Embed Size (px)
Citation preview

166 Biochimica et Biophysica Acta, 1164 (1993) 166-172 © 1993 Elsevier Science Publishers B.V. All rights reserved 0167-4838/93/$06.00
BBAPRO 34526
Oxidative stress response in yeast: purification and some properties of a membrane-bound glutathione
peroxidase from Hansenula mrakii
Linh-Thuoc Tran, Yoshiharu Inoue and Akira Kimura Research Institute for Food Science, Kyoto University, Uji, Kyoto (Japan)
(Received 4 February 1993)
Key words: Glutathione peroxidase; Glutathione S-transferase; Oxidative stress; (H. mrakii)
Giutathione peroxidase was purified from the total membrane fractions of a yeast, Hansenula mrakii IFO 0895. The purified enzyme gave a single protein band with a molecular mass of 28 kDa on SDS-PAGE. The enzyme showed activity to various lipid hydroperoxides and their methyl esters. The enzyme was also active toward phosphatidylcholine hydroperoxide and cholesterol hydroperoxide. Since the enzyme was not active on hydrogen peroxide, the enzyme was thought to be a kind of glutathione S-transferase, although the purified enzyme did not show the glutathione-conjugating activity with electrophilic compounds such as 1-chloro-2,4-dinitrobenzene and o-dinitrobenzene, which are used as the substrate of glutathione S-transferase in yeast. The glutathione peroxidase in H. mrakii was then suggested to be a novel type of glutathione peroxidase in substrate specificity and intracellular localization, being different from those of other sources purified so far.
Introduction
All aerobic organisms use molecular oxygen for res- piration or oxidation of nutrients to acquire the energy to live. The molecular oxygen is reduced to water through acceptance of four electrons. During the re- duction of molecular oxygen, several reactive oxygens are formed; i.e., acceptance of one, two and three electrons to form, respectively, superoxide radical ( 0 2), hydrogen peroxide ( H 2 0 2) and hydroxyl radical (HO ) . Such reactive oxygens have been reported to be causative agents in several degenerative diseases [1,2]. These reactive oxygens attack almost all cell compo- nents, DNA, protein and lipid membrane, and they sometimes cause lethal damage to the cells.
Lipid hydroperoxides are also one of the reactive oxygen species. Radiation [3], halocarbons [4,5], some drugs [6-8] and herbicides [9,10] have been known to be causative of oxidative stress being able to peroxidize the biological membrane in vivo. Among the reactive oxygens, HO" as well as perhydroxyl radical ( H O O ) can extract bis-allylic hydrogen atom of unsaturated fatty acid (LH) to form lipid alkyl radical (L) . The L is oxidized by molecular oxygen to generate a lipid
Correspondence to: Y. Inoue, Research Institute for Food Science, Kyoto University, Uji, Kyoto 611, Japan.
peroxy radical ( L O O ) , and the L O O thus formed reacts with LH to give lipid hydroperoxide (LOOH) and L. The radical chain reaction is then propagated. Occurrence of the lipid hydroperoxides in the biologi- cal membrane may be one of the major oxidative damages to the cells.
Both prokaryotic and eukaryotic cells have defensive mechanisms for such oxidative stresses. For example, Escherichia coli and Salmonella typhimurium cells have the oxyR-controlled regulon of hydrogen peroxide-in- ducible genes [11,12]. E. coli cells also have a soxRS- regulon that is induced by superoxide radical [13-16]. The eukaryotic microorganisms, such as yeast Saccha- romyces cerevisiae and mold Neurospora crassa, have superoxide dismutase to protect the cells against oxida- tive stress by superoxide radical [17,18]. In contrast to the voluminous studies of the defensive mechanism against reactive oxygens such as O~ and H 2 0 2 in bacterial cells [19-21], the studies of the adaptation or resistance to oxidative stress in yeast have not been so much advanced [22,23]. Furthermore, the defensive mechanism for the oxidative stress caused by lipid hydroperoxide has not been studied in detail in mi- croorgansims [24,26].
As the first step in this study, we screened several yeast strains for resistance against lipid hydroperoxide, and we found that Hansenula mrakii IFO 0895 could grow in a medium containing 4 mM linoleic acid hydro- peroxide in which any other yeast strains tested could

not grow [24]. The resistance was supposedly caused by a glutathione peroxidase which was induced when the yeast was cultured in the presence of lipid hydro- peroxide. Recently, we isolated lipid hydroperoxide- sensitive mutants from H. mrakii, and proved that the glutathione peroxidase was essential for the resistance against lipid hydroperoxide in this yeast [25]. In this report, we purified the glutathione peroxidase from the total membrane fractions of H. mrakii and some prop- erties of the enzyme were investigated.
Materials and Methods
Chemicals. Cholesterol, phosphatidylcholine, fatty acids and their methyl esters, and S-hexylglutathione agarose were purchased from Sigma (St. Louis, MO, USA). Lipid hydroperoxides were prepared by photo- sensitized oxidation method and were purified accord- ing to the method of Terao et al. [27,28]. The silver staining kit was obtained from Daiichi (Tokyo, Japan). Other reagents were all analytical grade.
Microorganism and culture. Hansenula mrakii IFO 0895 was obtained from the Institute for Fermentation, Osaka, Japan. The cells were cultured in nutrient medium (1.0% glucose, 0.5% peptone, 0.2% yeast ex- tract, 0.03% K2HPO4, 0.03% KH2PO4, 0.01% MgCI 2 (pH 5.5)) with reciprocal shaking at 30°C until station- ary phase. An appropriate amount of the culture was transferred to a 2-1itre Sakaguchi flask containing 1 litre of SD minimal medium (2.0% glucose, 0.67% yeast nitrogen base (pH 5.5)), and the cells were cul- tured at 30°C. When the optical density of the culture at 610 nm (OD610) reached approx. 1.0, 2 mM tert-butyl hydroperoxide was added to the medium and cultiva- tion was continued until OD610 reached 5.0. Cells were harvested by centrifugation at 5000 rpm for 10 min at 4°C, and washed once with 0.85% NaC1 solution.
Preparation of total membrane fractions. Cells (1 g as wet wt.) obtained as described above were suspended in 10 mM potassium phosphate buffer (pH 7.0) con- taining 0.2 mM phenylmethylsulfonyl fluoride (buffer A), and were disrupted by glass beads in a Braun homogenizer for 2 min at 0°C. Homogenates were centrifuged at 15 000 x g for 30 min at 4°C, and the resultant supernatants were again centrifuged at 200000 x g for 60 min. The precipitates were resus- pended in buffer A containing 2 M KCI and the concentration of protein was adjusted to 10 mg/ml. The suspension was gently stirred for 5 h at 4°C to remove loosely membrane-bound proteins, as well as to wash out the contaminating soluble proteins, and then centrifuged at 200000 x g for 60 min at 4°C. The resultant precipitates were used as the total membrane fractions.
Solubilization of glutathione peroxidase from total membrane fractions. Total membrane fractions as ob-
167
tained above were suspended in buffer A, and the detergent (0.5% CHAPS, 3-cholamidopropyldimethyl- ammonio-l-propane sulfonate) dissolved in the same buffer was added to the suspension to bring the con- centration of protein to be 10 mg/ml. The mixture was kept for 12 h at 4°C with gentle stirring to extract the proteins from the membrane, and the mixture was then centrifuged at 200 000 x g for 60 min at 4°C. Resultant supernatants (CHAPS extracts) were used as the source of glutathione peroxidase.
Purification of glutathione peroxidase. Cells (52 g as wet wt.) from 6-1itre culture were disrupted by glass beads in a Dyno-Mill for 15 rain at 0°C. The total membrane fractions were prepared as described above, and the enzyme was solubilized by 0.5% CHAPS. Un- less otherwise stated, all purification procedures were done at 0-4°C.
Blue-Sepharose CL-6B column chromatography. The enzyme solution after solubilized by CHAPS (56 ml, 11.9 mg protein) was applied onto a Blue-Sepharose CL-6B column (1.8 x 7.0 cm) equilibrated with buffer A containing 0.5% CHAPS (buffer B). The column was first washed with 80 ml of buffer B and then with 80 ml of the same buffer containing 0.2 M KCI. The enzyme adsorbed to the resin was eluted with buffer B contain- ing 0.5 M KCI. Fractions were collected at a flow rate of 2-ml portions per tube. The active fractions (frac- tions 88-102) were combined, concentrated by Amicon PM10 membrane, and dialyzed against buffer B.
Hydroxyapatite column chromatography. The dialysate (1.6 ml, 0.78 mg protein) was loaded onto a hydroxyapatite column (1.8 x 16 cm) equilibrated with buffer B. The enzyme was eluted with a linear gradient of potassium phosphate buffer (pH 7.0) containing 0.5% CHAPS (0.01 M-0.5 M, total volume 150 ml). The fractions were collected at a flow rate of 2-ml portions per tube. Active fractions (conductivity, 1.4- 2.3 mS) were pooled and dialyzed against buffer B.
S-Herylglutathione agarose column chromatography. The dialysate (11.5 ml, 0.11 mg protein) was applied onto an S-hexylglutathione agarose column (0.9 x 3.2 cm) equilibrated with buffer B. The column was washed thoroughly with buffer B, and then the enzyme was eluted with buffer B containing 1 M KCI. Fractions were collected at a flow rate of 2-ml portions per tube. The active fractions were pooled, concentrated by Am- icon PM10 membrane, and dialyzed against buffer B. The dialysate was used for the characterization of glutathione peroxidase.
Enzyme assay. Glutathione peroxidase activity was assayed by the following two methods:
Coupled enzymatic method. Glutathione peroxidase was assayed by measuring the oxidized glutathione using glutathione reductase as described by Flohe and Gunzler [29]. The reaction mixture (1.0 ml) containing 2.5 mM tert-butyl hydroperoxide, 10 mM glutathione,

168
50 mM potassium phosphate buffer (pH 7.0), 0.3 mM NADPH, 3.4 U / m l glutathione reductase and the en- zyme was incubated at 25°C, and decrease of ab- sorbance of the mixture at 340 nm was monitored. tert-Butyl hydroperoxide was replaced by equivalent amount of other peroxides when substrate specificity of the enzyme was examined.
DTNB method. For the investigation of substrate specificity of hydrogen donors, the enzyme activity was also assayed by determining the decrease of glu- tathione (reduced form) or other alternative sulfhydryl compounds using 5,5'-dithiobis-2-nitrobenzoic acid (DTNB) according to the method of Sedlak and Lind- say [30]. The reaction mixture (1.0 ml) containing 2.5 mM tert-butyl hydroperoxide, 10 mM glutathione (or an alternative sulflaydryl compounds), 50 mM potas- sium phosphate buffer (pH 7.0) and the enzyme. The reaction was carried out at 25°C, and during 10-min period 50 /xl of the reaction mixture was taken every l-rain interval. The solution was transferred into a cuvette containing 1.5 mM DTNB. Residual sulfhydryl compounds were determined by measuring the ab- sorbance at 412 nm using a millimolar absorption coef- ficient 13.0 cm -1.
In both methods, control without enzyme was always taken for every assay, and 1 U of the enzyme activity was defined as the amount of enzyme oxidizing 1 ~mol of glutathione per rain at 25°C.
Glutathione S-transferase activity was measured by spectrophotometric assays essentially according to the method of Habig et al. [31]. The reaction mixture (1.0 ml) contained 100 mM potassium phosphate buffer (pH 7.0), various concentrations of electrophilic com- pounds and glutathione (GSH) and enzyme. The con- centrations of electrophilic compound and GSH, and Ama x for the conjugated product were as follows: 1.0 mM 1-chloro-2,4-dinitrobenzene, 5.0 mM GSH, )tma x =
340 nm; 1.0 mM 1,2-dichloro-4-nitrobenzene, 1.0 mM G S H , Ama x ~--345 nm; 1.0 mM p-nitrobenzyl chloride, 5.0 mM GSH, Ama x =- 310 rim; 0.1 mM p-nitrophenethyl bromide, 5.0 mM GSH, Amax = 310 nm; 0.2 mM 4- nitropyridine-N-oxide, 5.0 mM GSH, ,h.ma x = 295 nm; 5.0 mM 1,2-epoxy-3-(p-nitrophenoxy)propane, 5.0 mM G S H , Ama x = 360 nm. When o-dinitrobenzene was used, the enzyme activity was assayed in the mixture (1.0 ml) containing 100 mM potassium phosphate buffer (pH 7.0), 2 mM o-dinitrobenzene, 2 mM GSH and enzyme at 25°C for 15 min as described by Kumagai et al. [32]. After the reaction, 50 tzl of acetic anhydride was added to the mixture to terminate the reaction. The released nitrite was measured by coupling with N-(1- naphthyl)ethylenediamine dihydrochloride and sul- fanilamide at 540 nm as described by Asaoka and Takahashi [33].
Protein was determined by the method of Lowry et al. [34], preceded by precipitating the protein with
trichloroacetic acid to avoid the interferance of deter- gent [35]. In the final preparation step of the enzyme purification, the protein was determined by measuring the absorbance at 280 nm.
Polyacrylamide gel electrophoresis. Polyacrylamide gel electrophoresis was carried out according to the method of Laemmli [36]. After electrophoresis, the gel was stained for proteins with silver using a kit (Silver Stain- ing Kit, Daiichi, Japan).
High-performance liquid chromatography (HPLC). The final preparation after S-hexylglutathione agarose column chromatography was dialyzed against excess amount of water. The dialysate was concentrated to 50 /zl (approx. 1 /zg protein) by centrifugation using an ultrafree-C3LGC (Millipore), and was mixed with an equivolume of 28% acetonitrile containing 0.1% triflu- oroacetic acid (final concentrations of acetonitrile and trifluoroacetic acid are 14% and 0.05%, respectively). The mixture was applied to HPLC (Model CCPE-II, Tosoh, Tokyo, Japan) equipped with Octadecyl-NPR column (Tosoh). The protein was eluted with a linear gradient of acetonitrile (14%-70%) containing 0.05% trifluoroacetic acid.
Results
Occurrence of glutathione peroxidase in the membrane of H. mrakii
Previously we have found that the glutathione per- oxidase was recovered from the insoluble fractions of the cell homogenates of H. mrakii [24]. Therefore, the glutathione peroxidase of H. mrakii was suggested to occur in a membrane-bound form. Since CHAPS has a high critical micellar concentration value and small micellar size that are preferable properties for the purification of enzyme, the detergent was chosen to solubilize the glutathione peroxidase from the total membrane fractions of H. mrakii. As shown in Table I, almost all glutathione peroxidase activity was remained
TABLE I
Recovery of glutathione peroxidase activity in membrane fractions Total activity of homogenates was taken as 100%. n.d., not detected.
Step Total activity Recovery (u) (%)
Homogenate 3.52 100 200000 x g
Supernatant 0.22 6.30 Pellet 3.30 93.7
2 M KCI, 200 000 x g Supernatant 0.40 11.4 Pellet 2.90 82.4
0.5% CHAPS, 200000x g Supernatant 2.90 82.4 Pellet n.d. -
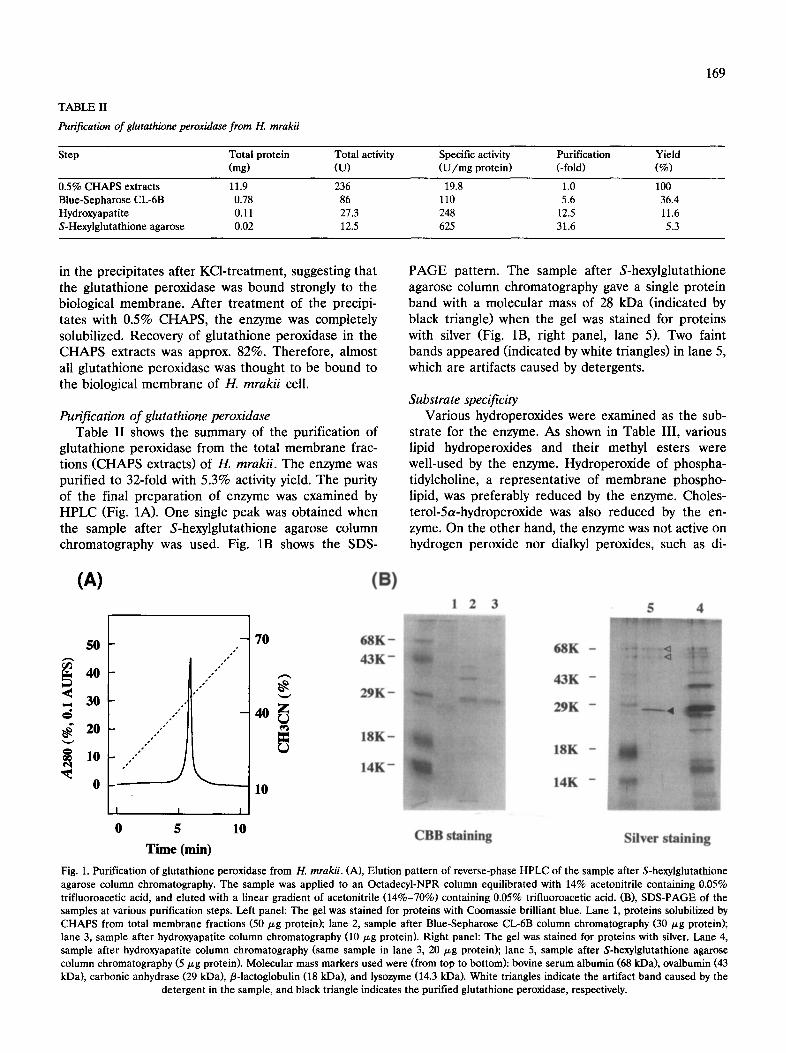
TABLE II
Purification of glutathione peroxidase from H. mrakii
169
Step Total protein Total activity Specific activity Purification Yield (mg) (U) (U/mg protein) (-fold) (%)
0.5% CHAPS extracts 11.9 236 19.8 1.0 100 Blue-Sepharose CL-6B 0.78 86 110 5.6 36.4 Hydroxyapatite 0.11 27.3 248 12.5 11.6 S-Hexylglutathione agarose 0.02 12.5 625 31.6 5.3
in the precipitates after KCl-treatment, suggesting that the glutathione peroxidase was bound strongly to the biological membrane. After treatment of the precipi- tates with 0.5% CHAPS, the enzyme was completely solubilized. Recovery of glutathione peroxidase in the CHAPS extracts was approx. 82%. Therefore, almost all glutathione peroxidase was thought to be bound to the biological membrane of H. mrakii cell.
Purification of glutathione peroxidase Table II shows the summary of the purification of
glutathione peroxidase from the total membrane frac- tions (CHAPS extracts) of H. mrakii. The enzyme was purified to 32-fold with 5.3% activity yield. The purity of the final preparation of enzyme was examined by HPLC (Fig. 1A). One single peak was obtained when the sample after S-hexylglutathione agarose column chromatography was used. Fig. 1B shows the SDS-
PAGE pattern. The sample after S-hexylglutathione agarose column chromatography gave a single protein band with a molecular mass of 28 kDa (indicated by black triangle) when the gel was stained for proteins with silver (Fig. 1B, right panel, lane 5). Two faint bands appeared (indicated by white triangles) in lane 5, which are artifacts caused by detergents.
Substrate specificity Various hydroperoxides were examined as the sub-
strate for the enzyme. As shown in Table III, various lipid hydroperoxides and their methyl esters were well-used by the enzyme. Hydroperoxide of phospha- tidylcholine, a representative of membrane phospho- lipid, was preferably reduced by the enzyme. Choles- terol-5a-hydroperoxide was also reduced by the en- zyme. On the other hand, the enzyme was not active on hydrogen peroxide nor dialkyl peroxides, such as di-
(A) (B) 1 2 3 5 4
5 0
4 0
.< 3 0
2 0
lO
0
o.
oo o. +
.o° /
/ / "
,o
/ / "
I I
5 10
Time (min)
70
10
6 8 K -
4 3 K -
2 9 K -
1 8 K -
1 4 K -
C B B s t a i n i n g
6 8 K -
4 3 K -
2 9 K -
1 8 K -
1 4 K -
S i l v e r s t a i n i n g
Fig. 1. Purification of glutathione peroxidase from H. mrakii. (A), Elution pattern of reverse-phase HPLC of the sample after S-hexylglutathione agarose column chromatography. The sample was applied to an Octadecyl-NPR column equilibrated with 14% acetonitrile containing 0.05% trifluoroacetic acid, and eluted with a linear gradient of acetonitrile (14%-70%) containing 0.05% trifluoroacetic acid. (B), SDS-PAGE of the samples at various purification steps. Left panel: The gel was stained for proteins with Coomassie brilliant blue. Lane 1, proteins solubilized by CHAPS from total membrane fractions (50/zg protein); lane 2, sample after Blue-Sepharose CL-6B column chromatography (30/~g protein); lane 3, sample after hydroxyapatite column chromatography (10/zg protein). Right panel: The gel was stained for proteins with silver. Lane 4, sample after hydroxyapatite column chromatography (same sample in lane 3, 20/zg protein); lane 5, sample after S-hexylglutathione agarose column chromatography (5/~g protein). Molecular mass markers used were (from top to bottom): bovine serum albumin (68 kDa), ovalbumin (43 kDa), carbonic anhydrase (29 kDa), fl-lactoglobulin (18 kDa), and lysozyme (14.3 kDa). White triangles indicate the artifact band caused by the
detergent in the sample, and black triangle indicates the purified glutathione peroxidase, respectively.
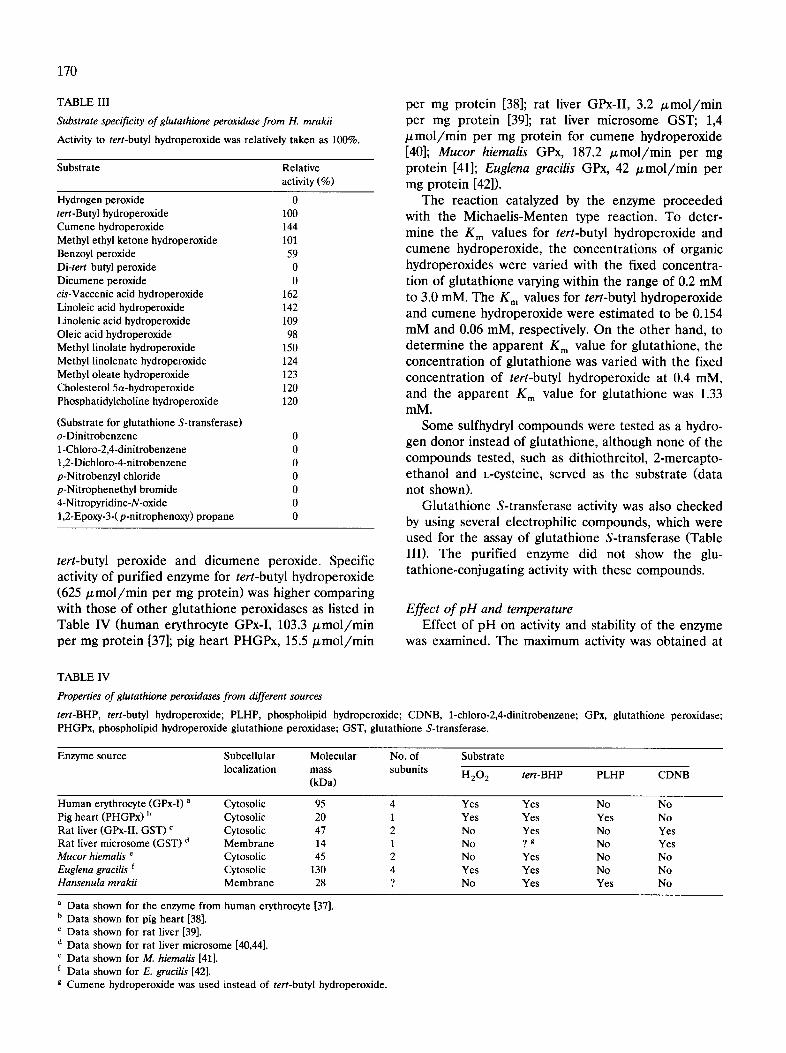
170
TABLE III
Substrate specificity of glutathione peroxidase from H. mrakii
Activity to tert-butyl hydroperoxide was relatively taken as 100%.
Substrate Relative activity (%)
Hydrogen peroxide 0 tert-Butyl hydroperoxide 100 Cumene hydroperoxide 144 Methyl ethyl ketone hydroperoxide 101 Benzoyl peroxide 59 Di-tert butyl peroxide 0 Dicumene peroxide 0 cis-Vaccenic acid hydroperoxide 162 Linoleic acid hydroperoxide 142 Linolenic acid hydroperoxide 109 Oleic acid hydroperoxide 98 Methyl linolate hydroperoxide 150 Methyl linolenate hydroperoxide 124 Methyl oleate hydroperoxide 123 Cholesterol 5a-hydroperoxide 120 Phosphatidylcholine hydroperoxide 120
(Substrate for glutathione S-transferase) o-Dinitrobenzene 0 1-Chloro-2,4-dinitrobenzene 0 1,2-Dichloro-4-nitrobenzene 0 p-Nitrobenzyl chloride 0 p-Nitrophenethyl bromide 0 4-Nitropyridine-N-oxide 0 1,2-Epoxy-3-(p-nitrophenoxy) propane 0
tert-butyl peroxide and dicumene peroxide. Specific activity of purified enzyme for tert-butyl hydroperoxide (625/~mol/min per mg protein) was higher comparing with those of other glutathione peroxidases as listed in Table IV (human erythrocyte GPx-I, 103.3/~mol/min per mg protein [37]; pig heart PHGPx, 15.5/zmol/min
per mg protein [38]; rat liver GPx-II, 3.2 ~mol/min per mg protein [39]; rat liver microsome GST; 1,4 /zmol/min per mg protein for cumene hydroperoxide [40]; Mucor hiemalis GPx, 187.2 /~mol/min per mg protein [41]; Euglena gracilis GPx, 42 /xmol/min per mg protein [42]).
The reaction catalyzed by the enzyme proceeded with the Michaelis-Menten type reaction. To deter- mine the g m values for tert-butyl hydroperoxide and cumene hydroperoxide, the concentrations of organic hydroperoxides were varied with the fixed concentra- tion of glutathione varying within the range of 0.2 mM to 3.0 mM. The K m values for tert-butyl hydroperoxide and cumene hydroperoxide were estimated to be 0.154 mM and 0.06 mM, respectively. On the other hand, to determine the apparent K m value for glutathione, the concentration of glutathione was varied with the fixed concentration of tert-butyl hydroperoxide at 0.4 mM, and the apparent K m value for glutathione was 1.33 mM.
Some sulfhydryl compounds were tested as a hydro- gen donor instead of glutathione, although none of the compounds tested, such as dithiothreitol, 2-mercapto- ethanol and L-cysteine, served as the substrate (data not shown).
Glutathione S-transferase activity was also checked by using several electrophilic compounds, which were used for the assay of glutathione S-transferase (Table III). The purified enzyme did not show the glu- tathione-conjugating activity with these compounds.
Effect of pH and temperature Effect of pH on activity and stability of the enzyme
was examined. The maximum activity was obtained at
TABLE IV
Properties of glutathione peroxidases from different sources
tert-BHP, tert-butyl hydroperoxide; PLHP, phospholipid hydroperoxide; CDNB, 1-chloro-2,4-dinitrobenzene; GPx, glutathione peroxidase; PHGPx, phospholipid hydroperoxide glutathione peroxidase; GST, glutathione S-transferase.
Enzyme source Subcellular Molecular No. of Substrate localization mass subunits
(kDa) H202 tert-BHP PLHP CDNB
Human erythrocyte (GPx-I) a Cytosolic 95 4 Yes Yes No No Pig heart (PHGPx) b Cytosolic 20 1 Yes Yes Yes No Rat liver (GPx-II, GST) c Cytosolic 47 2 No Yes No Yes Rat liver microsome (GST) d Membrane 14 1 No ? g No Yes Mucor hiemalis e Cytosolic 45 2 No Yes No No Euglena gracilis ~ Cytosolic 130 4 Yes Yes No No Hansenula mrakii Membrane 28 ? No Yes Yes No
a Data shown for the enzyme from human erythrocyte [37]. b Data shown for pig heart [38]. c Data shown for rat liver [39]. d Data shown for rat liver microsome [40,44]. e Data shown for M. hiemalis [41]. f Data shown for E. gracilis [42]. g Cumene hydroperoxide was used instead of tert-butyl hydroperoxide.

pH 7.0 and the half-maximum activity was obtained at pH 5.0 and pH 9.0, respectively. The enzyme was stable between pH 6.5 and 8.0 after the treatment of the enzyme at 4°C for 24 h in buffers with various pH values.
Effect of temperature on activity and stability was also investigated. Enzyme activity reached maximum when the assay was done at 50°C. The activity of the enzyme was retained after 10-min incubation below 45°C, but above 50°C the enzyme activity was lost.
Discussion
Glutathione peroxidase was purified to the homoge- nous state on SDS-PAGE from the total membrane fractions of H. mrakii IFO 0895. Some properties of the enzyme were summarized in Table IV in compari- son with glutathione peroxidases from other sources. Glutathione peroxidases purified and characterized so far can be divided into two groups; i.e., a selenium-de- pendent enzyme (glutathione peroxidase) and a sele- nium-independent enzyme (glutathione S-transferase). The glutathione peroxidase purified from H. mrakii (Hansenula-GPx) was different from the glutathione peroxidases so far purified in substrate specificity. The Hansenula-GPx could not reduce hydrogen peroxide, while the other glutathione peroxidases, except for the glutathione peroxidase from Mucor hiemalis [41], could use it as a substrate. Since the glutathione S-trans- ferase could not reduce hydrogen peroxide, the Hansenula-GPx was thought to be a kind of glutathione S-transferases. However, as shown in Table III, the Hansenula-GPx did not catalyze the conjugation of glutathione with electrophilic compounds such as 1- chloro-2,4-dinitrobenzene and o-dinitrobenzene, which are used as the substrate of glutathione S-transferase in yeast [32]. On the other hand, Hansenula-GPx could reduce phosphatidylcholine hydroperoxide and choles- terol-5a-hydroperoxide. Usually, the glutathione perox- idase and glutathione S-transferase were not active on phosphatidylcholine hydroperoxide and cholesterol hy- droperoxide, except for the phospholipid hydro- peroxide glutathione peroxidase (PHGPx) reported by Ursini et al. [38].
One of the characteristics of the Hansenula-GPx is that the enzyme is strongly bound to the biological membrane. Morgenstern et al. have purified the glu- tathione S-transferase from the membrane fractions of rat liver microsome, although the enzyme did not ac- tive on phospholipid hydroperoxide [40,43,44]. The PHGPx being active to phosphatidylcholine hydro- peroxide was purified from cytosolic fractions of pig heart [38]. We could not subject the Hansenula-GPx to a direct analysis of selenium because of its insufficient amount of the purified enzyme. We are now trying to purify the enzyme from a large amount of starting
171
material to analyze the existence of selenium. The result would clarify that the Hansenula-GPx is a new type of glutathione peroxidase or glutathione S-trans- ferase. However, even if the Hansenula-GPx contained selenium or not, the enzyme hereto purified could be distinguishable from the classical type of glutathione peroxidase and glutathione S-transferase so far puri- fied because of its substrate specificity and intracellular localization.
Glutathione peroxidase is thought to play an impor- tant role in the protection of the cells against oxidative stress such as peroxidation of membrane lipids. Since glutathione peroxidases have been purified from cy- tosolic fractions of the cell homogenates, the mem- brane-protective function of the enzyme was supposed to be associated with a preceding action of phospho- lipase [45]. Glutathione S-transferases have been puri- fied from cytoplasm as well as membrane fractions, however, the direct reduction of peroxidized mem- brane lipids by glutathione S-transferase has been de- nied [46,47]. On the other hand, PHGPx was proved to be able to reduce peroxidized membrane lipids, thus suggesting an alternative mechanism for protection of membrane from peroxidation through direct interac- tion of the enzyme with peroxidized membrane lipids. Although the PHGPx activity was also reported to be detected from the cell membranes, the PHGPx was, so far, purified from the cytosolic fractions [38,48]. From this stand of view, the Hansenula-GPx turns to be of physiologically interest. Because the enzyme was puri- fied from the membrane fractions and was active to- ward hydroperoxide of phosphatidylcholine, a repre- sentative of membrane phospholipid, it can serve as a proof of the physiological function of glutathione per- oxidase in the direct reduction of peroxidized mem- brane lipids.
References
1 Ames, B.N. (1983) Science 221, 1256-1264. 2 Cerutti, P.A. (1985) J. Biol. Chem. 251, 7816-7820. 3 Lai, E.K., Crossley, C., Shridhar, R., Misra, H.P., Janzen, E.G.
and McCay, P.B. (1986) Arch. Biochem. Biophys. 255, 156-160. 4 McCay, P.B., Lai, E.K., Poyer, J.L., DuBose, C.M. and Janzen,
E.G. (1984) J. Biol. Chem. 259, 2135-2141. 5 Poyer, J.L., McCay, P.B., Weddle, C.C. and Downs, P.E. (1981)
Biochem. Pharmacol. 30, 1517-1519. 6 Stuart, M.J., DeAlarcon, P.A. and Barvinchak, K.M. (1978) Am.
J. Hematol. 5, 295-303. 7 Hiraiwa, K., Oka, T. and Yagi, K. (1983) J. Biochem. 93, 1203-
1210. 8 Svingen, B.A., Powis, G., Appel, P.L. and Scott, M. (1981) Cancer
Res. 41, 3395-3399. 9 Bindoli, A., Valente, M. and Cavallinu, L. (1985) Biochem. Int.
10, 753-761. 10 Burk, R.F., Lawrence, R.A. and Lane, J.M. (1980) J. Clin. Invest.
65, 1024-1031. 11 Christman, C.F., Storz, G. and Ames, B.N. (1989) Proc. Natl.
Acad. Sci. USA 86, 3484-3488.

172
12 Storz, G., Tartaglia, L.A. and Ames, B.N. (1990) Science 248, 189-194.
13 Greenberg, J.T., Monach, P., Chou, J.H., Josephy, D. and Dem- pie, B. (1990) Proc. Natl. Acad. Sci. USA 87, 6181-6185.
14 Tsaneva, I.R. and Weiss, B. (1990) J. Bacteriol. 172, 4197-4205. 15 Nunoshiba, T., Hidalgo, E., Amabile Cuevas, C.F. and Demple,
B. (1992) J. Bacteriol. 174, 6054-6060. 16 Wu, J. and Weiss, B. (1991) J. Bacteriol. 173, 2864-2871. 17 Munkres, K.D. (1990) Free Radic. Biol. Med. 9, 29-38. 18 Munkres, K.D. (1990) Free Radic. Biol. Med. 9, 39-50. 19 Demple, B. and Amabile Cuevas, C.F. (1991) Cell 67, 837-839. 20 Storz, G. and Tartaglia, L.A. (1992) J. Nutr. 122, 627-630. 21 Farr, S.B. and Kogoma, T. (1991) Microbiol. Rev. 55, 561-585. 22 Collinson, L.P. and Dawes, I.W. (1992) J. Gen. Microbiol. 138,
329-335. 23 Jamieson, D.J. (1992) J. Bacteriol. 174, 6678-6681. 24 Inoue, Y., Ichiryu, T., Yoshikawa, K., Tran, L.-T., Murata, K. and
Kimura, A. (1990) Agric. Biol. Chem. 54, 3289-3293. 25 Inoue, Y., Tran, L.-T. and Kimura, A. (1993) J. Ferment. Bioeng.
75, 229-231. 26 Inoue, Y., Yoshikawa, K., Tran, L.-T. and Kimura, A. (1992)
World J. Microbiol. Biotechnol. 8, 296-300. 27 Terao, J., Asano, I. and Matsushita, S. (1985) Lipids 20, 312-317. 28 Terao, J. and Matsushita, S. (1981) Agric. Biol. Chem. 45, 601-
608. 29 Flohe, L. and Gunzler, W.A. (1984) Methods Enzymol. 105,
114-121. 30 Sedlak, J. and Lindsay, R.H. (1968) Anal. Biochem. 25, 192-205. 31 Habig, W.H., Pabst, M.J. and Jakoby, W.B. (1974) J. Biol. Chem.
249, 7130-7139. 32 Kumagai, H., Tamaki, H., Koshino, Y., Suzuki, H. and Tochikura,
T. (1988) Agric. Biol. Chem. 52, 1377-1382.
33 Asaoka, K. and Takahashi, K. (1983) J. Biochem. 94, 1685-1688. 34 Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J.
(1951) J. Biol. Chem. 193, 265-275. 35 Bensadoun, A. and Weistein, D. (1976) Anal. Biochem. 70, 241-
250. 36 Laemmli, U.K. (1970) Nature 277, 680-685. 37 Awasthi, Y.C., Beutler, E. and Srivastava, S.K. (1975) J. Biol.
Chem. 250, 5144-5149. 38 Ursini, F., Maiorino, M. and Gregolin, C. (1985) Biochim. Bio-
phys. Acta 839, 62-70. 39 Prohaska, J.R. and Ganther, H.E. (1977) Biochem. Biophys. Res.
Commun. 76, 437-445. 40 Morgenstern, R. and DePrierre, J.W. (1983) Eur. J. Biochem.
134, 591-597. 41 Aisaka, K., Uwajima, T. and Terada, O. (1983) Agric. Biol.
Chem. 47, 1107-1113. 42 Overbaugh, J.M. and Fall, R. (1985) Plant Physiol. 77, 437-442. 43 Morgenstern, R., Meijer, J., DePrierre, J.W. and Ernster, L.
(1980) Eur. J. Biochem. 104, 167-174. 44 Morgenstern, R., Guthenberg, C. and DePrierre, J.W. (1982)
Eur. J. Biochem. 128, 243-248. 45 Grosmann, A. and Wendel, A. (1983) Eur. J. Biochem. 135,
549-552. 46 Gibson, D.D., Hornbrook, K.R. and McCay, P.B. (1980) Biochim.
Biophys. Acta 620, 572-683. 47 Tan, K.H., Meyer, D.J., Belin, J. and Ketterer, B. (1984) Biochem.
J. 220, 243-252. 48 Roveri, A., Casasco, A., Maiorino, M., Dalan, P., Calligaro, F.
and Ursini, F. (1992) J. Biol. Chem. 267, 6142-6146.





























