Embed Size (px)
Citation preview

ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS
Vol. 286, No. 1, April, pp. 217-221, 1991
pH-Sensitive Control of Arginase by at Submicromolar Concentrations
Nicholas J. Kuhn,l Judith Talbot, and Simon Ward
Mn(ll) Ions
School of Biochemistry, University of Birmingham, Birmingham B15 2TT, United Kingdom
Received July 25, 1990, and in revised form November 2, 1990
The manganese dependence of arginase was reinves- tigated with extracts of mouse liver to see whether more physiological properties were displayed than have been reported for the purified enzyme. In a preincubation with Mn(I1) ions at 37°C the enzyme underwent a slow and reversible activation. At least 90-95% of the activation achieved was dependent on Mn2+. However, no Mn2+ was required for catalytic activity in the assay. The activation showed little dependence upon pH over the range 6.5- 9.5, whereas the catalytic activity increased la-fold in apparent accord with the titration curve of an ionizable group of pK,, 7.9. The Mn2+ dependence of arginase ac- tivation obeyed Michaelis-Menten kinetics, with Kd varying from 0.3 PM at pH 6.8 to 0.08 pM at pH 7.7. Free Mn2+ concentrations were established in these assays with a trimethylenediaminetetraacetate-Mn buffer. V,, increased about three-fold over this range. The calculated arginase activity at 0.05 pM Mn2+ increases about nine- fold over this physiological pH range. An enzyme model is proposed to explain these findings. The activity of ar- ginase at “physiological” [Mn2+] and the pronounced pH dependence conferred upon it are consistent with a re- cently revised role for the urea cycle in the control of bicarbonate and pH in the body. It appears possible that arginase loses Mn2+ sensitivity during the usual purifi- cation. 0 1991 Academic Press. Inc.
Mammalian hepatic arginase is one of a small group of enzymes specifically activated by the reversible binding of Mn2+ ions. This is of interest in the light of suggestions that Mn2+ may play not only a catalytic but also a regu- latory role in metabolism (l-3). Accordingly, the man- ganese-associated characteristics of this enzyme have been explored in normal rats and mice and in those made di- abetic with streptozotocin (4-7). A particular feature is the need to activate the enzyme, before assay, in a tem-
1 To whom correspondence should be addressed.
0003.9861/91 $3.00 Copyright 0 1991 by Academic Press, Inc. All rights of reproduction in any form reserved.
perature-, time-, and manganese-dependent preincuba- tion. However, as pointed out by Pace et al. (8) such Mn2+ activation of purified arginase displays two seemingly un- physiological features (4, 6). First, only about half the enzyme activity is reversibly dependent upon Mn2+, the remaining activity being irreversibly lost when tightly bound Mn2+ is removed. Second, the reversible Mn2+-de- pendent activity shows a Kd for Mn2+ of about 50 PM, which probably exceeds the free [Mn’+] at least lOO- fold (9).
It seemed possible that these features might reflect the purification procedure usually employed, which includes exposure of the enzyme to organic solvents and to high temperature. Although the enzyme is evidently robust, its regulatory properties might be less so. Therefore we have reexamined the manganese dependence of mouse liver ar- ginase under the mildest conditions, namely in high-speed supernatants of buffered saline extracts treated with Chelex to remove endogenous bivalent metal ions. The lack of interfering reactions and the high dilution at which assays can be performed ensure suitable conditions for kinetic assays. Known concentrations of free Mn2+ were established with the aid of suitable buffers.
MATERIALS AND METHODS
Enzyme preparation. Adult mouse liver was homogenized in 3 vol of 10 mM Te&NaOH/O.l M KC1 buffer, pH 7.0 at 0°C. This was cen- trifuged at 100,OOOg for 30 min, and the clear supernatant was stored in aliquots at -18’C. For use, portions (0.2 ml) were freshly thawed each day, diluted IO-fold with water, and treated for 15 min at 0°C with Chelex (0.2 g), which was then removed by centrifugation. This reduced the basal arginase activity from about 50 to 20-30% of the fully activated enzyme (see Fig 1). The Chelex itself was previously washed six times with deionized water, twice with 0.4 M Tes-NaOH buffer, pH 6.5, and finally once with water (6-7 ml each wash).
Preincubation and assay of arginase. For the standard procedure, enzyme (45 ~1) was preincubated for several hours at 37°C in 120 ~1
* Abbreviations used: Ac2N(CH2)aNAc2, trimethylenediaminetetraac- etate; Tes, 2-{ [2-hydroxy-l,l-bis(hydroxymethyl)ethyl]amino}-eth- anesulfonic acid.
217
218 KUHN, TALBOT, AND WARD
Incubation time Ih)
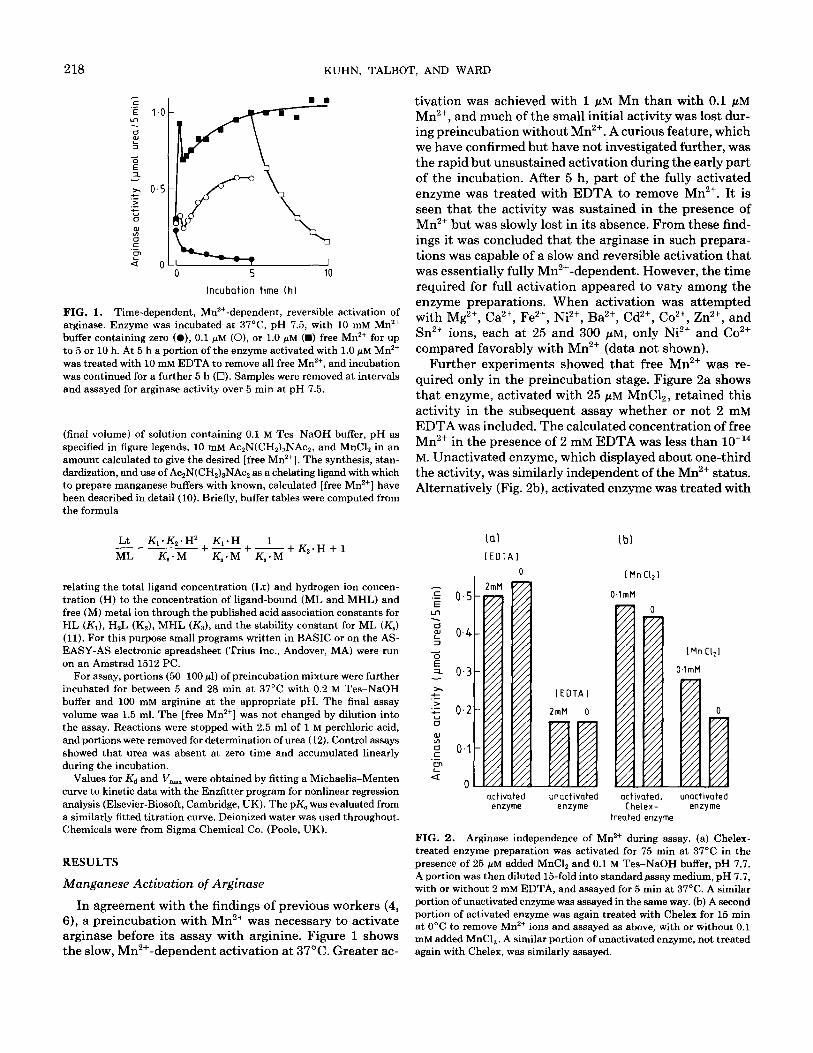
FIG. 1. Time-dependent, Mn ‘+-dependent, reversible activation of arginase. Enzyme was incubated at 37”C, pH 7.5, with 10 mM Mn*+ buffer containing zero (O), 0.1 pM (O), or 1.0 pM (D) free Mn*+ for up to 5 or 10 h. At 5 h a portion of the enzyme activated with 1.0 FM Mn*+ was treated with 10 mM EDTA to remove all free Mn’+, and incubation was continued for a further 5 h (0). Samples were removed at intervals and assayed for arginase activity over 5 min at pH 7.5.
(final volume) of solution containing 0.1 M Tes-NaOH buffer, pH as specified in figure legends, 10 mM Ac2N(CH2)aNAc2, and MnCl, in an amount calculated to give the desired [free Mn*+]. The synthesis, stan- dardization, and use of Ac2N(CH2)aNAc, as a chelating ligand with which to prepare manganese buffers with known, calculated [free Mn’+] have been described in detail (10). Briefly, buffer tables were computed from the formula
Lt K,.K,.H*+Ki*H I
ML= K.*M
1 -+Ka.H+l
K..M K.-M
relating the total ligand concentration (Lt) and hydrogen ion concen- tration (H) to the concentration of ligand-bound (ML and MHL) and free (M) metal ion through the published acid association constants for HL (Kl), H2L (K,), MHL (K3), and the stability constant for ML (K.) (11). For this purpose small programs written in BASIC or on the AS- EASY-AS electronic spreadsheet (Trius Inc., Andover, MA) were run on an Amstrad 1512 PC.
For assay, portions (50-100 ~1) of preincubation mixture were further incubated for between 5 and 28 min at 37°C with 0.2 M Tes-NaOH buffer and 100 mM arginine at the appropriate pH. The final assay volume was 1.5 ml. The [free Mn’+] was not changed by dilution into the assay. Reactions were stopped with 2.5 ml of 1 M perchloric acid, and portions were removed for determination of urea (12). Control assays showed that urea was absent at zero time and accumulated linearly during the incubation.
Values for Kd and V,,,,, were obtained by fitting a Michaelis-Menten curve to kinetic data with the Enzfitter program for nonlinear regression analysis (Elsevier-Biosoft, Cambridge, UK). The pK, was evaluated from a similarly fitted titration curve. Deionized water was used throughout. Chemicals were from Sigma Chemical Co. (Poole, UK).
RESULTS
Manganese Activation of Arginase
In agreement with the findings of previous workers (4, 6), a preincubation with Mn2+ was necessary to activate arginase before its assay with arginine. Figure 1 shows the slow, Mn2+-dependent activation at 37’C. Greater ac-
tivation was achieved with 1 PM Mn than with 0.1 pM Mn2+, and much of the small initial activity was lost dur- ing preincubation without Mn2+. A curious feature, which we have confirmed but have not investigated further, was the rapid but unsustained activation during the early part of the incubation. After 5 h, part of the fully activated enzyme was treated with EDTA to remove Mn2+. It is seen that the activity was sustained in the presence of Mn2+ but was slowly lost in its absence. From these find- ings it was concluded that the arginase in such prepara- tions was capable of a slow and reversible activation that was essentially fully Mn2+-dependent. However, the time required for full activation appeared to vary among the enzyme preparations. When activation was attempted with Mg2+, Ca2+, Fe2+, Ni2+ Ba2+, Cd2+, Co2+, Zn2+, and Sn2’ ions, each at 25 and 300 PM, only Ni2+ and Co2+ compared favorably with Mn2+ (data not shown).
Further experiments showed that free Mn2+ was re- quired only in the preincubation stage. Figure 2a shows that enzyme, activated with 25 PM MnC12, retained this activity in the subsequent assay whether or not 2 mM
EDTA was included. The calculated concentration of free Mn2+ in the presence of 2 mM EDTA was less than lo-l4 M. Unactivated enzyme, which displayed about one-third the activity, was similarly independent of the Mn2+ status. Alternatively (Fig. 2b), activated enzyme was treated with
(a) [EDTAI
0
[EOTAI
2mM 0
lb1
[Mn&l
O.lmM
[MnC121
O.lmM
activated unactivoted enzyme enzyme
activated, unactivated Chelex- enzyme
treated enzyme
FIG. 2. Arginase independence of Mn2+ during assay. (a) Chelex- treated enzyme preparation was activated for 75 min at 37°C in the presence of 25 NM added MnClz and 0.1 M Tes-NaOH buffer, pH 7.7. A portion was then diluted 15-fold into standard assay medium, pH 7.7, with or without 2 mM EDTA, and assayed for 5 min at 37’C. A similar portion of unactivated enzyme was assayed in the same way. (b) A second portion of activated enzyme was again treated with Chelex for 15 min at 0°C to remove Mn2+ ions and assayed as above, with or without 0.1 mM added MnCl,. A similar portion of unactivated enzyme, not treated again with Chelex, was similarly assayed.
ARGINASE CONTROL BY MI?+ AND pH 219
PH
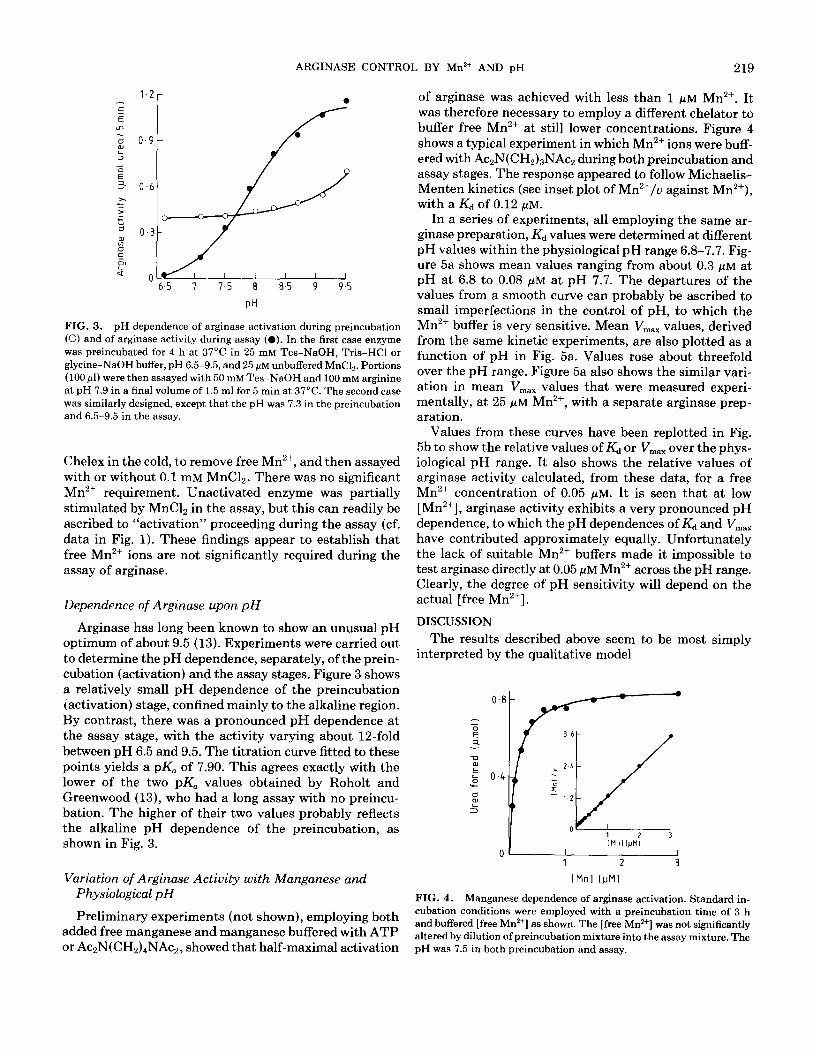
FIG. 3. pH dependence of arginase activation during preincubation (0) and of arginase activity during assay (0). In the first case enzyme was preincubated for 4 h at 37°C in 25 mM Tes-NaOH, Tris-HCl or glycine-NaOH buffer, pH 6.5-9.5, and 25 pM unbuffered MnC12. Portions (100 ~1) were then assayed with 50 mM Tes-NaOH and 100 mM arginine at pH 7.9 in a final volume of 1.5 ml for 5 min at 37°C. The second case was similarly designed, except that the pH was 7.3 in the preincubation and 6.5-9.5 in the assay.
Chelex in the cold, to remove free Mn’+, and then assayed with or without 0.1 mM MnC12. There was no significant Mn2+ requirement. Unactivated enzyme was partially stimulated by MnC12 in the assay, but this can readily be ascribed to “activation” proceeding during the assay (cf. data in Fig. 1). These findings appear to establish that free Mn2+ ions are not significantly required during the assay of arginase.
Dependence of Arginase upon pH
Arginase has long been known to show an unusual pH optimum of about 9.5 (13). Experiments were carried out to determine the pH dependence, separately, of the prein- cubation (activation) and the assay stages. Figure 3 shows a relatively small pH dependence of the preincubation (activation) stage, confined mainly to the alkaline region. By contrast, there was a pronounced pH dependence at the assay stage, with the activity varying about la-fold between pH 6.5 and 9.5. The titration curve fitted to these points yields a pK, of 7.90. This agrees exactly with the lower of the two pK, values obtained by Roholt and Greenwood (13), who had a long assay with no preincu- bation. The higher of their two values probably reflects the alkaline pH dependence of the preincubation, as shown in Fig. 3.
Variation of Arginase Activity with Manganese and Physiological pH
Preliminary experiments (not shown), employing both added free manganese and manganese buffered with ATP
of arginase was achieved with less than 1 PM Mn2+. It was therefore necessary to employ a different chelator to buffer free Mn2+ at still lower concentrations. Figure 4 shows a typical experiment in which Mn2+ ions were buff- ered with Ac2N(CH2)3NAc2 during both preincubation and assay stages. The response appeared to follow Michaelis- Menten kinetics (see inset plot of Mn’+/u against Mn’+), with a Kd of 0.12 PM.
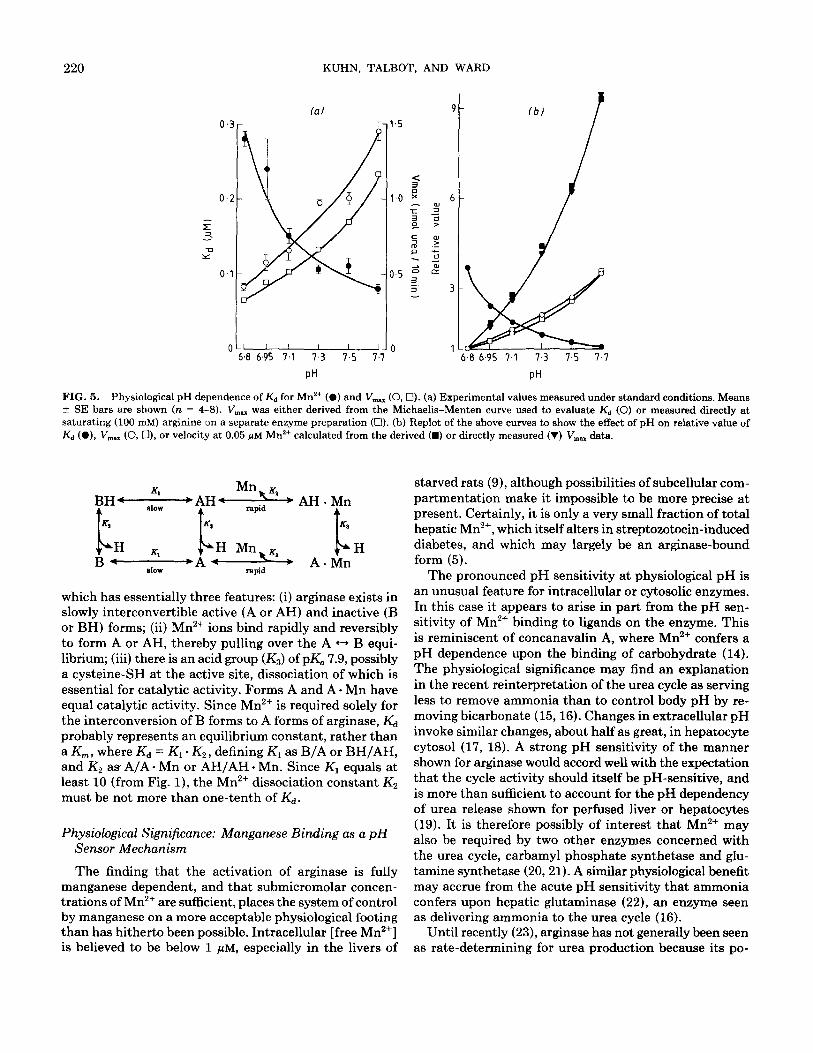
In a series of experiments, all employing the same ar- ginase preparation, Kd values were determined at different pH values within the physiological pH range 6.8-7.7. Fig- ure 5a shows mean values ranging from about 0.3 j.&M at pH at 6.8 to 0.08 PM at pH 7.7. The departures of the values from a smooth curve can probably be ascribed to small imperfections in the control of pH, to which the Mn2+ buffer is very sensitive. Mean V,,, values, derived from the same kinetic experiments, are also plotted as a function of pH in Fig. 5a. Values rose about threefold over the pH range. Figure 5a also shows the similar vari- ation in mean V,,, values that were measured experi- mentally, at 25 WM Mn’+, with a separate arginase prep- aration.
Values from these curves have been replotted in Fig. 5b to show the relative values of Kd or V,,, over the phys- iological pH range. It also shows the relative values of arginase activity calculated, from these data, for a free Mn2+ concentration of 0.05 PM. It is seen that at low [Mn’+], arginase activity exhibits a very pronounced pH dependence, to which the pH dependences of Kd and V,,,,, have contributed approximately equally. Unfortunately the lack of suitable Mn2+ buffers made it impossible to test arginase directly at 0.05 PM Mn2+ across the pH range. Clearly, the degree of pH sensitivity will depend on the actual [free Mn’+].
DISCUSSION
The results described above seem to be most simply interpreted by the qualitative model
l
36
0 1 [Mnl
I,,: 3
1 2 3
[Mnl (JJM)
FIG. 4. Manganese dependence of arginase activation. Standard in- cubation conditions were employed with a preincubation time of 3 h and buffered [free Mn*+] as shown. The [free Mn’+] was not significantly altered by dilution of preincubation mixture into the assay mixture. The
or Ac~N(CH~)~NAC~, showed that half-maximal activation pH was 7.5 in both preincubation and assay.
220 KUHN, TALBOT, AND WARD
0 1 6.8 6.95 7.1 7.3 7.5 7.7 6.8 6.95 7 1 7.3 7.5 7.7
PH PH
FIG. 5. Physiological pH dependence of K,+ for Mn2+ (0) and V,, (0,O). (a) Experimental values measured under standard conditions. Means + SE bars are shown (n = 4-8). V,, was either derived from the Michaelis-Menten curve used to evaluate Kd (0) or measured directly at saturating (100 mM) arginine on a separate enzyme preparation (Cl). (b) Replot of the above curves to show the effect of pH on relative value of Kd (a), V,,,.. (0, Cl), or velocity at 0.05 jtM Mn*+ calculated from the derived (m) or directly measured (V) V,., data.
l AH.Mn
K3
c H * A.Mn
which has essentially three features: (i) arginase exists in slowly interconvertible active (A or AH) and inactive (B or BH) forms; (ii) Mn2+ ions bind rapidly and reversibly to form A or AH, thereby pulling over the A t-, B equi- librium; (iii) there is an acid group (I&) of pK, 7.9, possibly a cysteine-SH at the active site, dissociation of which is essential for catalytic activity. Forms A and A. Mn have equal catalytic activity. Since Mn2+ is required solely for the interconversion of B forms to A forms of arginase, Kd probably represents an equilibrium constant, rather than a K,,, , where Kd = Kl . K2, defining Kl as B/A or BH/AH, and K2 as A/A * Mn or AH/AH * Mn. Since Kl equals at least 10 (from Fig. l), the Mn2+ dissociation constant K2 must be not more than one-tenth of Kd.
Physiological Significance: Manganese Binding as a pH Sensor Mechanism
The finding that the activation of arginase is fully manganese dependent, and that submicromolar concen- trations of Mn’+ are sufficient, places the system of control by manganese on a more acceptable physiological footing than has hitherto been possible. Intracellular [free Mn’+] is believed to be below 1 PM, especially in the livers of
starved rats (9), although possibilities of subcellular com- partmentation make it impossible to be more precise at present. Certainly, it is only a very small fraction of total hepatic Mn’+, which itself alters in streptozotocin-induced diabetes, and which may largely be an arginase-bound form (5).
The pronounced pH sensitivity at physiological pH is an unusual feature for intracellular or cytosolic enzymes. In this case it appears to arise in part from the pH sen- sitivity of Mn2+ binding to ligands on the enzyme. This is reminiscent of concanavalin A, where Mn2+ confers a pH dependence upon the binding of carbohydrate (14). The physiological significance may find an explanation in the recent reinterpretation of the urea cycle as serving less to remove ammonia than to control body pH by re- moving bicarbonate (l&16). Changes in extracellular pH invoke similar changes, about half as great, in hepatocyte cytosol (17, 18). A strong pH sensitivity of the manner shown for arginase would accord well with the expectation that the cycle activity should itself be pH-sensitive, and is more than sufficient to account for the pH dependency of urea release shown for perfused liver or hepatocytes (19). It is therefore possibly of interest that Mn2+ may also be required by two other enzymes concerned with the urea cycle, carbamyl phosphate synthetase and glu- tamine synthetase (20,21). A similar physiological benefit may accrue from the acute pH sensitivity that ammonia confers upon hepatic glutaminase (22), an enzyme seen as delivering ammonia to the urea cycle (16).
Until recently (23), arginase has not generally been seen as rate-determining for urea production because its po-

ARGINASE CONTROL BY Mn*+ AND pH 221
tential activity so far exceeds that of all other urea cycle enzymes (24). Yet when the total arginase activity of rat liver (35,000 gmol/min/g at pH 9.5) is corrected for pH, for subsaturation by Mn2+ (unknown at present), for an arginine concentration probably far below the K,,, of l-2 mM (24), and for the possible inhibition by branched chain amino acids (25), the resultant expressed activity is un- likely to exceed by much the rate of urea formation in the fed rat (about 1.1. pmol/min/g (26)). The position of arginase downstream of a branch point in metabolism, where arginine can serve both protein synthesis and urea formation, would also appear to make it a natural point of regulation. From a different viewpoint, arginase could be rate-determining through only a small fraction of the total being involved in the channeling of newly synthe- sized arginine, as demonstrated in hepatocytes (27).
ACKNOWLEDGMENT
We are grateful to Dr C. W. Wharton for critically reading the manu- script.
REFERENCES
1. Schramm, V. L. (1982) Trends Biochem. Sci. 7, 369-371.
2. Williams, R. J. P. (1982) FEBS Lett. 140, 3-10.
3. Schramm, V. L. (1986) in Manganese in Metabolism and Enzyme Function (Schramm, V. L., and Wedler, F. C., Eds.), pp. 109-132, Academic Press, London.
4. Hirsch-Kolb, H.. Kolb, H. J., and Greenberg, D. M. (1971) J. Biol. Chem. 246,395401.
5. Bond, J. S., Failla, M. I~., and Unger, D. F. (1983) J. Biol. Chem. 258,8004-8009.
6. Bond, J. S. (1986) in Manganese in Metabolism and Enzyme Func- tion (Schramm, V. L., and Wedler, F. C., Eds.), pp. 239-257, Aca- demic Press. London.
7. Spolarics, Z., and Bond, J. S. (1989) Arch. Biochem. Biophys. 274, 426-433.
8. Pace, C. N., Buonanno, A., and Simmons-Hansen, J. (1980) Anal. Biochem. 109, 261-265.
9. Ash, D. E., and Schramm, V. L. (1982) J. Biol. Chem. 257,9261- 9264.
10. Kuhn, N. J., Ward, S., and Leong, W. S. (1991) Eur. J. Biochem. 196, 2433250.
11. Anderegg, G. (1964) Helu. Chim. Acta 47, 1801-1814.
12. Archibald, R. M. (1945) J. Biol. Chem. 157, 507-517.
13. Roholt, 0. A., and Greenberg, D. M. (1956) Arch. Biochem. Biophys. 62, 454-470.
14. Sherry, A. D., Newman, A. D., and Gutz, C. G. (1975) Biochemistry 14, 2191-2196.
15. Atkinson, D. E., and Bourke, E. (1984) Trends Biochem. Sci. 9, 297-300.
16. Haussinger, D., Meijer, A. J., Gerok, W., and Sies, H. (1988) in pH Homeostasis, Mechanisms, and Control (Haussinger, D., Ed.), pp. 337-377, Academic Press, London.
1’7. Cohen, R. D., Henderson, R. M., Iles, R. A., and Smith, J. A. (1982) J. Physiol. 330, 69-80.
18. Kashiwagura, T., Deutsch, C. J., Taylor, J., Erecinska, M., and Wil- son, D. F. (1984) J. Biol. Chem. 259,237-243.
19. Bean, E. S., and Atkinson, D. E. (1984) J. Biol. Chem. 259, 1552- 1559.
20. Cohen, N. S. (1984) Arch. Biochem. Biophys. 232.38-46.
21. Wedler, F. C., and Toms, R. (1986) in Managanese in Metabolism and Enzyme Function (Schramm, V. L., and Wedler, F. C., Eds.), pp. 221-238, Academic Press, London.
22. Verhoeven, A. J., van Iwaarden, J. F., Joseph, S. K., and Meijer, A. J. (1983) Eur. J. Biochem. 133, 241-244.
23. Garganta, C. L., and Bond, J. S. (1986) Anal. Biochem. 154, 388- 394.
24. Raijman, L. (1976) in The Urea Cycle (Grisolia, S., Baguena, R., and Mayor, F., Eds.), pp. 2433254, Wiley, New York.
25. Carvajal, N., and Cederbaum, S. D. (1986) Biochem. Biophys. Acta 870, 181-184.
26. Schimke, R. (1962) J. Biol. Chem. 237,459-468.
27. Cheung, C. W., Cohen, N. S., and Raijman, L. (1989) J. Biol. Chem. 264,4038-4044.





























