Embed Size (px)
Citation preview

logy 544 (2006) 39–48www.elsevier.com/locate/ejphar
European Journal of Pharmaco
Paradoxical nifedipine facilitation of 45Ca uptake into rathippocampal synaptosomes
Joana Costa, M. Graça Lobo, Paulo Correia-de-Sá ⁎
Laboratório de Farmacologia/UMIB, Instituto de Ciências Biomédicas de Abel Salazar (ICBAS), Universidade do Porto,L. Prof. Abel Salazar, 2, 4099-003 Porto, Portugal
Received 27 November 2005; received in revised form 23 May 2006; accepted 19 June 2006Available online 27 June 2006
Abstract
Nifedipine has a high incidence of neurologic adverse reactions as compared with other dihydropyridine Cav1 (L-type) channel blockers usedfor treating cardiovascular diseases. The mechanism mediating neuronal excitation by nifedipine is still in debate. Nifedipine caused a dual role onveratridine-induced 45Ca uptake by rat hippocampal synaptosomes. In the nanomolar range (0.001–0.3 μM), nifedipine decreased 45Ca uptake in acadmium-sensitive manner. In contrast with nitrendipine (0.001–10 μM), nifedipine consistently facilitated 45Ca accumulation when used in lowmicromolar concentrations (0.3–10 μM). The cadmium-insensitive nifedipine facilitation became less evident upon increasing veratridineconcentration from 5 to 20 μM and was not detected when the synaptosomes where depolarised with 30 mM KCl. Na+ substitution by N-methyl-D-glucamine (132 mM) or blockade of Na+ currents with tetrodotoxin (1 μM) both prevented nifedipine excitation. The Na+/Ca2+-exchangerinhibitor, KB-R7943 (3–50 μM), did not reproduce nifedipine actions. Data suggest that tetrodotoxin-sensitive Na+ channels may operateparadoxical nifedipine facilitation of 45Ca uptake by rat hippocampal synaptosomes.© 2006 Elsevier B.V. All rights reserved.
Keywords: Nifedipine; Nitrendipine; 45Ca uptake; Tetrodotoxin-sensitive Na+ channel; Na+/Ca2+-exchanger; Hippocampal synaptosome
1. Introduction
The effects of nifedipine, a drug used commonly for treatinghypertension and angina, are usually attributed to its blockingaction on Cav1 (L-type) channels. Compared with other dihy-dropyridines, nifedipine has been reported to have a high incidenceof neurologic adverse reactions (e.g. anxiety, insomnia, dizziness,tremor, nervousness, confusion) (Testa et al., 1998). Due to itslipophylic properties, nifedipine can easily cross the blood–brainbarrier and low micromolar concentrations may be achieved in thebrain when high doses are administered to patients withcardiovascular diseases (Janicki et al., 1988). Recently, Hirasawaand Pittman (2003) found a paradoxical effect of nifedipine onminiature excitatory postsynaptic currents recorded from magno-cellular neurons of the supraoptic nucleus of the hypothalamus.These authors showed that nifedipine, but not other chemicallyrelated dihydropyridines such as nimodipine or nitrendipine,
⁎ Corresponding author. Tel.: +351 22 2062243; fax: +351 22 2062232.E-mail address: [email protected] (P. Correia-de-Sá).
0014-2999/$ - see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.ejphar.2006.06.040
caused a long-lasting increase in spontaneous glutamate release.Surprisingly, the effect of the drug was not through blockade ofCav1 (L-type) channels and was largely independent of Ca2+-induced changes in basal release probability. Moreover, whilstinvestigating the antiepileptic efficacy of nifedipine, Straub et al.(2000) observed that nifedipine (20–80 μM) transiently increasedepileptiform potentials before its suppressive activity in hippo-campal and neocortical slices. Interestingly, elevations of K+
concentration accentuated the depressive actions of nifedipine,particularly in the hippocampus where Cav1 (L-type) channels aredistributed all over the neurons (Wang et al., 2000). Thus, a majorunresolved question is the site and the mechanism of neuronalexcitation by nifedipine.
While trying to evaluate the ability of nifedipine tomodify 45Cauptake into rat hippocampal synaptosomes depolarised by the Na+
channel activator, veratridine, we made the incidental observationthat nifedipine in the micromolar range increased rather thandepressed 45Ca uptake in a cadmium-insensitive manner. Ca2+
homeostasis is controlled by the equilibrium between (1) Ca2+
influx through voltage-sensitive and/or receptor-operated channels,

Table 1Basal 45Ca uptake into rat hippocampal synaptosomes under control conditionsand in the presence of modulators of intracellular Ca2+ homeostasis
Drugs Concent. (mM) 45Ca uptake(nmol/mg of protein)
Control 3.57±0.39 (16)TTX 0.001 3.46±0.60 (6)Cd2+ 0.5 1.52±0.24 (16)a
Na+/NMDG+ 0/132 7.05±0.70 (8)a
Na+/Choline+ 0/132 9.40±0.84 (8)a
KB-R7943 0.003 3.16±0.30 (8)0.050 3.40±0.29 (8)
Synaptosomes were incubated in the absence (control) and in the presence of testdrugs for a period of 10 min before addition of 45CaCl2 (specific activity0.32 μCi/μmol of Ca2+). After a 3-min incubation period, 45Ca uptake wasterminated by adding ice-cold Tris-EGTA solution and followed by filtrationunder negative pressure through GF/C glass fibre filters. Data represent theaverage (±S.E.M.) of 45Ca uptake (nmol/mg of protein) into the rat hippocampalsynaptosomes. In parenthesis is shown the number of experiments, performed intriplicate. a P<0.05 (one-way ANOVA followed by Dunnett’s modified t-test)when compared to control.
40 J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
(2) Ca2+ efflux by the activity of theNa+/Ca2+ exchanger andCa2+-pumps located in the plasmamembrane, and (3) Ca2+ sequestrationby intracellular stores. Differential effects on each of thesemechanisms could explain the dual activity of nifedipine on 45Cauptake in the central nervous system. In view of the relevance ofincreased Ca2+ influx into hippocampal nerve terminals duringphysiological (e.g. learning and memory) and pathological (e.g.epileptiform attacks, ischemia) conditions, we designed experi-ments to study the pharmacology of nifedipine excitation underdifferent conditions of synaptosomal depolarisation.
Preliminary accounts of some of the results have alreadybeen published (Costa et al., 2004).
2. Methods
2.1. Preparation of the synaptosomal fraction of rat hippocampus
Rats of either sex (Charles River, Barcelona, Spain) were keptat a constant temperature (21 °C) and a regular light (06.30–19.30 h) dark (19.30–06.30 h) cycle with food and water adlibitum. Animal handling and experiments carried out followedthe guidelines of the International Council for Laboratory AnimalScience. Synaptosomes were prepared from rat hippocampalhomogenates as previously described (Cunha et al., 1992).Briefly, Wistar rats (150–200 g) were decapitated and the brainswere rapidly removed into ice-cold 0.32 M sucrose solutioncontaining 1 mM EDTA, 1 mg/ml bovine serum albumin and20 mM Tris-base, buffered to pH 7.4 with 1 M NaOH. The brainwas cut longitudinally and the two hippocampi (from 2–4 rats)dissected out were placed immediately in cold isotonic sucrosesolution (1:10, w/v) and were homogenised (four to ten up-and-down strokes, 800 rpm) in a Potter–Elvehjem homogeniser with aTeflon piston at 4 °C. The suspension was centrifuged at 3000 gduring 10 min, and the supernatant was collected and centrifugedat 14,000 g for 12min at 4 °C. The pellet was re-suspended in 1mlof a 45%v/v Percoll solutionmade up in a pre-incubationmedium[composition was (in mM) 140 NaCl, 5 KCl, 1.3 MgCl2, 1.2NaH2PO4, 1.2 CaCl2, 20 Tris-base, 1 EDTA, 10 glucose and1 mg/ml bovine serum albumin, pH 7.4]. After centrifugation at14,000 g for 2 min, the top layer was removed (synaptosomalfraction), washed in 1 ml of pre-incubation medium, and re-suspended in an incubation medium [containing (in mM) 132NaCl, 5KCl, 1.3MgCl2, 1.2NaH2PO4, 1.2CaCl2, and 10 glucosebuffered with 20 Tris-base, pH 7.4]. The synaptosomal proteinconcentrationwasmeasured according to themethod of Lowry, asmodified by Peterson (1977).
2.2. Determination of 45Ca uptake by hippocampal synaptosomes
Synaptosomes possess many properties of intact nerve ter-minals including metabolic activity, generation of restingpotential and capability of releasing neurotransmitters afterdepolarisation. In the present study, we determined the increasein [Ca2+]i by direct measurement of evoked 45Ca uptake into rathippocampal synaptosomes. Even though radioactively labelledcalcium loading do not resolve the very rapid synaptic eventsthat occur inmilliseconds (as compared with fluorescent dyes), it
can detect specifically intracellular Ca2+ changes that depend onthe entrance of 45Ca through the plasma membrane. Samples(50 μl) of the synaptosomal suspension (∼ 0.25 mg of protein)were pre-incubated in the absence and in the presence of testdrugs for a period of 10 min, at 37 °C. Synaptosomes wereincubated during 3 min with a solution containing 45CaCl2 (at afinal concentration of 0.32 μCi/μmol of Ca2+) in the presence ofdepolarising agents, KCl (15–30mM) or veratridine (1–20 μM).This was done to compensate for the slower reaction kinetics ofveratridine as compared to KCl (Sitges and Galindo, 2005).Moreover, veratridine (10 μM)-induced 45Ca uptake was fasterduring the first minute (4.98±0.23 nmol/mg of protein, n=4)followed by a slight increase until the third minute of incubation(5.75±0.18 nmol/mg of protein, n=5) (see e.g. Gonçalves et al.,1991). As this corresponds to the greatest period in whichstimulated 45Ca uptake was observed without much variabilityof non-specific 45Ca uptake (see Table 1) it was chosen for use inthe remaining experiments, unless otherwise indicated. In someexperiments 45Ca uptake was induced by L-glutamate (10–100μM) or by the nicotinic agonist, 1,1-dimetyl-4-diphenylpi-perazinium (DMPP, 1–10 μM). 45Ca uptake was terminated byadding 4 ml ice-cold Tris-EGTA solution [containing (in mM)120 NaCl, 5 KCl, 5 EGTA and 20 Tris-base, pH 7.4] andfollowed by filtration under negative pressure through GF/Cglass fibre filters. The filters were washed three times with 5 mlice-cold washing solution [containing (in mM) 132 NaCl, 5 KCl,1.3 MgCl2, 1.2 CaCl2, and 20 Tris-base, pH 7.4]. The radio-activity associated with the synaptosomes was determined byliquid scintillation spectrometry after addition of 3.5 ml ofPackard Insta Gel II scintillation cocktail. The net stimulated45Ca uptake is the difference between 45Ca uptake under stim-ulation conditions and without stimulation (basal conditions)(see Table 1). To avoid endogenous adenosine interference (seee.g., Morgan et al., 1987; Triggle, 2003), all the experimentswere performed in the presence of adenosine deaminase (2 U/ml). In each experiment, 45Ca uptake determinations wereperformed in triplicate.
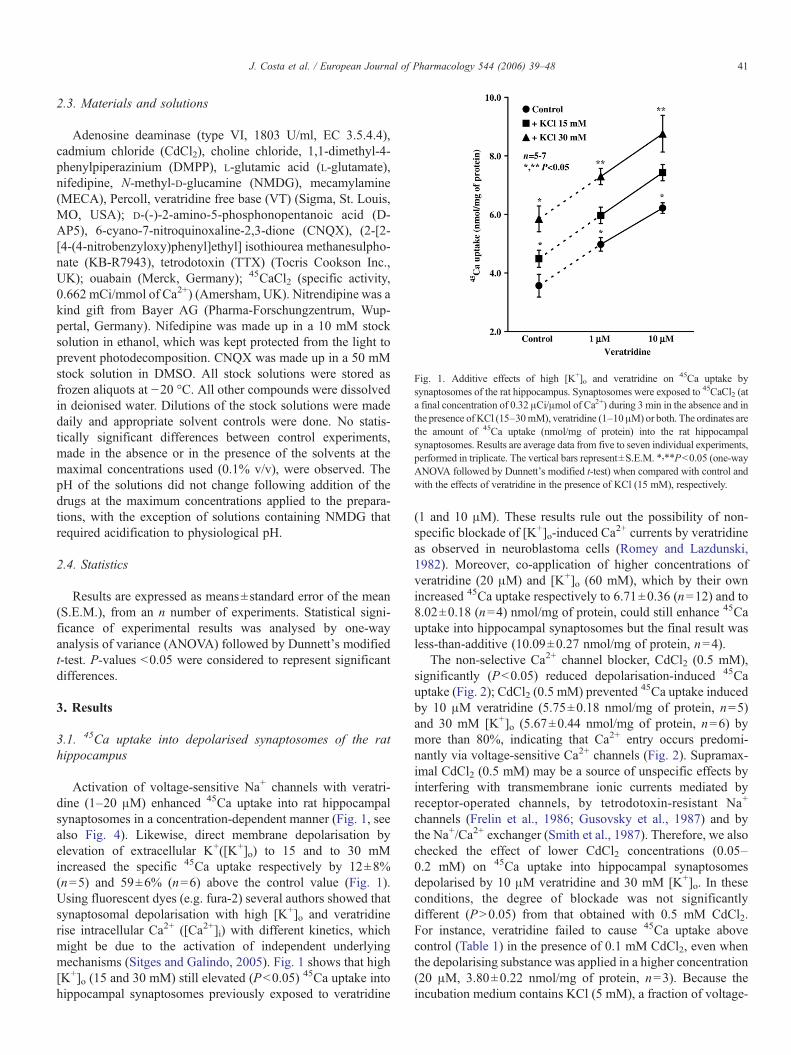
Fig. 1. Additive effects of high [K+]o and veratridine on 45Ca uptake bysynaptosomes of the rat hippocampus. Synaptosomes were exposed to 45CaCl2 (ata final concentration of 0.32 μCi/μmol of Ca2+) during 3 min in the absence and inthe presence of KCl (15–30mM), veratridine (1–10μM) or both. The ordinates arethe amount of 45Ca uptake (nmol/mg of protein) into the rat hippocampalsynaptosomes. Results are average data from five to seven individual experiments,performed in triplicate. The vertical bars represent±S.E.M. ⁎,⁎⁎P<0.05 (one-wayANOVA followed by Dunnett's modified t-test) when compared with control andwith the effects of veratridine in the presence of KCl (15 mM), respectively.
41J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
2.3. Materials and solutions
Adenosine deaminase (type VI, 1803 U/ml, EC 3.5.4.4),cadmium chloride (CdCl2), choline chloride, 1,1-dimethyl-4-phenylpiperazinium (DMPP), L-glutamic acid (L-glutamate),nifedipine, N-methyl-D-glucamine (NMDG), mecamylamine(MECA), Percoll, veratridine free base (VT) (Sigma, St. Louis,MO, USA); D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl] isothiourea methanesulpho-nate (KB-R7943), tetrodotoxin (TTX) (Tocris Cookson Inc.,UK); ouabain (Merck, Germany); 45CaCl2 (specific activity,0.662 mCi/mmol of Ca2+) (Amersham, UK). Nitrendipine was akind gift from Bayer AG (Pharma-Forschungzentrum, Wup-pertal, Germany). Nifedipine was made up in a 10 mM stocksolution in ethanol, which was kept protected from the light toprevent photodecomposition. CNQX was made up in a 50 mMstock solution in DMSO. All stock solutions were stored asfrozen aliquots at −20 °C. All other compounds were dissolvedin deionised water. Dilutions of the stock solutions were madedaily and appropriate solvent controls were done. No statis-tically significant differences between control experiments,made in the absence or in the presence of the solvents at themaximal concentrations used (0.1% v/v), were observed. ThepH of the solutions did not change following addition of thedrugs at the maximum concentrations applied to the prepara-tions, with the exception of solutions containing NMDG thatrequired acidification to physiological pH.
2.4. Statistics
Results are expressed as means±standard error of the mean(S.E.M.), from an n number of experiments. Statistical signi-ficance of experimental results was analysed by one-wayanalysis of variance (ANOVA) followed by Dunnett's modifiedt-test. P-values <0.05 were considered to represent significantdifferences.
3. Results
3.1. 45Ca uptake into depolarised synaptosomes of the rathippocampus
Activation of voltage-sensitive Na+ channels with veratri-dine (1–20 μM) enhanced 45Ca uptake into rat hippocampalsynaptosomes in a concentration-dependent manner (Fig. 1, seealso Fig. 4). Likewise, direct membrane depolarisation byelevation of extracellular K+([K+]o) to 15 and to 30 mMincreased the specific 45Ca uptake respectively by 12±8%(n=5) and 59±6% (n=6) above the control value (Fig. 1).Using fluorescent dyes (e.g. fura-2) several authors showed thatsynaptosomal depolarisation with high [K+]o and veratridinerise intracellular Ca2+ ([Ca2+]i) with different kinetics, whichmight be due to the activation of independent underlyingmechanisms (Sitges and Galindo, 2005). Fig. 1 shows that high[K+]o (15 and 30 mM) still elevated (P<0.05) 45Ca uptake intohippocampal synaptosomes previously exposed to veratridine
(1 and 10 μM). These results rule out the possibility of non-specific blockade of [K+]o-induced Ca
2+ currents by veratridineas observed in neuroblastoma cells (Romey and Lazdunski,1982). Moreover, co-application of higher concentrations ofveratridine (20 μM) and [K+]o (60 mM), which by their ownincreased 45Ca uptake respectively to 6.71±0.36 (n=12) and to8.02±0.18 (n=4) nmol/mg of protein, could still enhance 45Cauptake into hippocampal synaptosomes but the final result wasless-than-additive (10.09±0.27 nmol/mg of protein, n=4).
The non-selective Ca2+ channel blocker, CdCl2 (0.5 mM),significantly (P<0.05) reduced depolarisation-induced 45Cauptake (Fig. 2); CdCl2 (0.5 mM) prevented 45Ca uptake inducedby 10 μM veratridine (5.75±0.18 nmol/mg of protein, n=5)and 30 mM [K+]o (5.67±0.44 nmol/mg of protein, n=6) bymore than 80%, indicating that Ca2+ entry occurs predomi-nantly via voltage-sensitive Ca2+ channels (Fig. 2). Supramax-imal CdCl2 (0.5 mM) may be a source of unspecific effects byinterfering with transmembrane ionic currents mediated byreceptor-operated channels, by tetrodotoxin-resistant Na+
channels (Frelin et al., 1986; Gusovsky et al., 1987) and bythe Na+/Ca2+ exchanger (Smith et al., 1987). Therefore, we alsochecked the effect of lower CdCl2 concentrations (0.05–0.2 mM) on 45Ca uptake into hippocampal synaptosomesdepolarised by 10 μM veratridine and 30 mM [K+]o. In theseconditions, the degree of blockade was not significantlydifferent (P>0.05) from that obtained with 0.5 mM CdCl2.For instance, veratridine failed to cause 45Ca uptake abovecontrol (Table 1) in the presence of 0.1 mM CdCl2, even whenthe depolarising substance was applied in a higher concentration(20 μM, 3.80±0.22 nmol/mg of protein, n=3). Because theincubation medium contains KCl (5 mM), a fraction of voltage-

Fig. 2. Influence of cadmium (0.5 mM), tetrodotoxin (1 μM), KB-R7943(3 μM), and external Na+ replacement by NMDG (132 mM) or by choline(132 mM) on 45Ca uptake into rat hippocampal synaptosomes depolarised by(A) veratridine (10 μM) and (B) KCl (30 mM). Synaptosomes were pre-incubated with the test drugs for 10 min before addition of the 45Ca-containingsolutions. Incubation of the synaptosomal fraction with 45CaCl2 (specificactivity 0.32 μCi/μmol of Ca2+) was performed during 3 min in the presence of(A) veratridine (10 μM) and (B) KCl (30 mM). The ordinates are the amount of45Ca uptake (nmol/mg of protein) into the rat hippocampal synaptosomes. Theaverage 45Ca uptake in control conditions is also shown for comparison (dashedline). Each column represents average data from n number of experiments(shown in parenthesis), performed in triplicate. The vertical bars represent S.E.M. ⁎P<0.05 (one-way ANOVA followed by Dunnett's modified t-test) whencompared with the effects of (A) veratridine (10 μM) or (B) KCl (30 mM) alone.
42 J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
sensitive Ca2+ channels opens periodically thus contributing tobasal intracellular 45Ca levels, which may be reduced by CdCl2(0.5 mM) (see Table 1).
Tetrodotoxin, applied in a concentration (1 μM) that wasvirtually devoid of effect on basal 45Ca uptake (Table 1),completely prevented veratridine (10 μM)-induced synaptosomal45Ca entry but had no significant (P>0.05) effect on the uptake of
45Ca caused either by 15 mM or by 30 mM [K+]o (Fig. 2). Theseresults suggest that voltage-dependent Na+ channels are notinvolved in direct membrane depolarisation by high [K+]o.Likewise, substitution of extracellular Na+([Na+]o) either byN-methyl-D-glucamine (NMDG, 132 mM) or by choline(132 mM) prevented veratridine (10 μM)-induced 45Ca uptake,without significantly (P>0.05) modifying 45Ca entry elicited by30 mM [K+]o (Fig. 2). The magnitude of veratridine (10 μM)-induced rise in 45Ca uptake by hippocampal synaptosomesdepends on the concentration of externalNa+, as it gradually fadedout when [Na+]o was substituted by NMDG (data not shown).
As shown in Table 1, basal 45Ca uptake increased afterreplacement of extracellular Na+ by NMDG (132 mM) or bycholine (132 mM). This was not due to increases in Ca2+ influxthrough voltage-sensitive channels, since Na+-free solutionsmaintained 45Ca uptake induced by 30 mM [K+]o at almost thesame level as in the control solution. [Ca2+]i accumulationinduced by Na+-free media may be due to a reduction of cytosolicCa2+ extrusion through the Na+/Ca2+ exchanger in the absence ofNa+ in the incubation buffer. Nevertheless, selective inhibition ofthe Na+/Ca2+ exchanger (NCX1) with KB-R7943 (3 μM)(Iwamoto, 2004; but see e.g., Watano et al., 1996) failed tomodify basal 45Ca entry and to inhibit depolarization-induced45Ca uptake triggered by veratridine (10 μM) or by high [K+]o(Fig. 2). It was necessary a KB-R7943 concentration (50 μM)high above its IC50 (1.2–2.4 μM) value for inhibiting Na+/Ca2+
exchange in order to reduce 45Ca loading by veratridine (10 μM,37±3%, n=8), without affecting 45Ca uptake elicited by 30 mM[K+]o (data not shown).
3.2. Nifedipine, but not nitrendipine, causes a dual role on45Ca uptake into synaptosomes depolarised by veratridine
Nifedipine (0.001–10 μM) exerted a dual role on veratridine(10 μM)-induced 45Ca uptake into rat hippocampal synapto-somes (Fig. 3A). In the nanomolar range (<0.3 μM), nifedipinedecreased 45Ca uptake; the maximal inhibitory effect (33±4%below the control value, n=8) was observed with 10 nM ni-fedipine. When applied in higher concentrations (0.3–10 μM),nifedipine consistently facilitated the uptake of 45Ca reaching amaximum (31±5% above the control value, n=5) at 3-μMconcentration. A chemically related dihydropyridine, nitrendi-pine, dose-dependently inhibited veratridine (10 μM)-induced45Ca uptake when it was applied in the same concentrationrange (0.001–10 μM) (Fig. 3B). At the highest concentrationtested, nitrendipine (10 μM) reduced 45Ca uptake by 30±3%below the control value (n=6). In this range of concentrations,none of the dihydropyridines changed significantly (P>0.05)the basal 45Ca uptake (see Fig. 4).
Nifedipine (0.001–0.1 μM) inhibition was sensitive toblockade of voltage-sensitive Ca2+ channels by CdCl2 (0.5 mM)(Fig. 3A), even when this compound was used in a concentrationas low as 0.05 mM. Likewise, CdCl2 (0.5 mM) reduced theinhibitory action of nitrendipine in all range of concentrations(0.001–10 μM) (Fig. 3B). On the contrary, nifedipine (0.3–10 μM)-induced rise of 45Ca uptake was virtually unchanged(P>0.05) in the presence of CdCl2 (0.5 mM) (Fig. 3A).

43J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
3.3. The facilitatory role of nifedipine depends on Na+-loadinginto depolarised synaptosomes
Since Ca2+ entry through voltage-sensitive Ca2+ channelsmight not be responsible for the nifedipine facilitation of 45Cauptake into rat hippocampal synaptosomes depolarised by
Fig. 3. Concentration–response curves for the effects of (A) nifedipine (0.001–10 μM) and (B) nitrendipine (0.001–10 μM) on veratridine (10 μM)-induced 45Cauptake into rat hippocampal synaptosomes performed in the absence (open circles)and in the presence (filled circles) of cadmium (0.5 mM). Synaptosomes were pre-incubated with the test drugs for 10 min before addition of the 45Ca-containingsolutions. Incubation of the synaptosomal fraction with 45CaCl2 (specific activity0.32 μCi/μmol of Ca2+) was performed during 3 min in the presence of veratridine(10μM). The ordinates are the amount of 45Ca uptake (nmol/mg of protein) into therat hippocampal synaptosomes. The average 45Ca uptake in control conditions isalso shown for comparison (dashed line). Each point is the average of (A) three toeight and (B) four to ten individual experiments, performed in triplicate. Thevertical bars represent±S.E.M. ⁎P<0.05 (one-way ANOVA followed byDunnett's modified t-test) when compared with the effects of (A) nifedipine or(B) nitrendipine in the absence of cadmium (0.5 mM).
Fig. 4. Nifedipine (0.3–3 μM) facilitation of 45Ca uptake into hippocampalsynaptosomes depolarised by increasing concentrations of veratridine. Synapto-somes were pre-incubated with nifedipine (0.3–3 μM) for 10 min beforeaddition of the 45Ca-containing solutions. Exposure to 45CaCl2 (at a finalconcentration of 0.32 μCi/μmol of Ca2+) was performed during 3 min in theabsence and in the presence of veratridine (5–20 μM). The ordinates are theamount of 45Ca uptake (nmol/mg of protein) into the rat hippocampalsynaptosomes. Results are average data from four to six individual experiments,performed in triplicate. The vertical bars represent±S.E.M. ⁎,⁎⁎P<0.05 (one-way ANOVA followed by Dunnett's modified t-test) when compared withcontrol and with the effect of nifedipine in the presence of veratridine (5 μM),respectively. Please note that nifedipine was devoid of effect on 45Ca uptake intonon-depolarised synaptosomes of the rat hippocampus.
veratridine (10 μM), we investigated the role of nifedipine(0.3–3 μM) on Na+-dependent synaptosomal depolarisation. Themagnitude of nifedipine (0.3–3 μM) facilitation was inverselyrelated to the amount of Na+ loaded into hippocampal syn-aptosomes by veratridine (5–20μM) (Fig. 4), but this was not dueto saturation of the system since high [K+]o could still elevate45Ca uptake into synaptosomes exposed to veratridine (seeFig. 1). Furthermore, substitution of extracellular Na+ by NMDG(132 mM) or blockade of Na+ currents with tetrodotoxin (1 μM)prevented nifedipine-facilitation of 45Ca uptake induced byveratridine (10 μM) (Fig. 5A). Moreover, nifedipine (0.3–3 μM)failed to rise 45Ca uptake when synaptosomes were depolarisedby 30 mM [K+]o (Fig. 5B), i.e. nifedipine (0.3–3 μM) dose-dependently reduced 45Ca uptake elicited by high [K+]o, when itwas applied in a concentration (30 mM) that caused a similar netincrease of 45Ca entry as veratridine (10 μM) (see Fig. 2).Nifedipine (0.3–10 μM)-induced decrease of 45Ca uptake bysynaptosomes depolarised by 30 mM [K+]o was prevented byCdCl2 (0.5 mM) (Fig. 5B). At concentrations below 0.3 μM,nifedipine was devoid of effect on 45Ca uptake induced by high[K+]o depolarisation. Whether the requirement of high concen-trations of nifedipine to reduce [K+]o-induced
45Ca uptake meansthat KCl and veratridine activate different calcium channelsdeserves further investigations, as a number of Cav1 (L-type)

Fig. 5. Influence of cadmium (0.5 mM), tetrodotoxin (1 μM), and external Na+
replacement by NMDG (132 mM) on the effects of nifedipine (0.3–3 μM) on45Ca uptake into rat hippocampal synaptosomes depolarised by (A) veratridine(10 μM) and by (B) KCl (30 mM). Synaptosomes were pre-incubated with thetest drugs for 10 min before addition of the 45Ca-containing solutions.Incubation of the synaptosomal fraction with 45CaCl2 (specific activity 0.32 μCi/μmol of Ca2+) was performed during 3 min in the presence of (A) veratridine(10 μM) and (B) KCl (30 mM). The ordinates are the variation of 45Ca uptake(nmol/mg of protein) into the rat hippocampal synaptosomes from controlexperiments. Each column represents average data from (A) four to seven or (B)four to five individual experiments, performed in triplicate. The vertical barsrepresent S.E.M. ⁎P<0.05 (one-way ANOVA followed by Dunnett's modifiedt-test) when compared with the effects of nifedipine on synaptosomesdepolarised by (A) veratridine (10 μM) or (B) KCl (30 mM), respectively.
44 J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
specific agents have demonstrated activity in the micromolarrange at T-and N-type channels (see e.g. Triggle, 2003). Overall,these findings are in keeping with the indispensable role ofvoltage-sensitive Na+ channels for nifedipine-induced increasesof 45Ca uptake into depolarised synaptosomes.
Taking into account that when Na+/K+-ATPase is inhibited byouabain a considerable fraction of intracellular Na+ rise in a TTX-insensitive manner (see e.g. Galván and Sitges, 2004), we examine
whether pump inhibition could be involved in the mechanism ofnifedipine facilitation of 45Ca uptake into hippocampal synapto-somes. Exposure of synaptosomes to ouabain (5mM) induced a net45Ca uptake into hippocampal synaptosomes of 4.77±0.39 nmol/mg of protein (n=8), which was comparatively less than theincrease caused by 10 μM veratridine and by 30 mM [K+]o (seeFig. 2). In the presence of nifedipine (3 μM), the net increase of45Ca uptake induced by ouabain (5 mM) returned (P<0.05) to thecontrol level (3.57±0.78 nmol/mg of protein, n=5). It is alsoworth to note that the increase on basal 45Ca uptake resultingfrom omission of extracellular Na+ and replacement either byNMDG (132 mM) or by choline (132 mM) (Table 1) waspartially reduced by nifedipine (3 μM) to 4.52±0.35 (n=5) and7.34±0.31 (n=5) nmol/mg of protein, respectively. These find-ings rule out the possibility of relatively high concentrations(>10 μM) of nifedipine causing sequestration of intracellular45Ca due to inhibition of Na+/Ca2+ exchange in the absence ofextracellular Na+(Carvalho et al., 1986). In keeping with thishypothesis, KB-R7943 (3–50 μM) inhibition of Na+/Ca2+
exchange failed to mimic the facilitatory action of nifedipine(0.3–3 μM) (Table 1) and to increase 45Ca uptake induced byveratridine (10 μM) (Fig. 2).
3.4. Nifedipine inhibits receptor-operated 45Ca uptake intohippocampal synaptosomes
Released neurotransmitters from depolarised hippocampalnerve endings, such as glutamate and acetylcholine, may act onionotropic presynaptic receptors. In view of the possibility thatnifedipine-facilitation could be partly due to depolarisation-induced influx of Ca2+ through receptor-operated channels, weexamined the influence of nifedipine (3 μM) on 45Ca uptakeinduced by L-glutamate and by the nicotinic receptor agonist, 1,1-dimethyl-4-phenylpiperazinium (DMPP). L-glutamate (10–100 μM) and DMPP (1–10 μM) concentration-dependentlyincreased 45Ca uptake into the rat hippocampal synaptosomes(Fig. 6). Mecamylamine (100 μM), a nicotinic channel blocker,prevented 45Ca uptake induced by DMPP (10 μM, 4.71±0.14 nmol/mg of protein, n=7) (Fig. 6A). D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5, 50 μM) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μM), which antagonizerespectively ionotropic NMDA (N-methyl-D-aspartate) and non-NMDA glutamate receptors, partially blocked intracellular 45Caaccumulation induced by L-glutamate (Fig. 6B), when thiscompoundwas applied in a concentration (100μM) that increased45Ca uptake (5.60±0.79 nmol/mg of protein, n=4) by a similaramount to that observedwith 30mM [K+]o and 10μMveratridine(see Fig. 2). These results are compatible with the physiologicalroles of presynaptic NMDA, non-NMDA (AMPA/kainate) andnicotinic receptors as promoters of neurotransmitter release in thehippocampus (see for a review, Meir et al., 1999).
Since DMPP (10 μM) and L-glutamate (100 μM) actions wereboth resistant to blockade of Na+ channels by tetrodotoxin (1 μM)(Fig. 6), receptor-operated intracellular 45Ca accumulation is likelyto be independent on the influx of Na+ through tetrodotoxin-sensitive channels. CdCl2 (0.5 mM) completely prevented theuptake of 45Ca caused by L-glutamate (100 μM) (Fig. 6B), but it

Fig. 6. Receptor-operated 45Ca uptake into rat hippocampal synaptosomescaused by activation of (A) nicotinic and (B) glutamate receptors: effects oftetrodotoxin, cadmium and nifedipine. Synaptosomes were pre-incubated withmecamylamine (Meca, 100 μM), 6-cyano-7-nitroquinoxaline-2,3-dione(CNQX, 20 μM), D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5, 50 μM),tetrodotoxin (TTX, 1 μM), cadmium (0.5 mM), or nifedipine (3 μM) for 10 minbefore addition of the 45Ca-containing solutions. Incubation of the synaptosomalfraction with 45CaCl2 (specific activity 0.32 μCi/μmol of Ca2+) was performedduring 3 min in the presence of (A) 1,1-dimethyl-4-phenylpiperazinium (DMPP,1–10 μM) or (B) L-glutamate (10–100 μM). The ordinates are the amount of45Ca uptake (nmol/mg of protein) into the rat hippocampal synaptosomes. Theaverage 45Ca uptake in control conditions is also shown for comparison (dashedline). Each column represents average data from n number of experiments(shown in parenthesis), performed in triplicate. The vertical bars represent S.E.M.⁎P<0.05 (one-way ANOVA followed by Dunnett's modified t-test) whencompared with basal 45Ca uptake; ⁎⁎P<0.05 (one-way ANOVA followed byDunnett's modified t-test) when compared with the effects of (A) DMPP (10 μM)or (B) L-glutamate (100 μM) alone, respectively.
45J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
inhibited DMPP (10 μM)-induced 45Ca entry only by 60%(Fig. 6A). These observations suggest that voltage-gated Ca2+
channels are tonically activated during local depolarisation ofhippocampal nerve terminals by L-glutamate and DMPP. Apossible explanation for the residual 45Ca accumulation duringsimultaneous application of DMPP and CdCl2 (0.5 mM) is thecontinuous Ca2+ influx through the nicotinic pore (Wonnacott,1997). As shown in Fig. 6, nifedipine (3 μM) lowered 45Ca uptakeinduced by both L-glutamate (100 μM) and DMPP (10 μM),although to a lesser extent than that caused by CdCl2 (0.5 mM),indicating that voltage-sensitive Cav1 (L-type) channels areactivated by membrane-bound glutamate and nicotinic receptorsin parallel with nifedipine-insensitive channels. We cannot,however, exclude the possibility of CdCl2 (0.5 mM) and nifedipine(3 μM) affecting Ca2+ permeability through receptor-operatedchannels.
4. Discussion
It is generally accepted that synaptosomes represent asuitable preparation to study the function of nerve endingssuch as the accumulation of Ca2+ in response to depolarisationand the role of increases in intracellular Ca2+ in the couplingbetween stimulus and neurotransmitter release (see for review,Tareilus and Breer, 1995). High [K+]o and veratridine are themost common strategies to depolarise synaptosomal plasmamembrane as they allow to differentiate between presynapticNa+ channel-mediated responses (Adam-Vizi and Liget, 1986;see Results). Both compounds increased intracellular 45Caaccumulation in the rat hippocampus as well as in the humanbrain (Meder et al., 1997; Fink et al., 2002). Voltage-sensitiveCa2+ channels represent the most significant pathway by which45Ca is taken up by hippocampal synaptosomes depolarised byhigh [K+]o and veratridine, as their effects were almostcompletely (<80%) prevented by the non-selective blocker ofmembrane-bound Ca2+ channels, CdCl2 (0.1–0.5 v mM) (butsee e.g. Galindo and Sitges, 2004). Although it has been shown,that Cd2+ may block competitively tetrodotoxin-resistant Na+
channels (IC50=0.2 mM) (Frelin et al., 1986; Gusovsky et al.,1987), these channels account for only a small amount (<10%)of Na+ influx into brain synaptosomes elicited by agents thatcause opening of Na+ channels, such as veratridine (Gusovskyet al., 1987). Moreover, previous studies support the assumptionthat intracellular Ca2+ stores do not substantially participate inthe generation of pre-synaptic Ca2+ signals (Tareilus and Breer,1995; Okada et al., 1986; Mulkey and Zucker, 1991).
In the case of veratridine-induced increase in 45Ca uptake,others have shown that Na+ influx caused by veratridine may alsolead to intrasynaptosomal Ca2+ accumulation via inversion of theNa+/Ca2+ exchanger (Meder et al., 1997; Fink et al., 2002;Galindo and Sitges, 2004), a mechanism that can be inhibited byCd2+ (Smith et al., 1987). Here, we showed that veratridine-induced 45Ca uptake was partially reduced by the Na+/Ca2+
exchanger inhibitor, KB-R7943 (Meder et al., 1997), only whenthis compound was used in a concentration (50 μM) well abovethe IC50 value (1.2–2.4 μM) known to inhibit Na+-dependentincreases in [Ca2+]i in both native and transfected cells (Iwamoto,

46 J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
2004; Watano et al., 1996). Although we could not exclude aneffect of KB-R7943 (50 μM) on voltage-sensitive Na+ and Ca2+
(L-type) channels (Watano et al., 1996; Ouardouz et al., 2005), thelack of effect on high [K+]o-evoked increase in
45Ca uptake arguesagainst the possibility of KB-R7943 (50 μM) blocking Ca2+
channels. Based on the experiments with KB-R7943 and CdCl2(see above), the results suggest that veratridine-induced increasein 45Ca uptake is not substantially due to the operation of the Na+/Ca2+ exchanger but rather to a predominant Ca2+ influx throughvoltage-sensitive Ca2+ channels. This rationale agrees with datashowing that the turnover rate of Ca2+ by voltage-sensitive Ca2+
channels (107 Ca2+/s) (Hille, 1992) is much higher than that of theNa+/Ca2+ exchanger (∼5×103 Ca2+/s) (Hilgemann, 1996).
It is well established that Ca2+ is the trigger for fast actionpotential-evoked synaptic transmission and is required forcoupling nerve-induced excitation to cardiac and smooth musclecontraction. As a treatment for hypertension and angina agentsthat interfere with Ca2+ entry such as dihydropyridine channelblockers are commonly used. Drugs with core 1,4-dihydropyr-idine structures potently block Cav1 (L-type) channels, whichhave been implicated on muscle contraction and neurotransmit-ter release in several brain regions (Miller, 1987). Interestingly,Cav1 (L-type) channels may account for about 23% of all Ca2+
channels in the hippocampus (Reuter, 1995). Besides the well-known blocking action on Cav1 (L-type) channels, here weshowed that the dihydropyridine nifedipine (0.3–3 μM) exerts aparadoxical facilitatory effect on 45Ca uptake into hippocampalnerve terminals depolarised by veratridine. Nifedipine facilita-tion was not mimicked by the chemically related compound,nitrendipine, which differ only in the position (ortho or para) ofthe nitro group in the aryl ring. Surprisingly, the facilitatoryeffect of nifedipine had little to do with its action on voltage-sensitive Cav1 (L-type) channels, because non-selective block-ade of Ca2+ channels with CdCl2 did not prevent it.
Although the exact mechanism is unclear, the results in thisstudy rule out interference of nifedipine with several possiblemechanisms related to enhancement of 45Ca uptake. These me-chanisms include (1) direct depolarisation of the synaptosomalplasma membrane by increasing [K+]o, (2) activation of Ca2+-permeant ionotropic (e.g. glutamate, nicotinic) receptors, and (3)inhibition of the Na+/Ca2+ exchanger activity with KB-R7943. Inall these conditions, nifedipine (0.3–3 μM) consistently reduced,rather than increased, intracellular 45Ca accumulation. Nifedipineinhibition is most likely dependent on activation of voltage-sensitive Ca2+ channels because CdCl2 occluded it. Others havesuggested that the 1,4-dihydropyridine nucleus is a privilegedstructure or scaffold for molecules active at a diverse collection ofion channels (including Na+ and K+ channels) and pharmaco-logical receptors (for a review, see Triggle, 2003). For instance,dihydropyridines have been shown to block cation-selectiveligand-gated ion channels, including NMDA receptors (Skeenet al., 1994), nicotinic acetylcholine receptors (Donnelly-Robertset al., 1995) and 5-HT3A receptors (Hargreaves et al., 1996).Therefore, a possibility exists that inhibition of 45Ca uptake intodepolarised synaptosomes by low concentrations of nifedipinemay be partially due to activation of receptor-operated channelsby endogenously released transmitters. Nevertheless, this mech-
anism does not explain the paradoxical facilitatory effect ofnifedipine on intracellular 45Ca accumulation. In contrast withprevious studies (Galindo and Sitges, 2004; Ouardouz et al.,2005), we excluded the possibility of nifedipine interference withendogenous adenosine actions (see e.g. Morgan et al., 1987;Triggle, 2003), which could lead to disinhibition of Ca2+ entryand to an apparent increase in intracellular 45Ca accumulation.This was achieved by systematically adding to the incubationbuffer adenosine deaminase (2 U/ml), the enzyme that inactivatesadenosine into inosine.
The real novelty of the present study is the observation that, inaddition to the well-characterized interaction with the Cav1 (L-type) channel (Kd values in the nanomolar range), nifedipine canincrease 45Ca uptake into depolarised nerve terminals byinteracting with a non-related target (see also Zernig, 1990).Due to the high hydrophobicity of dihydropyridines (Pang andSperelakis, 1984), non-specific actions on membrane proteinscould be expected from these drugs in the micromolarconcentration range when high doses are administered to patientswith cardiovascular diseases (Janicki et al., 1988). The facilitatoryeffect of nifedipine is manifested only when synaptosomes aredepolarised by veratridine. Albeit the higher concentrations(20μM)used, Galindo and Sitges (2004) failed to observe a rise inthe internal concentrations of Na+ and Ca2+ in striatal synapto-somes depolarised by veratridine. Despite tissue differences,failure to observe nifedipine-induced modifications of ionicfluxes might be due to endogenous adenosine interference (seeabove) and to insufficient drug equilibration time, as these authorsfailed to detect inhibition of ionic fluxes when the time ofincubation was shortened from 6 to 3 min (compared with the 10-min incubation used in the present study). The involvement ofNa+ channel conductance was verified by the blockade ofnifedipine facilitation with tetrodotoxin, as well as by replacingextracellular Na+ either by NMDG or choline. Moreover,facilitation of 45Ca uptake by nifedipine becomes less evidentas one increases the concentration of veratridine (5–20 μM), thussuggesting that both drugs may interact to enhance Na+ channelpermeability. Blockade of Na+/K+-ATPase by nifedipine seemsunlikely, because it attenuated rather than increased intracellular45Ca accumulation caused by ouabain. It has been demonstratedthat under certain conditions (e.g. kinase A-mediated phosphor-ylation, plasma membrane perturbation) the tetrodotoxin-sensi-tive Na+ channel is transformed into one that is promiscuous inrespect to ion selectivity, permittingCa2+ to permeate as readily asNa+ upon cellular depolarisation (Santana et al., 1998). It remainsto be investigated if contribution of high-density Na+ channels toincrease intracellular Ca2+ is a general signalling mechanism inexcitable cells other than cardiomyocytes and whether this can bemodulated by nifedipine. Furthermore, different classes ofdihydropyridine antagonists have differential effects on mem-brane rigidity (Aiello et al., 1998), which may ultimately resultfrom very small molecular changes (reviewed by Triggle, 2003).In support of the nifedipine effect on Na+ conductance beingmediated through an action on membrane lipids, we found thatnifedipine (0.3–10 μM) is able to attenuate hypotonic haemolysisin a similar manner as the neuroleptic drug, chlorpromazine(Costa, Lobo and Correia-de-Sá, unpublished observations).

47J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
Therefore, a plausible mechanism for nifedipine-mediatedfacilitation of 45Ca uptake into depolarised synaptosomes is aconformational stabilization of the Na+ channel to a state alsofavouring Ca2+ permeability. A more complete understanding ofthis mechanism will require additional studies, including analysisat the single channel level.
Hirasawa and Pittman (2003) argue that potential side effectsof nifedipine could be linked to its action on miniatureexcitatory postsynaptic currents due to facilitation of spontane-ous glutamate release through a mechanism that is largelycalcium independent. Whether miniature synaptic activity isable to justify neuronal excitability caused by micromolarnifedipine is a matter of debate (Das et al., 2004). Although thiswas not examined explicitly, nifedipine rise of depolarisation-evoked Ca2+ uptake into nerve terminals of the hippocampus isin keeping with the idea that facilitation of transmitter releasemight be a major contributor for nifedipine CNS side effects.Since synaptosomes may be contaminated by glia particles, itsinterference on the paradoxical effect of nifedipine could not beruled out. On the other hand, nifedipine itself can reduceneuronal cell viability when applied in very high doses (10–100 μM) during certain amounts of time (Takahashi et al.,1999). Therefore, it will require further investigations to clarifywhether the facilitatory effect of nifedipine on synaptictransmission underlies some of its neurologic adverse effects(Testa et al., 1998). Nevertheless, our results suggest that the useof nifedipine to characterize Cav1 (L-type) channels inneuropharmacological studies should be interpreted withcaution given its multiple actions on isolated nerve endings,namely by interfering with Na+ channel conductance.
Acknowledgements
This research was supported, in part by the FCT projects(POCTI/36545/FCB/2000 and UMIB-215/94). We also thankMrs. M. Helena Costa e Silva, Suzete Liça and Belmira Costafor their excellent technical assistance.
References
Adam-Vizi, V., Liget, E., 1986. Calcium uptake of rat brain synaptosomes as afunction of membrane potential under different depolarization conditions.J. Physiol. (Lond.) 372, 363–377.
Aiello, M., Moran, O., Pisciotta, M., Gambale, F., 1998. Interaction betweendihydropyridines and phospholipid bilayers: a molecular dynamics simulation.Eur. Biophys. J. 27, 211–218.
Carvalho, C.A., Coutinho, O.P., Carvalho, A.P., 1986. Effects of Ca2+ channelblockers on Ca2+ translocation across synaptosomal membranes. J. Neurochem.47, 1774–1784.
Costa, J., Lobo,M.G., Correia-de-Sá, P., 2004. Nifedipine facilitates 45Ca uptakeinto rat hippocampal synaptosomes depolarized by veratridine independentlyof calcium channels. Fundam. Clin. Pharmacol. 18 (Suppl. 1) (P12.06.).
Cunha, R.A., Sebastião, A.M., Ribeiro, J.A., 1992. Ecto-5′-nucleotidase isassociated with cholinergic nerve terminals in the hippocampus but not inthe cerebral cortex of the rat. J. Neurochem. 59, 657–666.
Das, P., Bell-Horner, C.L., Huang, R.Q., Raut, A., Gonzales, E.B., Chen, Z.L.,Covey, D.F., Dillon, G.H., 2004. Inhibition of type A GABA receptors byL-type calcium channel blockers. Neuroscience 124, 195–206.
Donnelly-Roberts, D.L., Americ, S.P., Sullivan, J.P., 1995. Functionalmodulation of human “ganglionic-like” neuronal nicotinic acetylcholine
receptors (nAChRs) by L-type calcium channel antagonists. Biochem.Biophys. Res. Commun. 213, 657–662.
Fink, K., Meder, W.P., Clusmann, H., Göthert, M., 2002. Ca2+ entry via P/Q-typeCa2+ channels and the Na+/Ca2+ exchanger in rat and human neocorticalsynaptosomes. Naunyn-Schmiedeberg's Arch. Pharmacol. 366, 458–463.
Frelin, C., Cognard, C., Vigne, P., Lazdunski, M., 1986. Tetrodotoxin-sensitiveand tetrodotoxin-resistant Na+ channels differ in their sensitivity to Cd2+ andZn2+. Eur. J. Pharmacol. 122, 245–250.
Galindo, C.A., Sitges, M., 2004. Dihydropyridines mechanism of action instriatal isolated nerve endings: comparison with ω-agatoxin IVA. Neuro-chem. Res. 29, 659–669.
Galván, E., Sitges, M., 2004. Characterization of the participation of sodiumchannels on the rise in Na+ induced by 4-aminopyridine (4-AP) insynaptosomes. Neurochem. Res. 29, 347–355.
Gonçalves, M.L., Pinto, F., Ribeiro, J.A., 1991. Effect of adenosine on 45Cauptake by electrically stimulated rat brain synaptosomes. J. Neurochem. 56,1769–1773.
Gusovsky, F., McNeal, E.T., Daly, J.W., 1987. Stimulation of phosphoinositidebreakdown in brain synaptoneurosomes by agents that activate sodiuminflux: antagonism by tetrodotoxin, saxitoxin and cadmium. Mol.Pharmacol. 32, 479–487.
Hargreaves, A.C., Gunthorpe, M.J., Taylor, C.W., Lummis, S.C.R., 1996. Directinhibition of 5-hydroxytryptamine 3 receptors by antagonists of L-type Ca2+
channels. Mol. Pharmacol. 50, 1284–1294.Hilgemann, D.W., 1996. Unitary cardiac Na+/Ca2+ exchange current magnitudes
determined from channel-like noise and charge movements of ion transport.Biophys. J. 71, 759–768.
Hille, B., 1992. Ion Channels in Excitable Membranes. Sinauer Associates Inc.,Sunderland, MA, USA.
Hirasawa, M., Pittman, Q.J., 2003. Nifedipine facilitates neurotransmitterrelease independently of calcium channels. Proc. Natl. Acad. Sci. U. S. A.100, 6139–6144.
Iwamoto, T., 2004. Forefront of Na+/Ca2+ exchanger studies: molecularpharmacology of Na+/Ca2+ exchanger inhibitors. J. Pharm. Sci. 96, 27–32.
Janicki, P.K., Siembab, D., Paulo, E.A., Krzascik, P., 1988. Single-dose kineticsof nifedipine in rat plasma and brain. Pharmacology 36, 183–187.
Meder, W., Fink, K., Göthert, M., 1997. Involvement of different calciumchannels in K+- and veratridine-induced increases of cytosolic calciumconcentration in rat cerebral cortical synaptosomes. Naunyn-Schmiede-berg's Arch. Pharmacol. 356, 797–805.
Meir, A., Ginsburg, S., Butkevich, A., Kachalsky, S.G., Kaiserman, I., Ahdut,R., Demirgoren, S., Rahamimoff, R., 1999. Ion channels in presynapticnerve terminals and control of transmitter release. Physiol. Rev. 79,1019–1088.
Miller, J., 1987. Multiple Ca2+ channels and neuronal function. Science 235,46–52.
Morgan, P.F., Tamborska, E., Patel, J., Marangos, P.J., 1987. Interactionsbetween calcium channel compounds and adenosine systems in the brain ofthe rat. Neuropharmacology 26, 1693–1699.
Mulkey, R.M., Zucker, R.S., 1991. Action potentials must admit calcium toevoke transmitter release. Nature 350, 153–155.
Okada, M., Kazunori, M., Iwasaki, K., Fujiwara, M., 1986. Is the augmentationof K+- evoked intrasynaptosomal Ca2+ concentrations due to influx of Ca2+
in rat brain synaptosomes. J. Neurochem. 52, 1837–1842.Ouardouz, M., Zamponi, G.W., Barr, W., Kiedrowski, L., Stys, P.K., 2005.
Protection of ischemic rat spinal cord white matter: dual role of KB-R7943on Na+/Ca2+ exchange and L-type Ca2+ channels. Neuropharmacology 48,566–575.
Pang, D.C., Sperelakis, N., 1984. Uptake of calcium antagonistic drugs intomuscles as related to their lipid solubilities. Biochem. Pharmacol. 33,821–826.
Peterson, G.L., 1977. A simplification of the protein assay method of Lowry et al.,which is more generally applicable. Anal. Biochem. 83, 346–356.
Reuter, H., 1995. Measurements of exocytosis from single presynaptic nerveterminals reveal heterogeneous inhibition by Ca2+- channel blockers.Neuron 14, 773–779.
Romey, G., Lazdunski, M., 1982. Lipid-soluble toxins thought to be specific forNa+ channels block Ca2+ channels in neuronal cells. Nature 297, 79–88.

48 J. Costa et al. / European Journal of Pharmacology 544 (2006) 39–48
Santana, L.F., Gómez, A.M., Lederer, W.J., 1998. Ca2+ flux throughpromiscuous cardiac Na+ channels: slip-mode conductance. Science 279,1027–1033.
Sitges, M., Galindo, C.A., 2005.ω-Agatoxin-TK is a useful tool to study P-typeCa2+ channel-mediated changes in internal Ca2+ and glutamate release indepolarised brain nerve terminals. Neurochem. Int. 46, 53–60.
Skeen, G.A., White, H.S., Twyman, R.E., 1994. The dihydropyridine ni-trendipine reduces N-methyl-D-aspartate evoked currents of rodent corticalneurons through a direct interaction with the NMDA receptor associated ionchannel. J. Pharmacol. Exp. Sci. 271, 30–38.
Smith, J.B., Cragoe, E.J., Smith, L., 1987. Na+/Ca2+ antiport in cultured arterialsmooth muscle cells. J. Biol. Chem. 262, 11988–11994.
Straub, H., Köhling, R., Frieler, A., Grigat, M., Speckmann, E.-J., 2000.Contribution of L-type calcium channels to epileptiform activity inhippocampal and neocortical slices of guinea-pigs. Neuroscience 95, 63–72.
Takahashi, S., Shibata, M., Fukuuchi, Y., 1999. Role of sodium ion influx indepolarisation-induced neuronal cell death by high KCl or veratridine. Eur.J. Pharmacol. 372, 297–304.
Tareilus, E., Breer, H., 1995. Presynaptic calcium channels: pharmacology andregulation. Neurochem. Int. 26, 539–558.
Testa, M.A., Turner, R.R., Sominson, D.C., Krafcik, M.B., Calvo, C., Luque-Otero, M., 1998. Quality of life and calcium channel blockade withnifedipine GITS versus amlodipine patients in Spain. J. Hypertens. 16,1839–1847.
Triggle, D.J., 2003. 1,4-Dihydropyridines as calcium channel ligands andprivileged structures. Cell. Mol. Neurobiol. 23, 293–303.
Wang, G., Ding, S., Yunokuchi, K., 2000. Different distribution of nifedipine-andω-conotoxin GVIA-sensitive Ca2+ channels in rat hippocampal neurons.NeuroReport 11, 2419–2423.
Watano, T., Kimura, J., Morite, T., Nakanishi, H., 1996. A novel antagonist,No. 7943, of the Na+/Ca2+ exchange current in guinea-pig cardiac ven-tricular cells. Br. J. Pharmacol. 119, 555–563.
Wonnacott, S., 1997. Presynaptic nicotinic receptors. Trends Neurosci. 20,92–98.
Zernig, G., 1990. Widening potential for Ca2+ antagonists: non-L-type Ca2+
channel interaction. Trends Pharmacol. Sci. 11, 38–44.





























