Embed Size (px)
Citation preview

Nanoscale
PAPER
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article OnlineView Journal | View Issue
Department of Chemistry, Indian Institute
India. E-mail: [email protected].
26581102; Tel: +91-11-26591379
† Electronic supplementary informa10.1039/c3nr06586c
Cite this: Nanoscale, 2014, 6, 4588
Received 12th December 2013Accepted 4th February 2014
DOI: 10.1039/c3nr06586c
www.rsc.org/nanoscale
4588 | Nanoscale, 2014, 6, 4588–4597
Palladium–phosphorus/sulfur nanoparticles (NPs)decorated on graphene oxide: synthesis using thesame precursor for NPs and catalytic applications inSuzuki–Miyaura coupling†
Hemant Joshi, Kamal Nayan Sharma, Alpesh K. Sharma and Ajai Kumar Singh*
PdP2 and Pd4S nanoparticles (NPs) (size: �2–6 and 9–15 nm respectively) have been prepared for the first
time from a single source precursor complex [Pd(L)Cl2] (1) by its one pot thermolysis at 200 �C in TOP and
OA/ODE (1 : 1) respectively. These NPs were stirred with graphene oxide (GO) at room temperature to
prepare NP composites, GO–PdP2 and GO–Pd4S. The GO–PdP2 NPs have been synthesized for the first
time. The thioether ligand L prepared by reaction of 1,3-dibromo-2-propanol with the in situ generated
PhSNa reacts with [PdCl2(CH3CN)2] in CH3CN at 70 �C resulting in 1. The L and 1 have been
characterized by 1H and 13C{1H} NMR and HR-MS. The single crystal structure of 1 determined by X-ray
diffraction reveals nearly square planar geometry around the Pd metal centre. The catalytic activities of
two palladium nano-phases having phosphorus and sulphur respectively as a co-constituent for Suzuki–
Miyaura coupling have been found to be exceptionally different, as PdP2 nanoparticles (NPs) grafted on
graphene oxide (GO–PdP2) are significantly more efficient than Pd4S NPs grafted on GO. Without
grafting PdP2 and Pd4S both have low efficiency. This is the first report comparing the influence of P and
S on the catalytic activity of Pd NPs. TEM, SEM-EDX and powder-XRD have been used to authenticate all
NPs. The GO–PdP2 NPs have been found to be efficient catalysts for Suzuki–Miyaura coupling reactions
(yield up to 96% in 30 min) at room temperature to 80 �C. Their recyclability has been found up to 6
cycles. In contrast, GO–Pd4S NPs are little active in comparison with GO–PdP2 NPs. The size of NPs and
their distribution on GO appear to be key factors affecting the catalytic efficiency of the composite NPs.
Leaching of Pd from GO–PdP2 NPs contributes significantly to the catalysis as evidenced by the three
phase test, hot-filtration and recycling experiments. The catalysis is almost homogeneous.
Introduction
Nano-catalysts, nanoparticles (NPs) stabilized with diverseagents and implanted or dispersed on a support, are ofcontinuous attraction and covered extensively in several reviewarticles.1 Nano-catalysts have more catalytically active sitesresulting from enhanced surface to volume ratio and are moreefficient generally in comparison with traditional catalysts.Sometimes in the course of efficient catalysis NPs are in situgenerated2 (e.g. Pd chalcogenide2). Among the nano-catalysts,palladium and palladium based NPs3 have acquired an impor-tant place in the last decade. Various types of NPs of pure Pdhave been more studied for catalysis than those having otherelements. Highly branched palladium nanostructures havebeen reported for efficient hydrogenation of nitrobenzene to
of Technology Delhi, New Delhi 110016,
in; [email protected]; Fax: +91-11-
tion (ESI) available. See DOI:
aniline.3c Palladium hollow spheres synthesized using silicaspheres as templates have been reported as recyclable hetero-geneous catalysts for Suzuki coupling.3a The anchoring of theseNPs on various supports improves many times overall quality ofthe catalyst. Shape-controlled Pd NPs supported on powderedalumina have been designed for selective hydrogenation ofbutadiene to butene.3b The use of pre-formed palladium chal-cogenide NPs immobilized on a solid surface such as silica,alumina, polymer or graphene oxide (GO), as a heterogeneouscatalyst, for coupling reactions has been little explored. Amongthese supports GO4,5 emerged as extremely versatile carbonmaterial modifying favorably properties of anchored moleculeshas been envisaged very promising. The current interest in GOis because it is a low cost alternative to carbon nanotubes,6
important as hydrogen storage7 and semiconducting material.8
The structure and composition9 of nonstoichiometric andhygroscopic GO have contributed signicantly to the interest inthis material. GO prepared easily by controlled chemical orelectrochemical oxidation of graphite via its nitrate or hydrogensulfate salt10 forms intercalation compounds with cationic
This journal is © The Royal Society of Chemistry 2014

Paper Nanoscale
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
species via carboxylic and hydroxyl groups present on itssurface.11 The chemical, mechanical and thermal stability of GOalong with its high surface area (desirable for two-dimensionalsupport layers for NPs used as heterogeneous catalysts) havecontributed signicantly to attraction for it.12 However, thereare only a few examples of GO-based materials used in catal-ysis.13 Mastalir et al. have reported high catalytic activity andselectivity of the GO–nano-Pd system in liquid-phase hydroge-nation of alkynes.13b,c Ion exchange of GO with a Pd complex andits subsequent reduction to nano-sized crystallites by H2 haveresulted in a catalyst that has surpassed the activity ofcommercially available ‘supported Pd catalysts’ such as Pd onactivated carbon (Pd/C) and Pd graphimet.14 The role of GO asan active hydrogen donor in palladium catalyzed Ullmannreaction has been reported.15 The Pd NPs immobilized onpartially reduced GO have been reported for catalysis of Suzuki–Miyaura coupling reactions.16 Recently Pd17Se15 nanoparticlesgraed on graphene oxide have been reported for roomtemperature C–O coupling of phenols with aryl halides.17 Theinterest in nanocrystals of palladium chalcogenides18 is growingfast due to immense possibilities of their applications incatalysis2 and existing extensive applications as low resistanceohmic contacts of semiconducting electronic devices.18b
However, straight synthesis of high quality palladium chalco-genide NPs is little explored in comparison with those of othermetal chalcogenide semiconducting materials.19 The knownmethods of their preparation suffer from one or more of thefollowing limitations. (i) Size of NPs is in a wide range. (ii) Twosources are needed for palladium and chalcogen. (iii) Sophis-ticated physical techniques have to be invoked. (iv) Hightemperature is essential. The catalysts dependent on suchprocedures for generation of palladium–chalcogenide NPswould only be academic curios. The preparation of NPs bythermolysis of a single source precursor (SSP) appears to be apractical solution. SSPs may be used at moderate temperaturewith easy work-up and with them the issues related to air andmoisture instability of precursors (at higher temperature)become insignicant. The toxicity risks and contamination arereduced. Further in SSP routes there is an intrinsic control onreactivity and stoichiometry.20 Ligand designing (not alwayseasy) is another possible tool to control composition,morphology and the level of impurities in the nal product.Thermal decomposition of complexes containing Pd–chalcogenbond(s) into nanocrystals of palladium chalcogenides appearsto be easy, as syntheses of PdTe,19 Pd17Se15,5 Pd4Se and Pd7Se4via an appropriate Pd complex as a SSP have been reported.21
Encouraged by the catalytic potential of Pd17Se15 nanoparticlesgraed on graphene oxide, it was thought worthwhile to designGO–Pd–chalcogenide based other catalytic systems and exploretheir activity. The target for this purpose was Pd4S but surpris-ingly we obtained PdP2 in the adopted SSP route depending onthe solvent. To the best of our knowledge, there is not a singlereport on the synthesis of two types of NPs from the same SSPjust by changing the solvent of thermolysis. A further compositeof Go with any palladium phosphide is not known and thepresent one is the rst example. Herein we report for the rsttime designing of GOs anchored with PdP2 and Pd4S and
This journal is © The Royal Society of Chemistry 2014
catalysis of Suzuki reactions with both of them. Pd–NP catalystsfor C–C coupling reactions such as Mizoroki–Heck22 andSuzuki–Miyaura reactions23 have been reported.24,25 Pd NPsstabilized in ionic liquids,26 polymers,27 organic–inorganicuorinated hybrid materials,28 and glass–polymer composites29
and anchored on supports viz. carbon,30 silica,31 or alumina32
are known to be efficient catalysts for C–C coupling reactions.
ExperimentalPhysical measurement
A Bruker Spectrospin DPX 300 NMR spectrometer was used torecord 1H and 13C{1H} NMR spectra at 300.13 and 75.47 MHzrespectively. The chemical shis (ppm) are reported relativeto internal standard (tetramethylsilane) in both the cases. Allreactions were carried out at room temperature to 80 �C inglassware dried in an oven. X-ray diffraction data for crystalsof 1 were collected on a BRUKER AXS SMART-APEX CCDdiffractometer using Mo-Ka ¼ 0.71073 A radiation. Frameswere collected at T ¼ 298 K by u, 4, and 2q-rotations with afull quadrant data collection strategy (four domains eachwith 600 frames) at 10 s per frame with SMART. Themeasured intensities were reduced to F2 and corrected forabsorption with SADABS.33 Structure solution, renement,and data output were carried out with the SHELXTL packageby direct methods.34 Non-hydrogen atoms were renedanisotropically. For scanning electron microscopy (SEM)studies, a Carl ZEISS EVO5O instrument was used. Thesample was mounted on a circular metallic sample holderwith sticky carbon tape. The associated EDX system ModelQuanTax 200 based on the SDD technology and providing anenergy resolution of 127 eV at Mn Ka was used for estimatingelemental compositions. Powder X-ray diffraction patternswere recorded on a Bruker D8 advance diffractometer usingNi-ltered Cu-Ka radiation, a scan speed of 1 s and a scanstep of 0.05�. A transmission electron microscope, JEOL JEM200CX operated at 200 kV, was used for TEM studies. Thespecimens for such studies were prepared by dispersing thepowdered sample in ethanol by ultrasonic treatment. Fewdrops of the resulting solution were put on a porous carbonlm supported on a copper grid and dried in air. Estimationof palladium in NPs was carried out with an AA700 seriesatomic absorption spectrometer (Lab India). The surface areawas determined using Nova 2000e (Quantachrome Corp.)equipment by the Brunauer–Emmett–Teller (BET) method.The sample of nanoparticles was degassed at 120 �C for 4 hprior to the surface area measurements.
Chemicals and reagents
Trioctylphosphine (TOP), octadecane (ODE), oleic acid (OA),PdCl2, graphite akes, thiophenol, 1,3-dibromo-2-propanol andsodium borohydride procured from Sigma-Aldrich (USA) wereused as received. KMnO4, NaNO3, and H2O2 were procured fromFisher Scientic International, Inc. All the solvents (AR grade)viz. toluene, acetone, chloroform, acetonitrile and ethanol weredried and distilled before use by known standard procedures.35
Nanoscale, 2014, 6, 4588–4597 | 4589

Nanoscale Paper
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
Synthesis of 1,3-bis(phenylsulfanyl) propan-2-ol (L)
Thiophenol (0.441 g, 4.0 mmol) was added to sodium hydroxide(0.160 g, 4.0 mmol) dissolved in 30 mL of EtOH and reuxed for1 h. 1,3-Dibromo-2-propanol (0.436 g, 2 mmol) dissolved in 20mL of EtOH was added to it. The reaction mixture was furtherreuxed for 5 h. Thereaer, it was stirred overnight at roomtemperature and poured into cold water (30 mL). The L wasextracted with chloroform (4 � 25 mL). The extract was washedwith water (3 � 40 mL) and dried over anhydrous sodiumsulfate. The solvent was evaporated off under reduced pressureon a rotary evaporator resulting in L as a light yellow oil. Yield(0.47 g, 85%). 1H NMR (CDCl3, 25 �C, TMS); (d, ppm): 2.80 (s,1H, OH), 3.00–3.22 (m, 4H, CH2S), 3.80–3.82 (m, 1H, CHOH),7.16–7.23 (m, 6H, m-Ar, p-Ar), 7.25–7.32 (m, 4H, o-Ar); 13C{1H}NMR (CDCl3, 25 �C, TMS); (d, ppm): 39.8 (C5), 68.1 (C6), 126.5(C1), 128.9 (C2), 129.7 (C3), 135.0 (C4). HR-MS [M + Na]+ (m/z)299.0545; calc. value for C15H16NaOS2; 299.0535 (d: –3.3 ppm).
Synthesis of complex 1
[PdCl2(CH3CN)2] (0.129 g, 0.5 mmol) was added to a solution of L(0.227 g, 0.5 mmol) made in CH3CN (5 mL) and the mixture waskept on stirring at 70 �C. The reaction mixture was stirred for afurther 5 h, concentrated with a rotary evaporator and mixedwith diethyl ether. The precipitated orange coloured compoundwas ltered, washed with diethyl ether (10 mL) and dried invacuo to give [Pd(L)Cl2] (1) as a yellow solid. Single crystals of 1were grown by slow evaporation of its solution in an acetonitrileand methanol mixture (3 : 1). Yield (0.19 g, 84%). 1H NMR(DMSO-d6, 25 �C, TMS); (d, ppm): 3.02–3.19 (m, 4H, CH2S), 3.75(s, 1H, CHOH), 7.18–7.33 (m, 10H, ArH); 13C{1H} NMR (DMSO-d6, 25 �C, TMS); (d, ppm): 39.2 (C5), 66.3 (C6), 126.3 (C1), 128.7(C2), 129.5 (C3), 136.5 (C4). HR-MS [M + Na]+ (m/z) 474.8945; calc.value for C15H16Cl2NaOPdS2; 474.8946 (d: 0.3 ppm).
Synthesis of graphene oxide (GO)
GO was synthesized by the modied Hummers' procedure.10c 500mg of graphite powder and 2.0 g of sodium nitrate (NaNO3) wereput into cold (temp. <5 �C) concentrated H2SO4 (18 mL, 98%).The mixture was stirred continuously for 1 h keeping itstemperature �5 �C by cooling it in an ice bath. Potassiumpermanganate (3 g) was added gradually and the reactioncontinued for another 2 h keeping the temperature below 5 �C.Thereaer the mixture was heated for 30 min at 35 �C and 40 mLof deionised (DI) water was added slowly. The temperature of themixture was increased to 100 �C and kept at that level for 15 min.The mixture was cooled to room temperature and diluted with 70mL of DI water. Thereaer 10 mL of H2O2 (35%) was added. Thecolour of the suspension changed to bright yellow. The suspen-sion was ltered and washed successively with 400 mL of 5%HCltwice and 70 mL of DI water thrice. Finally, the precipitate wasdried in a vacuumdesiccator for at least 5 days before further use.
Synthesis of PdP2 nanoparticles
The slurry of 0.5 mmol of complex 1 made in 10 mmol of tri-octylphosphine (TOP) was taken in a three necked round bottom
4590 | Nanoscale, 2014, 6, 4588–4597
(r.b.) ask (100 mL) and heated to 100 �C to remove water andoxygen. The resulting nearly homogeneous brown solution wasrst heated to 200 �C under a N2 atmosphere and kept at thesame temperature for 60 min, affording a dark colloidal solu-tion. The solution was cooled and NPs of the PdP2 phase wereprecipitated by adding excess acetone. The NPs were centrifuged(5000 rpm), washed with acetone and dried in air at 60 �C.
Synthesis of Pd4S nanoparticles
A slurry of 0.5 mmol of complex 1 prepared in a 1 : 1 (v/v)OA–ODE mixture was taken in a three necked r.b. ask(100 mL). It was heated to 100 �C to remove water and oxygen.The resulting almost homogeneous brown solution was heatedto 200 �C under N2 and kept at that temperature for 60 min,affording a dark colloidal solution. The solution was cooled andNPs of the Pd4S phase were precipitated by adding excessacetone. They were centrifuged, washed twice with acetone(30 mL) and dried in air at 60 �C.
Synthesis of graphene oxide graed with PdP2 and Pd4Snanoparticles
The GO (100 mg) was completely dispersed in 20 mL of DI waterusing sonication. 40 mg of PdP2/Pd4S was dispersed in 20 mL oftoluene by sonication and added to the suspension of GO. Themixture was stirred for 24 h at room temperature. The precipi-tate was separated, washed with acetone and dried in a vacuumdesiccator. The composite so-obtained was labeled as GO–PdP2/GO–Pd4S NPs (Scheme 3).
Procedure for the Suzuki–Miyaura coupling reaction
An oven-dried ask was charged with aryl bromide (1.0 mmol),phenylboronic acid (1.3 mmol), K2CO3 (2.0 mmol), EtOH(2.0 mL), H2O (2.0 mL) and GO–PdP2 or GO–Pd4S NPs. The askwas placed on an oil bath at room temperature to 80 �C with anair condenser and the reactionmixture was stirred. The reactionmonitored by TLC was carried out until the maximum conver-sion of aryl halide to the product occurred. The mixture wasextracted with diethyl ether (2 � 20 mL), washed with water anddried over anhydrous Na2SO4. The solvent of the extract wascompletely removed with a rotary evaporator to obtain theproduct, which was further puried by column chromatographyon silica gel. All coupling products were authenticated by 1Hand 13C{1H} NMR spectra (Scheme 4).
Three-phase test
The 4-bromobenzoic acid-immobilized (as amide) silica (0.20 g)prepared by a standard procedure (details in the ESI†), phe-nylboronic acid (0.36 g, 3 mmol), 4-bromoacetophenone (0.20 g,1 mmol), K2CO3 (0.56 g, 4 mmol) and catalyst (0.5 mol%) weretaken in an EtOH (8mL) and water (4 mL)mixture. It was heatedat 80 �C for 12 h and thereaer cooled to room temperature andltered through a G-4 crucible. The residue was washed with20 mL of H2O followed by diethyl ether (50 mL). The ltrate andwashings were collected together and mixed with 50 mL ofwater. The resulting mixture was extracted with diethyl ether
This journal is © The Royal Society of Chemistry 2014

Paper Nanoscale
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
(50 mL). The solvent of the extract was evaporated off on a rotaryevaporator, and the residue was subjected to 1H NMR. The silicabased solid residue (le on ltration) was hydrolyzed with KOH(1.68 g dissolved in 10 mL of EtOH + 5 mL of H2O) at 90 �C for 3days. The resulting solution was neutralized with aqueous 20%(v/v) HCl, extracted with dichloromethane followed by ethylacetate. The solvent of the combined extract was evaporated off,and the resulting residue was analyzed by 1H NMR.
Fig. 1 Molecular structure of 1 with ellipsoids at the 30% probabilitylevel. Bond length (A): Pd(1)–S(1) 2.277(15), Pd(1)–S(2) 2.279(14), Pd(1)–Cl(1) 2.317(16), Pd(1)–Cl(2) 2.310(15); bond angle (�): S(1)–Pd(1)–S(2)100.41(5), Cl(1)–Pd(1)–S(1) 84.08(6), Cl(1)–Pd(1)–S(2) 175.22(5), Cl(2)–Pd(1)–S(1) 174.40(6), Cl(2)–Pd(1)–S(2) 84.12(6).
Results and discussion
The syntheses of 1,3-bis(phenylsulfanyl)propan-2-ol (L) and itsPd(II)-complex 1 are summarized in Scheme 1. The ligand wasfound to be soluble in common organic solvents. The solubilityof complex 1 was found to be good in DMF, DMSO, PhCH3,CH3CN, CHCl3 and CH2Cl2, moderate in THF and almostnegligible in CH3OH, diethyl ether and hexane.
NMR and mass spectra
The air stable ligand L and Pd-complex 1 were characterizedusing 1H and 13C{1H} NMR spectra and HR-MS. The 1H and 13C{1H} NMR spectra (see ESI, Fig. S1–S4†) of L and complex 1 havebeen found to be consistent with their molecular structures(Scheme 1). In 1H and 13C{1H} NMR spectra of complex 1 signalsof various protons and carbon atoms appear at somewhat lowerfrequencies relative to those of the free ligand which coordi-nates with palladium in a bidentate (S, S) mode. In the massspectrum of ligand L the peak appearing at 299.0545 may beascribed to the [M + Na] cation (see ESI, Fig. S5†). The massspectrum of complex 1 also shows a [M + Na] cation peak at474.89457 (see ESI, Fig. S6†).
Fig. 2 C–H/Cl interactions in 1.
Crystal structure
The single crystal structure of complex 1 has been solved. Thedetails of crystal data and renements are given in Table S1 ofESI.† Themolecular structure of 1 is given in Fig. 1 with selectedbond lengths and angles (more values are given in Table S2 ofESI†). The geometry around Pd in complex 1 is nearly squareplanar and the ligand is coordinated with Pd in a bidentate (S, S)mode forming a six membered chelate ring. The Pd–S bondlength of complex 1 is 2.277(15) A, consistent with previouslyreported values36 2.286(12) A for the [PdCl(O�, N, S) ligand]. ThePd–Cl bond lengths in 1 are also normal. Intermolecular C–H/Cl interactions are shown in Fig. 2 and values of such distancesare given in ESI, Table S3.†
Scheme 1 Synthesis of L and complex 1.
This journal is © The Royal Society of Chemistry 2014
Characterization of the GO–PdP2 and GO–Pd4S catalysts
The GO was characterized by TEM (Fig. 4) and PXRD (see ESI,Fig. S7†). The diffraction pattern of GO has a peak centered at2q ¼ 11.8�, corresponding to the [001] interlayer spacing of7.46 A.37 The PdP2 and Pd4S NPs and GO–PdP2 and GO–Pd4SNPs were characterized by TEM, SEM-EDX and powder XRD.The powder X-ray diffraction (PXRD) patterns of the presentlysynthesized NPs of PdP2 and GO–PdP2 are shown in the ESI(Fig. S8 and S9†). The comparison of the present PXRD patternwith those of standard phases suggests that the phase of thepresent PdP2 NPs has a monoclinic structure (JCPDS 77-1421).The morphologies of the present PdP2 and GO–PdP2 NPs wereexamined by TEM recorded at room temperature. The nano-structures (Fig. 3 and 5) obtained from 1 (in TOP) are spherical.The powder X-ray diffraction (PXRD) patterns reveal that Pd4Sand GO–Pd4S have a tetragonal structure (JCPDS 73-1387) andthe sharp nature of peaks indicates that their size is somewhatlarger compared to PdP2 nanoparticles (see ESI, Fig. S10 andS11†). In PXRD patterns of GO–PdP2 and GO–Pd4S NPs
Nanoscale, 2014, 6, 4588–4597 | 4591
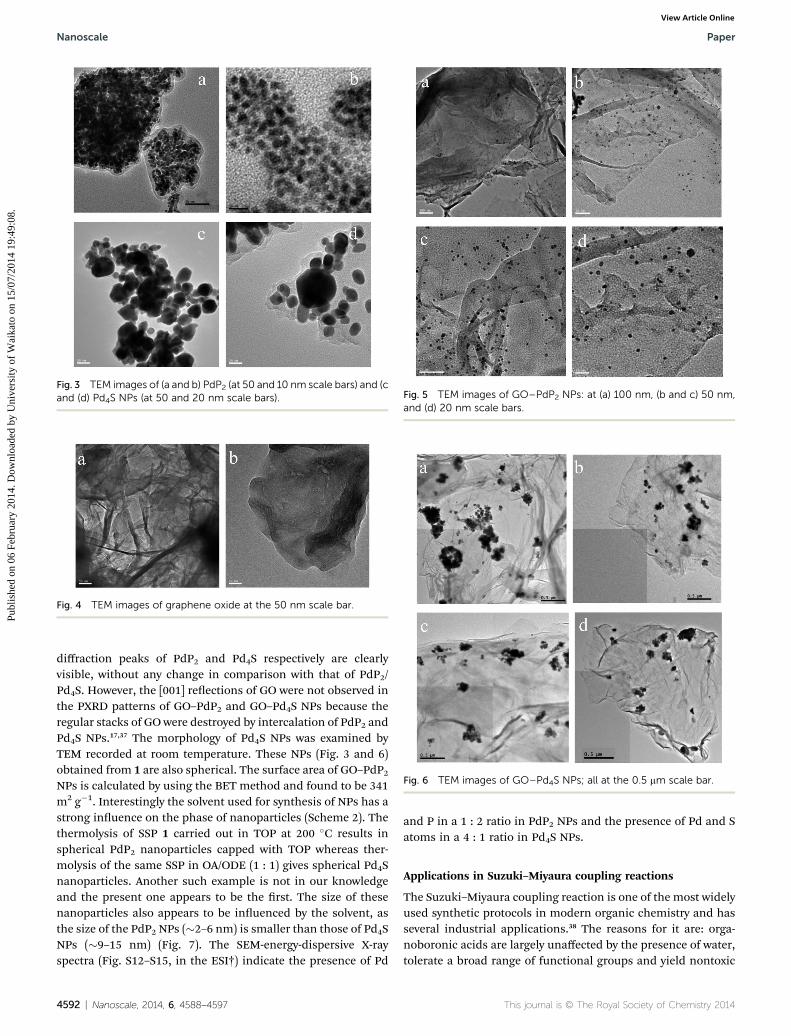
Fig. 3 TEM images of (a and b) PdP2 (at 50 and 10 nm scale bars) and (cand (d) Pd4S NPs (at 50 and 20 nm scale bars).
Fig. 4 TEM images of graphene oxide at the 50 nm scale bar.
Fig. 5 TEM images of GO–PdP2 NPs: at (a) 100 nm, (b and c) 50 nm,and (d) 20 nm scale bars.
Fig. 6 TEM images of GO–Pd4S NPs; all at the 0.5 mm scale bar.
Nanoscale Paper
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
diffraction peaks of PdP2 and Pd4S respectively are clearlyvisible, without any change in comparison with that of PdP2/Pd4S. However, the [001] reections of GO were not observed inthe PXRD patterns of GO–PdP2 and GO–Pd4S NPs because theregular stacks of GO were destroyed by intercalation of PdP2 andPd4S NPs.17,37 The morphology of Pd4S NPs was examined byTEM recorded at room temperature. These NPs (Fig. 3 and 6)obtained from 1 are also spherical. The surface area of GO–PdP2NPs is calculated by using the BET method and found to be 341m2 g�1. Interestingly the solvent used for synthesis of NPs has astrong inuence on the phase of nanoparticles (Scheme 2). Thethermolysis of SSP 1 carried out in TOP at 200 �C results inspherical PdP2 nanoparticles capped with TOP whereas ther-molysis of the same SSP in OA/ODE (1 : 1) gives spherical Pd4Snanoparticles. Another such example is not in our knowledgeand the present one appears to be the rst. The size of thesenanoparticles also appears to be inuenced by the solvent, asthe size of the PdP2 NPs (�2–6 nm) is smaller than those of Pd4SNPs (�9–15 nm) (Fig. 7). The SEM-energy-dispersive X-rayspectra (Fig. S12–S15, in the ESI†) indicate the presence of Pd
4592 | Nanoscale, 2014, 6, 4588–4597
and P in a 1 : 2 ratio in PdP2 NPs and the presence of Pd and Satoms in a 4 : 1 ratio in Pd4S NPs.
Applications in Suzuki–Miyaura coupling reactions
The Suzuki–Miyaura coupling reaction is one of the most widelyused synthetic protocols in modern organic chemistry and hasseveral industrial applications.38 The reasons for it are: orga-noboronic acids are largely unaffected by the presence of water,tolerate a broad range of functional groups and yield nontoxic
This journal is © The Royal Society of Chemistry 2014
Scheme 2 Synthesis of PdP2 and Pd4S NPs.
Scheme 3 Synthesis of GO–PdP2 and GO–Pd4S NPs.
Scheme 4 Suzuki–Miyaura coupling reaction.
Fig. 7 HRTEM images of (a) GO–PdP2 and (b) GO–Pd4S NPs at (a) 10nm and (b) 20 nm scale bars.
Table 1 Effect of different Pd loadings on reaction with GO–PdP2a
Pd mol% 1.0 0.5 0.25 0.05
Conversion (%) 98 96 87 65
a Reaction conditions: aryl or heteroaryl halide (1 mmol), boronic acid(1.3 equiv.), K2CO3 (2 equiv.), EtOH (2 mL), H2O (2 mL), temp. 80 �C.
Paper Nanoscale
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
byproducts. In recent years, for the catalysis of this reactiondone homogeneously, the research has also been focused on theheterogeneous catalytic systems.39,40 These systems show thesame or even higher activities than their homogeneous coun-terparts and do not require working under an inert atmosphere.Further they are very easy to recover, though the activities oendrop in recycling experiments.40,41
The GO–PdP2 and GO–Pd4S NPs were investigated for theSuzuki–Miyaura coupling reaction. The single source precursorcomplex 1 and these two composites were rst screened as a
This journal is © The Royal Society of Chemistry 2014
catalyst. Complex 1 is dissolved in the medium used for cataly-sis while GO–PdP2 and GO–Pd4S remain suspended in thesolvent when stirred but settle on keeping aside. For screeningpurposes catalytic activities of complex 1 and both the GOanchored NPs were studied at room temperature for Suzuki–Miyaura coupling of the 4-bromobenzaldehyde substratekeeping Pd loading at 0.5 mol%. The order of activity wasGO–PdP2 [ GO–Pd4S > complex 1. Aer 30 min, there wascomplete conversion with catalyst GO–PdP2, only 58% withGO–Pd4S and poor 13% with SSP complex 1. At room temper-ature, with catalyst GO–Pd4S 78% conversion reached aer 5 hand complex 1 showed only 27% yield aer 24 h stirring. Thus 1as a catalyst displayed the lowest activity. In comparison with4-bromobenzaldehyde, Suzuki–Miyaura coupling of deactivatedsubstrate, 4-bromoanisol, using a Pd loading of 1.0 mol% hasdemonstrated the sharp difference between activities as acatalyst of complex 1 and two NPs. The yield of the coupledproduct aer 4 h in the catalysis with GO–PdP2 and GO–Pd4Swas 88 and 56% respectively. It was only 7% with 1 at roomtemperature and for a reaction time of 4 h. The catalytic activityof PdP2/Pd4S is signicantly improved on composite formationwith GO. For pure PdP2 and Pd4S both the yields in 1 h forcoupling of the 4-bromobenzaldehyde substrate at 80 �C with0.5 mol% Pd loading are 48% and 29% respectively. Thusdetailed investigations have been made on catalysis of Suzukicoupling with GO–PdP2.
The effect of different Pd loadings on Suzuki–Miyaura C–Ccoupling reaction of 4-bromobenzaldehyde catalyzed with GO–PdP2 has been studied. The results are shown in Table 1. At80 �C with a Pd loading of 1.0 mol% the reaction was completedin 15 min with a yield of 98% of coupled product. On decreasingthe Pd loading to 0.5 mol% conversion was 96% in 30 min. Ondecreasing the loading of Pd to 0.25 mol%, the conversiondropped to 87% for 30 min. With 0.05% Pd at 80 �C, theconversion reached 65% of aryl bromide in 2 h. Aer completereaction using 1 mol% Pd, the catalyst was centrifuged and thesolution was analyzed for Pd by ame AAS. It showed theconcentration of leached Pd as 2.4 ppm. Perhaps leached Pdfrom catalyst GO–PdP2 also contributes to catalysis.
The scope of substrate GO–PdP2 and GO–Pd4S catalysedSuzuki–Miyaura reaction was investigated (Table 2). The reac-tions proceed well with a wide range of aryl bromides contain-ing electron withdrawing groups (1–6), and also give a very goodyield with those having electron-donating groups (9 and 10).
The recyclability of the catalyst GO–PdP2 was studied in thecoupling reaction of 4-bromobenzaldehyde with phenylboronicacid at 80 �C catalyzed with 0.5 mol% of Pd. The completion ofreaction was accomplished in 30 min in the rst two runs(Fig. 8). The activity moderately dropped in the next runs but
Nanoscale, 2014, 6, 4588–4597 | 4593
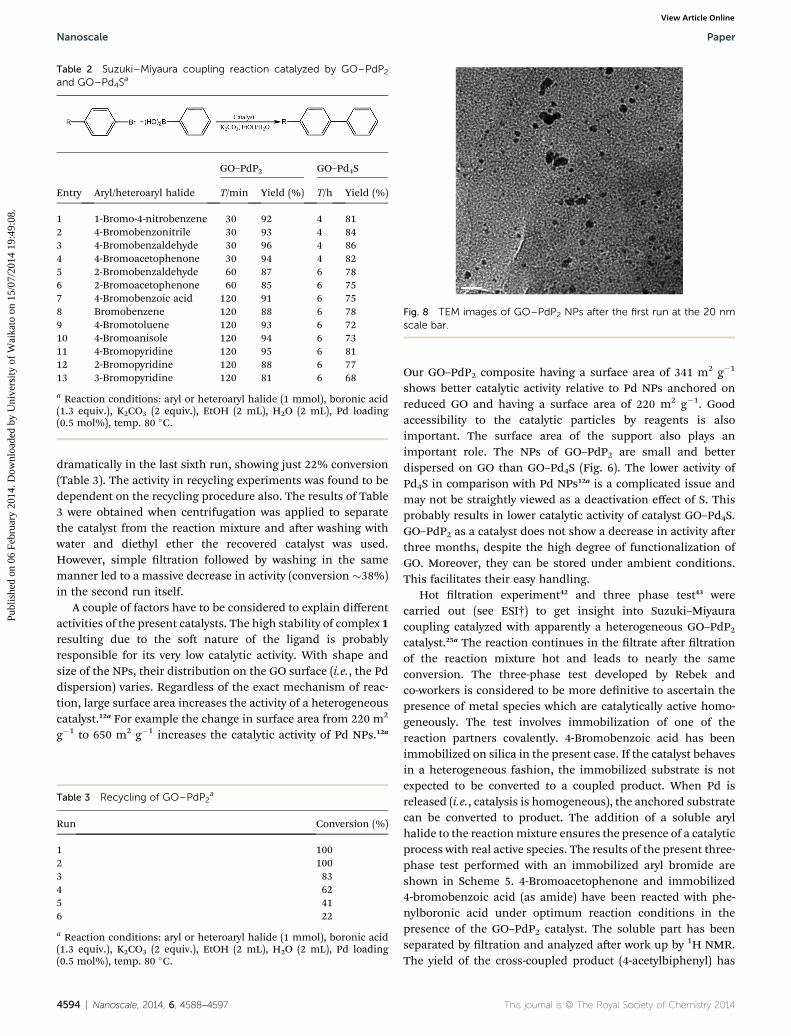
Table 2 Suzuki–Miyaura coupling reaction catalyzed by GO–PdP2and GO–Pd4S
a
Entry Aryl/heteroaryl halide
GO–PdP2 GO–Pd4S
T/min Yield (%) T/h Yield (%)
1 1-Bromo-4-nitrobenzene 30 92 4 812 4-Bromobenzonitrile 30 93 4 843 4-Bromobenzaldehyde 30 96 4 864 4-Bromoacetophenone 30 94 4 825 2-Bromobenzaldehyde 60 87 6 786 2-Bromoacetophenone 60 85 6 757 4-Bromobenzoic acid 120 91 6 758 Bromobenzene 120 88 6 789 4-Bromotoluene 120 93 6 7210 4-Bromoanisole 120 94 6 7311 4-Bromopyridine 120 95 6 8112 2-Bromopyridine 120 88 6 7713 3-Bromopyridine 120 81 6 68
a Reaction conditions: aryl or heteroaryl halide (1 mmol), boronic acid(1.3 equiv.), K2CO3 (2 equiv.), EtOH (2 mL), H2O (2 mL), Pd loading(0.5 mol%), temp. 80 �C.
Fig. 8 TEM images of GO–PdP2 NPs after the first run at the 20 nmscale bar.
Nanoscale Paper
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
dramatically in the last sixth run, showing just 22% conversion(Table 3). The activity in recycling experiments was found to bedependent on the recycling procedure also. The results of Table3 were obtained when centrifugation was applied to separatethe catalyst from the reaction mixture and aer washing withwater and diethyl ether the recovered catalyst was used.However, simple ltration followed by washing in the samemanner led to a massive decrease in activity (conversion �38%)in the second run itself.
A couple of factors have to be considered to explain differentactivities of the present catalysts. The high stability of complex 1resulting due to the so nature of the ligand is probablyresponsible for its very low catalytic activity. With shape andsize of the NPs, their distribution on the GO surface (i.e., the Pddispersion) varies. Regardless of the exact mechanism of reac-tion, large surface area increases the activity of a heterogeneouscatalyst.12a For example the change in surface area from 220 m2
g�1 to 650 m2 g�1 increases the catalytic activity of Pd NPs.12a
Table 3 Recycling of GO–PdP2a
Run Conversion (%)
1 1002 1003 834 625 416 22
a Reaction conditions: aryl or heteroaryl halide (1 mmol), boronic acid(1.3 equiv.), K2CO3 (2 equiv.), EtOH (2 mL), H2O (2 mL), Pd loading(0.5 mol%), temp. 80 �C.
4594 | Nanoscale, 2014, 6, 4588–4597
Our GO–PdP2 composite having a surface area of 341 m2 g�1
shows better catalytic activity relative to Pd NPs anchored onreduced GO and having a surface area of 220 m2 g�1. Goodaccessibility to the catalytic particles by reagents is alsoimportant. The surface area of the support also plays animportant role. The NPs of GO–PdP2 are small and betterdispersed on GO than GO–Pd4S (Fig. 6). The lower activity ofPd4S in comparison with Pd NPs12a is a complicated issue andmay not be straightly viewed as a deactivation effect of S. Thisprobably results in lower catalytic activity of catalyst GO–Pd4S.GO–PdP2 as a catalyst does not show a decrease in activity aerthree months, despite the high degree of functionalization ofGO. Moreover, they can be stored under ambient conditions.This facilitates their easy handling.
Hot ltration experiment42 and three phase test43 werecarried out (see ESI†) to get insight into Suzuki–Miyauracoupling catalyzed with apparently a heterogeneous GO–PdP2catalyst.25a The reaction continues in the ltrate aer ltrationof the reaction mixture hot and leads to nearly the sameconversion. The three-phase test developed by Rebek andco-workers is considered to be more denitive to ascertain thepresence of metal species which are catalytically active homo-geneously. The test involves immobilization of one of thereaction partners covalently. 4-Bromobenzoic acid has beenimmobilized on silica in the present case. If the catalyst behavesin a heterogeneous fashion, the immobilized substrate is notexpected to be converted to a coupled product. When Pd isreleased (i.e., catalysis is homogeneous), the anchored substratecan be converted to product. The addition of a soluble arylhalide to the reactionmixture ensures the presence of a catalyticprocess with real active species. The results of the present three-phase test performed with an immobilized aryl bromide areshown in Scheme 5. 4-Bromoacetophenone and immobilized4-bromobenzoic acid (as amide) have been reacted with phe-nylboronic acid under optimum reaction conditions in thepresence of the GO–PdP2 catalyst. The soluble part has beenseparated by ltration and analyzed aer work up by 1H NMR.The yield of the cross-coupled product (4-acetylbiphenyl) has
This journal is © The Royal Society of Chemistry 2014

Scheme 5 Two-phase test for Suzuki–Miyaura coupling.
Paper Nanoscale
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
been found to be �94%. The solid phase has been hydrolyzed,and the resulting products aer work up have been analyzed by1H NMR. Of the immobilized 4-bromobenzoic acid (as amide),�91% has been converted to the cross-coupled product(biphenyl-4-carboxylic acid).
These results suggest that catalysis progresses via a solublespecies. Most probably it is Pd(0) leached from the GO–PdP2catalyst. The recycled catalyst was efficient if centrifugation wasapplied for separation. This implies that probably the leachedPd(0) species are re-deposited on the support. This is in accor-dance with the low Pd level, shown by ame AAS studies onsupernatant liquid aer centrifugation. Thus PdP2 anchored ona support acts as a reservoir and only a small fraction of activecatalytic species is released from it into solution. These resultsare in accordance with the widespread belief that Pd NPsfunction as a reservoir catalytically active Pd(0). Only a smallamount of metal goes into solution as Pd(0) atoms and catalyzesthe reaction and aer completion of the transformation Pd(0)returns to a solid support if it is there (dissolution–re-deposi-tion mechanism).24,44,45 Probably on using ltration in place ofcentrifugation the re-deposition is much less and consequentlya distinct decrease in activity is observed. The good increase inthe catalytic activity of NPs when they are anchored on GOsupports their strong binding onto the surface. Thus the pres-ence of a support inuences the catalytic efficiency but does notensure heterogeneous catalysis absolutely. Homogeneous activespecies may be generated as in the case of GO–PdP2.
Conclusions
1,3-Bis(phenylsulfanyl)propan-2-ol (L) has been synthesized byreaction of 1,3-dibromo-2-propanol with the in situ generatedPhSNa. Its Pd(II) complex (1) is formed by reacting L with[PdCl2(CH3CN)2] in CH3CN at 70 �C. The L and 1 have beenauthenticated by 1H, 13C{1H} NMR and HRMS. The singlecrystal structure of 1 is solved. Using complex 1 as a SSPsynthesis of two NPs (PdP2 and Pd4S) has been achieved for therst time. In TOP PdP2 NPs are exclusively formed whereas inOA/ODE (1 : 1) NPs formed are of Pd4S. The size of the NPs isalso inuenced by the solvent used as TOP has given NPs ofsmall size. These NPs were further graed on graphene oxide.The resulting GO–PdP2 and the GO–Pd4S NPs were successfullyapplied as catalysts to the Suzuki–Miyaura coupling reaction.These novel apparently heterogeneous catalysts are easy toprepare and handle as they are stable in air. The GO–PdP2 NPsprepared for the rst time show higher catalytic activity than
This journal is © The Royal Society of Chemistry 2014
GO–Pd4S. Pd(0) leaching makes the catalytic process to moveand its low level makes GO–PdP2 NPs recyclable. Reuse of thecatalyst is accompanied by some loss in activity depending onthe recycling procedure. The catalysis with GO–PdP2 appears tobe almost homogeneous.
Acknowledgements
The authors thank the Council of Scientic and IndustrialResearch (CSIR), New Delhi, India for the project no. 01(2421)10/EMR-II and JRF/SRF to KNS and AS. Department of Scienceand Technology (India) is acknowledged for research project(SR/S1/IC-40/2010), nancial support for single crystal X-ray(FIST) and HR-TEM (NSIT) facilities at IIT Delhi. HJ thanksUniversity Grants Commission (India) for JRF.
Notes and references
1 (a) A. Eppler, G. Rupprechter, L. Guczi and G. A. Somorjai,J. Phys. Chem. B, 1997, 101, 9973; (b) A. Balanta, C. Godardand C. Claver, Chem. Soc. Rev., 2011, 40, 4973; (c) A. Fihri,M. Bouhrara, B. Nekoueishahraki, J. M. Basset andV. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181; (d)M. Kralik and A. Biffis, J. Mol. Catal. A: Chem., 2001, 177,113; (e) J. M. Thomas, B. F. G. Johnson, R. Raja, G. Sankarand P. A. Midgley, Acc. Chem. Res., 2003, 36, 20; (f)M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469.
2 (a) G. K. Rao, A. Kumar, J. Ahmed and A. K. Singh, Chem.Commun., 2010, 46, 5954; (b) K. N. Sharma, H. Joshi,V. V. Singh, P. Singh and A. K. Singh, Dalton Trans., 2013,42, 3908; (c) K. N. Sharma, H. Joshi, A. K. Sharma,O. Prakash and A. K. Singh, Organometallics, 2013, 32, 2443.
3 (a) S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem.Soc., 2002, 124, 7642; (b) L. Piccolo, A. Valcarcel, M. Bausach,C. Thomazeau, D. Uziob and G. Berhault, Phys. Chem. Chem.Phys., 2008, 10, 5504; (c) J. Watt, S. Cheong, M. F. Toney,B. Ingham, J. Cookson, P. T. Bishop and R. D. Tilley, ACSNano, 2010, 4, 396.
4 (a) P. M. Ajayan and B. I. Yakobson, Nature, 2006, 441, 818;(b) D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner,G. H. B. Dommett, G. Evmenenko, S. T. Nguyen andR. S. Ruoff, Nature, 2007, 448, 457; (c) R. K. Prud'homme,I. A. Aksay, D. H. Adamson and A. A. Abdala, US Pat.092432, 2007; (d) R. Ruoff, Nat. Nanotechnol., 2008, 3, 10.
5 M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp,A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car,R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19,4396.
6 (a) T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin,M. Herrera-Alonso, R. D. Piner, D. H. Adamson,H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen,I. A. Aksay, R. K. Prud'homme and L. C. Brinson, Nat.Nanotechnol., 2008, 3, 327; (b) T. Ramanathan,S. Stankovich, D. A. Dikin, H. Liu, H. Shen, S. T. Nguyenand L. C. Brinson, J. Polym. Sci., Part B: Polym. Phys., 2007,45, 2097; (c) P. G. Liu, K. C. Gong, P. Xiao and M. Xiao,
Nanoscale, 2014, 6, 4588–4597 | 4595

Nanoscale Paper
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
J. Mater. Chem., 2000, 10, 933; (d) F. M. Uhl and C. A. Wilkie,Polym. Degrad. Stab., 2004, 84, 215.
7 Y. Matsuo, K. Kume, T. Fukutsuda and Y. Sugie, Carbon,2003, 41, 2167.
8 (a) G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol.,2008, 3, 270; (b) C. Berger, Z. Song, X. Li, X. Wu, N. Brown,C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov,E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006,312, 1191; (c) M. Freitag, Nat. Nanotechnol., 2008, 3, 455;(d) X. Wang, L. Zhi and K. Mullen, Nano Lett., 2008, 8, 323.
9 (a) U. Hofmann, A. Frenzel, D. Wilm and E. Csalan, Kolloid-Z., 1932, 61, 297; (b) A. Clauss, R. Plass, H.-P. Boehm andU. Hofmann, Z. Anorg. Allg. Chem., 1957, 291, 205; (c)G. Ruess, Kolloid-Z., 1945, 110, 17; (d) R. J. Beckett andR. C. Cro, J. Phys. Chem., 1952, 56, 929; (e) T. Cassagneau,F. Guerin and J. H. Fendler, Langmuir, 2000, 16, 7318–7324;(f) T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon,1988, 26, 357.
10 (a) B. C. Brodie, Ann. Chim. Phys., 1860, 59, 466; (b)L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481; (c)W. S. Hummers and R. E. Offemann, J. Am. Chem. Soc.,1958, 80, 1339; (d) H.-P. Boehm and W. Scholz, Liebigs Ann.Chem., 1965, 691, 1; (e) H.-P. Boehm, M. Eckel andW. Scholz, Z. Anorg. Allg. Chem., 1967, 353, 236.
11 Y. Matsuo and T. Nakajima, Carbon, 1994, 32, 469.12 (a) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth
and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262; (b)Y. Q. He, N. N. Zhang, Y. Liu, J. P. Gao, M. C. Yi,Q. J. Gong and H. X. Qiu, Chin. Chem. Lett., 2012, 23, 41.
13 (a) G. I. Titelman, S. V. Karamanenko, Y. N. Novikov,E. V. Gorozhankin and E. Z. Golosman, USSR Patent SU1806005, 1993; (b) A. Mastalir, Z. Kiraly, A. Patzko,I. Dekany and P. L'Argentiere, Carbon, 2008, 46, 1631; (c)A. Mastalir, Z. Kiraly, M. Benko and I. Dekany, Catal. Lett.,2008, 124, 34.
14 (a) R. C. Cro, Aust. J. Chem., 1956, 9, 184; (b) L. B. Ebert,J. Mol. Catal., 1982, 15, 275; (c) J. M. Lalancette, (VentronCorp.). US Pat. 3847963, 1974; (d) A. Mastalir, F. Notheisz,M. Bartok, T. Haraszti, Z. Kiraly and I. Dekany, Appl.Catal., A, 1996, 144, 237.
15 J. Cheng, G. Zhang, J. Du, L. Tang, J. Xu and J. Li, J. Mater.Chem., 2011, 21, 3485.
16 S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall,ACS Catal., 2012, 2, 145.
17 H. Joshi, K. N. Sharma, A. K. Sharma, O. Prakash andA. K. Singh, Chem. Commun., 2013, 49, 7483.
18 (a) S. Eijsbouts, V. H. J. DeBeer and R. Prins, J. Catal., 1988,109, 217; (b) S. Dey and V. K. Jain, Platinum Met. Rev., 2004,48, 16; (c) H. Joshi, K. N. Sharma, V. V. Singh, P. Singh andA. K. Singh, Dalton Trans., 2013, 42, 2366; (d)K. N. Sharma, H. Joshi, A. K. Sharma, O. Prakash andA. K. Singh, Chem. Commun., 2013, 49, 9344.
19 (a) F. Gao, Q. Lu and D. Zhao, Nano Lett., 2003, 3, 85; (b)V. M. Huxter, T. Mirkovic, P. S. Nair and G. D. Scholes,Adv. Mater., 2008, 20, 2439; (c) Y. Du, B. Xu, T. Fu, M. Cai,F. Li, Y. Zhang and Q. Wang, J. Am. Chem. Soc., 2010, 132,1470.
4596 | Nanoscale, 2014, 6, 4588–4597
20 (a) P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart andS. R. P. Silva, J. Cryst. Growth, 1996, 167, 133; (b)M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010,110, 4417.
21 V. V. Singh, G. K. Rao, A. Kumar and A. K. Singh, DaltonTrans., 2012, 41, 1142.
22 (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000,100, 3009; (b) R. F. Heck, Acc. Chem. Res., 1979, 12, 146; (c)R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320;(d) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn.,1971, 44, 581; (e) N. J. Whitcombe, K. K. Hii andS. E. Gibson, Tetrahedron, 2001, 57, 7449.
23 (a) A. Suzuki and N. Miyaura, J. Chem. Soc., Chem. Commun.,1979, 866; (b) A. Suzuki, Pure Appl. Chem., 1994, 66, 213; (c)Y. Deng, L. Gong, A. Mi, H. Liu and Y. Jiang, Synthesis,2003, 337; (d) N. Miyaura, K. Yamada and A. Suzuki,Tetrahedron Lett., 1979, 3437; (e) N. Miyaura and A. Suzuki,Chem. Rev., 1995, 95, 2457.
24 L. Djakovich, K. Kohler and J. G. de Vries, The Role ofPalladium Nanoparticles as Catalysts for Carbon–CarbonCoupling Reactions in Nanoparticles and Catalysis, ed. D.Astruc, Wiley-VCH, Weinheim, Germany, 1st edn, 2008, p.303.
25 (a) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv.Synth. Catal., 2006, 348, 609; (b) L. X. Yin and J. Liebscher,Chem. Rev., 2007, 107, 133.
26 (a) L. Xu, W. Chen and J. Xiao, Organometallics, 2000, 19,1123; (b) R. R. Deshmukh, R. Rajagopal andK. V. Srinivasan, Chem. Commun., 2001, 1544; (c) X. Yang,Z. Fei, D. Zhao, W. H. Ang, Y. Li and P. J. Dyson, Inorg.Chem., 2008, 47, 3292.
27 (a) Y. Li, X. M. Hong, D. M. Collard and M. A. El-Sayed, Org.Lett., 2000, 2, 2385; (b) J. Hu and Y. B. Liu, Langmuir, 2005,21, 2121; (c) R. Narayanan and M. A. El-Sayed, J. Am. Chem.Soc., 2003, 125, 8340.
28 S. Niembro, A. Shar, A. Vallribera and R. Alibes, Org. Lett.,2008, 10, 3215.
29 K. Mennecke, R. Cecilia, T. N. Glasnov, S. Gruhl, C. Vogt,A. Feldhoff, M. A. L. Vargas, C. O. Kappe, U. Kunz andA. Kirschning, Adv. Synth. Catal., 2008, 350, 717.
30 (a) G. Marck, A. Villiger and R. Buchecker, TetrahedronLett., 1994, 35, 3277; (b) T. Tagata and M. Nishida, J.Org. Chem., 2003, 68, 9412; (c) R. K. Arvela andN. E. Leadbeater, Org. Lett., 2005, 7, 2101; (d) H. Sajiki,T. Kurita, A. Kozaki, G. Zhang, Y. Kitamura, T. Maegawaand K. Hirota, Synthesis, 2005, 537; (e) Z. Li, J. Liu,Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3,839; (f) Z. Li, J. Liu, C. Xia and F. Li, ACS Catal., 2013, 3,2440.
31 (a) F. Bigi, S. Coluccia, S. Maggi, G. Martra, A. Mazzacaniand G. Sartori, Res. Chem. Intermed., 2003, 39, 285; (b)R. B. Bedford, U. G. Singh, R. I. Walton, R. T. Williamsand S. A. Davis, Chem. Mater., 2005, 17, 701; (c) S. Jana,B. Dutta, R. Bera and S. Koner, Inorg. Chem., 2008, 47,5512; (d) N. Erathodiyil, S. Ooi, A. M. Seayad, Y. Han,S. S. Lee and J. Y. Ying, Chem. –Eur. J., 2008, 14,3118.
This journal is © The Royal Society of Chemistry 2014

Paper Nanoscale
Publ
ishe
d on
06
Febr
uary
201
4. D
ownl
oade
d by
Uni
vers
ity o
f W
aika
to o
n 15
/07/
2014
19:
49:0
8.
View Article Online
32 (a) R. L. Augustine and S. T. O'Leary, J. Mol. Catal., 1992, 72,229; (b) A. Biffis, M. Zecca andM. Basato, Eur. J. Inorg. Chem.,2001, 1131.
33 G. M. Sheldrick, SADABS V2.10, 2003.34 (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found.
Crystallogr., 1990, 46, 467; (b) G. M. Sheldrick, SHELXL-NTVersion 6.12, University of Gottingen, Germany, 2000.
35 B. S. Furniss, A. J. Hannaford, P. W. G. Smitha andA. R. Tatchell, Vogel's Textbook of Practical OrganicChemistry, ELBS, Longman Group UK Ltd., 5th edn, 1989.
36 P. Singh and A. K. Singh, Inorg. Chim. Acta, 2012, 387, 441.37 P. G. Liu, K. C. Gong, P. Xiao and M. Xiao, J. Mater. Chem.,
2000, 10, 933.38 J. Tsuji, Palladium Reagents and Catalysts: New Perspectives
for the 21st Century, Wiley, Chichester, UK, 1st edn, 2004.39 (a) C. R. LeBlond, A. T. Andrews, Y. Sun and J. R. Sowa, Org.
Lett., 2001, 3, 1555; (b) Y. Kitamura, S. Sako, T. Udzu,
This journal is © The Royal Society of Chemistry 2014
A. Tsutsui, T. Maegawa, Y. Monguchi and H. Sajiki, Chem.Commun., 2007, 5069.
40 (a) A. Arcadi, G. Cerichelli, M. Chiarini, M. Correa andD. Zorzan, Eur. J. Org. Chem., 2003, 2003, 4080; (b)M. Lysen and K. Kohler, Synthesis, 2006, 692.
41 H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67,2721.
42 R. A. Sheldon, M. Wallau, I. Arends and U. Schuchardt, Acc.Chem. Res., 1998, 31, 485.
43 J. Rebek and F. Gavina, J. Am. Chem. Soc., 1975, 97, 454.44 I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am.
Chem. Soc., 2001, 123, 10139.45 (a) M. A. De La Rosa, E. Velarde and A. Guzman, Synth.
Commun., 1990, 20, 2059; (b) M. T. Reetz and J. G. de Vries,Chem. Commun., 2004, 1559; (c) A. V. Gaikwad,A. Holuigue, M. B. Thathagar, J. E. ten Elshof andG. Rothenberg, Chem. –Eur. J., 2007, 13, 6908.
Nanoscale, 2014, 6, 4588–4597 | 4597





























