manoach! D,! Schoenfeld! DA,! .! .! .! Coyle! JT.! (1999).! A! placebo*controlled! trial! of! D*cycloserine! added! to! conventional! neuroleptics! in! patients! with!schizophrenia.!Archives!of!General!Psychiatry,!56(1):!21*27.!!
Goffman! E.! (1963).! Stigma:! Notes! on! the! management! of! spoiled! identity.! Englewood! Cliffs,! NJ:!Prentice*Hall.!!
Goldstein! L.! (1967).! Pathways! of! glutamine! deamination! and! their! control! in! the! rat! kidney.! The!American!Journal!of!Physiology,!213(4):!983*989.!!
Gomes! I,! Collins! A,! Lonjou! C,! Thomas! NS,! Wilkinson! J,! Watson! M! &! Morton! N.! (1999).! Hardy*Weinberg!quality!control.!Annals!of!Human!Genetics,!63(6):!535*538.!!

140!!
!!
Gonzalez*Maeso!J,!Ang!RL,!Yuen!T,!Chan!P,!Weisstaub!NV,!Lopez*Gimenez!JF,!.!.!.!Sealfon!SC.!(2008).!Identification! of! a! serotonin/glutamate! receptor! complex! implicated! in! psychosis.! Nature,!452(7183):!93*97.!!
Gottesman!II.!(1991).!Schizophrenia!genesis:!The!origin!of!madness.!New!York:!Freeman.!!
Gottesman! II!&!Shields! J.! (1967).!A!polygenic!theory!of!schizophrenia.!Proceedings!of! the!National!Academy!of!Sciences!of!the!United!States!of!America,!58(1):!199*205.!!
Grace! AA.! (2012).! Dopamine! system! dysregulation! by! the! hippocampus:! Implications! for! the!pathophysiology!and!treatment!of!schizophrenia.!Neuropharmacology,!62(3):!1342*1348.!!
Grimwood! S,! Slater! P,! Deakin! JF! &! Hutson! PH.! (1999).! NR2B*containing! NMDA! receptors! are! up*regulated!in!temporal!cortex!in!schizophrenia.!Neuroreport,!10(3):!461*465.!!
Groc! L,! Choquet! D! &! Chaouloff! F.! (2008).! The! stress! hormone! corticosterone! conditions! AMPAR!surface!trafficking!and!synaptic!potentiation.!Nature!Neuroscience,!11(8):!868*870.!!
Gross! H! &! Langner! E.! (1966).! Das! wirkungprofil! eines! chemischen! neuartigen! breitband*neuroleptikums! der! dibenzodiazepingruppe.! Wiener! Medizinische! Wochenschrift,! 116:! 814*816.!!
Grucza! RA,! Johnson! EO,! Krueger! RF,! Breslau! N,! Saccone! NL,! Chen! LS,! .! .! .! Bierut! LJ.! (2010).!Incorporating!age!at!onset!of!smoking!into!genetic!models!for!nicotine!dependence:!Evidence!for!interaction!with!multiple!genes.!Addiction!Biology,!15(3):!346*357.!!
Grueter! BA! &! Winder! DG.! (2005).! Group! II! and! III! metabotropic! glutamate! receptors! suppress!excitatory! synaptic! transmission! in! the! dorsolateral! bed! nucleus! of! the! stria! terminalis.!Neuropsychopharmacology,!30(7):!1302*1311.!!
Gu!Z,!Jiang!Q,!Fu!AK,!Ip!NY!&!Yan!Z.!(2005).!Regulation!of!NMDA!receptors!by!neuregulin!signaling!in!prefrontal!cortex.!The!Journal!of!Neuroscience,!25(20):!4974*4984.!!
Guengerich!FP.!(2008).!Cytochrome!P450!and!chemical!toxicology.!Chemical!Research!in!Toxicology,!21(1):!70*83.!!
Gunduz*Bruce!H,!McMeniman!M,!Robinson!DG,!Woerner!MG,!Kane!JM,!Schooler!NR!&!Lieberman!JA.!(2005).!Duration!of!untreated!psychosis!and!time!to!treatment!response!for!delusions!and!hallucinations.!The!American!Journal!of!Psychiatry,!162(10):!1966*1969.!!
Gurling!HM,!Kalsi!G,!Brynjolfson!J,!Sigmundsson!T,!Sherrington!R,!Mankoo!BS,!.! .! .!Curtis!D.!(2001).!Genomewide! genetic! linkage! analysis! confirms! the! presence! of! susceptibility! loci! for!schizophrenia,!on!chromosomes!1q32.2,!5q33.2,!and!8p21*22!and!provides!support!for!linkage!to!schizophrenia,!on!chromosomes!11q23.3*24!and!20q12.1*11.23.!American!Journal!of!Human!Genetics,!68(3):!661*673.!!!

141!!
!!
Gutierrez*Maldonado!J,!Caqueo*Urizar!A!&!Kavanagh!DJ.!(2005).!Burden!of!care!and!general!health!in! families! of! patients! with! schizophrenia.! Social! Psychiatry! and! Psychiatric! Epidemiology,!40(11):!899*904.!!
Guy!W.!(1976).!ECDEU!assessment!manual!for!psychopharmacology.!Rockville,!MD:!US!Department!of!Health,!Education,!and!Welfare.!!
Haak! LL.! (1999).! Metabotropic! glutamate! receptor! modulation! of! glutamate! responses! in! the!suprachiasmatic!nucleus.!Journal!of!Neurophysiology,!81(3):!1308*1317.!!
Hafner!H,!Maurer!K,!Loffler!W,!Fatkenheuer!B,!an!der!Heiden!W,!Riecher*Rossler!A,!.!.!.!Gattaz!WF.!(1994).! The! epidemiology! of! early! schizophrenia.! Influence! of! age! and! gender! on! onset! and!early!course.!The!British!Journal!of!Psychiatry,!Supplemental,!(23):!S29*S38.!!
Hafner! H,! Loffler!W,!Maurer! K,! Hambrecht!M! &! an! der! Heiden!W.! (1999).! Depression,! negative!symptoms,! social! stagnation! and! social! decline! in! the! early! course! of! schizophrenia.! Acta!Psychiatrica!Scandinavica,!100(2):!105*118.!!
Hagg! S,! Spigset! O,! Mjorndal! T! &! Dahlqvist! R.! (2000).! Effect! of! caffeine! on! clozapine!pharmacokinetics!in!healthy!volunteers.!British!Journal!of!Clinical!Pharmacology,!49(1):!59*63.!!
Hagger!C,!Buckley!P,!Kenny!JT,!Friedman!L,!Ubogy!D!&!Meltzer!HY.!(1993).!Improvement!in!cognitive!functions! and! psychiatric! symptoms! in! treatment*refractory! schizophrenic! patients! receiving!clozapine.!Biological!Psychiatry,!34(10):!702*712.!!
Hall*Flavin!DK,!Winner!JG,!Allen!JD,!Carhart!JM,!Proctor!B,!Snyder!KA,!.!.!.!Mrazek!DA.!(2013).!Utility!of!integrated!pharmacogenomic!testing!to!support!the!treatment!of!major!depressive!disorder!in!a!psychiatric!outpatient!setting.!Pharmacogenetics!and!Genomics,!23(10):!535*548.!!
Hamon!J,!Paraire!J!&!Velluz!J.!(1952).!Remarques!sur!l'action!du!4560!R.P!sur!l'agitation!maniaque.!Annales!Médico!Psychologiques,!110(13):!331*335.!!
Hanson!A!&!Levin!B.!(2013).!Mental!health!informatics.!Part!3!P!Competencies!and!strategies:!Types!of!data.!New!York,!NY:!Oxford!University!Press.!!
Hardy!GH.!(1908).!Mendelian!proportions!in!a!mixed!population.!Science,!28(706):!49*50.!!
Hashimoto! K.! (2010).! Glycine! transport! inhibitors! for! the! treatment! of! schizophrenia.! The! Open!Medicinal!Chemistry!Journal,!4:!10*19.!!
Harris!EW,!Ganong!AH!&!Cotman!CW.!(1984).!Long*term!potentiation!in!the!hippocampus!involves!activation!of!N*methyl*D*aspartate!receptors.!Brain!Research,!323(1):!132*137.!!
Harris!LW,!Sharp!T,!Gartlon!J,!Jones!DN!&!Harrison!PJ.!(2003).!Long*term!behavioural,!molecular!and!morphological! effects! of! neonatal! NMDA! receptor! antagonism.! The! European! Journal! of!Neuroscience,!18(6):!1706*1710.!!

142!!
!!
Harrison! PJ! &! Law! AJ.! (2006).! Neuregulin! 1! and! schizophrenia:! Genetics,! gene! expression,! and!neurobiology.!Biological!Psychiatry,!60(2):!132*140.!!
Harrow! M,! Grossman! LS,! Herbener! ES! &! Davies! EW.! (2000).! Ten*year! outcome:! Patients! with!schizoaffective! disorders,! schizophrenia,! affective! disorders! and!mood*incongruent! psychotic!symptoms.!The!British!Journal!of!Psychiatry,!177:!421*426.!!
Harvey! RJ! &! Yee! BK.! (2013).! Glycine! transporters! as! novel! therapeutic! targets! in! schizophrenia,!alcohol!dependence!and!pain.!Nature!Reviews,!Drug!Discovery,!12(11):!866*885.!!
Heresco*Levy! U.! (2003).! Glutamatergic! neurotransmission! modulation! and! the! mechanisms! of!antipsychotic!atypicality.!Progress!in!NeuroPPsychopharmacology!&!Biological!Psychiatry,!27(7):!1113*1123.!!
Heresco*Levy! U,! Javitt! DC,! Ermilov! M,! Mordel! C,! Horowitz! A! &! Kelly! D.! (1996).! Double*blind,!placebo*controlled,! crossover! trial! of! glycine! adjuvant! therapy! for! treatment*resistant!schizophrenia.!The!British!Journal!of!Psychiatry,!169(5):!610*617.!!
Hermans! E! &! Challiss! RA.! (2001).! Structural,! signalling! and! regulatory! properties! of! the! group! I!metabotropic! glutamate! receptors:! Prototypic! family! C! G*protein*coupled! receptors.! The!Biochemical!Journal,!359(3):!465*484.!!
Heston! LL.! (1966).! Psychiatric! disorders! in! foster! home! reared! children! of! schizophrenic!mothers.!The!British!Journal!of!Psychiatry,!112(489):!819*825.!!
Higgins!J.!(1966).!Effects!of!child!rearing!by!schizophrenic!mothers.!Journal!of!Psychiatric!Research,!4(3):!153*167.!!
Higgins! J.! (1976).! Effects! of! child! rearing! by! schizophrenic! mothers:! A! follow*up.! Journal! of!Psychiatric!Research,!13(1):!1*9.!!
Higuchi!M,!Single!FN,!Kohler!M,!Sommer!B,!Sprengel!R!&!Seeburg!PH.!(1993).!RNA!editing!of!AMPA!receptor! subunit! GluR*B:! A! base*paired! intron*exon! structure! determines! position! and!efficiency.!Cell,!75(7):!1361*1370.!!
Hippius!H.! (1996).!The! founding! of! the! CINP! and! the! discovery! of! clozapine.! In!D.!Healy! (Ed),!The!psychopharmacologists.!New!York,!NY:!Chapman!&!Hall.!!
Hirai! H,! Kirsch! J,! Laube! B,! Betz! H! &! Kuhse! J.! (1996).! The! glycine! binding! site! of! the! N*methyl*D*aspartate!receptor!subunit!NR1:!Identification!of!novel!determinants!of!co*agonist!potentiation!in!the!extracellular!M3*M4!loop!region.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!93(12):!6031*6036.!!
Hoggart!CJ,!Parra!EJ,!Shriver!MD,!Bonilla!C,!Kittles!RA,!Clayton!DG!&!McKeigue!PM.!(2003).!Control!of! confounding! of! genetic! associations! in! stratified! populations.! American! Journal! of! Human!Genetics,!72(6):!1492*1504.!!

143!!
!!
Hong!CJ,!Yu!YW,!Lin!CH,!Cheng!CY!&!Tsai!SJ.!(2001).!Association!analysis!for!NMDA!receptor!subunit!2B! (GRIN2B)! genetic! variants! and! psychopathology! and! clozapine! response! in! schizophrenia.!Psychiatric!Genetics,!11(4):!219*222.!!
Hopkins! CR.! (2011).! ACS! Chemical! Neuroscience! molecule! spotlight! on! RG1678.! ACS! Chemical!Neuroscience,!2(12):!685*686.!!
Horacek! J,! Libiger! C,! Hoschl! K,! Borzova! I,! Hendrychova! J.! (2001).! Clozapine*induced! concordant!agranulocytosis! in!monozygotic! twins.! International! Journal! of! Psychiatry! in! Clinical! Practice,!5(1):!71*73.!!
Hosking! L,! Lumsden! S,! Lewis! K,! Yeo! A,! McCarthy! L,! Bansal! A,! .! .! .! Xu! CF.! (2004).! Detection! of!genotyping! errors! by! Hardy*Weinberg! equilibrium! testing.! European! Journal! of! Human!Genetics,!12(5):!395*399.!!
Hovatta! I,! Varilo! T,! Suvisaari! J,! Terwilliger! JD,! Ollikainen! V,! Arajarvi! R,! .! .! .! Peltonen! L.! (1999).! A!genomewide! screen! for! schizophrenia!genes! in!an! isolated!Finnish! subpopulation,! suggesting!multiple!susceptibility!loci.!American!Journal!of!Human!Genetics,!65(4):!1114*1124.!!
Hoyer!C,!Kranaster!L,!Leweke!FM,!Klosterkotter!J,!Meyer*Lindenberg!A!&!Koethe!D.!(2010).!Familial!differential! treatment! response! in! schizophrenia! *! lessons! from! a! case! of! three! affected!siblings.!Pharmacopsychiatry,!43(5):!196*197.!!
Hu! G,! Schones! DE,! Cui! K,! Ybarra! R,! Northrup! D,! Tang! Q,! .! .! .! Zhao! K.! (2011).! Regulation! of!nucleosome!landscape!and!transcription!factor!targeting!at!tissue*specific!enhancers!by!BRG1.!Genome!Research,!21(10):!1650*1658.!!
Huettner! JE.! (2003).!Kainate! receptors!and!synaptic! transmission.!Progress! in!Neurobiology,!70(5):!387*407.!!
Hwang! R,! Souza! RP,! Tiwari! AK,! Zai! CC,!Muller! DJ,! Potkin! SG,! .! .! .! Kennedy! JL.! (2011).! Gene*gene!interaction! analyses! between!NMDA! receptor! subunit! and! dopamine! receptor! gene! variants!and!clozapine!response.!Pharmacogenomics,!12(2):!277*291.!!
Ichise!T,!Kano!M,!Hashimoto!K,!Yanagihara!D,!Nakao!K,!Shigemoto!R,!.!.!.!Aiba!A.!(2000).!mGluR1!in!cerebellar! purkinje! cells! essential! for! long*term! depression,! synapse! elimination,! and!motor!coordination.!Science,!288(5472):!1832*1835.!!
Idanpaan*Heikkila! J,! Alhava! E,! Olkinuora!M! &! Palva! IP.! (1997).! Agranulocytosis! during! treatment!with!clozapine.!European!Journal!of!Clinical!Pharmacology,!11:!193*198.!!
Insel! TR! &! Scolnick! EM.! (2006).! Cure! therapeutics! and! strategic! prevention:! Raising! the! bar! for!mental!health!research.!Molecular!Psychiatry,!11(1):!11*17.!!
International! HapMap! Consortium.! (2003).! The! international! HapMap! project.! Nature,! 426(6968):!789*796.!!

144!!
!!
International! Schizophrenia! Consortium,! Purcell! SM,!Wray! NR,! Stone! JL,! Visscher! PM,! O'Donovan!MC,! .! .! .!Sklar!P.! (2009).!Common!polygenic!variation!contributes!to!risk!of!schizophrenia!and!bipolar!disorder.!Nature,!460(7256):!748*752.!!
Ishimaru! M,! Kurumaji! A! &! Toru! M.! (1992).! NMDA*associated! glycine! binding! site! increases! in!schizophrenic!brains.!Biological!Psychiatry,!32(4):!379*381.!!
Jablensky! A,! Sartorius! N,! Ernberg! G,! Anker! M,! Korten! A,! Cooper! JE,! .! .! .! Bertelsen! A.! (1992).!Schizophrenia:! Manifestations,! incidence! and! course! in! different! cultures.! A! world! health!organization!ten*country!study.!Psychological!Medicine!Monograph,!Supplemental,!20:!S1*S97.!!
Janca!A,!Kastrup!M,!Katschnig!H,!Lopez*Ibor!JJ!Jr,!Mezzich!JE!&!Sartorius!N.!(1996).!The!World!Health!Organization!short!disability!assessment!schedule! (WHO!DAS*S):!A!tool! for! the!assessment!of!difficulties!in!selected!areas!of!functioning!of!patients!with!mental!disorders.!Social!Psychiatry!and!Psychiatric!Epidemiology,!31(6):!349*354.!!
Javitt! DC! &! Zukin! SR.! (1991).! Recent! advances! in! the! phencyclidine!model! of! schizophrenia.! The!American!Journal!of!Psychiatry,!148(10):!1301*1308.!!
Javitt!DC,!Sershen!H,!Hashim!A!&!Lajtha!A.!(1997).!Reversal!of!phencyclidine*induced!hyperactivity!by! glycine! and! the! glycine! uptake! inhibitor! glycyldodecylamide.! Neuropsychopharmacology,!17(3):!202*204.!
Javitt!DC.! (2004).!Glutamate!as!a! therapeutic! target! in!psychiatric!disorders.!Molecular!Psychiatry,!9(11):!984*97.!!
Javitt!DC,!Duncan!L,!Balla!A!&!Sershen!H.!(2005).!Inhibition!of!system!A*mediated!glycine!transport!in! cortical! synaptosomes! by! therapeutic! concentrations! of! clozapine:! Implications! for!mechanisms!of!action.!Molecular!Psychiatry,!10(3):!275*287.!!
Javitt!DC,!Silipo!G,!Cienfuegos!A,!Shelley!AM,!Bark!N,!Park!M,!.!.!.!Zukin!SR.!(2001).!Adjunctive!high*dose! glycine! in! the! treatment! of! schizophrenia.! The! International! Journal! of!Neuropsychopharmacology,!4(4):!385*391.!
Jernigan!TL,!Zisook!S,!Heaton!RK,!Moranville!JT,!Hesselink!JR!&!Braff!DL.!(1991).!Magnetic!resonance!imaging! abnormalities! in! lenticular! nuclei! and! cerebral! cortex! in! schizophrenia.! Archives! of!General!Psychiatry,!48(10):!881*890.!!
Jia!P,!Wang!L,!Fanous!AH,!Pato!CN,!Edwards!TL,! International!Schizophrenia!Consortium!&!Zhao!Z.!(2012).! Network*assisted! investigation! of! combined! causal! signals! from! genome*wide!association!studies!in!schizophrenia.!PLoS!Computational!Biology,!8(7):!e1002587.!!
Johnson! JA,!Gong! L,!Whirl*Carrillo!M,!Gage!BF,! Scott! SA,! Stein!CM,! .! .! .! Clinical! Pharmacogenetics!Implementation! Consortium.! (2011).! Clinical! Pharmacogenetics! Implementation! Consortium!guidelines!for!CYP2C9!and!VKORC1!genotypes!and!warfarin!dosing.!Clinical!Pharmacology!and!Therapeutics,!90(4):!625*629.!

145!!
!!
Johnson! JW!&!Ascher!P.! (1987).!Glycine!potentiates! the!NMDA! response! in! cultured!mouse!brain!neurons.!Nature,!325(6104):!529*531.!!
Johnstone!EC,!Crow!TJ,!Frith!CD,!Husband!J!&!Kreel!L.!(1976).!Cerebral!ventricular!size!and!cognitive!impairment!in!chronic!schizophrenia.!Lancet,!2(7992):!924*926.!!
Jones!EM,!Fernald!A,!Bell!GI!&!Le!Beau!MM.!(1995).!Assignment!of!SLC6A9!to!human!chromosome!band!1p33!by!in!situ!hybridization.!Cytogenetics!and!Cell!Genetics,!71(3):!211.!!
Joo! A,! Shibata! H,! Ninomiya! H,! Kawasaki! H,! Tashiro! N! &! Fukumaki! Y.! (2001).! Structure! and!polymorphisms!of!the!human!metabotropic!glutamate!receptor!type!2!gene!(GRM2):!Analysis!of!association!with!schizophrenia.!Molecular!Psychiatry,!6(2):!186*192.!!
Kalow!W.! (1962).! Pharmacogenetics:! Heredity! and! the! response! to! drugs.! Philadelphia,! USA:!WB!Saunders!&!Co.!!
Kalsi!G,!Kuo!PH,!Aliev!F,!Alexander!J,!McMichael!O,!Patterson!DG,!.!.!.!Riley!BP.!(2010).!A!systematic!gene*based!screen!of!chr4q22*q32! identifies!association!of!a!novel! susceptibility!gene,!DKK2,!with!the!quantitative!trait!of!alcohol!dependence!symptom!counts.!Human!Molecular!Genetics,!19(12):!2497*2506.!!
Kane! J,! Honigfeld! G,! Singer! J! &! Meltzer! H.! (1988).! Clozapine! for! the! treatment*resistant!schizophrenic.!A!double*blind!comparison!with!chlorpromazine.!Archives!of!General!Psychiatry,!45(9):!789*796.!!
Kargieman!L,!Santana!N,!Mengod!G,!Celada!P!&!Artigas!F.! (2007).!Antipsychotic!drugs! reverse! the!disruption! in! prefrontal! cortex! function! produced! by! NMDA! receptor! blockade! with!phencyclidine.! Proceedings! of! the! National! Academy! of! Sciences! of! the! United! States! of!America,!104(37):!14843*14848.!!
Karreman! M! &! Moghaddam! B.! (1996).! The! prefrontal! cortex! regulates! the! basal! release! of!dopamine! in! the! limbic! striatum:! An! effect! mediated! by! ventral! tegmental! area.! Journal! of!Neurochemistry,!66(2):!589*598.!!
Kasai! K,! Shenton!ME,! Salisbury! DF,! Hirayasu! Y,! Lee! CU,! Ciszewski! AA,! .! .! .! McCarley! RW.! (2003).!Progressive!decrease!of!left!superior!temporal!gyrus!gray!matter!volume!in!patients!with!first*episode!schizophrenia.!The!American!Journal!of!Psychiatry,!160(1):!156*164.!!
Kattenstroth!G,!Tantalaki!E,!Sudhof!TC,!Gottmann!K!&!Missler!M.!(2004).!Postsynaptic!N*methyl*D*aspartate!receptor!function!requires!alpha*neurexins.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!101(8):!2607*2612.!!
Kay! SR,! Fiszbein! A! &! Opler! LA.! (1987).! The! positive! and! negative! syndrome! scale! (PANSS)! for!schizophrenia.!Schizophrenia!Bulletin,!13(2):!261*276.!!

146!!
!!
Kelleher!RJ!3rd,!Geigenmuller!U,!Hovhannisyan!H,!Trautman!E,!Pinard!R,!Rathmell!B,!.!.!.!Margulies!D.!(2012).!High*throughput!sequencing!of!mGluR!signaling!pathway!genes!reveals!enrichment!of!rare!variants!in!autism.!PloS!One,!7(4):!e35003.!!
Kemether!EM,!Buchsbaum!MS,!Byne!W,!Hazlett!EA,!Haznedar!M,!Brickman!AM,!.!.!.!Bloom!R.!(2003).!Magnetic! resonance! imaging! of! mediodorsal,! pulvinar,! and! centromedian! nuclei! of! the!thalamus!in!patients!with!schizophrenia.!Archives!of!General!Psychiatry,!60(10):!983*991.!!
Kendler!KS.!(1983).!Overview:!A!current!perspective!on!twin!studies!of!schizophrenia.!The!American!Journal!of!Psychiatry,!140(11):!1413*1425.!!
Kennedy!JL,!Altar!CA,!Taylor!DL,!Degtiar!I!&!Hornberger!JC.!(2014).!The!social!and!economic!burden!of! treatment*resistant! schizophrenia:! A! systematic! literature! review.! International! Clinical!Psychopharmacology,!29(2):!63*76.!!
Kenny!EM,!Cormican!P,!Furlong!S,!Heron!E,!Kenny!G,!Fahey!C,!.!.!.!Morris!DW.!(2014).!Excess!of!rare!novel! loss*of*function! variants! in! synaptic! genes! in! schizophrenia! and! autism! spectrum!disorders.!Molecular!Psychiatry,!19(8):!872*879.!!
Kent!WJ,!Sugnet!CW,!Furey!TS,!Roskin!KM,!Pringle!TH,!Zahler!AM!&!Haussler!D.!(2002).!The!human!genome!browser!at!UCSC.!Genome!Research,!12(6):!996*1006.!!
Kety!SS.!(1959).!Biochemical!theories!of!schizophrenia.!II.!Science,!129(3363):!1590*1596.!!
Khakh!BS!&!Henderson!G.! (2000).!Modulation!of! fast! synaptic! transmission!by!presynaptic! ligand*gated!cation!channels.!Journal!of!the!Autonomic!Nervous!System,!81(1*3):!110*121.!!
Kharazia!VN,!Phend!KD,!Rustioni!A!&!Weinberg!RJ.! (1996).!EM!colocalization!of!AMPA!and!NMDA!receptor!subunits!at!synapses!in!rat!cerebral!cortex.!Neuroscience!Letters,!210(1):!37*40.!!
Kikuchi!G.!(1973).!The!glycine!cleavage!system:!Composition,!reaction!mechanism,!and!physiological!significance.!Molecular!and!Cellular!Biochemistry,!1(2):!169*187.!!
Kinney!GG,!Sur!C,!Burno!M,!Mallorga!PJ,!Williams!JB,!Figueroa!DJ,! .! .! .!Conn!PJ.!(2003).!The!glycine!transporter! type! 1! inhibitor! N*[3*(4'*fluorophenyl)*3*(4'*phenylphenoxy)propyl]sarcosine!potentiates!NMDA!receptor*mediated!responses! in!vivo!and!produces!an!antipsychotic!profile!in!rodent!behavior.!The!Journal!of!Neuroscience,!23(20):!7586*7591.!!
Kinon! BJ,! Liu*Seifert! H,! Adams! DH! &! Citrome! L.! (2006).! Differential! rates! of! treatment!discontinuation! in! clinical! trials! as! a!measure! of! treatment! effectiveness! for! olanzapine! and!comparator!atypical!antipsychotics!for!schizophrenia.! Journal!of!Clinical!Psychopharmacology,!26(6):!632*637.!
Kinon!BJ,!Zhang!L,!Millen!BA,!Osuntokun!OO,!Williams!JE,!Kollack*Walker!S,! .! .! .!HBBI!Study!Group.!(2011).!A!multicenter,!inpatient,!phase!2,!double*blind,!placebo*controlled!dose*ranging!study!of! LY2140023! monohydrate! in! patients! with! DSM*IV! schizophrenia.! Journal! of! Clinical!Psychopharmacology,!31(3):!349*355.!!

147!!
!!
Kinon!BJ,!Millen!BA!&!Downing!AC.! (2013).! LY2140023!monohydrate! in! the! treatment!of!patients!with!schizophrenia:!Results!of!2!clinical! trials!assessing!efficacy! in! treating!acutely! ill!patients!and! those! with! prominent! negative! symptoms.! Schizophrenia! Bulletin,! Supplemental,! 39(1):!S338.!!
Kleckner! NW! &! Dingledine! R.! (1988).! Requirement! for! glycine! in! activation! of! NMDA*receptors!expressed!in!xenopus!oocytes.!Science,!241(4867):!835*837.!!
Klosterkotter!J,!Hellmich!M,!Steinmeyer!EM!&!Schultze*Lutter!F.!(2001).!Diagnosing!schizophrenia!in!the!initial!prodromal!phase.!Archives!of!General!Psychiatry,!58(2):!158*164.!!
Kohlrausch! FB.! (2013).! Pharmacogenetics! in! schizophrenia:! A! review! of! clozapine! studies.! Revista!Brasileira!De!Psiquiatria,!35(3):!305*317.!!
Kolachana! BS,! Saunders! RC! &! Weinberger! DR.! (1995).! Augmentation! of! prefrontal! cortical!monoaminergic! activity! inhibits! dopamine! release! in! the! caudate! nucleus:! An! in! vivo!neurochemical!assessment!in!the!rhesus!monkey.!Neuroscience,!69(3):!859*868.!!
Komuro! H! &! Rakic! P.! (1993).! Modulation! of! neuronal! migration! by! NMDA! receptors.! Science,!260(5104):!95*97.!!
Kraepelin! E.! (1899).! Diagnosis! and! prognosis! of! dementia! praecox.! Allgemeine! Zeitschrift! Fur!Psychiatrie,!56:!254*263.!!
Kraguljac! NV,! White! DM,! Reid! MA! &! Lahti! AC.! (2013).! Increased! hippocampal! glutamate! and!volumetric!deficits!in!unmedicated!patients!with!schizophrenia.!JAMA!Psychiatry,!70(12):!1294*1302.!!
Kreyenbuhl! J,! Buchanan!RW,!Dickerson! FB,!Dixon! LB!&! Schizophrenia! Patient!Outcomes!Research!Team! (PORT).! (2010).! The! schizophrenia! patient! outcomes! research! team! (PORT):! Updated!treatment!recommendations!2009.!Schizophrenia!Bulletin,!36(1):!94*103.!!
Kubota! T,! Jibiki! I,! Kishizawa! S! &! Kurokawa! K.! (1996).! Clozapine*induced! potentiation! of! synaptic!responses! in! the! perforant! path*dentate! gyrus! pathway! in! chronically! prepared! rabbits.!Neuroscience!Letters,!211(1):!21*24.!!
Kubota!T,! Jibiki! I,! Enokido!F,!Nakagawa!H!&!Watanabe!K.! (2000).! Effects!of!MK*801!on!clozapine*induced! potentiation! of! excitatory! synaptic! responses! in! the! perforant! path*dentate! gyrus!pathway!in!chronically!prepared!rabbits.!European!Journal!of!Pharmacology,!395(1):!37*42.!!
Kuryatov!A,!Laube!B,!Betz!H!&!Kuhse!J.!(1994).!Mutational!analysis!of!the!glycine*binding!site!of!the!NMDA!receptor:!Structural!similarity!with!bacterial!amino!acid*binding!proteins.!Neuron,!12(6):!1291*1300.!!
Laborit! H! &! Huguenard! P.! (1951).! L'hibernation! artificielle! par! moyen! pharmacodynamiques! et!physiques.!La!Presse!Médical,!59:!1329.!!

148!!
!!
Lacro! JP,!Dunn! LB,!Dolder!CR,! Leckband! SG!&! Jeste!DV.! (2002).! Prevalence!of! and! risk! factors! for!medication! nonadherence! in! patients!with! schizophrenia:! A! comprehensive! review!of! recent!literature.!The!Journal!of!Clinical!Psychiatry,!63(10):!892*909.!!
Lahiri!DK!&!Nurnberger! JI! Jr.! (1991).!A! rapid!non*enzymatic!method! for! the!preparation!of!HMW!DNA!from!blood!for!RFLP!studies.!Nucleic!Acids!Research,!19(19):!5444.!!
Lahti!AC,!Weiler!MA,!Tamara*Michaelidis!BA,!Parwani!A!&!Tamminga!CA.!(2001).!Effects!of!ketamine!in!normal!and!schizophrenic!volunteers.!Neuropsychopharmacology,!25(4):!455*467.!!
Lander!ES,!Linton!LM,!Birren!B,!Nusbaum!C,!Zody!MC,!Baldwin!J,!.!.!.!International!Human!Genome!Sequencing!Consortium.!(2001).!Initial!sequencing!and!analysis!of!the!human!genome.!Nature,!409(6822):!860*921.!!
Larson! MK,! Walker! EF! &! Compton! MT.! (2010).! Early! signs,! diagnosis! and! therapeutics! of! the!prodromal! phase! of! schizophrenia! and! related! psychotic! disorders.! Expert! Review! of!Neurotherapeutics,!10(8):!1347*1359.!
Laube! B,! Hirai! H,! Sturgess! M,! Betz! H! &! Kuhse! J.! (1997).! Molecular! determinants! of! agonist!discrimination!by!NMDA!receptor!subunits:!Analysis!of!the!glutamate!binding!site!on!the!NR2B!subunit.!Neuron,!18(3):!493*503.!!
Laursen!TM,!Munk*Olsen!T!&!Vestergaard!M.!(2012).!Life!expectancy!and!cardiovascular!mortality!in!persons!with!schizophrenia.!Current!Opinion!in!Psychiatry,!25(2):!83*88.!!
Lee!SH,!Kubicki!M,!Asami!T,!Seidman!LJ,!Goldstein!JM,!Mesholam*Gately!RI,!.!.!.!Shenton!ME.!(2013).!Extensive!white!matter!abnormalities! in!patients!with! first*episode!schizophrenia:!A!diffusion!tensor!imaging!(DTI)!study.!Schizophrenia!Research,!143(2*3):!231*238.!!
Lee!YL,!Teitelbaum!S,!Wolff!MS,!Wetmur!JG!&!Chen!J.!(2010).!Comparing!genetic!ancestry!and!self*reported!race/ethnicity!in!a!multiethnic!population!in!New!York!City.!Journal!of!Genetics,!89(4):!417*423.!!
Lemonde! S,! Turecki! G,! Bakish! D,! Du! L,! Hrdina! PD,! Bown! CD,! .! .! .! Albert! PR.! (2003).! Impaired!repression! at! a! 5*hydroxytryptamine! 1A! receptor! gene! polymorphism! associated!with!major!depression!and!suicide.!The!Journal!of!Neuroscience,!23(25):!8788*8799.!!
Lemonde! S,! Du! L,! Bakish! D,! Hrdina! P! &! Albert! PR.! (2004).! Association! of! the! C(*1019)G! 5PHT1A!functional!promoter!polymorphism!with!antidepressant!response.!The!International!Journal!of!Neuropsychopharmacology,!7(4):!501*506.!
Lett!TA,!Tiwari!AK,!Meltzer!HY,!Lieberman!JA,!Potkin!SG,!Voineskos!AN,! .! .! .!Muller!DJ.! (2011).!The!putative! functional! rs1045881!marker!of!neurexin*1! in!schizophrenia!and!clozapine!response.!Schizophrenia!Research,!132(2*3):!121*124.!!

149!!
!!
Levin!ED,!Caldwell!DP!&!Perraut!C.!(2007).!Clozapine!treatment!reverses!dizocilpine*induced!deficits!of!pre*pulse! inhibition!of! tactile! startle! response.! Pharmacology,!Biochemistry,!and!Behavior,!86(3):!597*605.!!
Lewis! CM! &! Knight! J.! (2012).! Introduction! to! genetic! association! studies.! Cold! Spring! Harbor!Protocols,!2012(3):!297*306.!!
Lewis!SW,!Ford!RA,!Syed!GM,!Reveley!AM!&!Toone!BK.!(1992).!A!controlled!study!of!99mTc*HMPAO!single*photon!emission!imaging!in!chronic!schizophrenia.!Psychological!Medicine,!22(1):!27*35.!!
Li!ML,!Hu!XQ,!Li!F!&!Gao!WJ.!(2015).!Perspectives!on!the!mGluR2/3!agonists!as!a!therapeutic!target!for! schizophrenia:! Still! promising! or! a! dead! end?! Progress! in! NeuroPPsychopharmacology! &!Biological!Psychiatry,!60:!66*76.!!
Li! X! &! Francke! U.! (1995).! Assignment! of! the! gene! SLC1A2! coding! for! the! human! glutamate!transporter!EAAT2!to!human!chromosome!11!bands!p13*p12.!Cytogenetics!and!Cell!Genetics,!71(3):!212*213.!!
Lieberman! JA,! Kane! JM! &! Alvir! J.! (1987).! Provocative! tests! with! psychostimulant! drugs! in!schizophrenia.!Psychopharmacology,!91(4):!415*433.!!
Lieberman!JA,!Safferman!AZ,!Pollack!S,!Szymanski!S,!Johns!C,!Howard!A,!.!.!.!Kane!JM.!(1994).!Clinical!effects! of! clozapine! in! chronic! schizophrenia:! Response! to! treatment! and! predictors! of!outcome.!The!American!Journal!of!Psychiatry,!151(12):!1744*1752.!!
Lieberman!JA,!Bymaster!FP,!Meltzer!HY,!Deutch!AY,!Duncan!GE,!Marx!CE,!.!.!.!Csernansky!JG.!(2008).!Antipsychotic! drugs:! Comparison! in! animal! models! of! efficacy,! neurotransmitter! regulation,!and!neuroprotection.!Pharmacological!Reviews,!60(3):!358*403.!!
Life! Technologies.! (2011).! Ion! torrent! amplicon! sequencing! applicaiton! note.! Unpublished!manuscript.!!
Lin!MW,!Curtis!D,!Williams!N,!Arranz!M,!Nanko!S,!Collier!D,!.!.!.!Gill!M.!(1995).!Suggestive!evidence!for! linkage! of! schizophrenia! to! markers! on! chromosome! 13q14.1*q32.! Psychiatric! Genetics,!5(3):!117*126.!!
Lindenmayer! J! &! Apergi! F.! (1996).! The! relationship! between! clozapine! plasma! levels! and! clinical!response.!Psychiatric!Annals,!26(7):!406*412.!!
Lindstrom! LH.! (1988).! The! effect! of! long*term! treatment! with! clozapine! in! schizophrenia:! A!retrospective!study!in!96!patients!treated!with!clozapine!for!up!to!13!years.!Acta!Psychiatrica!Scandinavica,!77(5):!524*529.!!
Linn! GS,! Negi! SS,! Gerum! SV! &! Javitt! DC.! (2003).! Reversal! of! phencyclidine*induced! prepulse!inhibition!deficits!by!clozapine!in!monkeys.!Psychopharmacology,!169(3*4):!234*239.!!

150!!
!!
Lipska! BK,! Swerdlow! NR,! Geyer! MA,! Jaskiw! GE,! Braff! DL! &! Weinberger! DR.! (1995).! Neonatal!excitotoxic!hippocampal!damage!in!rats!causes!post*pubertal!changes!in!prepulse!inhibition!of!startle!and!its!disruption!by!apomorphine.!Psychopharmacology,!122(1):!35*43.!!
Liu!N,! Zhao!H,! Patki! A,! Limdi!NA!&!Allison!DB.! (2011).! Controlling! population! structure! in! human!genetic! association! studies!with! samples! of! unrelated! individuals.! Statistics! and! its! Interface,!4(3):!317*326.!!
Liu! W,! Downing! AC,! Munsie! LM,! Chen! P,! Reed! MR,! Ruble! CL,! .! .! .! Nisenbaum! LK.! (2012).!Pharmacogenetic!analysis!of!the!mGlu2/3!agonist!LY2140023!monohydrate!in!the!treatment!of!schizophrenia.!The!Pharmacogenomics!Journal,!12(3):!246*254.!!
Lohmueller! KE,! Pearce! CL,! Pike! M,! Lander! ES! &! Hirschhorn! JN.! (2003).! Meta*analysis! of! genetic!association! studies! supports! a! contribution! of! common! variants! to! susceptibility! to! common!disease.!Nature!Genetics,!33(2):!177*182.!!
Long! AD! &! Langley! CH.! (1999).! The! power! of! association! studies! to! detect! the! contribution! of!candidate!genetic!loci!to!variation!in!complex!traits.!Genome!Research,!9(8):!720*731.!!
Luby! ED,! Cohen! BD,! Rosenbaum! G,! Gottlieb! JS! &! Kelley! R.! (1959).! Study! of! a! new!schizophrenomimetic!drug;!sernyl.!AMA!Archives!of!Neurology!and!Psychiatry,!81(3):!363*369.!!
Luisada! PV.! (1978).! The! phencyclidine! psychosis:! Phenomenology! and! treatment.! NIDA! Research!Monograph,!(21):!241*253.!!
Lundorf!MD,!Buttenschon!HN,!Foldager!L,!Blackwood!DH,!Muir!WJ,!Murray!V,! .! .! .!Mors!O.!(2005).!Mutational! screening! and! association! study! of! glutamate! decarboxylase! 1! as! a! candidate!susceptibility! gene! for! bipolar! affective! disorder! and! schizophrenia.! American! Journal! of!Medical!Genetics!Part!B,!135B(1):!94*101.!!
Lynex! CN,! Carr! IM,! Leek! JP,! Achuthan! R,! Mitchell! S,! Maher! ER,! .! .! .! Markham! AF.! (2004).!Homozygosity!for!a!missense!mutation!in!the!67!kDa!isoform!of!glutamate!decarboxylase!in!a!family!with!autosomal!recessive!spastic!cerebral!palsy:!Parallels!with!stiff*person!syndrome!and!other!movement!disorders.!BMC!Neurology,!4(1):!20.!!
Ma! Q! &! Lu! AY.! (2011).! Pharmacogenetics,! pharmacogenomics,! and! individualized! medicine.!Pharmacological!Reviews,!63(2):!437*459.!!
Malaspina!D,!Harlap! S,! Fennig! S,! Heiman!D,!Nahon!D,! Feldman!D!&! Susser! ES.! (2001).! Advancing!paternal!age!and!the!risk!of!schizophrenia.!Archives!of!General!Psychiatry,!58(4):!361*367.!!
Malhotra! AK,! Adler! CM,! Kennison! SD,! Elman! I,! Pickar! D! &! Breier! A.! (1997).! Clozapine! blunts! N*methyl*D*aspartate!antagonist*induced!psychosis:!A!study!with!ketamine.!Biological!Psychiatry,!42(8):!664*668.!!

151!!
!!
Malhotra!AK,!Correll!CU,!Chowdhury!NI,!Muller!DJ,!Gregersen!PK,!Lee!AT,! .! .! .!Kennedy! JL.! (2012).!Association! between! common! variants! near! the! melanocortin! 4! receptor! gene! and! severe!antipsychotic!drug*induced!weight!gain.!Archives!of!General!Psychiatry,!69(9):!904*912.!!
Mallolas! J,! Hurtado! O,! Castellanos! M,! Blanco! M,! Sobrino! T,! Serena! J,! .! .! .! Davalos! A.! (2006).! A!polymorphism!in!the!EAAT2!promoter!is!associated!with!higher!glutamate!concentrations!and!higher!frequency!of!progressing!stroke.!The!Journal!of!Experimental!Medicine,!203(3):!711*717.!!
Mandich!P,!Schito!AM,!Bellone!E,!Antonacci!R,!Finelli!P,!Rocchi!M!&!Ajmar!F.!(1994).!Mapping!of!the!human! NMDAR2B! receptor! subunit! gene! (GRIN2B)! to! chromosome! 12p12.! Genomics,! 22(1):!216*218.!!
Marcelis!M,!Navarro*Mateu!F,!Murray!R,!Selten!JP!&!Van!Os!J.!(1998).!Urbanization!and!psychosis:!A!study!of!1942*1978!birth!cohorts!in!the!Netherlands.!Psychological!Medicine,!28(4):!871*879.!!
Martin! S,! Henley! JM,! Holman! D,! Zhou! M,! Wiegert! O,! van! Spronsen! M,! .! .! .! Krugers! HJ.! (2009).!Corticosterone!alters!AMPAR!mobility!and!facilitates!bidirectional!synaptic!plasticity.!PloS!One,!4(3):!e4714.!!
Martinez*Hernandez!A,!Bell!KP!&!Norenberg!MD.!(1977).!Glutamine!synthetase:!Glial!localization!in!brain.!Science,!195(4284):!1356*1358.!!
Martucci! L!&!Kennedy! JL.! (2010).!Predicting!negative! symptom!change!during!drug! treatment.!US!Patent!7,794,934.!!
Masellis! M,! Paterson! AD,! Badri! F,! Lieberman! JA,! Meltzer! HY,! Cavazzoni! P! &! Kennedy! JL.! (1995).!Genetic!variation!of!5*HT2A!receptor!and!response!to!clozapine.!Lancet,!346(8982):!1108.!!
Massey! PV,! Johnson! BE,! Moult! PR,! Auberson! YP,! Brown! MW,! Molnar! E,! .! .! .! Bashir! ZI.! (2004).!Differential! roles! of! NR2A! and! NR2B*containing! NMDA! receptors! in! cortical! long*term!potentiation!and!long*term!depression.!The!Journal!of!Neuroscience,!24(36):!7821*7828.!!
Mata! I,!Madoz! V,! Arranz!MJ,! Sham! P!&!Murray! RM.! (2001).! Olanzapine:! Concordant! response! in!monozygotic!twins!with!schizophrenia.!The!British!Journal!of!Psychiatry,!178(1):!86.!!
Mathalon!DH,!Sullivan!EV,!Lim!KO!&!Pfefferbaum!A.!(2001).!Progressive!brain!volume!changes!and!the!clinical!course!of!schizophrenia!in!men:!A!longitudinal!magnetic!resonance!imaging!study.!Archives!of!General!Psychiatry,!58(2):!148*157.!!
Mathieu!G,! Groisman! IJ! &!Godard! B.! (2013).! Next! generation! sequencing! in! psychiatric! research:!What! study! participants! need! to! know! about! research! findings.! The! International! Journal! of!Neuropsychopharmacology,!16(9):!2119*2127.!!
Matsui! T,! Sekiguchi! M,! Hashimoto! A,! Tomita! U,! Nishikawa! T! &! Wada! K.! (1995).! Functional!comparison!of!D*serine!and!glycine!in!rodents:!The!effect!on!cloned!NMDA!receptors!and!the!extracellular!concentration.!Journal!of!Neurochemistry,!65(1):!454*458.!!

152!!
!!
Matys! V,! Kel*Margoulis! OV,! Fricke! E,! Liebich! I,! Land! S,! Barre*Dirrie! A,! .! .! .! Wingender! E.! (2006).!TRANSFAC! and! its! module! TRANSCompel:! Transcriptional! gene! regulation! in! eukaryotes.!Nucleic!Acids!Research,!Database!Issue,!34:!D108*D110.!!
Mayer! ML,! Westbrook! GL! &! Guthrie! PB.! (1984).! Voltage*dependent! block! by! Mg2+! of! NMDA!responses!in!spinal!cord!neurones.!Nature,!309(5965):!261*263.!!
Mayer! ML,! Vyklicky! L! Jr! &! Clements! J.! (1989).! Regulation! of! NMDA! receptor! desensitization! in!mouse!hippocampal!neurons!by!glycine.!Nature,!338(6214):!425*427.!!
Maziade!M,!Raymond!V,!Cliche!D,! Fournier! JP,!Caron!C,!Garneau!Y,! .! .! .! Simard!C.! (1995).! Linkage!results!on!11Q21*22!in!Eastern!Quebec!pedigrees!densely!affected!by!schizophrenia.!American!Journal!of!Medical!Genetics,!60(6):!522*528.!!
McBain! CJ! &! Mayer! ML.! (1994).! N*methyl*D*aspartic! acid! receptor! structure! and! function.!Physiological!Reviews,!74(3):!723*760.!!
McCarley! RW,!Wible! CG,! Frumin!M,! Hirayasu! Y,! Levitt! JJ,! Fischer! IA! &! Shenton!ME.! (1999).! MRI!anatomy!of!schizophrenia.!Biological!Psychiatry,!45(9):!1099*1119.!!
McClellan! JM,! Susser! E!&! King!MC.! (2007).! Schizophrenia:! A! common! disease! caused! by!multiple!rare!alleles.!The!British!Journal!of!Psychiatry,!190:!194*199.!!
McEvoy!JP,!Lieberman!JA,!Stroup!TS,!Davis!SM,!Meltzer!HY,!Rosenheck!RA,!.!.!.!CATIE!Investigators.!(2006).! Effectiveness! of! clozapine! versus! olanzapine,! quetiapine,! and! risperidone! in! patients!with!chronic!schizophrenia!who!did!not!respond!to!prior!atypical!antipsychotic!treatment.!The!American!Journal!of!Psychiatry,!163(4):!600*610.!!
McGlashan!TH.! (1988).!A!selective!review!of!recent!North!American! long*term!followup!studies!of!schizophrenia.!Schizophrenia!Bulletin,!14(4):!515*542.!!
McGrath!J,!Saha!S,!Welham!J,!El!Saadi!O,!MacCauley!C!&!Chant!D.!(2004).!A!systematic!review!of!the!incidence! of! schizophrenia:! The! distribution! of! rates! and! the! influence! of! sex,! urbanicity,!migrant!status!and!methodology.!BMC!Medicine,!2:!13.!!
McGrath! J,! Saha! S,! Chant!D!&!Welham! J.! (2008).! Schizophrenia:!A! concise! overview!of! incidence,!prevalence,!and!mortality.!Epidemiologic!Reviews,!30:!67*76.!!
McGurk!SR.!(1999).!The!effects!of!clozapine!on!cognitive!functioning!in!schizophrenia.!The!Journal!of!Clinical!Psychiatry,!Supplemental,!60(S12):!S24*S29.!!
McIlwain!ME,!Harrison!J,!Wheeler!AJ!&!Russell!BR.!(2011).!Pharmacotherapy!for!treatment*resistant!schizophrenia.!Neuropsychiatric!Disease!and!Treatment,!7:!135*149.!!
Meister!A.!(1974).!Glutamine!synthetase!of!mammals.!In!PD!Boyer!(Ed.),!The!enzymes!(Vol!10).!New!York:!Academic!Press.!!

153!!
!!
Melone!M,!Vitellaro*Zuccarello! L,!Vallejo*Illarramendi!A,!Perez*Samartin!A,!Matute!C,!Cozzi!A,! .! .! .!Conti! F.! (2001).! The! expression! of! glutamate! transporter! GLT*1! in! the! rat! cerebral! cortex! is!down*regulated!by!the!antipsychotic!drug!clozapine.!Molecular!Psychiatry,!6(4):!380*386.!!
Meltzer!HY,!Matsubara!S!&!Lee!JC.!(1989a).!Classification!of!typical!and!atypical!antipsychotic!drugs!on! the! basis! of! dopamine!D*1,!D*2! and! serotonin*2! pKi! values.! The! Journal! of! Pharmacology!and!Experimental!Therapeutics,!251(1):!238*246.!!
Meltzer!HY,!Bastani!B,!Kwon!KY,!Ramirez!LF,!Burnett!S!&!Sharpe!J.!(1989b).!A!prospective!study!of!clozapine! in! treatment*resistant! schizophrenic! patients.! I.! Preliminary! report.!Psychopharmacology,!Supplemental,!99:!S68*S72.!!
Meltzer! HY.! (1992).! Treatment! of! the! neuroleptic*nonresponsive! schizophrenic! patient.!Schizophrenia!Bulletin,!18(3):!515*542.!!
Meltzer!HY.! (1995a).! The! role! of! serotonin! in! schizophrenia! and! the!place!of! serotonin*dopamine!antagonist!antipsychotics.!Journal!of!Clinical!Psychopharmacology,!Supplemental,!15(1,!S1):!2S*3S.!!
Meltzer!HY!&!Okayli!G.!(1995b).!Reduction!of!suicidality!during!clozapine!treatment!of!neuroleptic*resistant!schizophrenia:!Impact!on!risk*benefit!assessment.!The!American!Journal!of!Psychiatry,!152(2):!183*190.!!
Meltzer!HY,!Bobo!WV,!Roy!A,!Jayathilake!K,!Chen!Y,!Ertugrul!A,!.!.!.!Small!JG.!(2008).!A!randomized,!double*blind!comparison!of!clozapine!and!high*dose!olanzapine!in!treatment*resistant!patients!with!schizophrenia.!The!Journal!of!Clinical!Psychiatry,!69(2):!274*285.!!
Mendel!G.!(1866).!Versuche!über!pflanzen*hybriden.!Verhandlungen!Des!Naturforschenden!Vereines!in!Brünn!(Abhandlungen),!4:!3*47.!!
Meshul!CK,!Bunker!GL,!Mason!JN,!Allen!C!&!Janowsky!A.!(1996).!Effects!of!subchronic!clozapine!and!haloperidol!on!striatal!glutamatergic!synapses.!Journal!of!Neurochemistry,!67(5):!1965*1973.!!
Meyer! JM.! (2001).! Individual! changes! in! clozapine! levels! after! smoking! cessation:! Results! and! a!predictive!model.!Journal!of!Clinical!Psychopharmacology,!21(6):!569*574.!!
Miyamoto!S,!Duncan!GE,!Marx!CE!&!Lieberman!JA.!(2005).!Treatments!for!schizophrenia:!A!critical!review! of! pharmacology! and! mechanisms! of! action! of! antipsychotic! drugs.! Molecular!Psychiatry,!10(1):!79*104.!!
Miyoshi!K,!Honda!A,!Baba!K,!Taniguchi!M,!Oono!K,!Fujita!T,! .! .! .!Tohyama!M.! (2003).!Disrupted*in*schizophrenia! 1,! a! candidate! gene! for! schizophrenia,! participates! in! neurite! outgrowth.!Molecular!Psychiatry,!8(7):!685*694.!!
Moghaddam! B! &! Javitt! D.! (2012).! From! revolution! to! evolution:! The! glutamate! hypothesis! of!schizophrenia!and!its!implication!for!treatment.!Neuropsychopharmacology,!37(1):!4*15.!!

154!!
!!
Mohn! AR,! Gainetdinov! RR,! Caron! MG! &! Koller! BH.! (1999).! Mice! with! reduced! NMDA! receptor!expression!display!behaviors!related!to!schizophrenia.!Cell,!98(4):!427*436.!!
Moniz! E.! (1937).! Prefrontal! leucotomy! in! the! treatment! of!mental! disorders.! Am! J! Psychiatry,! 93:!1379*1385.!!
Monn!JA,!Britton!TC,!Valli!MJ,!Massey!SM,!Henry!SS,!De!Dios!A,!.!.!.!Lindstrom!T.!(2005).!Discovery!of!LY2140023,! a! peptide! prodrug! of! the! mGlu2/3! receptor! agonist! LY404039.!Neuropharmacology,!49:!89.!!
Monyer! H,! Sprengel! R,! Schoepfer! R,! Herb! A,! Higuchi! M,! Lomeli! H,! .! .! .! Seeburg! PH.! (1992).!Heteromeric! NMDA! receptors:! Molecular! and! functional! distinction! of! subtypes.! Science,!256(5060):!1217*1221.!!
Moore! JH.! (2004).! Computational! analysis! of! gene*gene! interactions! using! multifactor!dimensionality!reduction.!Expert!Review!of!Molecular!Diagnostics,!4(6):!795*803.!!
Moore!TA,!Buchanan!RW,!Buckley!PF,!Chiles!JA,!Conley!RR,!Crismon!ML,!.! .! .!Miller!AL.!(2007).!The!Texas! medication! algorithm! project! antipsychotic! algorithm! for! schizophrenia:! 2006! update.!The!Journal!of!Clinical!Psychiatry,!68(11):!1751*1762.!!
Morel! BA.! (1860).!Traité! des! maladies! mentales.![Treatise! on! mental! diseases].! Paris,! France:!Masson.!
Morris! RG,! Anderson! E,! Lynch! GS! &! Baudry! M.! (1986).! Selective! impairment! of! learning! and!blockade! of! long*term! potentiation! by! an! N*methyl*D*aspartate! receptor! antagonist,! AP5.!Nature,!319(6056):!774*776.!!
Mortensen!PB,!Norgaard*Pedersen!B,!Waltoft!BL,!Sorensen!TL,!Hougaard!D,!Torrey!EF!&!Yolken!RH.!(2007).!Toxoplasma!gondii!as!a!risk!factor!for!early*onset!schizophrenia:!Analysis!of!filter!paper!blood!samples!obtained!at!birth.!Biological!Psychiatry,!61(5):!688*693.!!
Mortensen!PB,!Pedersen!CB,!Westergaard!T,!Wohlfahrt!J,!Ewald!H,!Mors!O,!.!.! .!Melbye!M.!(1999).!Effects!of! family!history!and!place!and!season!of!birth!on! the! risk!of! schizophrenia.! The!New!England!Journal!of!Medicine,!340(8):!603*608.!!
Mortimer!AM.!(2007).!Symptom!rating!scales!and!outcome!in!schizophrenia.!The!British!Journal!of!Psychiatry,!Supplemental,!50:!S7*S14.!!
Motsinger! AA,! Ritchie! MD! &! Reif! DM.! (2007).! Novel! methods! for! detecting! epistasis! in!pharmacogenomics!studies.!Pharmacogenomics,!8(9):!1229*1241.!!
Mouri!A,!Noda!Y,!Enomoto!T!&!Nabeshima!T.!(2007).!Phencyclidine!animal!models!of!schizophrenia:!Approaches! from! abnormality! of! glutamatergic! neurotransmission! and! neurodevelopment.!Neurochemistry!International,!51(2*4):!173*184.!!

155!!
!!
Mueller!HT,!Haroutunian!V,!Davis!KL!&!Meador*Woodruff! JH.! (2004).!Expression!of! the! ionotropic!glutamate! receptor! subunits! and! NMDA! receptor*associated! intracellular! proteins! in! the!substantia!nigra!in!schizophrenia.!Molecular!Brain!Research,!121(1*2):!60*69.!!
Mullis!K,!Faloona!F,!Scharf!S,!Saiki!R,!Horn!G!&!Erlich!H.!(1986).!Specific!enzymatic!amplification!of!DNA! in! vitro:! The! polymerase! chain! reaction.! Cold! Spring! Harbor! Symposia! on! Quantitative!Biology,!51(1):!263*273.!!
Murray!C!&!Lopez!A.! (Eds.).! (1996).!The!global!burden!of!disease:!A!comprehensive!assessment!of!mortality,! injuries,! and! risk! factors! in! 1990! and! projected! to! 2020! (Vol! 1).! Cambridge,! MA:!Harvard!University!Press.!!
Myles*Worsley!M,!Tiobech!J,!Browning!SR,!Korn!J,!Goodman!S,!Gentile!K,!.!.!.!Middleton!FA.!(2013).!Deletion! at! the! SLC1A1! glutamate! transporter! gene! co*segregates! with! schizophrenia! and!bipolar!schizoaffective!disorder!in!a!5*generation!family.!American!Journal!of!Medical!Genetics!Part!B,!Neuropsychiatric!Genetics,!162B(2):!87*95.!!
Nabeshima!T,!Fukaya!H,!Yamaguchi!K,!Ishikawa!K,!Furukawa!H!&!Kameyama!T.!(1987).!Development!of! tolerance! and! supersensitivity! to! phencyclidine! in! rats! after! repeated! administration! of!phencyclidine.!European!Journal!of!Pharmacology,!135(1):!23*33.!!
Nagai! Y,! Ohnuma! T,! Karibe! J,! Shibata! N,! Maeshima! H,! Baba! H,! .! .! .! Arai! H.! (2009).! No! genetic!association!between!the!SLC1A2!gene!and!Japanese!patients!with!schizophrenia.!Neuroscience!Letters,!463(3):!223*227.!!
Nakamura!K,!Kawasaki!Y,!Takahashi!T,!Furuichi!A,!Noguchi!K,!Seto!H!&!Suzuki!M.! (2012).!Reduced!white! matter! fractional! anisotropy! and! clinical! symptoms! in! schizophrenia:! A! voxel*based!diffusion!tensor!imaging!study.!Psychiatry!Research,!202(3):!233*238.!!
Nakatsu! Y,! Tyndale! RF,! DeLorey! TM,! Durham*Pierre! D,! Gardner! JM,! McDanel! HJ,! .! .! .! Sikela! JM.!(1993).!A!cluster!of!three!GABAA!receptor!subunit!genes!is!deleted!in!a!neurological!mutant!of!the!mouse!p!locus.!Nature,!364(6436):!448*450.!
Nakazawa! K,! Zsiros! V,! Jiang! Z,! Nakao! K,! Kolata! S,! Zhang! S! &! Belforte! JE.! (2012).! GABAergic!interneuron!origin!of!schizophrenia!pathophysiology.!Neuropharmacology,!62(3):!1574*1583.!!
Nanko! S,! Gill! M,! Owen! M,! Takazawa! N,! Moridaira! J! &! Kazamatsuri! H.! (1992).! Linkage! study! of!schizophrenia! with! markers! on! chromosome! 11! in! two! Japanese! pedigrees.! The! Japanese!Journal!of!Psychiatry!and!Neurology,!46(1):!155*159.!!
Neale!BM!&!Sham!PC.!(2004).!The!future!of!association!studies:!Gene*based!analysis!and!replication.!American!Journal!of!Human!Genetics,!75(3):!353*362.!!
Neeman!G,!Blanaru!M,!Bloch!B,!Kremer!I,!Ermilov!M,!Javitt!DC!&!Heresco*Levy!U.!(2005).!Relation!of!plasma! glycine,! serine,! and! homocysteine! levels! to! schizophrenia! symptoms! and!medication!type.!The!American!Journal!of!Psychiatry,!162(9):!1738*1740.!!

156!!
!!
Newman*Tancredi! A,! Chaput! C,! Verriele! L! &!Millan!M.! J.! (1996).! Clozapine! is! a! partial! agonist! at!cloned,!human!serotonin!5*HT1A!receptors.!Neuropharmacology,!35(1):!119*121.!!
Niciu!MJ,! Kelmendi! B! &! Sanacora! G.! (2012).! Overview! of! glutamatergic! neurotransmission! in! the!nervous!system.!Pharmacology,!Biochemistry,!and!Behavior,!100(4):!656*664.!!
Nielsen!DM,!Ehm!MG!&!Weir!BS.!(1998).!Detecting!marker*disease!association!by!testing!for!Hardy*Weinberg!disequilibrium!at!a!marker!locus.!American!Journal!of!Human!Genetics,!63(5):!1531*1540.!!
Ninan! I,! Jardemark!KE!&!Wang!RY.! (2003).!Differential!effects!of!atypical!and!typical!antipsychotic!drugs!on!N*methyl*D*aspartate*!and!electrically!evoked!responses!in!the!pyramidal!cells!of!the!rat!medial!prefrontal!cortex.!Synapse,!48(2):!66*79.!!
Noda!Y,!Kamei!H,!Mamiya!T,!Furukawa!H!&!Nabeshima!T.!(2000).!Repeated!phencyclidine!treatment!induces! negative! symptom*like! behavior! in! forced! swimming! test! in! mice:! Imbalance! of!prefrontal! serotonergic! and! dopaminergic! functions.! Neuropsychopharmacology,! 23(4):! 375*387.!!
Noda! Y,! Yamada! K,! Furukawa! H! &! Nabeshima! T.! (1995).! Enhancement! of! immobility! in! a! forced!swimming! test! by! subacute! or! repeated! treatment! with! phencyclidine:! A! new! model! of!schizophrenia.!British!Journal!of!Pharmacology,!116(5):!2531*2537.!!
Nothen! MM,! Rietschel! M,! Erdmann! J,! Oberlander! H,! Moller! HJ,! Nober! D! &! Propping! P.! (1995).!Genetic! variation!of! the!5*HT2A! receptor!and! response! to! clozapine.! Lancet,!346(8979):!908*909.!!
Nowak!L,!Bregestovski!P,!Ascher!P,!Herbet!A!&!Prochiantz!A.! (1984).!Magnesium!gates!glutamate*activated!channels!in!mouse!central!neurones.!Nature,!307(5950):!462*465.!!
Nudmamud*Thanoi!S!&!Reynold!GP.! (2004).!The!NR1!subunit!of! the!glutamate/NMDA!receptor! in!the! superior! temporal! cortex! in! schizophrenia! and! affective! disorders.! Neuroscience! Letters,!372(1*2):!173*177.!!
Numakawa!T,!Yagasaki!Y,!Ishimoto!T,!Okada!T,!Suzuki!T,!Iwata!N,!.!.!.!Hashimoto!R.!(2004).!Evidence!of! novel! neuronal! functions! of! dysbindin,! a! susceptibility! gene! for! schizophrenia.! Human!Molecular!Genetics,!13(21):!2699*2708.!!
Nyholt!DR.! (2004).!A!simple!correction! for!multiple! testing! for!single*nucleotide!polymorphisms! in!linkage!disequilibrium!with!each!other.!American!Journal!of!Human!Genetics,!74(4):!765*769.!!
Odom*White!A!&!de!Leon!J.!(1996).!Clozapine!levels!and!caffeine.!The!Journal!of!Clinical!Psychiatry,!57(4):!175*176.!!
Ohnuma!T,!Augood!SJ,!Arai!H,!McKenna!PJ!&!Emson!PC.!(1998).!Expression!of!the!human!excitatory!amino! acid! transporter! 2! and! metabotropic! glutamate! receptors! 3! and! 5! in! the! prefrontal!

157!!
!!
cortex! from! normal! individuals! and! patients! with! schizophrenia.! Molecular! Brain! Research,!56(1*2):!207*217.!!
Ohtsuki!T,!Sakurai!K,!Dou!H,!Toru!M,!Yamakawa*Kobayashi!K!&!Arinami!T.!(2001).!Mutation!analysis!of!the!NMDAR2B!(GRIN2B)!gene!in!schizophrenia.!Molecular!Psychiatry,!6(2):!211*216.!!
Olney! JW! &! Farber! NB.! (1995).! Glutamate! receptor! dysfunction! and! schizophrenia.! Archives! of!General!Psychiatry,!52(12):!998*1007.!!
Olney! JW,! Newcomer! JW! &! Farber! NB.! (1999).! NMDA! receptor! hypofunction! model! of!schizophrenia.!Journal!of!Psychiatric!Research,!33(6):!523*533.!!
Omkumar! RV,! Kiely! MJ,! Rosenstein! AJ,! Min! KT! &! Kennedy! MB.! (1996).! Identification! of! a!phosphorylation!site! for!calcium/calmodulin*dependent!protein!kinase! II! in! the!NR2B!subunit!of! the! N*methyl*D*aspartate! receptor.! The! Journal! of! Biological! Chemistry,! 271(49):! 31670*31678.!!
Ong!M,!Carreira!S,!Goodall!J,!Mateo!J,!Figueiredo!I,!Rodrigues!DN,!.!.!.!de!Bono!JS.!(2014).!Validation!and! utilisation! of! high*coverage! next*generation! sequencing! to! deliver! the! pharmacological!audit!trail.!British!Journal!of!Cancer,!111(5):!828*836.!!
O'Shea!RD.! (2002).!Roles! and! regulation!of! glutamate! transporters! in! the! central! nervous! system.!Clinical!and!Experimental!Pharmacology!&!Physiology,!29(11):!1018*1023.!!
Ossowska! K,! Pietraszek! M,! Wardas! J,! Nowak! G! &! Wolfarth! S.! (1999).! Chronic! haloperidol! and!clozapine! administration! increases! the! number! of! cortical! NMDA! receptors! in! rats.! NaunynPSchmiedeberg's!Archives!of!Pharmacology,!359(4):!280*287.!!
Overall!EJ!&!Gorham!DR.! (1962).!The!Brief!Psychiatric!Rating!Scale.!Psychological!Reports!10:!799*812.!!
Owen! RR! Jr,! Beake! BJ,!Marby! D,! Dessain! EC!&! Cole! JO.! (1989).! Response! to! clozapine! in! chronic!psychotic!patients.!Psychopharmacology!Bulletin,!25(2):!253*256.!!
Ozeki! Y,! Tomoda! T,! Kleiderlein! J,! Kamiya! A,! Bord! L,! Fujii! K,! .! .! .! Sawa! A.! (2003).! Disrupted*in*schizophrenia*1! (DISCP1):! Mutant! truncation! prevents! binding! to! NudE*like! (NUDEL)! and!inhibits! neurite! outgrowth.! Proceedings! of! the! National! Academy! of! Sciences! of! the! United!States!of!America,!100(1):!289*294.!!
Palego!L,!Biondi!L,!Giannaccini!G,!Sarno!N,!Elmi!S,!Ciapparelli!A,!.!.!.!Dell'Osso!L.!(2002a).!Clozapine,!norclozapine!plasma!levels,!their!sum!and!ratio! in!50!psychotic!patients:! Influence!of!patient*related! variables.! Progress! in!NeuroPPsychopharmacology!&!Biological! Psychiatry,! 26(3):! 473*480.!!
Palmer! BA,! Pankratz! VS! &! Bostwick! JM.! (2005).! The! lifetime! risk! of! suicide! in! schizophrenia:! A!reexamination.!Archives!of!General!Psychiatry,!62(3):!247*253.!!

158!!
!!
Park!S,! Jung!SW,!Kim!BN,!Cho!SC,!Shin!MS,!Kim!JW,! .! .! .!Kim!HW.!(2013).!Association!between!the!GRM7! rs3792452! polymorphism! and! attention! deficit! hyperacitiveity! disorder! in! a! Korean!sample.!Behavioral!and!Brain!Functions,!9(9):!1*8.!
Parker! JG,! Zweifel! LS,! Clark! JJ,! Evans! SB,! Phillips! PE! &! Palmiter! RD.! (2010).! Absence! of! NMDA!receptors! in! dopamine! neurons! attenuates! dopamine! release! but! not! conditioned! approach!during!pavlovian!conditioning.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!107(30):!13491*13496.!!
Parliament!of!Canada.!(2011).!Canadian!Human!Rights!Act!Amendment!(Genetic!Characteristics).!Bill!C*445:! The! Government! of! Canada.! http://www.parl.gc.ca/content/hoc/Bills/412/Private/C*445/C*445_1/C*445_1.pdf!
Parliament!of!Canada.!(2013).!Genetic!Nondiscrimination!Act.!Bill!S*201:!The!Government!of!Canada.!http://www.parl.gc.ca/content/hoc/Bills/412/Private/S*201/S*201_1/S*201_1.pdf!!
Parsey!RV,!Olvet!DM,!Oquendo!MA,!Huang!YY,!Ogden!RT!&!Mann!JJ.!(2006).!Higher!5*HT1A!receptor!binding! potential! during! a! major! depressive! episode! predicts! poor! treatment! response:!Preliminary!data!from!a!naturalistic!study.!Neuropsychopharmacology,!31(8):!1745*1749.!!
Paschou!P,!Drineas!P,!Lewis!J,!Nievergelt!CM,!Nickerson!DA,!Smith!JD,!.!.!.!Ziv!E.!(2008).!Tracing!sub*structure! in!the!European!American!population!with!PCA*informative!markers.!PLoS!Genetics,!4(7):!e1000114.!!
Patil!ST,!Zhang!L,!Martenyi!F,!Lowe!SL,!Jackson!KA,!Andreev!BV,!.!.!.!Schoepp!DD.!(2007).!Activation!of!mGlu2/3!receptors!as!a!new!approach!to!treat!schizophrenia:!A!randomized!phase!2!clinical!trial.!Nature!Medicine,!13(9):!1102*1107.!!
Patnala!R,!Clements!J!&!Batra!J.!(2013).!Candidate!gene!association!studies:!A!comprehensive!guide!to!useful!in!silico!tools.!BMC!Genetics,!14(39):!1*11.!
Pearce! IA,! Cambray*Deakin! MA! &! Burgoyne! RD.! (1987).! Glutamate! acting! on! NMDA! receptors!stimulates!neurite!outgrowth!from!cerebellar!granule!cells.!FEBS!Letters,!223(1):!143*147.!!
Peduzzi! P,! Concato! J,! Feinstein! AR! &! Holford! TR.! (1995).! Importance! of! events! per! independent!variable! in! proportional! hazards! regression! analysis.! II.! Accuracy! and! precision! of! regression!estimates.!Journal!of!Clinical!Epidemiology,!48(12):!1503*1510.!!
Peduzzi!P,!Concato!J,!Kemper!E,!Holford!TR!&!Feinstein!AR.!(1996).!A!simulation!study!of!the!number!of! events! per! variable! in! logistic! regression! analysis.! Journal! of! Clinical! Epidemiology,! 49(12):!1373*1379.!!
Pennacchio!LA,!Ahituv!N,!Moses!AM,!Prabhakar!S,!Nobrega!MA,!Shoukry!M,!.!.!.!Rubin!EM.!(2006).!In!vivo! enhancer! analysis! of! human! conserved! non*coding! sequences.! Nature,! 444(7118):! 499*502.!!

159!!
!!
Perkins!DO,!Gu!H,! Boteva! K!&! Lieberman! JA.! (2005).! Relationship! between!duration!of! untreated!psychosis!and!outcome!in!first*episode!schizophrenia:!A!critical!review!and!meta*analysis.!The!American!Journal!of!Psychiatry,!162(10):!1785*1804.!!
Perneger! TV.! (1998).! What's! wrong! with! Bonferroni! adjustments.! BMJ! (Clinical! Research! Ed),!316(7139):!1236*1238.!!
Phillips!KA,!Veenstra!DL,!Oren!E,!Lee!JK!&!Sadee!W.!(2001).!Potential!role!of!pharmacogenomics!in!reducing!adverse!drug!reactions:!A!systematic!review.!Jama,!286(18):!2270*2279.!!
Phillips!PC.!(2008).!Epistasis!*!The!essential!role!of!gene!interactions!in!the!structure!and!evolution!of!genetic!systems.!Nature!Reviews,!Genetics,!9(11):!855*867.!!
Pickar!D,!Owen!RR,!Litman!RE,!Konicki!E,!Gutierrez!R!&!Rapaport!MH.! (1992).!Clinical!and!biologic!response!to!clozapine!in!patients!with!schizophrenia.!Crossover!comparison!with!fluphenazine.!Archives!of!General!Psychiatry,!49(5):!345*353.!!
Pilowsky!LS,!Bressan!RA,!Stone!JM,!Erlandsson!K,!Mulligan!RS,!Krystal!JH!&!Ell!PJ.!(2006).!First!in!vivo!evidence! of! an! NMDA! receptor! deficit! in! medication*free! schizophrenic! patients.! Molecular!Psychiatry,!11(2):!118*119.!!
Pin! JP! &! Duvoisin! R.! (1995).! The! metabotropic! glutamate! receptors:! Structure! and! functions.!Neuropharmacology,!34(1):!1*26.!!
Pin!JP,!Galvez!T!&!Prezeau!L.!(2003).!Evolution,!structure,!and!activation!mechanism!of!family!3/C!G*protein*coupled!receptors.!Pharmacology!&!Therapeutics,!98(3):!325*354.!!
Pinard!E,!Alanine!A,!Alberati!D,!Bender!M,!Borroni!E,!Bourdeaux!P,!.!.!.!Zimmerli!D.!(2010).!Selective!GlyT1! inhibitors:! Discovery! of! [4*(3*fluoro*5*trifluoromethylpyridin*2*yl)piperazin*1*yl][5*methanesulfonyl*2*((S)*2,2,2*trifluoro*1*methylethoxy)phenyl]methanone! (RG1678),! a!promising!novel!medicine!to!treat!schizophrenia.!Journal!of!Medicinal!Chemistry,!53(12):!4603*4614.!!
Pique*Regi!R,!Degner! JF,! Pai!AA,!Gaffney!DJ,!Gilad!Y!&!Pritchard! JK.! (2011).!Accurate! inference!of!transcription! factor! binding! from! DNA! sequence! and! chromatin! accessibility! data.! Genome!Research,!21(3):!447*455.!!
Plomen! R,! DeFries! JC,!McClearn! GE! &! Rutter!M.! (1997).!Behavioral! genetics! (3rd! ed).! New! York:!Freeman.!!
Potkin! SG,! Costa! J,! Roy! S,! Sramek! J,! Jin! Y! &! Gulasekaram! B.! (1992).! Glycine! in! the! treatment! of!schizophrenia!*!Theory!and!preliminary!results.! In!HY!Meltzer!(Ed),!Novel!antipsychotic!drugs.!New!York:!Raven!Press!Ltd.!!
Price!AL,!Butler!J,!Patterson!N,!Capelli!C,!Pascali!VL,!Scarnicci!F,!.!.!.!Hirschhorn!JN.!(2008).!Discerning!the!ancestry!of!European!Americans!in!genetic!association!studies.!PLoS!Genetics,!4(1):!e236.!!

160!!
!!
Pritchard! JK.,!Stephens!M,!Rosenberg!NA!&!Donnelly!P.! (2000).!Association!mapping! in!structured!populations.!American!Journal!of!Human!Genetics,!67(1):!170*181.!!
Puckett!C,!Gomez!CM,!Korenberg!JR,!Tung!H,!Meier!TJ,!Chen!XN!&!Hood!L.!(1991).!Molecular!cloning!and!chromosomal! localization!of!one!of!the!human!glutamate!receptor!genes.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!88(17):!7557*7561.!!
Pulver!AE,!Lasseter!VK,!Kasch!L,!Wolyniec!P,!Nestadt!G,!Blouin!JL,!.!.!.!Chen!H.!(1995).!Schizophrenia:!A! genome! scan! targets! chromosomes! 3p! and! 8p! as! potential! sites! of! susceptibility! genes.!American!Journal!of!Medical!Genetics,!60(3):!252*260.!!
Purcell!S,!Neale!B,!Todd*Brown!K,!Thomas!L,!Ferreira!MA,!Bender!D,!.!.!.!Sham!PC.!(2007).!PLINK:!A!tool! set! for! whole*genome! association! and! population*based! linkage! analyses.! American!Journal!of!Human!Genetics,!81(3):!559*575.!!
Purkayastha! S,! Ford! J,! Kanjilal! B,!Diallo! S,!Del!Rosario! Inigo! J,!Neuwirth! L,! .! .! .! Banerjee!P.! (2012).!Clozapine!functions!through!the!prefrontal!cortex!serotonin!1A!receptor!to!heighten!neuronal!activity! via! calmodulin! kinase! II*NMDA! receptor! interactions.! Journal! of! Neurochemistry,!120(3):!396*407.!!
Pycock! CJ,! Kerwin! RW! &! Carter! CJ.! (1980).! Effect! of! lesion! of! cortical! dopamine! terminals! on!subcortical!dopamine!receptors!in!rats.!Nature,!286(5768):!74*76.!!
Qin!S,!Zhao!X,!Pan!Y,!Liu!J,!Feng!G,!Fu!J,! .! .! .!He!L.!(2005).!An!association!study!of!the!N*methyl*D*aspartate! receptor! NR1! subunit! gene! (GRIN1)! and! NR2B! subunit! gene! (GRIN2B)! in!schizophrenia!with!universal!DNA!microarray.!European!Journal!of!Human!Genetics,!13(7):!807*814.!!
Reicher!A,!Maurer!K,!Loffler!W,!Fatkenheuer!B,!van!der!Heiden!W,!Munk*Jorgenson!P,!.!.!Hafner!H.!(1990).!Gender!differences!in!age!at!onset!and!course!of!schizophrenic!disorders.!In!H!Hafner!&!W!Gattaz!(Eds),!Search!for!the!causes!of!schizophrenia.!Berlin:!Springer*Verlag.!!
Remington!G,!Saha!A,!Chong!SA!&!Shammi!C.!(2005).!Augmentation!strategies!in!clozapine*resistant!schizophrenia.!CNS!Drugs,!19(10):!843*872.!!
Riley!BP!&!McGuffin!P.!(2000).!Linkage!and!associated!studies!of!schizophrenia.!American!Journal!of!Medical!Genetics,!97(1):!23*44.!!
Riley! B! &! Kendler! KS.! (2006).! Molecular! genetic! studies! of! schizophrenia.! European! Journal! of!Human!Genetics,!14(6):!669*680.!!
Risch!N!&!Merikangas!K.!(1996).!The!future!of!genetic!studies!of!complex!human!diseases.!Science,!273(5281):!1516*1517.!!
Ritchie!MD,! Hahn! LW,! Roodi! N,! Bailey! LR,! Dupont!WD,! Parl! FF! &!Moore! JH.! (2001).!Multifactor*dimensionality!reduction!reveals!high*order!interactions!among!estrogen*metabolism!genes!in!sporadic!breast!cancer.!American!Journal!of!Human!Genetics,!69(1):!138*147.!!

161!!
!!
Ritchie!MD!&!Motsinger!AA.! (2005).!Multifactor!dimensionality! reduction! for!detecting!gene*gene!and! gene*environment! interactions! in! pharmacogenomics! studies.! Pharmacogenomics,! 6(8):!823*834.!!
Rodriguez*Moreno!A.! (2006).!The!role!of!kainate!receptors! in!the!regulation!of!excitatory!synaptic!transmission!in!the!hippocampus.!Revista!De!Neurologia,!42(5):!282*287.!!
Rorick*Kehn!LM,!Johnson!BG,!Burkey!JL,!Wright!RA,!Calligaro!DO,!Marek!GJ,!.!.!.!Schoepp!DD.!(2007).!Pharmacological!and!pharmacokinetic!properties!of!a!structurally!novel,!potent,!and!selective!metabotropic! glutamate! 2/3! receptor! agonist:! In! vitro! characterization! of! agonist! (*)*(1R,4S,5S,6S)*4*amino*2*sulfonylbicyclo[3.1.0]*hexane*4,6*dicarboxylic! acid! (LY404039).! The!Journal!of!Pharmacology!and!Experimental!Therapeutics,!321(1):!308*317.!!
Rosenheck!R,!Evans!D,!Herz!L,!Cramer!J,!Xu!W,!Thomas!J,!.!.!.!Charney!D.!(1999).!How!long!to!wait!for!a! response! to! clozapine:! A! comparison! of! time! course! of! response! to! clozapine! and!conventional! antipsychotic! medication! in! refractory! schizophrenia.! Schizophrenia! Bulletin,!25(4):!709*719.!!
Rosenthal!D,!Werder! PH,! Kety! SS,! Schulsinger! F,!Welner! J,!Ostergaard! L,! .! .! .! Kety! SS.! (1968).!The!transmission!of!schizophrenia.!Oxford:!Pergamon!Press.!!
Ross! JS! &! Cronin! M.! (2011).! Whole! cancer! genome! sequencing! by! next*generation! methods.!American!Journal!of!Clinical!Pathology,!136(4):!527*539.!!
Rothstein!JD,!Dykes*Hoberg!M,!Pardo!CA,!Bristol!LA,!Jin!L,!Kuncl!RW,!.!.!.!Welty!DF.!(1996).!Knockout!of! glutamate! transporters! reveals! a! major! role! for! astroglial! transport! in! excitotoxicity! and!clearance!of!glutamate.!Neuron,!16(3):!675*686.!!
Rubin! P,! Holm! S,! Friberg! L,! Videbech! P,! Andersen! HS,! Bendsen! BB,! .! .! .! Hemmingsen! R.! (1991).!Altered! modulation! of! prefrontal! and! subcortical! brain! activity! in! newly! diagnosed!schizophrenia!and!schizophreniform!disorder.!A!regional!cerebral!blood!flow!study.!Archives!of!General!Psychiatry,!48(11):!987*995.!!
Sadee! W.! (2012).! The! relevance! of! "missing! heritability"! in! pharmacogenomics.! Clinical!Pharmacology!and!Therapeutics,!92(4):!428*430.!!
Sadee!W.!(2013).!Gene*gene*environment!interactions!between!drugs,!transporters,!receptors,!and!metabolizing!enzymes:!Statins,!SLCO1B1,!and!CYP3A4!as!an!example.!Journal!of!Pharmaceutical!Sciences,!102(9):!2924*2929.!
Saha! S,! Chant! D! &! McGrath! J.! (2007).! A! systematic! review! of! mortality! in! schizophrenia:! Is! the!differential!mortality!gap!worsening!over! time?!Archives!of!General!Psychiatry,!64(10):!1123*1131.!
Sala! C,! Rudolph*Correia! S! &! Sheng! M.! (2000).! Developmentally! regulated! NMDA! receptor*dependent! dephosphorylation! of! cAMP! response! element*binding! protein! (CREB)! in!hippocampal!neurons.!The!Journal!of!Neuroscience,!20(10):!3529*3536.!!

162!!
!!
Salmi!P,!Chergui!K!&!Fredholm!BB.! (2005).!Adenosine*dopamine! interactions!revealed! in!knockout!mice.!Journal!of!Molecular!Neuroscience,!26(2*3):!239*244.!!
Sams*Dodd!F.!(1995).!Distinct!effects!of!d*amphetamine!and!phencyclidine!on!the!social!behaviour!of!rats.!Behavioural!Pharmacology,!6(1):!55*65.!!
Sams*Dodd! F.! (1998).! Effects! of! continuous! D*amphetamine! and! phencyclidine! administration! on!social! behaviour,! stereotyped! behaviour,! and! locomotor! activity! in! rats.!Neuropsychopharmacology,!19(1):!18*25.!!
Sanger! F,! Nicklen! S! &! Coulson! AR.! (1977).! DNA! sequencing! with! chain*terminating! inhibitors.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!74(12):!5463*5467.!!
Santos! SD,! Carvalho! AL,! Caldeira! MV! &! Duarte! CB.! (2009).! Regulation! of! AMPA! receptors! and!synaptic!plasticity.!Neuroscience,!158(1):!105*125.!!
Sasieni! PD.! (1997).! From! genotypes! to! genes:! Doubling! the! sample! size.! Biometrics,! 53(4):! 1253*1261.!!
Schene!AH,!van!Wijngaarden!B!&!Koeter!MW.!(1998).!Family!caregiving! in!schizophrenia:!Domains!and!distress.!Schizophrenia!Bulletin,!24(4):!609*618.!!
Schiller! J,! Schiller! Y! &! Clapham!DE.! (1998).! NMDA! receptors! amplify! calcium! influx! into! dendritic!spines!during!associative!pre*!and!postsynaptic!activation.!Nature!Neuroscience,!1(2):!114*118.!!
Schizophrenia!Linkage!Collaborative!Group.!(1996).!Additional!support!for!schizophrenia!linkage!on!chromosomes!6!and!8:!A!multicenter!study.!American!Journal!of!Medical!Genetics,!67(6):!580*594.!
Schizophrenia!Working! Group! of! the! Psychiatric! Genomics! Consortium.! (2014).! Biological! insights!from!108!schizophrenia*associated!genetic!loci.!Nature,!511(7510):!421*427.!!
Schmitt! A,! Zink!M,!Muller! B,!May! B,! Herb! A,! Jatzko! A,! .! .! .! Henn! FA.! (2003).! Effects! of! long*term!antipsychotic! treatment! on! NMDA! receptor! binding! and! gene! expression! of! subunits.!Neurochemical!Research,!28(2):!235*241.!!
Schork!NJ,!Murray!SS,!Frazer!KA!&!Topol!EJ.!(2009).!Common!vs.!rare!allele!hypotheses!for!complex!diseases.!Current!Opinion!in!Genetics!&!Development,!19(3):!212*219.!!
Schotte!A,!Janssen!PF,!Gommeren!W,!Luyten!WH,!Van!Gompel!P,!Lesage!AS,!.!.!.!Leysen!JE.!(1996).!Risperidone! compared! with! new! and! reference! antipsychotic! drugs:! In! vitro! and! in! vivo!receptor!binding.!Psychopharmacology,!124(1*2):!57*73.!!
Schumacher!J,!Schulze!TG,!Wienker!TF,!Rietschel!M!&!Nothen!MM.!(2000).!Pharmacogenetics!of!the!clozapine!response.!Lancet,!356(9228):!506*507.!!

163!!
!!
Schumacher! J,! Jamra! RA,! Freudenberg! J,! Becker! T,! Ohlraun! S,! Otte! AC,! .! .! .! Cichon! S.! (2004).!Examination! of! G72! and! D*amino*acid! oxidase! as! genetic! risk! factors! for! schizophrenia! and!bipolar!affective!disorder.!Molecular!Psychiatry,!9(2):!203*207.!!
Schwieler!L,!Engberg!G!&!Erhardt!S.!(2004).!Clozapine!modulates!midbrain!dopamine!neuron!firing!via!interaction!with!the!NMDA!receptor!complex.!Synapse,!52(2):!114*122.!!
Schwieler! L! &! Erhardt! S.! (2003).! Inhibitory! action! of! clozapine! on! rat! ventral! tegmental! area!dopamine! neurons! following! increased! levels! of! endogenous! kynurenic! acid.!Neuropsychopharmacology,!28(10):!1770*1777!!
Schwieler!L,!Linderholm!KR,!Nilsson*Todd!LK,!Erhardt!S!&!Engberg!G.!(2008).!Clozapine!interacts!with!the!glycine!site!of!the!NMDA!receptor:!Electrophysiological!studies!of!dopamine!neurons!in!the!rat!ventral!tegmental!area.!Life!Sciences,!83(5*6):!170*175.!!
Seeman! MV! &! Lang! M.! (1990).! The! role! of! estrogens! in! schizophrenia! gender! differences.!Schizophrenia!Bulletin,!16(2):!185*194.!!
Seeman! P! &! Lee! T.! (1975).! Antipsychotic! drugs:! Direct! correlation! between! clinical! potency! and!presynaptic!action!on!dopamine!neurons.!Science,!188(4194):!1217*1219.!!
Seeman! P.! (1987).! Dopamine! receptors! and! the! dopamine! hypothesis! of! schizophrenia.! Synapse,!1(2):!133*152.!!
Seppala!NH,! Leinonen!EV,! Lehtonen!ML!&!Kivisto! KT.! (1999).! Clozapine! serum! concentrations! are!lower! in! smoking! than! in! non*smoking! schizophrenic! patients.! Pharmacology! &! Toxicology,!85(5):!244*246.!!
Serretti!A,!Drago!A!&!De!Ronchi!D.!(2007).!HTR2A!gene!variants!and!psychiatric!disorders:!A!review!of! current! literature! and! selection! of! SNPs! for! future! studies.! Current! Medicinal! Chemistry,!14(19):!2053*2069.!!
Setsirichok! D,! Piroonratana! T,! Assawamakin! A,! Usavanarong! T,! Limwongse! C,!Wongseree!W,! .! .! .!Chaiyaratana!N.! (2012).! Small! ancestry! informative!marker! panels! for! complete! classification!between! the! original! four! HapMap! populations.! International! Journal! of! Data! Mining! and!Bioinformatics,!6(6):!651*674.!!
Shan! D,! Yates! S,! Roberts! RC! &! McCullumsmith! RE.! (2012).! Update! on! the! neurobiology! of!schizophrenia:!A!role!for!extracellular!microdomains.!Minerva!Psichiatrica,!53(3):!233*249.!!
Shank! RP!&!Aprison!MH.! (1981).! Present! status! and! significance! of! the! glutamine! cycle! in! neural!tissues.!Life!Sciences,!28(8):!837*842.!!
Shao!L,! Shuai!Y,!Wang! J,! Feng!S,! Lu!B,! Li! Z,! .! .! .! Zhong!Y.! (2011).! Schizophrenia! susceptibility!gene!dysbindin! regulates! glutamatergic! and! dopaminergic! functions! via! distinctive!mechanisms! in!drosophila.!Proceedings!of!the!National!Academy!of!Sciences!of!the!United!States!of!America,!108(46):!18831*18836.!!

164!!
!!
Shapiro!M.! (2001).! Plasticity,! hippocampal!place! cells,! and! cognitive!maps.! Archives!of!Neurology,!58(6):!874*881.!!
Shastry!P,!Basu!A!&!Rajadhyaksha!MS.!(2001).!Neuroblastoma!cell!lines:!A!versatile!in!vitro!model!in!neurobiology.!The!International!Journal!of!Neuroscience,!108(1*2):!109*126.!!
Shaw!SH,!Kelly!M,!Smith!AB,!Shields!G,!Hopkins!PJ,!Loftus! J,! .! .! .!DeLisi!LE.! (1998).!A!genome*wide!search!for!schizophrenia!susceptibility!genes.!American!Journal!of!Medical!Genetics,!81(5):!364*376.!!
Shendure! J!&! Ji!H.! (2008).!Next*generation!DNA! sequencing.!Nature!Biotechnology,! 26(10):! 1135*1145.!!
Sigwald! J,!Bouttier!D,!Raymond!C!&!Pichot!C.! (1959).!Four!cases!of! facio*bucco*linguo*masticatory!dyskinesia! of! prolonged! development! following! treatment! with! neuroleptics.! Revue!Neurologique,!100:!751*755.!!
Singh!SP!&!Singh!V.!(2011).!Meta*analysis!of!the!efficacy!of!adjunctive!NMDA!receptor!modulators!in!chronic!schizophrenia.!CNS!Drugs,!25(10):!859*885.!!
Sipos!A,!Rasmussen!F,!Harrison!G,!Tynelius!P,!Lewis!G,!Leon!DA!&!Gunnell!D.! (2004).!Paternal!age!and! schizophrenia:! A! population! based! cohort! study.! BMJ! (Clinical! Research! Ed),! 329(7474):!1070.!
Smith!KE,!Borden!LA,!Hartig!PR,!Branchek!T!&!Weinshank!RL.! (1992).!Cloning!and!expression!of! a!glycine!transporter!reveal!colocalization!with!NMDA!receptors.!Neuron,!8(5):!927*935.!!
Solanki! RK,! Singh! P! &! Munshi! D.! (2009).! Current! perspectives! in! the! treatment! of! resistant!schizophrenia.!Indian!Journal!of!Psychiatry,!51(4):!254*260.!!
Solberg! N! &! Krauss! S.! (2013).! Luciferase! assay! to! study! the! activity! of! a! cloned! promoter! DNA!fragment.!Methods!in!Molecular!Biology,!977:!65*78.!!
Solinas! M,! Ferre! S,! You! ZB,! Karcz*Kubicha! M,! Popoli! P! &! Goldberg! SR.! (2002).! Caffeine! induces!dopamine! and! glutamate! release! in! the! shell! of! the! nucleus! accumbens.! The! Journal! of!Neuroscience,!22(15):!6321*6324.!!
Sommer!B,!Keinanen!K,!Verdoorn!TA,!Wisden!W,!Burnashev!N,!Herb!A,!.!.!.!Seeburg!PH.!(1990).!Flip!and!flop:!A!cell*specific!functional!switch!in!glutamate*operated!channels!of!the!CNS.!Science,!249(4976):!1580*1585.!!
Song! L! &! Crawford! GE.! (2010).! DNase*seq:! A! high*resolution! technique! for! mapping! active! gene!regulatory!elements!across! the!genome!from!mammalian!cells.!Cold!Spring!Harbor!Protocols,!2010(2):!1*13.!

165!!
!!
Spurney!CF,!Baca!SM,!Murray!AM,!Jaskiw!GE,!Kleinman!JE!&!Hyde!TM.!(1999).!Differential!effects!of!haloperidol!and!clozapine!on!ionotropic!glutamate!receptors! in!rats.!Synapse,!34(4):!266*276.!doi:2*2!!
St! Clair! D,! Blackwood! D,! Muir! W,! Carothers! A,! Walker! M,! Spowart! G,! .! .! .! Evans! HJ.! (1990).!Association!within! a! family! of! a! balanced! autosomal! translocation!with!major!mental! illness.!Lancet,!336(8706):!13*16.!!
St! Clair! D,! Xu!M,!Wang! P,! Yu! Y,! Fang! Y,! Zhang! F,! .! .! .! He! L.! (2005).! Rates! of! adult! schizophrenia!following!prenatal!exposure!to!the!Chinese!famine!of!1959*1961.!Jama,!294(5):!557*562.!!
Stahl!SM.!(2007).!Beyond!the!dopamine!hypothesis!to!the!NMDA!glutamate!receptor!hypofunction!hypothesis!of!schizophrenia.!CNS!Spectrums,!12(4):!265*268.!!
Steck!H.! (1954).!Le!syndrome!extrapyramidal!et!diencephalique!au! largactil!et!au!serpasil.!Annales!Médico!Psychologique,!112:!737*743.!!
Stefansson!H,!Sarginson!J,!Kong!A,!Yates!P,!Steinthorsdottir!V,!Gudfinnsson!E,!.!.!.!St!Clair!D.!(2003).!Association!of! neuregulin! 1!with! schizophrenia! confirmed! in! a! Scottish! population.! American!Journal!of!Human!Genetics,!72(1):!83*87.!!
Stern! RG,! Kahn! RS,! Davidson! M,! Nora! RM! &! Davis! KL.! (1994).! Early! response! to! clozapine! in!schizophrenia.!The!American!Journal!of!Psychiatry,!151(12):!1817*1818.!!
Stowell! JN! &! Craig! AM.! (1999).! Axon/dendrite! targeting! of!metabotropic! glutamate! receptors! by!their!cytoplasmic!carboxy*terminal!domains.!Neuron,!22(3):!525*536.!!
Straub! RE,! Jiang! Y,!MacLean! CJ,!Ma! Y,!Webb!BT,!Myakishev!MV,! .! .! .! Kendler! KS.! (2002).! Genetic!variation! in! the! 6p22.3! gene!DTNBP1,! the! human! ortholog! of! the!mouse! dysbindin! gene,! is!associated!with!schizophrenia.!American!Journal!of!Human!Genetics,!71(2):!337*348.!!
Straub!RE,!Lipska!BK,!Egan!MF,!Goldberg!TE,!Callicott!JH,!Mayhew!MB,!.!.! .!Weinberger!DR.!(2007).!Allelic! variation! in! GAD1! (GAD67)! is! associated! with! schizophrenia! and! influences! cortical!function!and!gene!expression.!Molecular!Psychiatry,!12(9):!854*869.!!
Sturgeon!RD,!Fessler!RG!&!Meltzer!HY.!(1979).!Behavioral!rating!scales!for!assessing!phencyclidine*induced! locomotor! activity,! stereotyped! behavior! and! ataxia! in! rats.! European! Journal! of!Pharmacology,!59(3*4):!169*179.!!
Sumiyoshi! T,!Anil!AE,! Jin!D,! Jayathilake!K,! Lee!M!&!Meltzer!HY.! (2004).! Plasma!glycine!and! serine!levels! in! schizophrenia! compared! to! normal! controls! and! major! depression:! Relation! to!negative!symptoms.!The!International!Journal!of!Neuropsychopharmacology,!7(1):!1*8.!!
Sun!T,!Hu!G!&!Li!M.! (2009).!Repeated!antipsychotic! treatment!progressively!potentiates! inhibition!on! phencyclidine*induced! hyperlocomotion,! but! attenuates! inhibition! on! amphetamine*induced! hyperlocomotion:! Relevance! to! animal! models! of! antipsychotic! drugs.! European!Journal!of!Pharmacology,!602(2*3):!334*342.!!

166!!
!!
Susser! ES!&! Lin! SP.! (1992).! Schizophrenia! after! prenatal! exposure! to! the! Dutch! hunger!winter! of!1944*1945.!Archives!of!General!Psychiatry,!49(12):!983*988.!!
Suzuki!T,!Remington!G,!Arenovich!T,!Uchida!H,!Agid!O,!Graff*Guerrero!A!&!Mamo!DC.!(2011).!Time!course! of! improvement! with! antipsychotic! medication! in! treatment*resistant! schizophrenia.!British!Journal!of!Psychiatry,!199(4):!275*280.!!
Suzuki! T,! Remington!G,!Mulsant! BH,!Uchida!H,! Rajji! TK,! Graff*Guerrero! A,! .! .! .!Mamo!DC.! (2012).!Defining! treatment*resistant! schizophrenia! and! response! to! antipsychotics:! A! review! and!recommendation.!Psychiatry!Research,!197(1*2):!1*6.!!
Swerdlow! NR,! Bakshi! V,! Waikar! M,! Taaid! N! &! Geyer! MA.! (1998).! Seroquel,! clozapine! and!chlorpromazine! restore! sensorimotor! gating! in! ketamine*treated! rats.! Psychopharmacology,!140(1):!75*80.!!
Szatkowski!M!&!Attwell!D.! (1994).!Triggering!and!execution!of!neuronal!death! in!brain! ischaemia:!Two!phases!of!glutamate!release!by!different!mechanisms.!Trends!in!Neurosciences,!17(9):!359*365.!!
Szczepankiewicz!A,! Skibinska!M,! Suwalska!A,!Hauser! J!&!Rybakowski! JK.! (2009).!No!association!of!three! GRIN2B! polymorphisms! with! lithium! response! in! bipolar! patients.! Pharmacological!Reports,!61(3):!448*452.!!
Takahashi! T,! Svoboda! K! &! Malinow! R.! (2003).! Experience! strengthening! transmission! by! driving!AMPA!receptors!into!synapses.!Science,!299(5612):!1585*1588.!!
Takahashi! M,! Kakita! A,! Futamura! T,! Watanabe! Y,! Mizuno! M,! Sakimura! K,! .! .! .! Nawa! H.! (2006).!Sustained! brain*derived! neurotrophic! factor! up*regulation! and! sensorimotor! gating!abnormality! induced! by! postnatal! exposure! to! phencyclidine:! Comparison! with! adult!treatment.!Journal!of!Neurochemistry,!99(3):!770*780.!!
Talbot! K,! Eidem! WL,! Tinsley! CL,! Benson! MA,! Thompson! EW,! Smith! RJ,! .! .! .! Arnold! SE.! (2004).!Dysbindin*1! is! reduced! in! intrinsic,! glutamatergic! terminals! of! the! hippocampal! formation! in!schizophrenia.!The!Journal!of!Clinical!Investigation,!113(9):!1353*1363.!!
Tandon!R!&!Fleischhacker!WW.!(2005).!Comparative!efficacy!of!antipsychotics! in!the!treatment!of!schizophrenia:!A!critical!assessment.!Schizophrenia!Research,!79(2*3):!145*155.!!
Tandon! R.! (2011).! Antipsychotics! in! the! treatment! of! schizophrenia:! An! overview.! The! Journal! of!Clinical!Psychiatry,!Supplemental,!72(S1):!4*8.!!
Taylor!SF!&!Tso!IF!(2014).!GABA!abnormalities!in!schizophrenia:!A!methodological!review!of!in!vivo!studies.!Schizophrenia!Research,!advanced!online!publication.!!
Theisen! FM,!Gebhardt! S,! Haberhausen!M,!Heinzel*Gutenbrunner!M,!Wehmeier! PM,! Krieg! JC,! .! .! .!Hebebrand!J.!(2005).!Clozapine*induced!weight!gain:!A!study!in!monozygotic!twins!and!same*sex!sib!pairs.!Psychiatric!Genetics,!15(4):!285*289.!!

167!!
!!
Thierry*Mieg! D! &! Thierry*Mieg! J.! (2006).! AceView:! A! comprehensive! cDNA*supported! gene! and!transcripts!annotation.!Genome!Biology,!Supplemental,!7(S1):!S12*S14.!!
Thomson!AM.! (1990).!Glycine! is! a! coagonist! at! the!NMDA! receptor/channel! complex.! Progress! in!Neurobiology,!35(1):!53*74.!!
Tian!L,!Stefanidakis!M,!Ning!L,!Van!Lint!P,!Nyman*Huttunen!H,!Libert!C,! .! .! .!Gahmberg!CG.!(2007).!Activation!of!NMDA!receptors!promotes!dendritic!spine!development!through!MMP*mediated!ICAM*5!cleavage.!The!Journal!of!Cell!Biology,!178(4):!687*700.!!
Tienari!PJ!&!Wynne!LC.! (1994).!Adoption!studies!of!schizophrenia.!Annals!of!Medicine,!26(4):!233*237.!!
Tingley! WG,! Ehlers! MD,! Kameyama! K,! Doherty! C,! Ptak! JB,! Riley! CT! &! Huganir! RL.! (1997).!Characterization!of!protein!kinase!A!and!protein!kinase!C!phosphorylation!of!the!N*methyl*D*aspartate! receptor!NR1!subunit!using!phosphorylation! site*specific!antibodies.! The! Journal!of!Biological!Chemistry,!272(8):!5157*5166.!!
Torrey! E.! (2013).! Surviving! schizophrenia:! A!manual! for! families,! patients,! and! providers! (5th! ed).!New!York,!NY:!Harper!Perrenial.!!
Tran!B,!Brown!AM,!Bedard!PL,!Winquist!E,!Goss!GD,!Hotte!SJ,! .! .! .!Dancey!JE.! (2013).!Feasibility!of!real!time!next!generation!sequencing!of!cancer!genes!linked!to!drug!response:!Results!from!a!clinical!trial.!International!Journal!of!Cancer,!132(7):!1547*1555.!!
Tsai! GE,! Yang! P,! Chung! LC,! Lange! N!&! Coyle! JT.! (1998).! D*serine! added! to! antipsychotics! for! the!treatment!of!schizophrenia.!Biological!Psychiatry,!44(11):!1081*1089.!!
Tsai!GE,!Yang!P,!Chung!LC,!Tsai!IC,!Tsai!CW!&!Coyle!JT.!(1999).!D*serine!added!to!clozapine!for!the!treatment!of!schizophrenia.!The!American!Journal!of!Psychiatry,!156(11):!1822*1825.!!
Tsunoka!T,!Kishi!T,!Ikeda!M,!Kitajima!T,!Yamanouchi!Y,!Kinoshita!Y,!.!.!.!Iwata!N.!(2009).!Association!analysis! of! group! II! metabotropic! glutamate! receptor! genes! (GRM2! and! GRM3)! with! mood!disorders! and! fluvoxamine! response! in! a! Japanese! population.! Progress! in! NeuroPPsychopharmacology!&!Biological!Psychiatry,!33(5):!875*879.!!
Tsunoka!T,!Kishi!T,!Kitajima!T,!Okochi!T,!Okumura!T,!Yamanouchi!Y,!.!.!.!Iwata!N.!(2010).!Association!analysis!of!GRM2!and!HTR2A!with!methamphetamine*induced!psychosis!and!schizophrenia!in!the! Japanese! population.! Progress! in! NeuroPPsychopharmacology! &! Biological! Psychiatry,!34(4):!639*644.!!
Tu! JC,! Xiao! B,! Naisbitt! S,! Yuan! JP,! Petralia! RS,! Brakeman! P,! .! .! .! Worley! PF.! (1999).! Coupling! of!mGluR/Homer! and! PSD*95! complexes! by! the! shank! family! of! postsynaptic! density! proteins.!Neuron,!23(3):!583*592.!!
United!Kingdom!of!Great!Britain.!(2010).!Equality!Act!of!2010:!Government!of!the!United!Kingdom.!http://www.legislation.gov.uk/ukpga/2010/15/pdfs/ukpga_20100015_en.pdf!

168!!
!!
United!States!of!America.! (2008).!Genetic! Information!Nondiscrimination!Act!of!2008! (GINA).!H.R.,!493:! Government! of! the! United! States! of! America.! http://www.gpo.gov/fdsys/pkg/BILLS*110hr493enr/pdf/BILLS*110hr493enr.pdf!
van!den!Oord!EJ!&!Sullivan!PF.! (2003).!False!discoveries!and!models! for!gene!discovery.! Trends! in!Genetics,!19(10):!537*542.!!
Van!den!Pol!AN.! (1994).!Metabotropic!glutamate!receptor!mGluR1!distribution!and!ultrastructural!localization!in!hypothalamus.!The!Journal!of!Comparative!Neurology,!349(4):!615*632.!!
Vasudeva! S,! Sekhar! CK! &! Rao! PG.! (2013).! Caregivers! burden! of! patients! with! schizophrenia! and!bipolar!disorder:!A!sectional!study.!Indian!Journal!of!Psychological!Medicine,!35(4):!352*357.!!
Vesell!ES.!(1978).!Twin!studies!in!pharmacogenetics.!Human!Genetics,!Supplemental,!(1)(1):!19*30.!!
Vink! JM,! Smit! AB,! de! Geus! EJ,! Sullivan! P,! Willemsen! G,! Hottenga! JJ,! .! .! .! Boomsma! DI.! (2009).!Genome*wide!association!study!of!smoking!initiation!and!current!smoking.!American!Journal!of!Human!Genetics,!84(3):!367*379.!!
Visscher! PM,! Hill! WG! &! Wray! NR.! (2008).! Heritability! in! the! genomics! era:! Concepts! and!misconceptions.!Nature!Reviews,!Genetics,!9(4):!255*266.!!
Vogel! F.! (1959).! Moderne! problem! der! humangenetik.! Ergebnisse! der! Inneren! Medizin! und!Kinderheilkunde,!12:!52*125.!!
Voineskos! AN,! Foussias! G,! Lerch! J,! Felsky! D,! Remington! G,! Rajji! TK,! .! .! .! Mulsant! BH.! (2013).!Neuroimaging!evidence!for!the!deficit!subtype!of!schizophrenia.! JAMA!Psychiatry,!70(5):!472*480.!!
Vojvoda! D,! Grimmell! K,! Sernyak! M! &! Mazure! CM.! (1996).! Monozygotic! twins! concordant! for!response!to!clozapine.!Lancet,!347(8993):!61.!!
Volavka!J,!Czobor!P,!Sheitman!B,!Lindenmayer!JP,!Citrome!L,!McEvoy!JP,!.! .! .!Lieberman!JA.!(2002).!Clozapine,!olanzapine,! risperidone,!and!haloperidol! in! the! treatment!of!patients!with!chronic!schizophrenia! and! schizoaffective! disorder.! The! American! Journal! of! Psychiatry,! 159(2):! 255*262.!!
Wang!F,!Chen!H,! Steketee! JD!&!Sharp!BM.! (2007).!Upregulation!of! ionotropic! glutamate! receptor!subunits!within!specific!mesocorticolimbic!regions!during!chronic!nicotine!self*administration.!Neuropsychopharmacology,!32(1):!103*109.!!
Wang!J,!Zhuang!J,!Iyer!S,!Lin!X,!Whitfield!TW,!Greven!MC,!.!.!.!Weng!Z.!(2012).!Sequence!features!and!chromatin! structure! around! the! genomic! regions! bound!by! 119!human! transcription! factors.!Genome!Research,!22(9):!1798*1812.!!

169!!
!!
Ward!LD!&!Kellis!M.!(2012).!HaploReg:!A!resource!for!exploring!chromatin!states,!conservation,!and!regulatory!motif! alterations!within! sets!of! genetically! linked!variants.!Nucleic!Acids!Research,!Database!Issue,!(40):!D930*D934.!!
Wassef! A,! Baker! J! &! Kochan! LD.! (2003).! GABA! and! schizophrenia:! A! review! of! basic! science! and!clinical!studies.!Journal!of!Clinical!Psychopharmacology,!23(6):!601*640.!!
Watkins,! J! &! Collingridge! G.! (1994).! Phenylglycine! derivatives! as! antagonists! of! metabotropic!glutamate!receptors.!Trends!in!Pharmacological!Sciences,!15(9):!333*342.!!
Watkins!JC!&!Evans!RH.!(1981).!Excitatory!amino!acid!transmitters.!Annual!Review!of!Pharmacology!and!Toxicology,!21:!165*204.!!
Weickert!CS,! Straub!RE,!McClintock!BW,!Matsumoto!M,!Hashimoto!R,!Hyde!TM,! .! .! .! Kleinman! JE.!(2004).! Human! dysbindin! (DTNBP1)! gene! expression! in! normal! brain! and! in! schizophrenic!prefrontal!cortex!and!midbrain.!Archives!of!General!Psychiatry,!61(6):!544*555.!!
Weinberg!W.!(1908).!Über!den!nachweis!der!vererbung!beim!menschen.!Württemberg,!64:!369*382.!!
Weinberger! DR.! (1987).! Implications! of! normal! brain! development! for! the! pathogenesis! of!schizophrenia.!Archives!of!General!Psychiatry,!44(7):!660*669.!!
Weir! B.! (1996).! Genetic! data! analysis! II:! Methods! for! discrete! population! genetic! data!(2nd!ed).!Sunderland,!MA:!Sinauer!Associates.!!
Whitlock!JR,!Heynen!AJ,!Shuler!MG!&!Bear!MF.!(2006).!Learning! induces! long*term!potentiation!in!the!hippocampus.!Science,!313(5790):!1093*1097.!!
Williams!NM,!Preece!A,!Spurlock!G,!Norton!N,!Williams!HJ,!Zammit!S,!.!.!.!Owen!MJ.!(2003).!Support!for!genetic!variation!in!neuregulin!1!and!susceptibility!to!schizophrenia.!Molecular!Psychiatry,!8(5):!485*487.!!
Williams!NM,!Preece!A,!Morris!DW,!Spurlock!G,!Bray!NJ,!Stephens!M,! .! .! .!O'Donovan!MC.! (2004).!Identification! in! 2! independent! samples! of! a! novel! schizophrenia! risk! haplotype! of! the!dystrobrevin!binding!protein!gene!(DTNBP1).!Archives!of!General!Psychiatry,!61(4):!336*344.!!
Wilson! JF,! Weale! ME,! Smith! AC,! Gratrix! F,! Fletcher! B,! Thomas! MG,! .! .! .! Goldstein! DB.! (2001).!Population!genetic!structure!of!variable!drug!response.!Nature!Genetics,!29(3):!265*269.!!
Wilson!WH.!(1996).!Time!required!for!initial!improvement!during!clozapine!treatment!of!refractory!schizophrenia.!The!American!Journal!of!Psychiatry,!153(7):!951*952.!!
Winner! JG,! Carhart! JM,! Altar! CA,! Allen! JD! &! Dechairo! BM.! (2013).! A! prospective,! randomized,!double*blind! study! assessing! the! clinical! impact! of! integrated! pharmacogenomic! testing! for!major!depressive!disorder.!Discovery!Medicine,!16(89):!219*227.!!

170!!
!!
Wittke*Thompson! JK,! Pluzhnikov! A! &! Cox! NJ.! (2005).! Rational! inferences! about! departures! from!Hardy*Weinberg!equilibrium.!American!Journal!of!Human!Genetics,!76(6):!967*986.!!
Wolosker!H,!Blackshaw!S!&!Snyder!SH.!(1999).!Serine!racemase:!A!glial!enzyme!synthesizing!D*serine!to! regulate! glutamate*N*methyl*D*aspartate! neurotransmission.! Proceedings! of! the! National!Academy!of!Sciences!of!the!United!States!of!America,!96(23):!13409*13414.!!
World! Medical! Association! (WMA).! (2013).! WMA! Declaration! of! Helsinki:! Ethical! principles! for!medical! research! involving! human! subjects.! Retrieved! November! 6th,! 2014,! from!http://www.wma.net/en/30publications/10policies/b3/index.html!!
Wright! IC,! Rabe*Hesketh! S,! Woodruff! PW,! David! AS,! Murray! RM! &! Bullmore! ET.! (2000).! Meta*analysis!of!regional!brain!volumes!in!schizophrenia.!The!American!Journal!of!Psychiatry,!157(1):!16*25.!!
Wyatt!RJ!&!Henter! I.! (2001).!Rationale!for!the!study!of!early! intervention.!Schizophrenia!Research,!51(1):!69*76.!!
Xia!Z,!Dudek!H,!Miranti!CK!&!Greenberg!ME.!(1996).!Calcium!influx!via!the!NMDA!receptor!induces!immediate!early!gene!transcription!by!a!MAP!kinase/ERK*dependent!mechanism.!The!Journal!of!Neuroscience,!16(17):!5425*5436.!!
Xu!Z!&!Taylor!JA.!(2009).!SNPinfo:!Integrating!GWAS!and!candidate!gene!information!into!functional!SNP! selection! for! genetic! association! studies.! Nucleic! Acids! Research,! Web! Server! issue,! 37:!W600*W605.!!
Yamamori!H,!Hashimoto!R,!Fujita!Y,!Numata!S,!Yasuda!Y,!Fujimoto!M,!.!.!.!Takeda!M.!(2014).!Changes!in!plasma!D*serine,!L*serine,!and!glycine!levels!in!treatment*resistant!schizophrenia!before!and!after!clozapine!treatment.!Neuroscience!Letters,!582:!93*98.!!
Yamamoto!BK!&!Cooperman!MA.!(1994).!Differential!effects!of!chronic!antipsychotic!drug!treatment!on!extracellular!glutamate!and!dopamine!concentrations.!The! Journal!of!Neuroscience,!14(7):!4159*4166.!!
Yang! HC,! Liu! CM,! Liu! YL,! Chen! CW,! Chang! CC,! Fann! CS,! .! .! .! Hwu! HG.! (2013).! The! DAO! gene! is!associated! with! schizophrenia! and! interacts! with! other! genes! in! the! Taiwan! Han! Chinese!population.!PloS!One,!8(3):!e60099.!!
Yoo!HJ,!Cho!IH,!Park!M,!Yang!SY!&!Kim!SA.!(2012).!Family!based!association!of!GRIN2A!and!GRIN2B!with!Korean!autism!spectrum!disorders.!Neuroscience!Letters,!512(2):!89*93.!!
Yuan!HY,!Chen! JJ,! Lee!MT,!Wung! JC,!Chen!YF,!Charng!MJ,! .! .! .!Chen!YT.! (2005).!A!novel! functional!VKORC1! promoter! polymorphism! is! associated! with! inter*individual! and! inter*ethnic!differences!in!warfarin!sensitivity.!Human!Molecular!Genetics,!14(13):!1745*1751.!!

171!!
!!
Yuen!EY,!Liu!W,!Karatsoreos,!IN,!Ren!Y,!Feng!J,!McEwen!BS!&!Yan!Z.!(2011).!Mechanisms!for!acute!stress*induced! enhancement! of! glutamatergic! transmission! and! working!memory.! Molecular!Psychiatry,!16(2):!156*170.!!
Zafra! F,! Aragon! C,! Olivares! L,! Danbolt! NC,! Gimenez! C! &! Storm*Mathisen! J.! (1995).! Glycine!transporters!are!differentially!expressed!among!CNS!cells.! The! Journal!of!Neuroscience,!15(5,!2):!3952*3969.!!
Zai!CC,!Manchia!M,!De!Luca!V,!Tiwari!AK,!Chowdhury!NI,!Zai!GC,!.!.!.!Kennedy!JL.!(2012).!The!brain*derived! neurotrophic! factor! gene! in! suicidal! behaviour:! A! meta*analysis.! The! International!Journal!of!Neuropsychopharmacology,!15(8):!1037*1042.!!
Zhang!C,!Li!Z,!Wu!Z,!Chen!J,!Wang!Z,!Peng!D,! .! .! .!Fang!Y.! (2014).!A!study!of!N*methyl*D*aspartate!receptor! gene! (GRIN2B)! variants! as! predictors! of! treatment*resistant! major! depression.!Psychopharmacology,!231(4):!685*693.!!
Zhao!C,!Sun!T!&!Li!M.!(2012).!Neural!basis!of!the!potentiated!inhibition!of!repeated!haloperidol!and!clozapine! treatment! on! the! phencyclidine*induced! hyperlocomotion.! Progress! in! NeuroPPsychopharmacology!&!Biological!Psychiatry,!38(2):!175*182.!!
Zheng!X,!Zhang!L,!Wang!AP,!Bennett!MV!&!Zukin!RS.! (1997).!Ca2+! influx!amplifies!protein!kinase!C!potentiation!of!recombinant!NMDA!receptors.!The!Journal!of!Neuroscience,!17(22):!8676*8686.!!
Zuk! O,! Hechter! E,! Sunyaev! SR! &! Lander! ES.! (2012).! The! mystery! of! missing! heritability:! Genetic!interactions! create!phantom!heritability.! Proceedings!of! the!National!Academy!of! Sciences!of!the!United!States!of!America,!109(4):!1193*1198.!!
Zweifel!LS,!Argilli!E,!Bonci!A!&!Palmiter!RD.!(2008).!Role!of!NMDA!receptors!in!dopamine!neurons!for!plasticity!and!addictive!behaviors.!Neuron,!59(3):!486*496.!!
!





























