Embed Size (px)
Citation preview

pH-Controlled Polymer Surface Segregation
Richard L. Thompson,*,† Sarah J. Hardman,† Lian R. Hutchings,†
Amilcar Pillay Narrainen,†,§ and Robert M. Dalgliesh‡
Department of Chemistry, Durham UniVersity, Science Site, Durham DH1 3LE, U.K.,and ISIS, Rutherford Appleton Laboratories, Chilton, Didcot, OX11 0QX, U.K.
ReceiVed October 28, 2008. ReVised Manuscript ReceiVed December 15, 2008
A new approach to promoting and controlling polymer surface functionalization with acidic or basic polar functionalgroups is demonstrated and evaluated. Blended polymer films were annealed under pH-buffered conditions, and polarend-functional groups were found to promote surface segregation of the functional polymers. Surface segregation ofcarboxylic acid (COOH)-functionalized polystyrene increases dramatically with increasing pH from 1.9 to 9.4, whereasthe opposite behavior is seen for amine (NH2)-functionalized polystyrene. Neutron reflectometry and nuclear reactionanalysis were used to obtain surface excess values for the functional polymers. Subsequent SCFT analysis of thecomposition versus depth profiles indicates that the affinity of each functional group for the polymer surface changesby about 3kBT over this pH range.
Introduction
Control over polymer surface functionality is highly desirablefor a diverse range of applications, from wetting and adhesionto cell tissue culture.1,2 Recent advances made by ourselves3-9
and others10,11 have shown that multiple end-functional groupson polymer chains can confer significant changes to the surfaceproperties of polymer blends. It is relatively easy to achieve thisusing polymers that are end-functionalized with low-surface-energy groups such as fluorocarbons3-8,10,12 and siloxanes13,14
because these spontaneously segregate to film surfaces, reducingtheir surface energy. However, it is less trivial to enrich a surfacewith polar functional groups. This problem arises because thepolar functional groups normally have a significantly highersurface energy than the polymer matrix; therefore, their surfacesegregation is inhibited. However, by immersing the polymer ina polar liquid, the criteria for surface segregation are reversed,and it is possible to induce the adsorption of polar functional
groups at the polymer-liquid interface. Koberstein et al.15,16
found that by immersing a pure functionalized polymer in waterfor prolonged periods of time local reorganization could resultin a polar functionalized surface. Because of the fact that onlylocal reorganization was possible at ambient temperatures, onlyfunctionalities that were already at or adjacent to the film surfacecould contribute to changes in surface composition. Therefore,the architecture of the functionalized molecules had to beingeniously designed to ensure that polar groups were initiallyclose to the film surface. More recently, we have shown thatsurface segregation of polar end-functionalized polymers ispossible in blends by annealing blend films above their glass-transition temperature while immersed in a polar nonsolvent.17
This strategy allows the surface to be enriched by any functionalgroup that can diffuse to the surface and so does not requirecomplex polymer architectures or high concentrations offunctional groups. However, both approaches are limited in thenumber of functional groups per unit area that can be broughtto a film surface, and neither can discriminate between differentkinds of polar functionality that could be simultaneously presentin a polymer blend. In an attempt to overcome these limitations,increase the efficiency of surface segregation, and offer controlover the nature of polar functionalities that are drawn to a polymersurface, we have explored the influence of pH on the surfacesegregation of acid (COOH)- and base (NH2)-functionalizedpolymers.
In a series of influential papers, Whitesides et al.18-20
demonstrated that when aqueous solutions were placed onCOOH-functionalized surfaces the contact angle of the solutionhad a strong dependence on pH. Contact angles decreased withincreasing pH on COOH-functionalized polyethylene. Theincrease in wettability was due to the increase in deprotonation
* Corresponding author. E-mail: [email protected]. Tel: +44191 3342139. Fax: +44 191 3844737.
† Durham University.‡ ISIS.§ Present address: Materials Science Centre, The University of Manchester,
Grosvenor Street, Manchester M1 7HS, U.K.(1) Lee, J. H.; Jung, H. W.; Kang, I. K.; Lee, H. B. Biomaterials 1994, 15,
705–711.(2) Goddard, J. M.; Hotchkiss, J. H. Prog. Polym. Sci. 2007, 32, 698–725.(3) Thompson, R. L.; Narrainen, A. P.; Eggleston, S. M.; Ansari, I. A.;
Hutchings, L. R.; Clarke, N. J. Appl. Polym. Sci. 2007, 105, 623–628.(4) Narrainen, A. P.; Hutchings, L. R.; Ansari, I.; Thompson, R. L.; Clarke,
N. Macromolecules 2007, 40, 1969–1980.(5) Ansari, I. A.; Clarke, N.; Hutchings, L. R.; Pillay-Narrainen, A.; Terry,
A. E.; Thompson, R. L.; Webster, J. R. P. Langmuir 2007, 23, 4405–4413.(6) Pillay-Narrainen, A.; Hutchings, L. R.; Feast, W. J.; Thompson, R. L.;
Ansari, I. A.; Clarke, N. Macromol. Symp. 2006, 231, 103–109.(7) Narrainen, A. P.; Hutchings, L. R.; Ansari, I. A.; Clarke, N.; Thompson,
R. L. Soft Matter 2006, 2, 126–128.(8) Hutchings, L. R.; Narrainen, A. P.; Eggleston, S. M.; Clarke, N.; Thompson,
R. L. Polymer 2006, 47, 8116–8122.(9) Hutchings, L. R.; Narrianen, A. P.; Thompson, R. L.; Clarke, N.; Ansari,
L. Polym. Int. 2008, 57, 163–170.(10) Li, H.; Zhang, Y. M.; Zhang, H.; Xue, M. Z.; Liu, Y. G. J. Polym. Sci.,
Part A: Polym. Chem. 2006, 44(12), 3853–3858.(11) Hirao, A.; Sugiyama, K.; Yokoyama, H. Prog. Polym. Sci. 2007, 32,
1393–1438.(12) Narrainen, A. P.; Hutchings, L. R.; Ansari, I. A.; Clarke, N.; Thompson,
R. L. Soft Matter 2006, 2, 126–128.(13) Cai, Y. H.; Gardner, D.; Caneba, G. T. J. Adhes. Sci. Technol. 1999, 13,
1017–1027.(14) Lee, H.; Archer, L. A. Macromolecules 2001, 34, 4572–4579.
(15) Koberstein, J. T. J. Polym. Sci., Part B: Polym. Phys. 2004, 42, 2942–2956.
(16) O’Rourke-Muisener, P. A. V.; Jalbert, C. A.; Yuan, C. G.; Baetzold, J.;Mason, R.; Wong, D.; Kim, Y. J.; Koberstein, J. T. Macromolecules 2003, 36,2956–2966.
(17) Narrainen, A. P.; Clarke, N.; Eggleston, S. M.; Hutchings, L. R.; Thompson,R. L. Soft Matter 2006, 2(11), 981–985.
(18) Holmes-Farley, S. R.; Reamey, R. H.; McCarthy, T. J.; Deutch, J.;Whitesides, G. M. Langmuir 1985, 1(6), 725–740.
(19) Holmesfarley, S. R.; Bain, C. D.; Whitesides, G. M. Langmuir 1988, 4,921–937.
(20) Bain, C. D.; Whitesides, G. M. Langmuir 1989, 5, 1370–1378.
3184 Langmuir 2009, 25, 3184-3188
10.1021/la803583f CCC: $40.75 2009 American Chemical SocietyPublished on Web 02/04/2009
of COOH as the pH of the contact fluid was increased. Here, weexplore the converse situation whereby the pH of a contactingfluid is exploited to control the surface characteristics of a polymerblend. Understanding the response of polymer blend surfaces topH is not trivial, not the least of which because the elevatedtemperatures required to overcome the glass transition will alsoalter the hydrophilicity of polar groups with respect to thecontacting solution.21
Experimental SectionBlend films comprising 10 wt % end-functional deuterium-labeled
polystyrene in hydrogenous polystyrene (Polymer Laboratories, U.K.,Mw ) 3065 kg/mol, Mw/Mn ) 1.05) were prepared by codissolvingthe polymers in toluene and then spin coating them onto siliconblocks. The amine and carboxylic acid end-functional polymers weredPS-NH2 (Polymer Source, Canada; Mw ) 4.9 kg/mol, Mw/Mn )1.04) and dPS-2COOH (Mw ) 27.7 kg/mol, Mw/Mn ) 1.31). ThedPS-2COOH polymer had two carboxylic acid functional groups atone end of the polymer chain and was prepared in-house as describedpreviously.17 Prior to use, the silicon blocks were cleaned in toluene,followed by permanganic acid, and then thoroughly rinsed withdeionized water. The polymer films (1.8 µm thick) were completelyimmersed in pH-buffered aqueous solutions and then annealed inan autoclave at 122 °C for 2 h. The samples were allowed to coolto 70 °C (i.e., well below the glass-transition temperature, 103 °C)before being removed from the buffered solutions, rinsing in deionizedwater, and drying at room temperature. By quenching to below theglass-transition temperature while maintaining the pH-controlledenvironment, the samples were kinetically trapped in their equilibriumconformation at this pH and could not revert back to the preferredconformation for the sample in air.
Surface segregation of the deuterium-labeled polymers wasinvestigated by neutron reflectometry using the CRISP reflectometerat the ISIS neutron source, Chilton, U.K. Reflectivity data werecollected over a range of scattering vectors from 0.06 < Q/nm-1 <2.5, which encompassed the full range over which useful data couldbe obtained from the critical edge to the background. Followingnormalization and background subtraction, data fitting was carriedout using a maximum entropy (model-independent) fitting algorithm22
over 0.15 < Q/nm-1 < 1.5. Fits were found to be very stable withrespect to starting parameters. In addition to the maximum entropyfitting method, a simple model for the composition versus depthprofile given by eq 1 was used.
φ(x)) φbulk +φs - φbulk
2 (1+ erf(xs - x
ws)) (1)
Here, φ is volume fraction dPS as a function of depth x belowthe film surface (x ) 0). The surface and bulk volume fractions ofdPS are given by φs and φbulk, respectively, and xs defines the spatialextent of the adsorbed layer whose interface with the subphase ischaracterized by the width ws. A slightly more complex model inwhich the surface excess layer was followed by a layer that wasdepleted with respect to φbulk was also considered
φ(x)) φdep +φs - φdep
2 (1+ erf(xs - x
ws))+
φbulk - φdep
2 (1+ erf(x- xdep
wdep)) (2)
where φdep, xdep, and wdep are the volume fraction, maximum depth,and lower interface width of the depleted region, respectively.
Finally, nuclear reaction analysis (NRA) experiments were carriedout on representative samples of the blended films following neutronreflectometry. For these experiments, the samples were irradiated
with a beam of 0.7 MeV 3He+ ions at 83° to the sample normal. Theenergy of backscattered protons resulting from the reaction between3He and 2H within the polymer was analyzed to determine thecomposition versus depth profile of the deuterated functional polymer.NRA has an inherently poorer depth resolution than neutronreflectometry and is therefore less well suited than neutronreflectometry to determine the surface excess of low-molecular-weight, weakly adsorbing polymers. However, the analysis of NRAdata is unambiguous and can be used to validate the neutronreflectometry results. This technique is described in greater detailelsewhere.23-25
Results
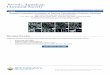
Typical neutron reflectivity data and fits are shown as RQ4
versus Q in Figure 1. The broad peak in the reflectivity foundin all data sets in the range of 0.1 < Q/nm-1 < 1.0 arises fromthe excess perdeuterated polymer at the film surface and wouldbe absent if the functional polymer had not adsorbed to the filmsurfaces. It is evident from the increase in the size of this peakin the raw data that the surface excess increases with increasingpH for dPS-2COOH and decreasing pH for dPS-NH2. Derivedconcentration profiles are shown in Figure 2 and demonstratethat pH exerts a strong influence on the near-surface concentrationof the adsorbed functional polymers. The surface excess, z* ()∫0
∞ [φ(x) - φbulk] dx), was calculated by numerically integratingthe derived volume fraction profiles. The uncertainty in the z*
(21) Carey, D. H.; Ferguson, G. S. J. Am. Chem. Soc. 1996, 118, 9780–9781.(22) Sivia, D. S.; Hamilton, W. A.; Smith, G. S. Physica B 1991, 173, 121–
138.
(23) Payne, R. S.; Clough, A. S.; Murphy, P.; Mills, P. J. Nucl. Instrum. MethodsPhys. Res., Sect. B 1989, 42, 130–134.
(24) Geoghegan, M. MeV Ion Beam Profiling of Polymer Surfaces andInterfaces. In Polymer Surfaces and Interfaces; Richards, R. W., Peace, S. K.,Eds.; John Wiley & Sons Ltd.: Chichester, U.K., 1999; Vol. III, pp 43-73.
(25) Chaturvedi, U. K.; Steiner, U.; Zak, O.; Krausch, G.; Schatz, G.; Klein,J. Appl. Phys. Lett. 1990, 56, 1228–1230.
Figure 1. Neutron reflectivity data and fits for (a) dPS-NH2 and (b)dPS-2COOH end-functionalized polymers in an hPS matrix. The insetsshow the same data in the R(Q) vs Q format.
pH-ResponsiVe Blends Langmuir, Vol. 25, No. 5, 2009 3185
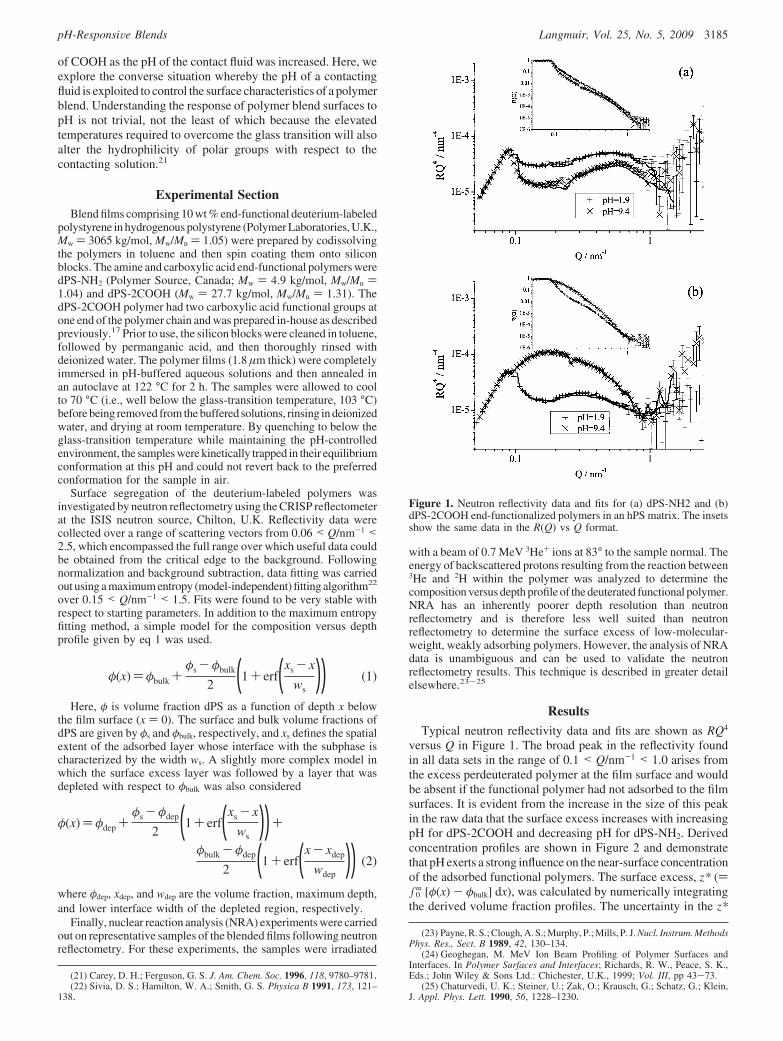
values is dominated by the uncertainty in φbulk adjacent to thesurface, which was estimated from the standard deviation in φ(x)over the range of 20 < x/nm < 30. Some small oscillations areapparent in the derived concentration profiles of the functionalpolymer adjacent to the surface. This is almost certainly due to“ringing”, an artifact of the maximum entropy fitting procedure.22
In some cases, a broader dip in φ(x) is apparent over a range of20-40 nm. To explore the significance of this broader depletionand to test the uniqueness of the maximum entropy fits to theneutron data, the analysis was repeated using the modelconcentration profiles defined by eqs 1 and 2. Previously, thesimple error function model (eq 1) has been used successfullyto describe equilibrium concentration profiles of adsorbed end-functional polymers at film surfaces.5,26,27 In fitting our reflec-tometry data, φs, xs, and ws were the adjustable parameters.Although the quality of the fits to the reflectometry data areslightly poorer than those obtained by the more flexible maximumentropy model, the fits are still very good, and the derived surfaceexcess values are also in excellent agreement. Derived concen-tration profiles and fits for the three fitting methods are shownin Figure 3 and clearly show that all methods yield consistentresults. Interestingly, using the more complex model (eq 2) inwhich φdep was also an adjustable parameter did not provide anyimprovement over the best fit that could be obtained with the
simpler model. The calculated reflectivities using these models(inset, Figure 3) are shown offset from each other and theexperimental data because they were indistinguishable to theeye.
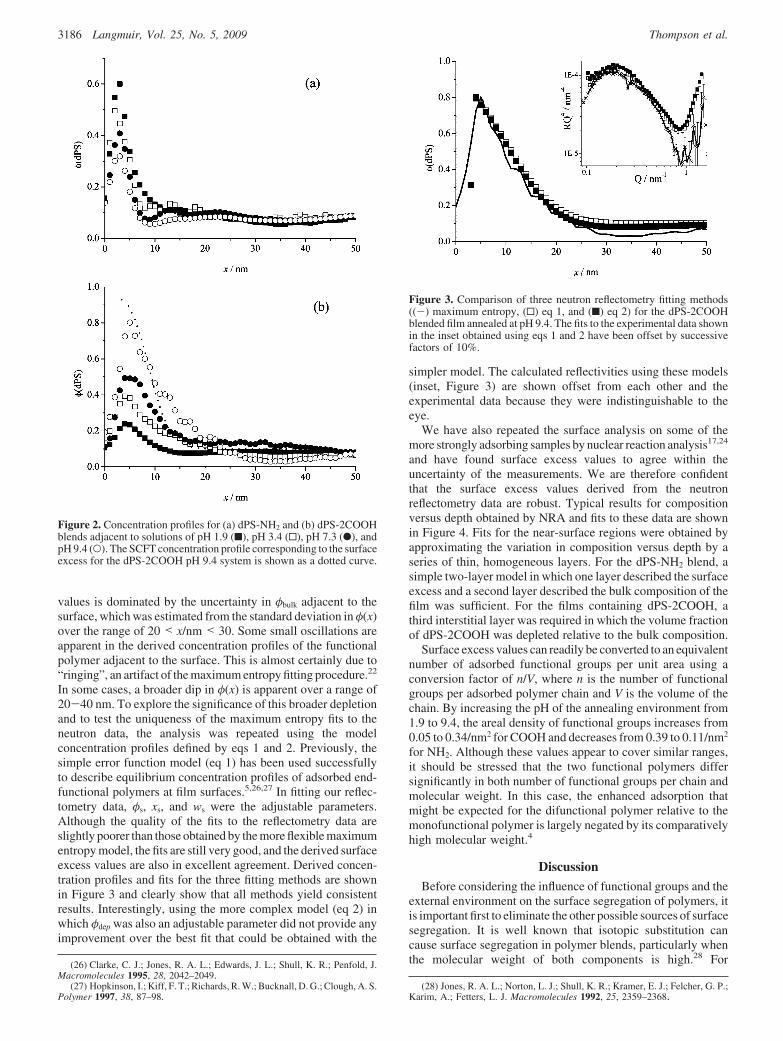
We have also repeated the surface analysis on some of themore strongly adsorbing samples by nuclear reaction analysis17,24
and have found surface excess values to agree within theuncertainty of the measurements. We are therefore confidentthat the surface excess values derived from the neutronreflectometry data are robust. Typical results for compositionversus depth obtained by NRA and fits to these data are shownin Figure 4. Fits for the near-surface regions were obtained byapproximating the variation in composition versus depth by aseries of thin, homogeneous layers. For the dPS-NH2 blend, asimple two-layer model in which one layer described the surfaceexcess and a second layer described the bulk composition of thefilm was sufficient. For the films containing dPS-2COOH, athird interstitial layer was required in which the volume fractionof dPS-2COOH was depleted relative to the bulk composition.
Surface excess values can readily be converted to an equivalentnumber of adsorbed functional groups per unit area using aconversion factor of n/V, where n is the number of functionalgroups per adsorbed polymer chain and V is the volume of thechain. By increasing the pH of the annealing environment from1.9 to 9.4, the areal density of functional groups increases from0.05 to 0.34/nm2 for COOH and decreases from 0.39 to 0.11/nm2
for NH2. Although these values appear to cover similar ranges,it should be stressed that the two functional polymers differsignificantly in both number of functional groups per chain andmolecular weight. In this case, the enhanced adsorption thatmight be expected for the difunctional polymer relative to themonofunctional polymer is largely negated by its comparativelyhigh molecular weight.4
Discussion
Before considering the influence of functional groups and theexternal environment on the surface segregation of polymers, itis important first to eliminate the other possible sources of surfacesegregation. It is well known that isotopic substitution cancause surface segregation in polymer blends, particularly whenthe molecular weight of both components is high.28 For(26) Clarke, C. J.; Jones, R. A. L.; Edwards, J. L.; Shull, K. R.; Penfold, J.
Macromolecules 1995, 28, 2042–2049.(27) Hopkinson, I.; Kiff, F. T.; Richards, R. W.; Bucknall, D. G.; Clough, A. S.
Polymer 1997, 38, 87–98.(28) Jones, R. A. L.; Norton, L. J.; Shull, K. R.; Kramer, E. J.; Felcher, G. P.;
Karim, A.; Fetters, L. J. Macromolecules 1992, 25, 2359–2368.
Figure 2. Concentration profiles for (a) dPS-NH2 and (b) dPS-2COOHblends adjacent to solutions of pH 1.9 (9), pH 3.4 (0), pH 7.3 (b), andpH 9.4 (O). The SCFT concentration profile corresponding to the surfaceexcess for the dPS-2COOH pH 9.4 system is shown as a dotted curve.
Figure 3. Comparison of three neutron reflectometry fitting methods((-) maximum entropy, (0) eq 1, and (9) eq 2) for the dPS-2COOHblended film annealed at pH 9.4. The fits to the experimental data shownin the inset obtained using eqs 1 and 2 have been offset by successivefactors of 10%.
3186 Langmuir, Vol. 25, No. 5, 2009 Thompson et al.
polystyrene systems, this becomes significant when the molecularweight of both polymers approaches 103 kg/mol. Because ouradditives are more than an order of magnitude below thisthreshold, their blends are too far from the coexistence point forthe overall molecular weight to induce surface segregation ofone component or the other. The large difference in the molecularweight of the blend components has itself been shown to causesignificant surface segregation of the lower-molecular-weightcomponent.29 However, this effect appears to be very small whenmeasured for blends that are of comparable molecular weightand composition to those that we have studied here.30 Above all,it should be noted that whereas these influences may make smallcontributions to the measured surface excesses their contributionswill be systematic in nature and will not have any pH dependence.
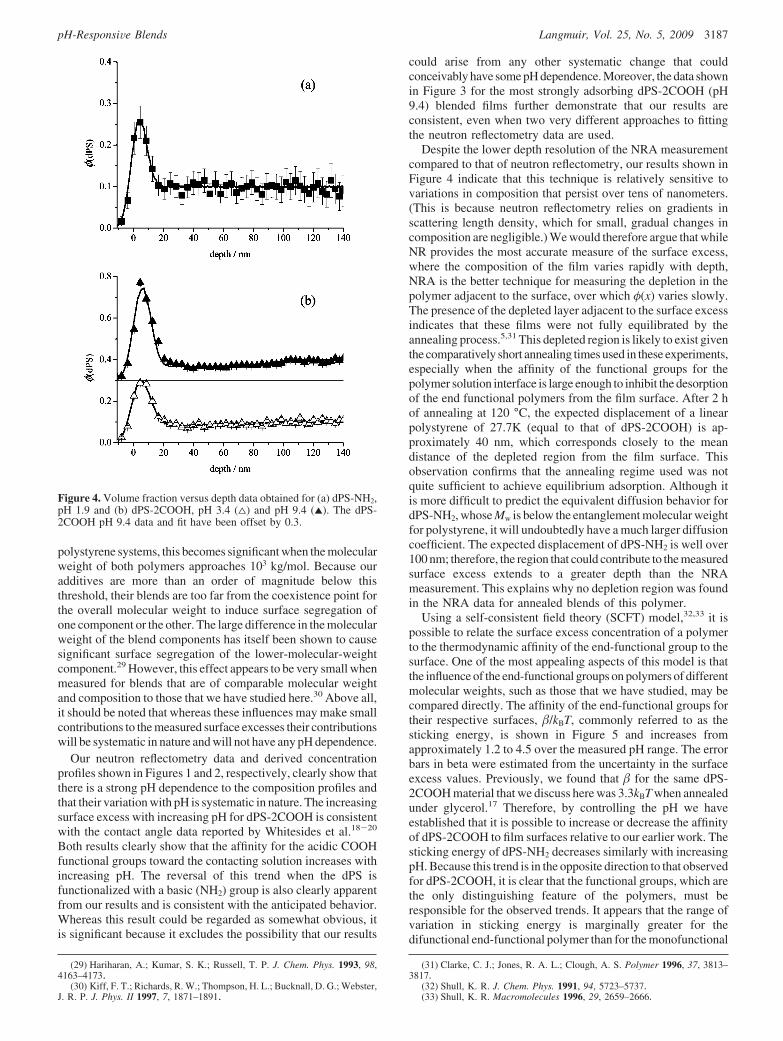
Our neutron reflectometry data and derived concentrationprofiles shown in Figures 1 and 2, respectively, clearly show thatthere is a strong pH dependence to the composition profiles andthat their variation with pH is systematic in nature. The increasingsurface excess with increasing pH for dPS-2COOH is consistentwith the contact angle data reported by Whitesides et al.18-20
Both results clearly show that the affinity for the acidic COOHfunctional groups toward the contacting solution increases withincreasing pH. The reversal of this trend when the dPS isfunctionalized with a basic (NH2) group is also clearly apparentfrom our results and is consistent with the anticipated behavior.Whereas this result could be regarded as somewhat obvious, itis significant because it excludes the possibility that our results
could arise from any other systematic change that couldconceivably have some pH dependence. Moreover, the data shownin Figure 3 for the most strongly adsorbing dPS-2COOH (pH9.4) blended films further demonstrate that our results areconsistent, even when two very different approaches to fittingthe neutron reflectometry data are used.
Despite the lower depth resolution of the NRA measurementcompared to that of neutron reflectometry, our results shown inFigure 4 indicate that this technique is relatively sensitive tovariations in composition that persist over tens of nanometers.(This is because neutron reflectometry relies on gradients inscattering length density, which for small, gradual changes incomposition are negligible.) We would therefore argue that whileNR provides the most accurate measure of the surface excess,where the composition of the film varies rapidly with depth,NRA is the better technique for measuring the depletion in thepolymer adjacent to the surface, over which φ(x) varies slowly.The presence of the depleted layer adjacent to the surface excessindicates that these films were not fully equilibrated by theannealing process.5,31 This depleted region is likely to exist giventhe comparatively short annealing times used in these experiments,especially when the affinity of the functional groups for thepolymer solution interface is large enough to inhibit the desorptionof the end functional polymers from the film surface. After 2 hof annealing at 120 °C, the expected displacement of a linearpolystyrene of 27.7K (equal to that of dPS-2COOH) is ap-proximately 40 nm, which corresponds closely to the meandistance of the depleted region from the film surface. Thisobservation confirms that the annealing regime used was notquite sufficient to achieve equilibrium adsorption. Although itis more difficult to predict the equivalent diffusion behavior fordPS-NH2, whose Mw is below the entanglement molecular weightfor polystyrene, it will undoubtedly have a much larger diffusioncoefficient. The expected displacement of dPS-NH2 is well over100 nm; therefore, the region that could contribute to the measuredsurface excess extends to a greater depth than the NRAmeasurement. This explains why no depletion region was foundin the NRA data for annealed blends of this polymer.
Using a self-consistent field theory (SCFT) model,32,33 it ispossible to relate the surface excess concentration of a polymerto the thermodynamic affinity of the end-functional group to thesurface. One of the most appealing aspects of this model is thatthe influence of the end-functional groups on polymers of differentmolecular weights, such as those that we have studied, may becompared directly. The affinity of the end-functional groups fortheir respective surfaces, �/kBT, commonly referred to as thesticking energy, is shown in Figure 5 and increases fromapproximately 1.2 to 4.5 over the measured pH range. The errorbars in beta were estimated from the uncertainty in the surfaceexcess values. Previously, we found that � for the same dPS-2COOH material that we discuss here was 3.3kBT when annealedunder glycerol.17 Therefore, by controlling the pH we haveestablished that it is possible to increase or decrease the affinityof dPS-2COOH to film surfaces relative to our earlier work. Thesticking energy of dPS-NH2 decreases similarly with increasingpH. Because this trend is in the opposite direction to that observedfor dPS-2COOH, it is clear that the functional groups, which arethe only distinguishing feature of the polymers, must beresponsible for the observed trends. It appears that the range ofvariation in sticking energy is marginally greater for thedifunctional end-functional polymer than for the monofunctional
(29) Hariharan, A.; Kumar, S. K.; Russell, T. P. J. Chem. Phys. 1993, 98,4163–4173.
(30) Kiff, F. T.; Richards, R. W.; Thompson, H. L.; Bucknall, D. G.; Webster,J. R. P. J. Phys. II 1997, 7, 1871–1891.
(31) Clarke, C. J.; Jones, R. A. L.; Clough, A. S. Polymer 1996, 37, 3813–3817.
(32) Shull, K. R. J. Chem. Phys. 1991, 94, 5723–5737.(33) Shull, K. R. Macromolecules 1996, 29, 2659–2666.
Figure 4. Volume fraction versus depth data obtained for (a) dPS-NH2,pH 1.9 and (b) dPS-2COOH, pH 3.4 (4) and pH 9.4 (2). The dPS-2COOH pH 9.4 data and fit have been offset by 0.3.
pH-ResponsiVe Blends Langmuir, Vol. 25, No. 5, 2009 3187
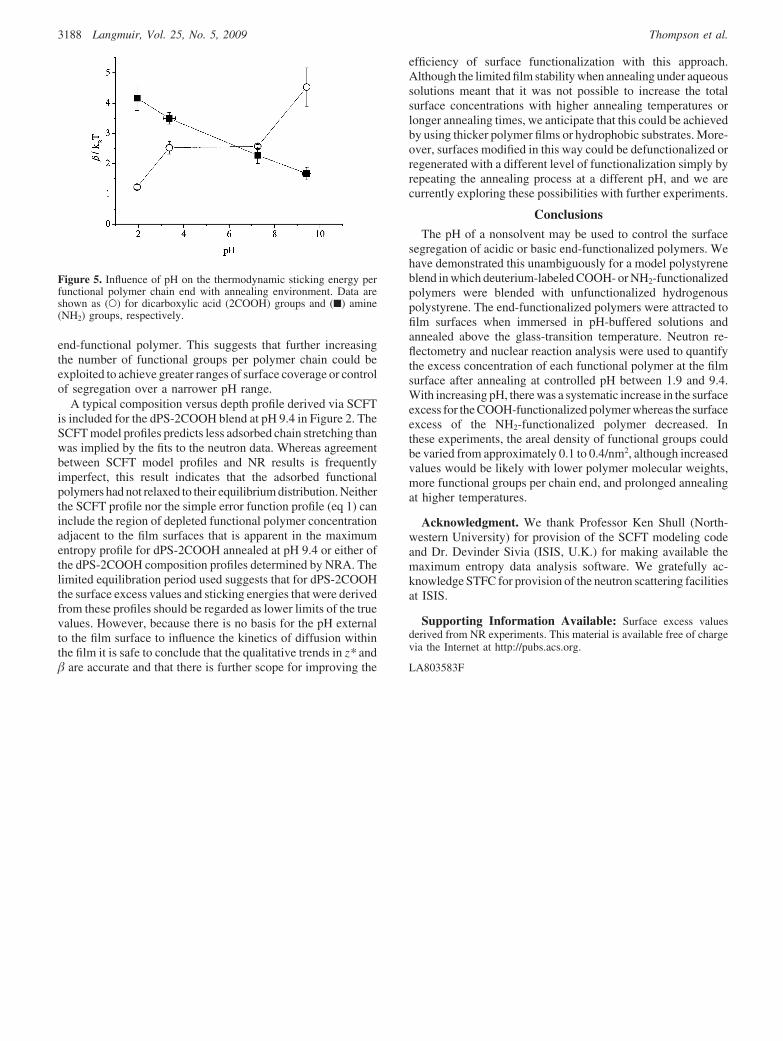
end-functional polymer. This suggests that further increasingthe number of functional groups per polymer chain could beexploited to achieve greater ranges of surface coverage or controlof segregation over a narrower pH range.
A typical composition versus depth profile derived via SCFTis included for the dPS-2COOH blend at pH 9.4 in Figure 2. TheSCFT model profiles predicts less adsorbed chain stretching thanwas implied by the fits to the neutron data. Whereas agreementbetween SCFT model profiles and NR results is frequentlyimperfect, this result indicates that the adsorbed functionalpolymers had not relaxed to their equilibrium distribution. Neitherthe SCFT profile nor the simple error function profile (eq 1) caninclude the region of depleted functional polymer concentrationadjacent to the film surfaces that is apparent in the maximumentropy profile for dPS-2COOH annealed at pH 9.4 or either ofthe dPS-2COOH composition profiles determined by NRA. Thelimited equilibration period used suggests that for dPS-2COOHthe surface excess values and sticking energies that were derivedfrom these profiles should be regarded as lower limits of the truevalues. However, because there is no basis for the pH externalto the film surface to influence the kinetics of diffusion withinthe film it is safe to conclude that the qualitative trends in z* and� are accurate and that there is further scope for improving the
efficiency of surface functionalization with this approach.Although the limited film stability when annealing under aqueoussolutions meant that it was not possible to increase the totalsurface concentrations with higher annealing temperatures orlonger annealing times, we anticipate that this could be achievedby using thicker polymer films or hydrophobic substrates. More-over, surfaces modified in this way could be defunctionalized orregenerated with a different level of functionalization simply byrepeating the annealing process at a different pH, and we arecurrently exploring these possibilities with further experiments.
Conclusions
The pH of a nonsolvent may be used to control the surfacesegregation of acidic or basic end-functionalized polymers. Wehave demonstrated this unambiguously for a model polystyreneblend in which deuterium-labeled COOH- or NH2-functionalizedpolymers were blended with unfunctionalized hydrogenouspolystyrene. The end-functionalized polymers were attracted tofilm surfaces when immersed in pH-buffered solutions andannealed above the glass-transition temperature. Neutron re-flectometry and nuclear reaction analysis were used to quantifythe excess concentration of each functional polymer at the filmsurface after annealing at controlled pH between 1.9 and 9.4.With increasing pH, there was a systematic increase in the surfaceexcess for the COOH-functionalized polymer whereas the surfaceexcess of the NH2-functionalized polymer decreased. Inthese experiments, the areal density of functional groups couldbe varied from approximately 0.1 to 0.4/nm2, although increasedvalues would be likely with lower polymer molecular weights,more functional groups per chain end, and prolonged annealingat higher temperatures.
Acknowledgment. We thank Professor Ken Shull (North-western University) for provision of the SCFT modeling codeand Dr. Devinder Sivia (ISIS, U.K.) for making available themaximum entropy data analysis software. We gratefully ac-knowledge STFC for provision of the neutron scattering facilitiesat ISIS.
Supporting Information Available: Surface excess valuesderived from NR experiments. This material is available free of chargevia the Internet at http://pubs.acs.org.
LA803583F
Figure 5. Influence of pH on the thermodynamic sticking energy perfunctional polymer chain end with annealing environment. Data areshown as (O) for dicarboxylic acid (2COOH) groups and (9) amine(NH2) groups, respectively.
3188 Langmuir, Vol. 25, No. 5, 2009 Thompson et al.