Embed Size (px)
Citation preview

BLBK098-Handerson March 26, 2009 22:2
Part I
Systems Theory andCooperative Bacteria
COPYRIG
HTED M
ATERIAL

BLBK098-Handerson March 26, 2009 22:2

BLBK098-Handerson March 26, 2009 22:2
Chapter 1
Systems thinking in biologyRobert M. Seymour
1.1 Introduction
The aim of this volume is to bring those interestedin the periodontal diseases up to date with ad-vances in related areas of biology and physiologyas they pertain to the cellular and molecular foun-dations of these complex conditions. A further aimis to introduce the reader to the major advance inbiology in the 21st century: systems biology. Sys-tems biology is a relatively new movement in thebiological sciences that aims to reverse the decades-old paradigm of molecule-based research in whichmore and more detailed properties of smaller andsmaller components of biological systems are iso-lated and dissected. While certainly not undervalu-ing the spectacular successes of this relentlessly re-ductionist approach, systems biology is pursuinga reverse, integrationist agenda, in which whole-system properties are ‘built-up’ from smaller com-ponent processes. These emergent, whole system,properties are considered the most significant bi-ological phenomena, and the ones most likely toyield payoffs in areas such as clinical applicationand drug discovery/design. Systems biology is in-evitably a multidisciplinary endeavour, involvingthe active participation not only of experimen-tal biologists and medically orientated scientistsbut also physicists, mathematicians, engineers andcomputer scientists. The close integration of math-ematical or computer model-building with hypoth-esis generation and experimentation is a hallmark.
Here we review some characteristic themes fromthis young, but already vast, research domain. It isthe mainstream future of the biological sciences.
1.2 The molecular revolution
The molecular revolution that began in the 1950shas, by definition, revolutionised the biological sci-
ences and still occupies the vigorous mainstreamof research effort in almost all the major areasof the life sciences. This effort has resulted in thecomplete sequencing of the genomes of a growingnumber of organisms (see Chapters 13–15 for de-tails on genome sequencing of oral bacteria), iden-tification of proteins, their structure and functionalproperties, and their interactions in both intra- andinter-cellular signalling pathways, the mapping ofmetabolic processes, and the many knock-on ef-fects of these developments for drug discovery,clinical applications and comparative methods inecological and epidemiological studies.
This ongoing push, which continues to ex-pand on the back of ever-improving technologies,such as DNA microchips, is generating an ever-increasing tidal wave of data. The major analyticalresponse to this tsunami has been the developmentof the discipline of bioinformatics – in essence thesystematic application of statistical and computa-tional methodologies for data mining and data cu-ration. Again, this is a thriving research area thatcontinues to offer significant rewards to biologists(see Chapters 2 and 13–15). In addition, data min-ing of bacterial communities in the oral cavity is arapidly growing area, which is described in detailin Chapter 4.
Nevertheless, the successes gained and momen-tum built up by the molecular revolution havetended to swamp some doubts about where, ul-timately, all this reductionist effort will lead.Although there are still university departmentsnamed, for historical reasons, ‘Anatomy’ or ‘Phys-iology’, these have long since ceased to have anycontact with classical, whole-organism biology,and nowadays are staffed almost exclusively byscientists trained in the reductionist methodolo-gies and pursuing a reductionist research agenda.Doubts persist about what it is, in the end, we are
3

BLBK098-Handerson March 26, 2009 22:2
4 Periodontal medicine and systems biology
trying to understand about organisms. The answermust surely be something like: We want to knowwhat makes them tick – how and why do theydo what they do? This very complex question hasmany layers of meaning, and many avenues of re-sponse. Is the reductionist, molecular focus currentin the biological sciences today in danger of notseeing the wood for the trees? Has the turnoverfrom whole-organism biology to the total domi-nance of the molecular focus resulted in the loss ofmuch potentially valuable expertise?
1.3 Mathematical models in biology
One doubt about the molecular focus has beenits tendency to emphasise structure over function,and the piecemeal over the big picture. Method-ologies, such as in vitro cell cultures, inevitablyhave tended to work with isolated biological com-ponents, often in artificial environments very re-mote from anything encountered in vivo, andused to identify specific molecular components in-volved in simple input–response structures. A pic-ture of functional properties of cells is then inferredfrom the results of many such component-by-component analyses. This approach has led to, forexample, an appreciation of many of the details,and the impressive level of complexity found inintra-cellular signalling pathways, as illustrated inFig. 1.1.
Nevertheless, physiology has never died. Func-tional, dynamic properties of large-scale systemshave been investigated successfully in parallel withthe developing molecular revolution. Much of thiswork has been achieved with the judicious useof mathematical models. The systematic use ofmathematical models in disparate areas of biol-ogy ranging from enzyme kinetics to ecology andpopulation genetics has a long history (Murray,2001). However, much of this is concerned withequilibrium properties of the modelled systems,rather than specific dynamic features, for exam-ple Lotka–Volterra-type models of ecosystem dy-namics, and Fisher–Wright models in populationgenetics (Ewens, 1979). A classic and paradigmaticexample of a non-equilibrium process model is theHodgkin–Huxley equations describing the devel-opment and transmission of a membrane poten-tial along the squid axon (Hodgkin and Huxley,1952). Much successful mathematical modellinghas built on this paradigm example, which con-
cerns itself with electrophysiological phenomena.Similarly, the investigation of intra-cellular cal-cium dynamics is a significant area of researchthat has benefited from the extensive developmentof mathematical models (e.g. Sneyd et al., 1993;Hofer, 1999; Kummer et al., 2000). Extensivesummaries of mathematical models in various ar-eas of biology can be found in Keener and Sneyd(1998) and Murray (2001).
These modelling efforts, like the molecular revo-lution, have largely proceeded piecemeal, focusingon small, isolated systems, reflecting the prevail-ing implicit assumption that if we collect enoughdetail about individual structures and processes, awhole view of the ‘wood’ will emerge. However,over the past decade or so there has been a grow-ing realisation amongst biologists and researchersin related disciplines that, in spite of the powerand sophistication of bioinformatic tools, the hugeand growing datasets now available are no longeramenable to researchers’ collective intuition, dueto their scale and depth. Furthermore, this scaleand depth makes the systematic inference of func-tional properties of whole biological systems ofinterest extremely problematic due to the presenceof feedbacks and other non-linear interaction ef-fects.
1.4 From structure to function:systems biology
Living systems are maintained by the continuousflow of matter and energy, and thus any biologicalsystem will inevitably be a subsystem of a largerone. The biologist therefore typically has to dealwith an open, multi-level and multi-componentsystem, the perceived nature of which evolves withour increasing understanding. A key feature ofsuch a system is the interactions (or coupling inengineering terminology) among its components,in which a variety of spatial and temporal scalesmay exist. These interactions may be strong orweak, unidirectional or bidirectional, dependingon the current state of the system, and often gen-erate emergent properties through nonlinear in-teractions. Some of these multi-level, hierarchicalfeatures are illustrated in Fig. 1.2.
For example, to begin to represent a cell andits wide range of functions, we have to integrateindividual models for relevant gene expression,metabolic and signalling pathways, as well as the
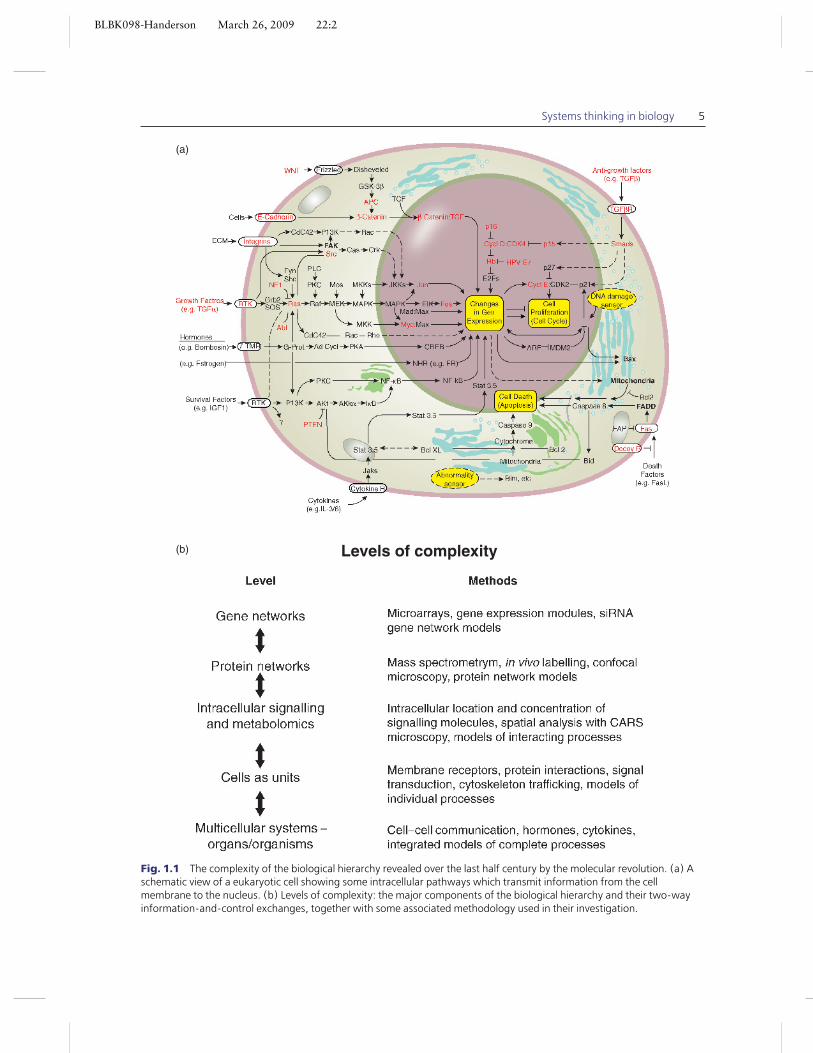
BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 5
(a)
(b) Levels of complexity
–
Fig. 1.1 The complexity of the biological hierarchy revealed over the last half century by the molecular revolution. (a) Aschematic view of a eukaryotic cell showing some intracellular pathways which transmit information from the cellmembrane to the nucleus. (b) Levels of complexity: the major components of the biological hierarchy and their two-wayinformation-and-control exchanges, together with some associated methodology used in their investigation.

BLBK098-Handerson March 26, 2009 22:2
6 Periodontal medicine and systems biology
associated biophysical processes for intracellular,extracellular and intercellular transport, etc. At thenext scale up, a multi-cellular, tissue or organismlevel model has to be formed by integrating dif-ferent kinds of cell functions and cell–cell commu-nication in their intra- and extra-cellular environ-ments. This is typical of the ‘bottom-up’ approachimplicit in the reductionist, molecular revolutionparadigm, and contrasts with the classical, physi-ological ‘top-down’ approach, which tends to startfrom the system as a whole.
In response to this now very apparent complex-ity, the last 10 years or so have seen the growth ofa new paradigm in the biological sciences: systemsbiology. This is, broadly speaking, a move to bringa systems engineering perspective to the analysisand understanding of the integrated functioning ofbiological systems (Anand et al., 2000; Csete andDoyle, 2002; Alon, 2007; Boogerd et al., 2007).Thus, complex biological systems are conceivedas being ‘engineered’ by natural selection in orderto perform whatever functional roles they areperceived to have. Such ‘natural engineering’ isof course less systematic than the engineeringthat lies behind the design of complex humanartefacts such as an automobile or jumbo jet.Nevertheless, there is anticipation that naturalselection will have discovered and utilised somebasic engineering design principles, for examplethe use of modular design structures, robustnessthrough redundancy and the deployment of nested
control systems. If these design principles can beuncovered in natural systems, then a deeper, morefunctionally based understanding can be achievedthan is possible with the piecemeal methods of themolecular revolution.
The main ‘ideological’ goal of systems biology isto bring approaches from engineering disciplinesinto the problem-solving methodology of biolo-gists. These approaches are not grouped aroundthe peculiarities of any particular system (e.g. acell type or organ such as the heart or the liver)but around individual biological problems whosetreatment can be useful for other, possibly as yetunforeseen, problems. Biological systems on what-ever hierarchical level of interest (be they organ-isms in populations, organs in organisms, cells,transcription factors, genes or single molecules) areusing the common criteria of modularity, processand functionality. These can be formulated in thetechnical terminology of ‘modules’ and ‘protocols’and find mathematical expression in control the-ory (Sontag, 1998), or conditional probability dis-tributions and graphical models used to describenetworks. Such concepts are critical if datasets areto be analysed under functional criteria. The em-phasis then is on ‘generic’ properties – featuresthat apply across a wide range of systems andcircumstances. A schematic of this new analyti-cal paradigm, and methods associated with it, isshown in Fig. 1.2. Paradigmatic issues that arisefrom this perspective are:
Equilibrium/non-equilibrium Stochastic effects
Temporal scales
Model simplification
Networkreconstruction
Comparativegenomics
Model dialogue
Spatial scales
Network topology/dynamics
Complex biological system
Whole system behaviour
Data validation
Control structures
Hierarchicalorganisations
Fig. 1.2 Schematic of components of acomplex biological system from the point ofview of ‘systems thinking’. This emphasisesnetworks, dynamics, control structures, modelsand integrated behaviour of the whole systememerging from lower-level interactions. Thewhole model construct must be both informedby, and capable of, validation against datausing methods relevant to the varioushierarchical level (as indicated in Fig. 1.1b).

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 7
� At what (conceptual) hierarchical level are bi-ological systems predictable, and what func-tional features are robust enough to be pre-dictable?
� How does this robustness and predictability‘emerge’ from possibly non-predictable lowerlevel phenomena?
� Are these predictable features generic proper-ties of large classes of systems, or are they non-generic, highly optimised, and therefore highlyevolved for specific roles in specific contexts?
The challenge posed by these objectives is consid-erable, and will require the efforts of many disci-plines: experimental and theoretically minded bi-ologists, engineers (especially control engineers),computer scientists (software engineers, bioinfor-maticians) and mathematical modellers, as well asthe utilisation of the fast-developing in vivo imag-ing technologies coming out of the biophysics com-munity. Some of the challenges to be faced in thisendeavour are:
� To link the biological task of data collectionand analysis with mathematical and computa-tional modelling to enable the construction andanalysis of integrated systems.
� To synthesise individual biological processesanalysed with widely varying techniques acrossa range of scales into a constituent whole.
� To generate an experimental and theoreticalscaffold that integrates the function of geneand protein networks with intra-cellular sig-nalling and inter-cellular interactions medi-ated by direct cell–cell communication, surfacemembrane receptors and soluble factors suchas cytokines and hormones.
� To elucidate the molecular, cellular and evo-lutionary bases for coordinated function ofmulticellular systems using data derived fromgenomics, proteomics, protein–protein inter-actions, transcriptional regulation, intracellu-lar signalling mechanisms and metabolomicsthrough to cell–environment and cell–cellinteractions.
1.5 Systematic simplification
The ‘bottom-up’ approach to systems biology re-quires that biological modelling is scaled fromrelatively small components to the whole systemlevel (Fig. 1.2). This can produce extremely com-
plex models. The major success to date of this ap-proach is the systems model of the heart (Noble,2002), which has developed over several decades,building on a modification of the Hodgkin–Huxleymodel (Hodgkin and Huxley, 1952), and proceed-ing with gradual and well-supported increases incomplexity. Currently, the model involves somehundreds of equations, largely representing elec-trophysiological and electromechanical processes,and is linked to sophisticated computer visual-isations, particularly of solid geometries. It hasmore than proved its worth in aiding the under-standing of cardiac arrhythmia, with consequencesfor drug design and testing (Noble and Colatsky,2000).
Despite its complexity and the huge effort it hastaken to build, the model covers only a part of theheart’s mechanical, electrical and chemical prop-erties. This reveals the scale of the systems biol-ogy challenge. The model has been the seed forthe Physiome Project (Hunter et al., 2002), whichcollects and catalogues physiological models andsupports community access to these models. Theproject also provides web-accessible databases ofbiological data that potentially can be linked tomodels. In a similar vein is the UCL BeaconProject, which aims at systems modelling of liverphysiology and glucose homeostasis.
Nevertheless, with few exceptions most mathe-matical models are stand-alone models of isolatedbiological processes, with limited ambition.Sometimes these models are provisional, andembed contested hypotheses concerning structureor function. One key reason for such small-scalemodelling is the often insuperable difficulty ofvalidation. However, some very successful modelshave been ambitiously large scale and also wellvalidated. An important example is the modelbacterium of Denis Bray and colleagues, whichsuccessfully simulates chemosensitivity and theoperation of flagella (Bray et al., 1998; Levinet al., 1998). Thus, chemotactic signalling in Es-cherichia coli has the property that bacterial cellscan detect chemical gradients ranging over fiveorders of magnitude in concentration, and Bray’smodel showed how this low-threshold responseand wide dynamic range can be achieved witha collection of single surface receptor moleculesthat communicate laterally with one moleculeinfluencing its neighbours as a function of its ownactivity. The extent of this lateral activity spreadcan adapt to external circumstances.

BLBK098-Handerson March 26, 2009 22:2
8 Periodontal medicine and systems biology
Given the ambition of systems biology to cap-ture functional properties at higher levels thatemerge from molecular mechanisms, it will of ne-cessity have to deal with large, composite andcomplex models, built systematically from smallercomponents spanning several hierarchical levels(Fig. 1.2). Such constructs may perform well assimulations of the real system, but to do so imposesan enormous burden on data gathering. Neverthe-less, some successes are already apparent in this ap-proach when coupled with large-scale, dedicated,high-throughput experimental work, for examplein imaging (Nelson et al., 2002) and metabolomics(Maher et al., 2003).
Representing all aspects of a biological systemin the smallest possible detail is clearly not feasi-ble. In modelling, we cannot hope to recreate theworld as an isomorphic in silico image of itself(Finkelstein et al., 2004) therefore judicious sim-plification is required in model construction. Thisis particularly true when attempting to link differ-ent processes at different spatio-temporal scales,such as gene and protein networks. For example,to represent biochemical networks involving manydifferent proteins, we could model the interactionsbetween proteins using simple stimulus–responsefunctions, while retaining the complexity of theinteraction network. Alternatively, we could focuson a few proteins and model the complex trans-formational processes involved in their interactionin great detail. Such simplification choices have atleast two major facets:
� Choosing a modelling scheme that allows suffi-cient descriptive power at the relevant concep-tual level(s) (Fig. 1.2), while having the flexi-bility to link to other component models andallowing contextualisation in terms of knownor obtainable data.
� Choosing a level of detail in terms of the num-ber and complexity of interactions to model ex-plicitly within the descriptive scheme; whetherand how to model space, and determining therelevant timescale(s).
Some work has been invested in analysing ‘sys-tematic simplification’ procedures, in particularusing a common simplification methodology onall parts of the system (e.g. Hetherington et al.,2006). This approach has had significant success inmetabolic control analysis, in which useful (equi-librium) models have been constructed using onlythe stoichiometries of constituent reactions, inter-
preted using flux balance analysis or elementarymode analysis (Fell, 1997; Schuster et al., 1999;Edwards et al., 2001; Holzhutter, 2004; Hornberget al., 2007).
One popular systematic simplification strategyis to replace smooth input–output responseprofiles with two-state (OFF–ON) functions.Sometimes three states are used (DOWN–0–UP).This is common in modelling gene networks inwhich a gene produces a transcription factor thatbinds to other genes (and possibly itself). Thestate of the receiving gene is then determined by‘integrating’ all its arriving inputs (commonly,using a weighted summation with a thresholdresponse). Examples of the use of this modellingstrategy include investigating the evolution of thesex-determination gene network in Drosophila(MacCarthy et al., 2003), and the role of per-turbation experiments in reverse engineering thestructure of gene networks (MacCarthy et al.,2005). In a similar vein, such methods havebeen applied to biochemical networks (Glass andKauffmann, 1973), and to network interactions inthe innate immune response (Kaufman et al., 1985;Kaufman and Thomas, 1987), and also analysedmore abstractly (Snoussi and Thomas, 1993;Thomas, 1998; Thomas and Kaufman, 2001).
A detailed statistical analysis of the sensitivity ofthe resilience of some features of the output of acomplex model under similar systematic simplifi-cation procedures is given in Hetherington et al.(2006). They show that, although the detailedquantitative behaviour of the complex model isoften not preserved under simplification, neverthe-less, simplified models can perform well in predict-ing which parameters have significant control overvarious system properties.
1.6 Multiple stable states
Perhaps the central concept of systems biology –indeed, of complex systems more generally – isthat of a network of interactions. The actors (ornodes) in these networks can be genes, proteins,cells or organisms (Fig. 1.1), and the mediatinginteractions can be of various kinds, includingtranscription factors, phosphorylation and de-phosphorylation of proteins, binding of ligandsto cell–membrane receptors, and changes ofphenotypic response to environmental cues. Thekey point is that networks describe information

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 9
transfer (by whatever mechanism) between nodes(of whatever type), and this information induceschanges in the properties or behaviour of thenodes, which in turn has knock-on consequencesvia network interactions. Most biological systemscan be described in terms of one or more suchnetworks. However, biological networks aredynamic structures, with behaviour that changesover time. They therefore exhibit adaptive be-haviour in response to their external environment(Bhalla and Iyengar, 1999; Alon, 2007).
As indicated earlier, homeostatic mechanismsoften allow biological systems – modelled asnetworks – to remain at or near equilibrium (orstationary) states. An equilibrium state is said tobe (locally) stable if relevant homeostatic mech-anisms will lead to a return to the equilibriumafter a suitably ‘small’ perturbation away from it(a large enough perturbation can always destroyany state – indeed, the whole system). Features ofstable equilibrium states are therefore of great in-terest in understanding system function. However,complex systems can have more than one possiblestable equilibrium state, and in these circumstancesit is often very unclear which state the systemwill assume under what conditions (Laurent andKellershohn, 1999). Furthermore, it is possiblethat a system will remain in a particular stable stateeven after the stimulus that originally pushed itthere is removed (Henderson and Seymour, 2003).
This issue of multistationarity is potentially im-portant in relation to possible pathological states,in which one stable equilibrium may be regardedas ‘healthy’, but an alternative, less accessible,stable state is pathological, associated with diseasesymptoms. This point of view is developed byHenderson and colleagues in relation to the early-onset cytokine response of the innate immunesystem, for example in the context of rheumatoidarthritis and systemic inflammatory responsesyndrome (SIRS) (Henderson et al., 1998; Wilsonet al., 1998; Seymour and Henderson, 2001; Jitet al., 2005). Similarly, for the adaptive immune re-sponse, the development of T-helper cell differenti-ation into Th-1 or Th-2 phenotypes in response toactivation of T-cell receptors by cytokines – whichin turn activate the transcription factors T-bet orGata-3 – can be viewed as alternative, irreversiblestable states of a network (Yates et al., 2004).
In Fig. 1.3 a simple network model of cytokineproduction and response by an immune system cell(e.g. monocyte/macrophage) is illustrated. This
model features up to five (unspecified) cytokinesthat can bind to specific cell–membrane receptors.These cytokines are implicated in periodontaldisease. Receptors are subject to (stimulated)internalisation, with consequent loss of boundligand. Binding of the various cytokine ligands totheir receptors results in a cell response that eitherincreases or decreases the production of any givencytokine, depending on a threshold-type responseto the (integrated) input of all the cytokines. Inthis model, these integrated threshold responses(which can be multiplicative as well as additive)are determined randomly, leading to randominteraction networks. Different random choicesof thresholds lead to different network instances.The model can result in unconstrained cytokineproduction – a clearly pathological outcome – or aconstrained stable state, and sometimes also stablecycles. As is illustrated in Fig. 1.3, multiple stablestates are possible in this model, but even with fivecytokines, there are a small number of such states(no more than five), with most networks yieldingonly one. This suggests that multiple stable statesmay in fact be less frequently found than onemight naively expect. Chronic pathologies arisingfrom the immune response being stuck in the‘wrong’ stable state are certainly possible, butperhaps to be expected only in individuals withuntypical cytokine network structures.
1.7 Regulatory networks, modules, motifsand control structures
Networks are formally represented by nodesand edges, representing pathways of informationflow between nodes. Edges may be directed,representing a specified direction of informationflow, or undirected, in which case informationflow is equally possible in either direction. Thein-degree of a node is the number of incomingedges into the node, and the out-degree is thenumber of outgoing edges. In an undirectednetwork, these concepts coincide. For a largenetwork, the distribution of (in- or out-) degreed over the nodes of the network has been thesubject of much study. For example, recentstudies of transcription networks in E. coli andSaccharomyces cerevisiae have revealed a roughlyexponential in-degree distribution, but a powerlaw out-degree distribution (Guelzim et al., 2002;Regulon DB, see http://regulondb.ccg.unam.mx/).
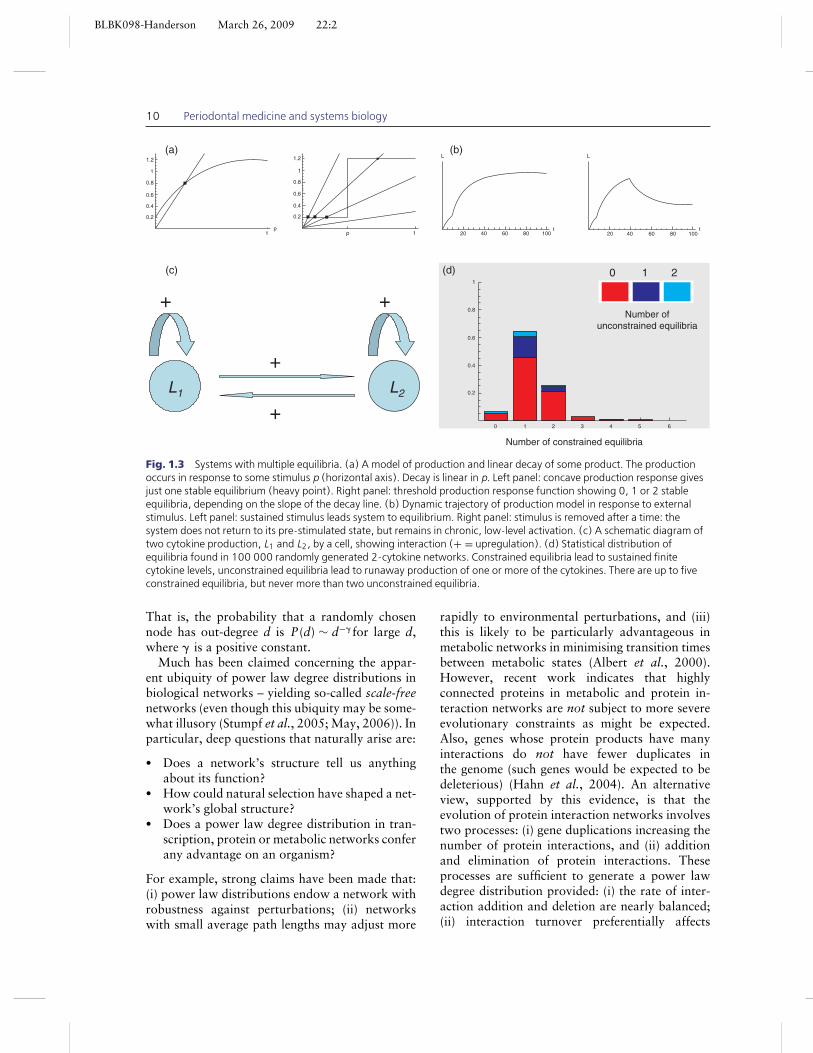
BLBK098-Handerson March 26, 2009 22:2
10 Periodontal medicine and systems biology
1.2
1
0.8
0.6
0.4
0.2
1p
1.2
0.8
0.6
0.4
0.2
p 1 20
L L
40
1
0.8
0 1 2
Number of
Number of constrained equilibria
unconstrained equilibria0.6
0.4
0.2
0 1 2 3 4 5 6
60 80 100 20 40 60 80 100t t
1
+
+
++
L1 L2
(a)
(c) (d)
(b)
Fig. 1.3 Systems with multiple equilibria. (a) A model of production and linear decay of some product. The productionoccurs in response to some stimulus p (horizontal axis). Decay is linear in p. Left panel: concave production response givesjust one stable equilibrium (heavy point). Right panel: threshold production response function showing 0, 1 or 2 stableequilibria, depending on the slope of the decay line. (b) Dynamic trajectory of production model in response to externalstimulus. Left panel: sustained stimulus leads system to equilibrium. Right panel: stimulus is removed after a time: thesystem does not return to its pre-stimulated state, but remains in chronic, low-level activation. (c) A schematic diagram oftwo cytokine production, L1 and L2, by a cell, showing interaction (+ = upregulation). (d) Statistical distribution ofequilibria found in 100 000 randomly generated 2-cytokine networks. Constrained equilibria lead to sustained finitecytokine levels, unconstrained equilibria lead to runaway production of one or more of the cytokines. There are up to fiveconstrained equilibria, but never more than two unconstrained equilibria.
That is, the probability that a randomly chosennode has out-degree d is P(d) ∼ d−� for large d,where � is a positive constant.
Much has been claimed concerning the appar-ent ubiquity of power law degree distributions inbiological networks – yielding so-called scale-freenetworks (even though this ubiquity may be some-what illusory (Stumpf et al., 2005; May, 2006)). Inparticular, deep questions that naturally arise are:
� Does a network’s structure tell us anythingabout its function?
� How could natural selection have shaped a net-work’s global structure?
� Does a power law degree distribution in tran-scription, protein or metabolic networks conferany advantage on an organism?
For example, strong claims have been made that:(i) power law distributions endow a network withrobustness against perturbations; (ii) networkswith small average path lengths may adjust more
rapidly to environmental perturbations, and (iii)this is likely to be particularly advantageous inmetabolic networks in minimising transition timesbetween metabolic states (Albert et al., 2000).However, recent work indicates that highlyconnected proteins in metabolic and protein in-teraction networks are not subject to more severeevolutionary constraints as might be expected.Also, genes whose protein products have manyinteractions do not have fewer duplicates inthe genome (such genes would be expected to bedeleterious) (Hahn et al., 2004). An alternativeview, supported by this evidence, is that theevolution of protein interaction networks involvestwo processes: (i) gene duplications increasing thenumber of protein interactions, and (ii) additionand elimination of protein interactions. Theseprocesses are sufficient to generate a power lawdegree distribution provided: (i) the rate of inter-action addition and deletion are nearly balanced;(ii) interaction turnover preferentially affects

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 11
highly connected proteins, and (iii) some addedinteractions add new proteins to the network(Wagner, 2003). The important and complex issueof the evolution of ‘robust design’ in organismsand its relation to regulatory networks is analysedin detail in a recent book (Wagner, 2007).
Gene duplication and subsequent divergence isa common mechanism for the generation of diver-sity, even though this process does not appear tobe important in all circumstances, for example indevelopmental mechanisms in multicellular organ-isms, which are largely controlled by highly con-served Hox genes acting within ‘modules’ such asarthropod limbs (Carroll, 2001). Thus, the greatdiversity of arthropod limb types is due to dif-ferences in the regulation of Hox genes along theanterior–posterior axis. More generally, the greatvariety of cell types within multicellular organismsis due ultimately to differences in gene expression,which can number in the hundreds of thousands.However, greater gene number itself is not a ma-jor driver of ‘complexity’, as, for example, mea-sured by number of cell-types within an organism.Instead, the differences in gene expression are of-ten controlled by a relatively small number of reg-ulatory proteins. Thus, only about 3–5% of theproteins encoded by organisms’ genomes are tran-scription regulators. For example, in the bacteriumBacillus subtilis (∼4100 genes) a small number ofregulators control the differential expression ofseveral hundred genes during sporulation, whilein yeast (S. cerevisiae, ∼6200 genes) a small setof transcription factors orchestrates the regula-tion of genes involved in cell-type differences. Inmetazoans, cell-type differences (e.g. between mus-cle and neural tissue) and body-region identityare typically regulated by a few proteins, whereaspattern formation within tissues is regulated bya larger set of proteins (Carroll, 2001). Regula-tory evolution creates new combinations of geneexpression and therefore enables increases in theinformation content of genomes, and the genera-tive potential of development without expansionof gene number. However, we do not understandwhy the actual complexity realised in evolution isfar less than appears to be possible genetically.
These and similar considerations have generatedconsiderable interest in ‘modular design’ – eitherin structure or function or both – as a mechanismfor generating diversity, robustness to componentfailure and adaptability (Csete and Doyle, 2002).A module is a structural/functional unit relatively
separable from its surrounding structure. Animportant property bestowed on evolving organ-isms by modularity is the ability to dissociatedevelopmental processes in one part of the bodyfrom another. Modularity facilitates change byconferring on organisms a greater ability to escapeinternal constraints, that is it confers robustnessand evolvability (Wagner, 1996; Wagner andAltenberg, 1996). Several mechanisms can giverise to modularity: symbiosis (e.g. mitochondria inthe eukaryotic cell), developmental segmentation,connectivity-sensitive growth and response tovariation (Barabasi and Albert, 1999; Holland,1999; Watson and Pollack, 2000; Lipson et al.,2002). Indeed, simple models show that mod-ularity can arise spontaneously in evolutionarysystems in response to environmental variationand selection (Lipson et al., 2002). Engineeringdesign methods based on evolutionary simulation(genetic algorithms) could well benefit fromevolving to variable, rather than stationary, fitnesscriteria, as a problem-independent method ofinducing modularity.
Little is known about the design principles oftranscriptional regulation networks that controlgene expression in cells. For example, can suchnetworks be broken down into basic buildingblocks? In taking a systems approach to under-standing transcriptional regulatory networks,much emphasis has been placed on decomposingthese networks into simpler subnetworks, oftencalled motifs (Lee et al., 2002; Shen-Orr et al.,2002; Alon, 2007). Such motifs are sometimesconstrued as just small-scale patterns that occurmore often than chance would predict, but alsocan sometimes be construed as representingspecific ‘control modules’ with specific dynamicalproperties (Fig. 1.4). The motif structure thenallows an easily interpretable view of the entireknown transcriptional network of the organism.This approach may help to define the basic com-putational elements of many biological networks.
Figure 1.4 shows some of the proposed dynamiccontrol motifs that are thought to be important ingene regulatory networks. Also shown is the out-put associated with three of these, obtained from adynamic model of gene transcription which incor-porates transcriptional delay. These are the auto-regulation loop, the two-component loop and thefeed-forward loop. Each of these has very specificdynamical properties. Thus, the auto-regulationloop, in which the transcription factor produced

BLBK098-Handerson March 26, 2009 22:2
12 Periodontal medicine and systems biology
Auto-regulation
Regulator chain
Single-inputmotif
pA
pA
p0
p0 A
pA
B
p0 A pA B pB
C
D
p1 p2
pB
B
p0
A B C A B C D
Multi-inputmotif
A
A
Multi-component loop Feed-forward loop 0.5
0.4
0.3
0.2
0.1
100 200 300 400 500
0.5
0.4
0.3
0.2
0.1
10
8
6
4
2
100 200 300 400
Constantactivation
Transcription
TF
P
500
100 200 300 400 500
50
40
30
20
10
100 200 300 400 500
50
40
30
20
10
100 200 300 400 500
10
8
6
4
2
20 40 60 80 100 120 140
10
8
6
4
2
20 40 60 80 100 120 140
25
20
15
10
5
20 40 60 80 100 120 140
10
8
6
4
2
20 40 60 80 100 120 140
25
20
15
10
5
20 40 60 80 100 120 140
(a)
(c)
(b)
(d)
(e)
Fig. 1.4 Control modules and motifs in transcription networks derived from a dynamic model of transcriptionincorporating transcriptional delay. (a) Some control motifs which have been found in the transcriptional network of E. coli.Genes are squares, their protein products (transcription factors) are ovals. Dashed arrows: production of a protein by agene. Solid arrows: binding of protein to gene leading to up- or down-regulation of the gene. (b) Dynamics associated withthe auto-regulation motif. Left panels: external stimulus level of the gene. Right panels: protein-production response. Toppair: low-level stimulation produces no response. Bottom pair: high-level stimulation produces full response. (c) Similarstimulus–response graphs for the feed-forward loop. Two proteins are involved – one for each gene. A significant responseoccurs only when the stimulus is sustained for long enough. (d) Response graph for a two-component loop. The twoprotein products (one for each gene) oscillate once the system is activated (constant stimulus not shown). (e)Post-transcriptional protein modification such as that found in the NF-�B system. This can lead to cyclic behaviour:Horizontal straight line is the constant stimulus; two oscillating curves are the transcription factor and the protein product.
by a gene (in response to some background levelof externally induced activation) binds to its parentgene and upregulates further production of itself,turns itself on only when background activationreaches some threshold level. In the feed-forwardloop, an external protein factor activates a pairof linked genes; the transcription factor producedby the first of these binds to the second and up-regulates (or downregulates) the protein producedby the second gene, with some time delay betweenthe two responses. In the two-component loop, thetranscription factors produced by a pair of genesbind to the non-parent gene, enhancing (or sup-pressing) its output. This leads to cyclic dynamics.
These and other regulated responses are thoughtto control the coordinated output of large genenetworks composed of linked dynamic motifs.
Dynamic control structures of course canoperate on many different levels in the biologicalhierarchy (Fig. 1.2) and indeed on more thanone level simultaneously. Thus, in the examplesof Fig. 1.4, both proteins and genes are activein control. However, this is construed as takingplace in the nucleus over a small spatial range.Intracellular protein signalling pathways are moreextensive in spatial scale, but generally operate ona fast timescale compared to that of gene transcrip-tion. One well-known interaction between the

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 13
time-scale of gene transcription and that ofprotein–protein signalling occurs in the NF-�Bpathway. In this pathway, an initiating event, suchas the binding of tumour necrosis factor (TNF) toits cell-surface receptor, leads to a chain of proteinmodifications culminating in the detachment ofNF-�B from its inhibitor I-�B, which is degradedvia ubiquitination. NF-�B is then free to migrateinto the nucleus, where it initiates transcription ofrelevant genes producing, amongst other products,the inhibitor protein I-�B. This latter then migratesout of the nucleus, and proceeds to bind to freeNF-�B, thereby inhibiting its activity (Hoffmannet al., 2002; Lipniacki et al., 2004; Nelson et al.,2004; Krishna et al., 2007). Although the detailsof all the steps in the NF-�B network are extremelycomplex, and not fully understood, nevertheless,this basic feedback loop is known to induce cyclesof production and suppression of NF-�B. This is anow classic example of dynamic feedback control.
Control analysis of other signalling pathways isalso an active area of research, e.g. the MAPK/ERKsignalling pathway (Marshall, 1995; Hornberget al., 2005a,b; Orton et al., 2005; Bluthgen, 2006;Santos et al., 2007).
1.8 Ecological models: quorum sensingand biofilms
Ecological communities have always been recog-nised as complex systems, in which there is a hier-archy of energy transfer from primary producers(photosynthesising plants) through herbivores, upto top predators, and down again to detritivores(Lindeman, 1942; Begon et al., 1990; Higashi andBurns, 1991). In this context, study of the dy-namics of populations has a long history datingfrom the classic work of Lotka and Volterra in the1920s and 1930s on pair-wise population dynam-ics. Interactions, either within or between species,in an ecological community can be conceptualisedas one of several possible classic forms: competi-tion (for a common resource), predator–prey; par-asitism, mutualism/symbiosis, or commensalisms.The equilibrium structure of ecological systemscan often be described by specifying what kindsof interactions are dominant (e.g. May, 1973).
Nevertheless, the properties and behaviour ofecological communities are still poorly under-stood. This is because there are so many factorsrelevant to a comprehensive understanding of
the function of any community: the physicaland chemical environment and its influence,succession and community development throughtime and space, mechanisms of communication(molecular or other forms of signalling) andinteraction (competition, mutualism, etc.), andthe role of disturbance in promoting diversity.Increasingly, the concept of a community asa network of trophic interactions, which hasemergent, whole-system, properties, is coming toprominence (Patten, 1991; Kondoh, 2003; May,2006). This is particularly true of communities ofmicroorganisms, for example those forming gutand oral consortia in mammalian hosts, and themany examples of multi-species biofilms, whichhave only fairly recently become the targets ofintensive research (Hooper et al., 1999; Horneret al., 2003; Handelsman, 2004; Rainey et al.,2005; IWA Task Group, 2006). The relevance forperiodontal disease is obvious.
One particularly intriguing form of communityinteraction that has come to light through thestudy of microorganisms is quorum sensing. Thiswas first demonstrated in the bacterium Vibrio fis-cheri and its host, the squid Euperymna scolopes,in which the bacteria colonise the squid light-organand emit light at night, providing counter illumina-tion that enables the host to avoid detection by itspredators who attack from below in moonlit wa-ters (Visick and McFall-Ngai, 2000; Visick et al.,2000). In quorum sensing, bacteria sense their ownpopulation density and express genes accordingly.Population density is detected by the accumula-tion of species-specific acyl homoserine lactones(AHLs). In V. fisheri, quorum sensing is used toregulate genes responsible for light emission. How-ever, it is now known that quorum sensing is alsowidely used by pathogenic bacteria as a mecha-nism for evading the host immune response. Thus,invading bacteria switch on virulence genes onlywhen their population density is sufficiently high,when they are in a position to overwhelm the host’sdefences (Prichard et al., 2003). This kind of co-ordinated response – essentially through coalitionformation – is certain to have significant implica-tions for whole-system community dynamics in awide range of contexts. For a more detailed discus-sion of the role of quorum sensing in oral bacterialbiofilms the reader should refer to Chapter 12.
Not least of these is the interface between bac-teriology and immunology through the interactionof animal – especially mammal – hosts with their

BLBK098-Handerson March 26, 2009 22:2
14 Periodontal medicine and systems biology
endogenous communities of ‘commensal’ micro-organisms. This interaction is now thought to behighly dynamic, involving much molecular ‘cross-talk’, which, in particular, is important to nor-mal gut development. Indeed, the gut of humanneonates contains large numbers of facultativeanaerobes, which decline in number during wean-ing as obligate anaerobes begin to take over. Overtime, the gut community evolves to a stable climaxcommunity in which obligate anaerobes predom-inate. In parallel to this shift in microbial com-munity composition is a marked morphologicalmaturation of the host gut, along with changesin the host immune system (Hooper, 2005). Thedynamics of microbial community development iseven faster in the oral context, occurring after eachtoothbrushing session.
The possible role of pathogenic bacteria in theevolution of host characteristics has been hypothe-sised by Seymour et al. (2004). They considered theevolution of the human ABO blood-group systemby constructing an epidemiological model involv-ing a virus, which may pick up terminal glycanson its envelope during invasion and transmissionbetween hosts (Preece et al., 2002), thereby biasingthe next host immunological response, and oppor-tunistic bacteria that adapt to the characteristics ofhost epithelial–mucosal surfaces. These forces gen-erate differential responses between hosts whichare able to maintain the commonly observed ABOfrequencies in human populations. This modelplaces the selective maintenance of the ABOpolymorphism firmly within a larger communitycontext.
Studies of interactions between the host im-mune system and microorganisms have generatedsome novel hypotheses. For example, one radicalhypothesis has it that the evolution of the adaptiveimmune system in vertebrates occurred in parallelwith, and in order to orchestrate, their strategyof hosting large consortia of resident microbes,presumably to exploit their nutrient-processing ca-pacity (Ruby et al., 2004; McFall-Ngai, 2007). Asystem-level model of such adaptive orchestrationof a commensal microorganism community hasrecently been developed (Seymour, 2005). Here,the host is modelled as a network of ‘sites’ in whichbacteria can grow and reproduce, with variableconfigurations of possible migration between sites.These sites are favourable to different species ofbacteria, so that, other things being equal, each siteis dominated by its resident specialist and, if mi-
gration is allowed, the whole system is dominatedby one specialist species. However, if a simplemodel of an adaptive immune response is imposed,which acts to suppress excessive growth in a site-and species-specific manner, then very diversecommunities of microorganisms can be sustained,with diversity at a hierarchy of abundancescales.
Network interactions are one, rather schematic,way of representing the influence of space oncommunity structure. Space, especially on sessilecommunities such as biofilms, allows the devel-opment of heterogeneous structures by preventingdirect interaction between component parts ofa system. This is an important mechanism forgenerating diversity and neutralising competitiveexclusion (Johnson and Seinen, 2002). In thecontext of biofilm formation, there is now a hugemodelling literature, both spatial and non-spatial(reviewed in IWA Task Group on Biofilm Model-ing, 2006). The complex effects of spatial structurein a simple spatial, agent-based-model (ABM)of a growth–predation model is illustrated inFig. 1.5.
Ecological communities exhibit all the systemcomplexity of physiological systems (Fig. 1.2),even though they are not teleological in the samesense, that is physiological mechanisms are presentfor a purpose: as control mechanisms to main-tain an organism within its domain of tolerance.For example, the glucose homeostasis system inmammals orchestrates the efficient delivery of en-ergy to the various tissues in the face of the di-verse demands that the organism encounters. Incontrast, the mechanisms that sometimes main-tain an ecological community in a (quasi) sta-ble state are not selected for this high-level pur-pose: the stable state is an emergent property ofthese mechanisms and in itself has no function.Of course, the evolution of interaction mecha-nisms always takes place and is shaped withinan ecological setting, and these mechanisms maytherefore play a role in maintaining this context –the mechanisms themselves are adapted. But thewhole-community system is not subject to natu-ral selection, and hence is not adapted for anypurpose.
This feature of ecological systems makes theiranalysis particularly difficult because our under-standing cannot easily be directed by a ‘top-down’view based on what the system is ‘for’, i.e. whatits evolved function is. Instead, communities are

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 15
c
b
2000
1500
1000
500
20
1400 (f)
(c)(b)
(e)(d)
(a)
1200
1000
800
600
400
200
20 40 60 80 100t
40 60 80 100t
Fig. 1.5 A simple spatial community. (a, d) Light blue background is the substrate, represented on a 50 × 50 lattice.Red-coloured lattice squares indicate the presence of a base-level (pioneer) organism growing into the available space.(b, e) The red organism is a necessary substrate over which individuals on a higher trophic level can grow. These individualsmove from square to square in search of the red organism. Density is shown as brown to purple on the light blue substrate,with darker colours representing higher density. The red organism is not shown. (c, f) Measure of the availability of redsubstrate to the higher trophic level over 100 time steps. Top row: growth of the red organism is too low for the higher-levelindividuals to thrive. Bottom row: vigorous growth of the red organism promotes a thriving community of the higher-levelindividuals.
essentially ‘bottom-up’ constructs, with ‘emergent’whole-system properties. Microbial communitiesin particular are set to yield a rich harvest of emer-gent properties, the uncovering of which will un-doubtedly lead to greater understanding and con-trol of health and disease.
1.9 Conclusions
The molecular revolution, beginning in the 1950s,which largely displaced classical physiology andwhole-organism biology, has come full circle withthe recent rise of the systems biology movement.This approach aims to reverse the trend towardever more refined reductionism, and instead in-tegrate the detailed knowledge that the molecu-lar revolution has provided, to refocus on howwhole system behaviour ‘emerges’ from detailedmolecular information. This approach can be ap-plied at many levels of the biological hierarchy(Fig. 1.1), although most current work tends tofocus on the cell-as-system, with its major compo-nents conceived as networks of interacting com-ponents: gene regulatory networks, metabolic net-works, protein signalling networks.
Systems thinking in biology is inherently multi-disciplinary, requiring the input of engineers and
computer scientists as much as biologists. Thisis because whole system behaviour requires dy-namic modelling, involving many temporal andspatial scales, stochastic effects, modular designand a hierarchy of orchestrating control struc-tures (Fig. 1.2). The modular and object-oriented‘design’ of many biological systems provides theflexibility for ‘plugability’, which facilitates theinter-operability of coupled components. Thus,mathematical/computational models of these sys-tems must exhibit analogous properties:
� multi-scaling: temporal and spatial betweendifferent components
� encapsulation: information exchange deter-mines a network of inputs and outputs betweencomponent models
� heterogeneity: different modelling paradigmsand computational environments can be em-ployed for different components.
Major issues for the systems modeller are: Howdo we decide what is the relevant scale on whichto build models? What is the best strategy forhandling the trade-off between quantitative mod-els (lots of detailed data) versus qualitative mod-els (capturing the phenomena)? These sorts of is-sues and their biological counterparts make the

BLBK098-Handerson March 26, 2009 22:2
16 Periodontal medicine and systems biology
traditional, hypothesis-driven paradigm of empir-ical biological research problematic. Indeed, somecommentators believe that biology is in the throesof a ‘neo-Baconian crisis’, in which induction, andhypothesis generation from the ever-increasingmountain of high-throughput data, is becomingincreasingly problematic.
It seems, therefore, that in the new systems biol-ogy era, the old hypothetico-deductive paradigmmay be more of an obstacle to progress thanan aid. Instead, a more ‘objectives’ driven ap-proach is more appropriate, in the sense that theaim is to characterise the mechanisms underlyingwhole-system ‘emergent’ behaviour, and in so do-ing to understand the nature and scope of suchbehaviour. The premature formulation of oftensimplistic hypotheses does not help this process.In summary, systems biology should be not onlya provider of models and simulations but also asource of understanding and a toolkit that com-plements and guides experimentation.
References
Albert, R., Jeong, H. and Barabasi, A.L. (2000) Errorand attack tolerance in complex networks. Nature,406, 378–382.
Alon, U. (2007) An Introduction to Systems Biology.Chapman & Hall, London.
Anand, R., Asthagiri, A.R. and Lauffenburger, D. (2000)Bioengineering models of cell signaling. Annual Re-view of Biomedical Engineering, 2, 31–53.
Barabasi, A.-L. and Albert, R. (1999) Emergence ofscaling in random networks. Science, 286, 509–512.
Begon, M., Harper, J.L. and Townsend, C.R. (1990)Ecology: Individuals, Populations and Communities,2nd edn. Blackwell, Oxford.
Bhalla, U. and Iyengar, R. (1999) Emergent propertiesof networks of biological signalling pathways. Science,283, 381–387.
Bluthgen, N. (2006) Systems-Biological Approach toRas-Mediated Signal Transduction. Logos Verlag,Berlin.
Boogerd, F.C., Bruggeman, F.J., Hofmeyr, J.S. and West-erhoff, H.V. (eds) (2007) Systems Biology: Philosoph-ical Foundations. Elsevier, North Holland.
Bray, D., Levin, M.D. and Morton-Firth, C.J. (1998)Receptor clustering as a cellular mechanism to controlsensitivity. Nature, 393, 85–88.
Carroll, S.B. (2001) Chance and necessity: the evolutionof morphological complexity and diversity. Nature,409, 477–492.
Csete, M.E. and Doyle, J.C. (2002) Reverse engineeringbiological complexity. Science, 295, 1678–1682.
Edwards, J.S., Ibarra, R.U. and Palsson, B.O. (2001) Insilico predictions of Escherichia coli metabolic capa-bilities are consistent with experimental data. NatureBiotechnology, 19, 125–130.
Ewens, W.J. (1979) Mathematical Population Genetics.Springer, Berlin.
Fell, D. (1997) Understanding the Control ofMetabolism. Portland Press, London.
Finkelstein, A., Hetherington, J., Li, L., et al. (2004)Computational challenges of systems biology. IEEEComputer, 37(5), 26–33.
Glass, L. and Kauffmann, S.A. (1973) The logical anal-ysis of continuous, non-linear biochemical controlnetworks. Journal of Theoretical Biology, 39, 103–129.
Guelzim, N., Bottani, S., Bourgine, P. and Kepes, F.(2002) Topological and causal structure of the yeasttranscriptional regulatory network. Nature Genetics,31(3), 60–63.
Hahn, M., Conant, G.C. and Wagner, A. (2004) Molec-ular evolution in large genetic networks: connectivitydoes not equal importance. Journal of Molecular Evo-lution, 58, 203–211.
Handelsman, J. (2004) Metagenomics: application of ge-nomics to uncultured microorganisms. Microbiologyand Molecular Biology Reviews, 68, 699–778.
Henderson, B., Seymour, R.M. and Wilson, M. (1998)The cytokine network in infectious diseases. Journalof Immunology and Immunopharmacology, 18(1–2),7–14.
Henderson, B. and Seymour, R.M. (2003) Microbialmodulation of cytokine networks. In: Bacterial Eva-sion of Host Immune Responses (eds B. Hender-son and P.C.F. Oyston). Cambridge University Press,Cambridge, pp. 223–242.
Hetherington, J.P.J., Warner, A. and Seymour, R.M.(2006) Simplification and its consequences in biolog-ical modelling: conclusions from a study of calciumoscillations in hepatocytes. Journal of the Royal Soci-ety, Interface, 3, 319–331.
Higashi, M. and Burns, T.P. (eds) (1991) TheoreticalStudies of Ecosystems: the Network Perspective. Cam-bridge University Press, Cambridge.
Hodgkin, A. and Huxley, A. (1952) A quantitative de-scription of membrane current and its application toconduction and excitation in nerve. Journal of Physi-ology, 117, 500–544.
Hofer, T. (1999) Model of intercellular calcium os-cillations in hepatocytes: synchronization of het-erogeneous cells. Biophysical Journal, 77, 1244–1256.
Hoffmann, A., Levchenko, A., Scott, M.L. and Balti-more, D. (2002) The I-�B – NF-�B signaling module:temporal control and selective gene activation. Sci-ence, 298, 1241–1245.
Holland, P.W.H. (1999) The future of evolutionary de-velopmental biology. Nature, 402, C41–C44.

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 17
Holzhutter, H.G. (2004) The principle of flux minimiza-tion and its application to estimate stationary fluxesin metabolic networks. European Journal of Biochem-istry, 271, 2905–2922.
Hooper, L.V. (2005) Resident bacteria as inductive sig-nals in mammalian gut development. In: The Influ-ence of Cooperative Bacteria on Animal Host Biology(eds M.J. McFall-Ngai, B. Henderson and E.G. Ruby).Cambridge University Press, Cambridge.
Hooper, L.V., Xu, J., Falk, P.G., Midtvedt, T. and Gor-don, J.I. (1999) A molecular sensor that allows a gutcommensal to control its nutrient foundation in acompetitive ecosystem. Proceedings of the NationalAcademy of Sciences of the USA, 96, 9833–9838.
Hornberg, J.J., Binder, B., Bruggeman, F.J., Schoe-berl, B., Heinrich, R. and Westerhoff, H.V. (2005)Control of MAPK signalling: from complexityto what really matters. Oncogene, 24, 5533–5542.
Hornberg, J.J., Bruggeman, F.J., Binder, B., et al.(2005) Principles behind multifarious control ofsignal transduction. ERK phosphorylation andkinase/phosphatase control. FEBS Journal, 272,244–258.
Hornberg, J.J., Bruggeman, F.J., Bakker, B.M. andWesterhoff, H.V. (2007) Metabolic control analysisto identify optimal drug targets. Progress in DrugResearch, 64, 173–189.
Horner-Devine, M.C., Carney, K.M. and Bohannan,B.J.M. (2003) An ecological perspective on bacterialbiodiversity. Proceedings of the Royal Society ofLondon B, 271, 113–122.
Hunter, P., Robbins, P. and Noble, D. (2002) The IUPSHuman Physiome Project. Pflugers Archiv, 445 (1),1–9.
IWA Task Group on Biofilm Modeling (2006) Mathe-matical Modeling of Biofilms. Scientific and TechnicalReport No. 18. IWA Publishing, London.
Jit, M., Henderson. B., Stevens, M. and Seymour,R.M. (2005) TNF� neutralisation in cytokine-drivendiseases: a mathematical model to account for thera-peutic success in rheumatoid arthritis but therapeuticfailure in systemic inflammatory response syndrome.Rheumatology, 44, 323–331.
Johnson, C.R. and Seinen, I. (2002) Selection forrestraint in competitive ability in spatial competitionsystems. Proceedings of the Royal Society of LondonB, 269, 655–663.
Kaufman, M. and Thomas, R. (1987) Model analysisof the bases of multistationarity in the humoralimmune response. Journal of Theoretical Biology,129, 141–162.
Kaufman, M., Urbain, J. and Thomas, R. (1985)Towards a logical analysis of the immune response.Journal of Theoretical Biology, 114, 527–561.
Keener, J. and Sneyd, J. (1998) Mathematical Physiol-ogy. Springer, Berlin.
Kondoh, M. (2003) Foraging adaptation and the rela-tionship between food web complexity and stability.Science, 299, 1388–1391.
Krishna, S., Jensen, M.H. and Sneppen, K. (2007)Minimal model of spiky oscillations in NF-kBsignaling. Proceedings of the National Academy ofSciences of the USA, published online July, 2006; doi:10.1073/pnas.0604085103.
Kummer, U., Olsen, L., Dixon, C., Green, A., Bornberg-Bauer, E. and Baier, G. (2000) Switching fromsimple to complex oscillations in calcium signaling.Biophysical Journal, 79, 1188–1195.
Laurent, M. and Kellershohn, N. (1999) Multistability:a major means of differentiation and evolution inbiological systems. Trends in Biochemical Sciences,24, 418–422.
Lee T.I., Rinaldi, N.J., Robert, F., et al. (2002) Tran-scriptional regulatory networks in Saccharomycescerevisiae. Science, 298, 799–804.
Levin, M.D., Morton-Firth, C.J., Abouhamad, W.N.,Bourret, R.B. and Bray, D. (1998) Origins of indi-vidual swimming behaviour in bacteria. BiophysicalJournal, 74, 175–181.
Lindeman, R.L. (1942) The trophic-dynamic aspect ofecology. Ecology, 23, 399–418.
Lipniacki, T., Paszek, P., Brasier, A.R., Luxon, B.A.and Kimmel, M. (2004) Mathematical model ofNFkappaB regulatory module. Journal of TheoreticalBiology, 228, 195–215.
Lipson, H., Pollack, J.B. and Suh, N.P. (2002) Onthe origin of modular variation. Evolution, 56(8),1549–1556.
MacCarthy, T., Seymour, R.M. and Pomiankowski, A.(2003) The evolutionary potential of the Drosophilasex determination gene network. Journal of Theoret-ical Biology, 225, 461–468.
MacCarthy, T., Pominakowski, A. and Seymour,R.M. (2005) Using large-scale perturbations in genenetwork reconstruction. BMC Bioinformatics, 6, 11.
Maher, A.D., Kuchel, P.W., Ortega, F., de Autauri, P.,Cantelles, J. and Cascante, M. (2003) Mathematicalmodelling of the urea cycle: A numerical investigationinto substrate channelling. European Journal ofBiochemistry, 270, 3953–3961.
Marshall, C.J. (1995) Specificity of receptor tyrosinekinase signalling: transient versus sustained extra-cellular signal-regulated kinase activation. Cell, 80,179–185.
May, R.M. (1973) Stability and Complexity of ModelEcosystems. Princeton University Press, Princeton.
May, R.M. (2006) Network structure and the biologyof populations. Trends in Ecology & Evolution,21(7), 394–399.
McFall-Ngai, M. (2007) Care for the community.Nature, 445(11), 153.
Murray, J.D. (2001) Mathematical Biology. Springer,Berlin.

BLBK098-Handerson March 26, 2009 22:2
18 Periodontal medicine and systems biology
Nelson, G., Paroan, L., Spiller, D.G., et al. (2002)Multi-parameter analysis of the kinetics of NF-kappaB signalling and transcription in single livingcells. Journal of Cell Science, 115, 1137–1148.
Nelson, D.E., Ihekwaba, A.E.C., Elliott, M., et al. (2004)Oscillations in NF-kappaB signaling control the dy-namics of gene expression. Science, 306, 704–708.
Noble, D. (2002) Modeling the heart: From genes tocells to whole organ. Science, 295, 1678–1682.
Noble, D. and Colatsky, T. (2000) A return to rationaldrug discovery: Computer-based models of cells,organs and systems in drug target identification.Emerging Therapeutic Targets, 4, 39–49.
Orton, R.J., Sturm, O.E., Vyshemirsky, V., Alder, M.,Gilbert, D.R. and Kolch, W. (2005) Computationalmodelling of the receptor-tyrosine-kinase-activatedMAPK pathway. Biochemistry Journal, 392, 249–261.
Patten, B.C. (1991) Network ecology: indirect de-termination of the life-environment relationship inecosystems. In: Theoretical Studies of Ecosystems:The Network Perspective (eds M. Higashi and T.P.Burns). Cambridge University Press, Cambridge.
Preece, A.F., Strahan, K.M., Devitt, J., Yamamoto, F.and Gustafsson, K. (2002) Expression of ABO or re-lated antigenic carbohydrates on viral envelopes leadsto neutralization in the presence of serum containingspecific natural antibodies and complement. Blood,99, 2477–2482.
Prichard, D., Hooi, D., Watson, E., et al. (2003)Bacterial quorum sensing signalling molecules asimmune modulators. In: Bacterial Evasion of HostImmune Responses (eds B. Henderson and P.C.Oyston). Cambridge University Press, Cambridge.
Rainey, P.B., Brockhurst, M.A., Buckling, A., Hodg-son, D.J. and Kassen, R. (2005) The use of modelPseudomonas fluorescens populations to study thecauses and consequences of microbial diversity. In:Soil Biodiversity and Ecosystem Function (eds R.Bardgett, D. Hopkins and M. Usher). CambridgeUniversity Press, Cambridge.
Ruby, E.G., Henderson, B. and McFall-Ngai, M. (2004)We get by with a little help from our (little) friends.Science, 303, 1305–1307.
Santos, S.D., Verveer, P.J. and Bastiaens, P.I. (2007)Growth factor-induced MAPK network topologyshapes Erk response determining PC-12 cell fate.Nature Cell Biology, 9, 324–330.
Schuster, S., Dandeker, T. and Fell, D.A. (1999)Detection of elementary flux modes in biochemicalnetworks: a promising tool for pathway analysis andmetabolic engineering. Trends in Biotechnology, 17,53–60.
Seymour, R.M. (2005) Commensal diversity and theimmune system: modelling the host-as-network.In: The Influence of Cooperative Bacteria onAnimal Host Biology (eds M.J. McFall-Ngai,
B. Henderson and E.G. Ruby). Cambridge UniversityPress, Cambridge.
Seymour, R.M. and Henderson, B. (2001) Pro-inflammatory–anti-inflammatory cytokine dynamicsmediated by cytokine-receptor dynamics in mono-cytes. Mathematical Medicine and Biology, 18(2),159–192.
Seymour, R.M., Allan, M.J., Pomiankowski, A. andGustafsson, K. (2004) Evolution of the human ABOpolymorphism by two complementary selective pres-sures. Proceedings of the Royal Society of London B,271, 1065–1072.
Shen-Orr, S.S., Milo, R., Mangan, S., and Alon, U.(2002) Network motifs in the transcriptional regu-lation network of Escherichia coli. Nature Genetics,31, 64–68.
Snoussi, E.H. and Thomas, R. (1993) Logical identifi-cation of all steady states. Bulletin of MathematicalBiology, 55, 973–991.
Sneyd, J., Girard, S. and Clapham, D. (1993) Calciumwave propagation by calcium induced calcium release;an unusual excitable system. Bulletin of MathematicalBiology, 55, 315–344.
Sontag, E.D. (1998) Mathematical Control Theory:Deterministic Finite Dimensional Systems, 2nd edn.Springer, New York.
Stumpf, M.P.H., Wiuf, C. and May, R.M. (2005)Subnets of scale-free networks are not scale free:Sampling properties of networks. Proceedings of theNational Academy of Sciences of the USA, 102(12),4221–4224.
Thomas, R. (1998) Laws for the dynamics of regulatorynetworks. International Journal of DevelopmentalBiology, 42, 479–485.
Thomas, R. and Kaufman, M. (2001) Multistation-arity, the basis of cell differentiation and memory.I. Structural conditions of multistationarity andother non-trivial behaviour. II. Logical analysis ofregulatory networks in terms of feedback circuits.Chaos, 14, 170–179.
Visick, K.L. and McFall-Ngai, M. (2000) An exclusivecontract: Specificity in the Vibrio-fischeri–Euprymnascolopes partnership. Journal of Bacteriology, 182,1779–1787.
Visick, K.L., Foster, J., Doino, J., McFall-Ngai, M. andRuby, E.G. (2000) Vibrio-fischeri lux genes play animportant role in colonization and development ofthe host light organ. Journal of Bacteriology, 182,4578–4586.
Wagner, A. (2003) How the global structure of proteininteraction networks evolves. Proceedings of theRoyal Society of London B, 270, 457–466.
Wagner, A. (2007) Robustness and Evolvability in Liv-ing Systems. Princeton University Press, Princeton, NJ.
Wagner, G.P. (1996) Homologues, natural kinds, andthe evolution of modularity. American Zoologist, 36,36–43.

BLBK098-Handerson March 26, 2009 22:2
Systems thinking in biology 19
Wagner, G.P. and Altenberg, L. (1996) Complex adap-tations and the evolution of evolvability. Evolution,50, 967–976.
Watson, R.A. and Pollack, J.B. (2000) Symbiotic com-bination as an alternative to sexual recombinationin genetic algorithms. In: Proceedings in ParallelProblem Solving in Nature, Vol. VI (eds Rothlauf, F.et al.). Springer, Mannheim.
Wilson, M., Seymour, R.M. and Henderson, B. (1998)Bacterial perturbation of cytokine networks. Infectionand Immunity, 66(6), 2401–2409.
Yates, A., Callard, R. and Stark, J. (2004) Combiningcytokine signalling with T-bet and Gata-3 regulationin Th1 and Th2 differentiation: a model for cellulardecision making. Journal of Theoretical Biology, 231,181–196.

BLBK098-Handerson March 26, 2009 22:2





























