Embed Size (px)
Citation preview

11084 ~ The Journal of Neurouienie, October 26, 2016.36(43):11 0 84-1 1 096
Cellular/Molecular
Oxidation of KCNB 1 Potassium Channels CausesNeurotoxicity and Cognitive Impairment in a Mouse Modelof Traumatic Brain Injury
Wei Yu, Randika Parakramaweera, Shavonne Teng, Manasa Gowda, Yashsavi Sharad, Smita Thaklcer-Varia,
Janet Alder, and Federico SesdDepartment of Neuroscience and Cell Biology, Rutgers University, Robert Wood Johnson Medical School, Piscataway, New Jersey 08854
The delayed rectifier potassium (K ')channel KCNBl (Kv2.1), which conducts a major somatodendrltic current in cortexand hippocam-
pus, isknown toundergo oxidation in the brain, but whether this can cause neurodegeneration and cognitive impairment is not known.
Here, we used transgenic mice harboring human KCNB1 wild-type (Tg-WT) or a nonoxidable C73A mutant (Tg-C73A) in cortex and
~ippocampus to deterniine whether oxidized KCNB 1 channels affect brain function. Animals were subjected to moderate traumatic bran
injury (TBT), a condition characterized by extensive oxidative stress. Dasatinib, a Food and Drug Administration—approved inhibitor of
Src tyrosine kinases, was used to impinge on the proapoptotic signaling pathway activated by oxidized KCNBl channels. Thus, typical
lesions of brain injury, namely, inflammation (astrocytosis), neurodegeneration, and cell death, were markedly reduced in Tg-C73A and
dasatinib-treated non-Tg animals. Accordingly, Tg-C73A mice and non-Tg mice treated with dasatinib exhibited improved behavioral
outcomes in motor (rotarod) and cognitive (Morris water maze) assays compared to controls. Moreover, the activity of Src kinases, along
with oxidative stress, were significantly diminished in Tg-C73A brains. Together, these data demonstrate that oxidation of KCNB 1
channels is a contributing mechanism to cellular and behavioral deficits in vertebrates and suggest a new therapeutic approach to TBI.
Key words: aging; dasatinib; Kv2.1; oxidative stress; ROS; Src kinases
Significance Statement
This study provides the first experimental evidence that oxidation of a K-} channel constitutes a mechanism of neuronal and
;,ognitive impairment in vertebrates. Speafically, the interaction of KCNB1 channels with reactive oxygen species plays a major
role in the etiology of mouse model of traumaric brain injury (TBI), a condition associated with extensive oxidative stress. In
addition, a Food and Drug Administration—approved drag ameliorates the outcome of TBI in mouse, by directly impinging on the
toxic pathway activated in response to oxidation of the KCNB i channel.'These findings elucidate a basic mechanism of neurotox-
i~ty in vertebrates and might lead to a new therapeutic approach to TBI ni humans, whi~.h, despite significant efforts, is a condition
that remains without effective pharmacological treatments.
IntroductionIon channels are versatile proteins that generate and modulateelectricity across biological membranes. Since electricity is an
Received July 73, 2016; revised Aug. 25, 2016; accepted Sept.1, 2016Author contribrtions: l.A., S.T: V., and F.S. designed researdi; W.Y., RP., S.T., M.&, Y.S., and F.S. performed
research; W.Y., R.P., and f.S. analned data; F.S. wmte the paper.Thawakwas wppated by National5dence Foundatbn Gent 145b675 and NIH Leant 1R21N5096619 01 ro F.S. The
ffiy1.2cassettewaza Idnd g~tGom Dr. PimGmnl. Wethank Drs. Madure and Qienforhelpwith theZehsAxb Imagerond
Shuang liu tacdkal reading ofthis manuscript We thank Nickkurato forhelpng with the geiwtypirg.
The authors declare no compMing financial interests.Correspondence should beaddressedto Dr. Federko5esti,DepartmentofNeuroscienceandCell Bblogy,Autgers
University, Ruled Wuud lohuwn Medial Ahuol, 683 Hues lene Wes[, Piscnleway, NJ 08854. [email protected] u.
D01:10.1523/1NEUROSCI1273-161016(opyrightm2016theauthors 0274b414/16/3611084-13515.00/0
essential ingredient of life, ion channels are found in all organ-isms, from prokaryotes to eukatyotes to archaeans, in virtuallyany type of cell (Hine, 2001). A growing number of ion channels,including the potassium {K +) channels, are reported to interactwith reactive oacygen species (ROS), either in cell signaling mech-anisms or as a side effect of aging and disease (Patel and Sesti,2016). Hence, o~cidative modification of K+ channels has thepotential to constitute a widespread mechanism of vulnerabilitybut a strong causative link between these modifications and be-havioral and functional impairment has still to be established forvertebrates. One channel known to undergo oxidation is the de-layed rectifier voltage-gated K+ channel KCNB1 (Kv2.1), whichcazries a major somatodendritic current that regulates high-frequenryfiring ofneurons inthe cortex and hippocampus (Mu-

Yu et al. • Oxidation of KCN61 in TBI
oligomerizarian
—~—~do-
~~~N ~ -~KCN61
Sr c
5ti~ 9
MROQIONORIA ~
APOPTosisFlgure 1. Predkted model of KfN61-oxidatlon-mediated apoptosh. Oxidative stress in-duces the forma8on of KCI~1 oligomers that accumulate in the plasma membrane. The pres-enceoftheseproteinag~egatesleadstotheactivadon ofSrcanddownsheamJNKkinases.Thelatteract to despbilize mitod~ondda,resulting to the release ofmore ROS (whkh may furtherwstain KCN61 oligomedzadon in a sort of aura-catatytic process) and apoptosis.
rakoshi and Trimmer, 1999; Du et al., 2000; Cotella et al., 2012).In vitro studies showed that oxidantscross-link KCNBI subunits,giving rise to oligomers that do not conduct current. In addition,oxidized KCNB1 channels are poorly endocytosed and tend tobuild up in the plasma membrane (Wu et al., 2013). TheseKCNB1 protein aggregates favor the activation of Src tyrosinekinases and downstream Jun N-terniinal kinases (JNK) that leadto apoptosis and release of more ROS by presumably destabiliz-ing mitochondria (Fig. 1). KCNBl oligomers have been detectedin the brains of aged mice and, in larger quantities, in the brain ofthe 3x-Tg-AD mouse model of Alzheimer's disease, which ex-press~s abnormal amounts of ROS (Oddo et al., 2003; Smith eta1., 2005; Sensi et al., 2008; Yao et al., 2009; Chou et al., 2011;McManus et al., 2011; Cotella et al., 2012). This body of evidenceargues that oxidized KCNBl channels may affect cortical and/orhippocampal excitability and, when oxidation is elevated, causeneuronal death. indeed, Frazzini et al. (2016) showed recentlythat KCNBI oligomers promote hyperexcitability in cultured3~I'g-AD primary hippocampal neurons, but whether oxidizedICCNB 1 channels affect brain function is not known. To deter-mine the role that oxidation of KCNBI plays for the pathophys-iology of the brain, we constructed a transgenic mouse thatexpresses a redox-resistant variant of human KCNB 1 (C73A),which we showed previously does not oligomerize and thas con-ducts normally (Cotella et al., 2012). To expose the channels tocontrolled and reproducible conditions of o~cidative stress, wesubjected the animals to mild to moderate traumatic braininjury—which is associated with extensive oxidation during thesecondary injury (Corps et al., 2015; Hiebert et al., 2015; Shen etal., 2016)—using the lateral fluid percussion (LFP) method(McIntosh et al., 1989; Alder et al., 2011; Roth et al., 2014). LFPproduces both focal and diffuse brain injury and is associatedwith extensive axonal damage and cell death that result in long-term neuromotar and cognitive deficits (Smith et al., 1991; Mi-yazaki et x1.,1992; Hamm et al.> 1993; Hicks et x1.,1993; Dixon et
1. Ne~eui., October 26,2016.36(43):11084-11096.11085
a1.,1999). These deficits reflect the outcomes observed in humanvictims of TBI and the model has therefore been used extensivelyto identify potential therapeutic treatments (Thompson et al.,2005; Masel and DeWitt, 2010).
Here, we show that genetic suppression of KCNB1 oxidationis protective in TBI. Compared to nontransgenic mice or trans-genic. mice expressing the wild-type channel, Tg-C73A animalsexhibit significantly improved sensorimotor and learning andmemory outcomes along with reduced inflammation, neurode-generation, and cell death. Moreover, pharmacological impinge-ment onthe pathway activated by KCNB 1 oxidation recapitulatesthe effects of decreasing KCNBi oxidation by genetic means.Thus, specific Src tyrosine kinase inhibitor dasatinib, a Food andDrug Administration (FDA)-approved drug sold under the com-mercial name of Sprycel, currently used to treat certain forms ofleukemia, ameliorates inflammation, neurodegeneration, andneuronal death and improves behavioral deficiency following theLFP injury.
Materials and MethodsConstruction of Tg-WT and Tg-C73A transgenic mice. Transgenic micewere constructed by the Ccnomc Editing Corc Pacility at Rutgers usingpronuclear injection. cDNA encoding human KCNBI tagged to the hu-man influenza hemag~utinin (HA) tag in the C tern~inus (Cotella et al.,2012) was inserted in the mouse Thy1.2 cassette using a XhoI restrictionsite. Constructs were linearized at an EcoR V site, for injection. Weobtained3 Tg-WT and 4 Tg-C73A founders.
Bfochemistry. The detailed biochemical procedure was described pre-viously (Cotella et al., 2012). Briefly, frozen, half sagittal brains of eithersex were homogenized with a bass tissue grinder in lysis buffer (0.32 Msucrose, 5 mmt Tris-Cl pH 6.8, 0.5 mrn EDTA, i mht PMSP, and proteaseinhibitor cocktail set I, Calbiochem). Samples were centrifuged at 2000rpm for 10 min, and the supernatant used for biochemical analysis. Pro-teincontent was quantified with die Bradford colorimetric assay (Sigma)apd dissolved in Laemmli buffer with or without reducing agents. Pro-teins were resolved by 8% SDS-PAGE and transferred to a PVDP mem-brane that was incubated in a 596 solution of nonfat milk in Tween20-PBS (PBST) for 2 h at room temperature. After overnight incubationat 4°C with the primary antibody (anti-KWl.I NeuroMab clone K89/34,UC Davis/NIH; anti-HA H6908, Sigma; anti-actin MAB1501, Milli-pore), the membrane was washed for 20 min and incubated at roomtemperature with the appropriate secondary antibody. To detect acti-vated Src tyrosine kinases, brain lysates were incubated at 4°C overnightin the presence ofanti-Src antibody (catalog #2108, Cell Signaling Tech-nology).Then, protein A agarose beads (30 µl of 5096 bead slurry) wereadded and ittcubated for 2 h at 4°C. Samples were centrifuged For 30 s at4°C, and the pellet was washed five times in cell ]psis buffer. The pelletwasresuspended with 50 µ12X SDS Laemmli buffer, heated at 100°C for 10min, and centrifuged for 1 min at 14,000 X g. The sample was loaded on8% SDS-PAGE gel and immunoblotted with either anti-Src antibody oranti-P-Src-antibody (catalog #2101, Cell Signaling Technology). Theblots were washed in PBST for 20 min and incubated for 5 min withchemilumiuescence substrates and exposed. Densitometric analysis wasperformed using Image) (NIH) software.
Lat+~erral fluid percussion injury. All experimental protocols itrvolvinganimals were approved by the Rutgers University Institutional MimalCare and tTse Committee. LFP brain injury involves the clisplacement ofneural tissue by a rapid Quid pulse to the brain and has been described indetail previously (Alder et al., 2011). For surgery, 3-month-old maleswere anesthetized with 4-5% isoflurane in 10096 OZ and placed in amouse stereotaxic frame. Mice were maintained at 2% isoflurane, andrespiration was moniWrecl 4hroughoul the procedure. The silo of injurywas located halfway between lambda and bregma, and betweenthe sag-ittal suture and the lateral ridge over the right hemisphere. A 3 mm thinplastic disc was fixed with Loctite glue (444 Tak Pak, Henkel) onto theskull. Using a trephine (3 mm outer diameter), a craniotomy was per-formed, kceping the dura intact A rigid Luer lock needle hub (3 mm

i308b • 1. PJeurosci., Oitober 26, 2016.3b(43):11084-11096
inside diameter) was secured to fine skull overthe opening with ryanoacrylate adhesive anddental acrylic (Bader Schein). The skull sutures anwere sealed with the cyanoacrylate co ensurethat the fluid bolus from the injury remainedwithin cranial cavity and the hub was filled non-~'gwith saline. After a GO min period of recovery,the animals were reanes(heticed and conn~cteclto the fluid percussion injury device (CastomDesign and Fabrication, Virginia Commnn- -~g_~wealth University) through the Lixer-loc fittingof the hub. Once a normal rreathing patternresixmed, before sensitivity to stimulation, a~•1.5 atan pulse (^'15 ms) was generateddxruugli dxe LPP device. Upon return of die Tg-C73Rrighting reflex (4 -10 min for moderate injury),the huh and dental acrylic were removed. Thescalp incision was sealed with 3M Vetbond an
(Thermo Fisher Scientific) and the animalswere returned to normal housing conditions.At this moderate level of injury, ~~10% of ani-tz~als died as a reseilt of the i~ijui•y within tineacute posttraumatic period (15 min), generallyfram respiratory failure and puLYxo~iary edema(Li£shitz, 2003). This is a normal and anrici- 7g-W7
~ateci feahire of the TBI model because it mim-icshuman TBl (Uomeniconi et aL, 2G07). Micethzrt undergo the surgical procedures but thatwere uninjured served as die sham controls.Assignment of mice to the LFP or sham groupwas done in a random manner.
l3rugc~dministration. Dasatinib (LC Labora-tories)was given urtraperitoneally at 25 rng/kg.Dasatinib was diluted iu vehicle solution (50 mmt NaAc, pH 5.0) from <200 mg/ml stock in dimethyl sulfate. Each mouse was subjected to a dailydose of either vehicle or tlzsatieaib salutioti via an iatraperitoneai injeotion skarting from the day of surgery (2 h after injury}.
Behrxvioresi cusays. For the Morris water maze (MWM), mice wereacclimated to the paradigm and tested for baseline response using <visible platform test 4 d before injury. The animals were placed. incircular pool of water containing nontoxic white paint and a clear plat•form for escape. To assess learning, mice were trained using a hiddexplatfo~~m fixed in one of four yuatirants for 6 consecutive d~iys starting a'~2 d postinjury (dpi; four trials/day). Black and wtuite distal cues werfplaced on the walls. T'he quadrant iii which the iuous~ was ylaced wa:pseudorandomly varied throughout training, and the time to locate therl~ttform was recorded. Maximum trial time was CO s, and the mouseremained or was placed on the platform for 15 s and. warned for 10 mirbetween trials. To assess memory retention, the day after the last trainingsession (8 dpi), the anin-~als were subjected to a GO s probe trial with theplatform removed and the time spent in the target quadrant was mea-sured (Longhi et al., 2405). DaCa were recorded, using avideo-Crackinfsystem (EthoVision XT, Noldus Information Technology).
Vestibulotrtotor rotarod test. A separate set of mice was used for motortesting. Mice were acclimated to the rotarod device three rimes per daywith 1 h intertrial intervals for the 2 d before the surgery. Balance andi~iotu3• function were measured un a 36 mui outer diameter rutatuzg runwhose velociTy increased from 4 to 40 rpm over a maximum 180 s inter-vai. Each trial ended when the animal fell off the rotaruci. Ac 1, 7, and 21dpi, each subject underwent three trials a day with 1 h intertrial interval:on the xotarad device. The same mice were used £or each rime point. Theaverage latency to fall of injured mice was recorded and was compared tothat of sham mice.
Ire¢»euyep)eulvcketraist~y. Mice were perfused tratiscardially wide 0.9%saline followed by 4% paraforcnaldehyde at either 7, 14 and 21 dpi. Thebrains were cryoprotected in 30% sucrose, and 2d µm frozen sectionswere prepared throughout the site of injury on the cortex and the hip-pacampus in a 1.20 series so that the same set of tissue samples could beused far expression of different makers. Foe activated Caspase-3;
Yu et al. ~ Oxidation of RCHQi in TSI
COR'CEX HIPPOCAMPUS ~
H-HA anti-KCNB1 anti-HA anti-KCN61
ti-HA
"~
v
Tg-C73A non-Tg
anti-KCN61
K t~
F ,
Tg-WT Tg-C73A
KCNBI, and HA irnmunohistochexzustry, sections were prekreated with0.01 M citrate buffer, and then anticleaved Caspase-3 antibody (1:1000,catalog #4bGl, Ceti Sigualiug Tectuiolagy) oi• anti-KCNBl antibody (1:100) aranti-HA antiUody (8µg/ml) was applied overnight, followed byapplic~rinn of the appropriate secondary conjugated antibody, ForFluoro-Jade C (FLJC) staining, sections were pretreated with 1% NaOHand 0.06% KMnO~, and then 0.0005% Fluoro-Jade C (catalog #AG325,Millipore)l0.0001% was applied for 10 min. For filial fibrillary acidicprotein (GFAP), slides were incubated overnight with anti-GFAP anti-body at 4°C (1:500, catalog #MAB3402, Millipore). Slides were then in-cubated in 2° goat anti-moose antibody (1:500, Alexa Fluor 594). Allslides were mounted in Vectashielci Antiftde Mounting iVfediurn withDAPI rnountipg buffer (Vector Laboratories) and stored at 4°C. Stainingwas visualised nn a 7.eiss Axinrhot microscope at 40X or with a 7,eissAxio Imager M1 at 100X (images in Pig. 2C). Positive cells oxi the ipsi-lateral hemisphere were counted in corona) sections representing a1:20 series throughout the site of injury at the cortex and inclusive ofthe entire length of the hippocampus. For the cortex, a total of sixfields af~view at 4Qx (three most dorsal along the surface of the cortexstarting from midline and moving laterally and three just ventral tothose fields) were counted oi~ the side ipsilateral to the injury. Theipsilateral hippocampus including CA1 and CA3 as well as the dentategyros was used for quantitati.on of cells in the hippocatnpas.
Preparation of hippocampal neuronuC cultures. The detailed procedurewas described previously (Thakker-Varia et al., 2001). Briefly, hip-pocampiwere obtained from time-mated emUryonic day 16 (E16) micekilled by COZ asphyaciation. Hippocampal tissme from individual em-bryos was mechanically triturated in Neurobasal medium containingB27 (Tnvitrogen) and glutamine and. plated in two 35 mm poly-n-lysine-coated Petri dishes at •350,flt?0 cells/dish (1.5 ml medium/dish). Cul-tures were maintained in Neurobasal medium at 37°C in a 95% air/5%COZ hurnidifieci iucuUator and coxitaineci virtually ~>ure neurons. Tailsamples from individual embryos were processed for genotyping.
Eleccrophysiology. Data were recorded with an Axopatch 200B ampli-
fier (Molecular Devices), a personal computer (Dell), and Clarnpex soft-
Figure 2. hKCNB1 is e~epressed in cortex and hippaampus. A, Representative images of corona) sefions ofi the cortex andhippocampus of the indicated genotypes stained wfth an antibody that recognhes the NA epitope tag of transgenic KCN61(anti-NA) and sections stained with an antibody that detects both endogenous and transgenic KCNBt (anti-KCNB1). B, Represen-tativeimage of acortical section of a Tg-C73A brain probed with anti-KCN61 and DNA stain DAPI (blue color). C, Representativeimages of single hippocampai neurons of the indicated genotypes stained with anti-NA or antl-KCN61. Scale bars:A, 200µm; 8,50µm; f, 10µm.
Yu et al. • Oxidation of KCN61 in TBI
A anti-KCN61
Q'c w
total KCNBi(anti-KCN81) ~ 0.5
actin ~~ ~~ ~ OA-~~ ~P
~~0~1 ~x
C Tg-WT Tg-C73A
non-Tg $ ; ~ j
~If
I
B
1. Neurosci., Odo6er 26, 201G • 3b(43):11084-11096.11087
anti-HA Statistical unalysis. Quantitative data arepresented as mean ± SEM. The level of signif-icance,assumed atrye 95g'o confidence limit or
~ ~'~ Beater (p < 0.05), was calculated using Stu-§ dent's t-test (http:(/studentsttest.com) and
o.s one-way ANOVA with a Tukey's post hoc test~ (http:/lastatsa.com/OneWay_Anova_with-
o.o _TukeyHSD). Forr~NOVA tests with (i}more
~~~^~p s~ than three independent samples or (2) com-~~ ~0 parisonF between sampie~ from different
E groups of data ("longitudinal"), only the pvalue is indicated in the figure, while thep value
~ ~ •o and the statistical significance of pairwise com-0.8 parisons are indicated in the legend (*p < 0.050.6 uid ~*p < O.oi in all fgures).
a
~°~ C ~ S'
coc /~~i ,tom
hKCN81 ~ ~(an6•HA)
actin rr~ ~ ~~~
D a000 x i
~. saoo
2000
,ateVm, ~1T1V~
0 -40 40 80
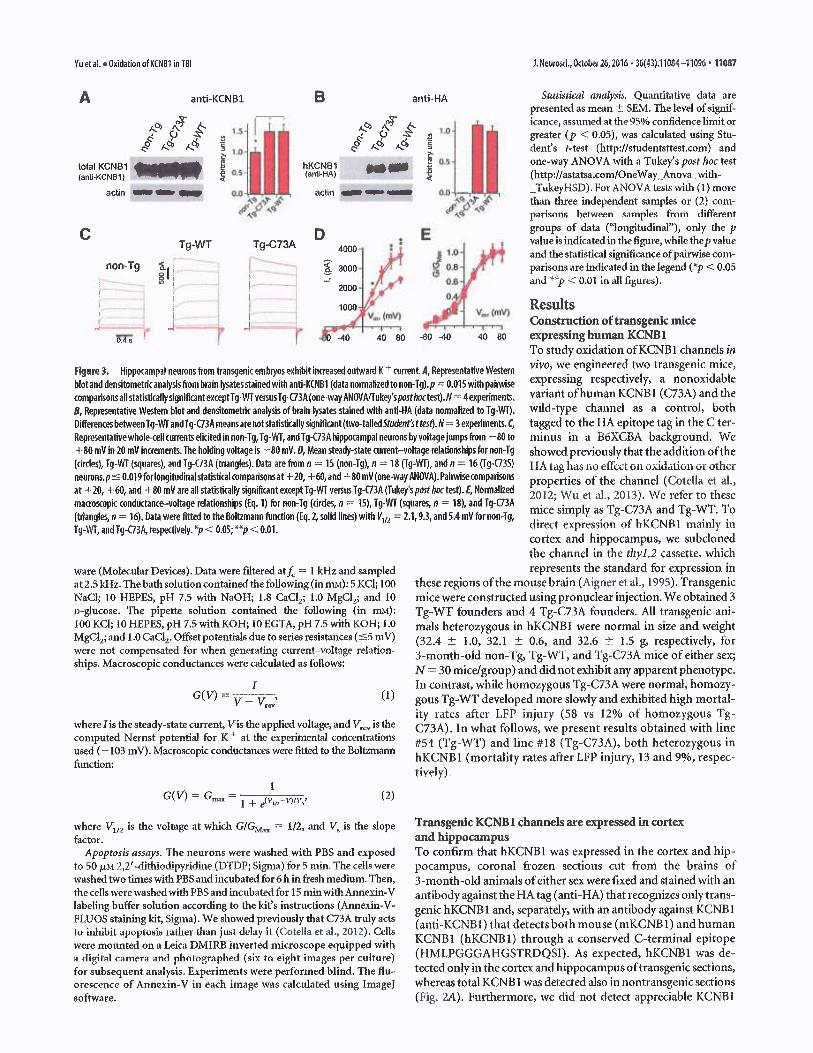
Figure 3. Nippaampa! neurons from transgenic embryos exhibit increased ouhvard K + curreblot and densitomeMc analysis from brain lysates stained with anti-KCN61(data normalized to noncomparisonsall statisticallysignificantexceptTg-1NTvetsusTg-C73A (one-way ANOVA/fukey's postB, Representative Western blot and densitomeMc analysis of brain tysates stained with anti-NADiflerencesbetween Tg-Wf andTg-C73A meansare notstatistkallysignificant(two-tailed Student sRepresentafivewhole~cell currents elicited innon-Tg, Tg-WT, and Tg-C73A hippocampal neurons b+80 mV in 20 mU increments. The holding voltage is —80 mV. D, Mean steady-state current—vol(circles), Tg-WT (squares), and Tg-C73A (triangles). Data arP from n = 15 (non-Tg), n = 78 (Tgneurons.p { 0.019 for longitudinal statistical comparisons at +20, +60, and +80 mV (one-way Aat +20, +60, and +80 m4 are all staNsticaliy significant except Tg-WT versus Tg-C73A (Tukey'smaa~oscopic conductance—voltage relationships (Eq.1) for non-Tg (drdes, n = 15), Tg-WT (squ(triangles, n =16). Data were fitted to the Bol~mann function (Eq. 2, wlid Ifnes) with V,n = 2.1Ty-YdT, and Ty-C73A, respectively. *p ~ 0.05; **v < 0.01.
ware (Molecular Devices). Data were filtered at f~ = 1 kHz and sampledat 2.5 kHz. The bath solution contained the following (in m.~t): 5 KC1;100NaCI; 10 HEPFS, pH 7.5 with NaOH; 1.8 CaC12; 1.0 MgCIZ; and 10n-glucose. The pipetke solution contained the following (in mM):1001CC1; 10 HEPES, pH 7.5 with KOH;10 EGTA, pH 7.5 with KOH; 1.0MgCIZ; and 1.0 CaC12.Offset potentials due to series resistances (~5 mV)were not compensated for when generating current—voltage relation-ships. Macroscopic conductances were calculated as follows:
G« V — V ~1~revs
where I is the steady-state current, V is the applied voltage, and V~~, is thecomputed Nernst potential for K+ at the es~erimental concentrationsused (-103 mV). Macroscopic conductances were fitted to the Boltzmannfunctiom:
1G(V) =Grote — 1 +
e~~~rz_~"'s, l2)
where V1~2 is the voltage at which G/GMT = 1/2, and Vs is the sloYefactor.
Apoptosis assays. The neurons were washed with PBS and exposedt~ 5~ µM 2,2'-ciithiodipyridine (DTDP; Sigma) for 5 min.'The cells werewashed two times with PBS and incubated for 6 h in fresh medium. Then,the cells were washed with PBS and incubated for 15 min with Annesin-Vlabeling buffer solution according to the kit's instructions (Annexin-V-FLUOS staining kit, Sigma). We showed previously that C73A truly actsto inluUit. apoptosis rather than just delay it (Cotella et ai., 2012). Cellswere mounted on a Leica DMIRB inverted microscope equipped witha digital camera and photographed (six to eight images per culture)for subsequent analysis. Experiments were performed blind. The flu-orescence of Annexin-V in each image was calculated using Image)software.
vm, (mV) ResultsConstruction of transgenic mice
-ao -ao ao so expressing human KCNB1To study o~dation of KCNB1 channels in
nt.A,RepresentatrreWestem vivo, we engineered two transgenic mice,
-Tg).p = 0.015 with pairwise expressing respectively, a nono~dable
hoctest).N=4experiments. variant ofhurnanKCNBI(C73A)andthe
(data normalized to Tg-Wf). ~'~d-type channel as a control, both
ttes~.N= 3experiments.C, tagged to the HA epitope tag in the C ter-yvoitagejumpstrom —80 to minus in a B6hCBA background. Werage reladonshipsfor non-Tg showed previously that the addition ofthe-Wt), and n =16 (fg-C73S) I IA tag has no effect on oxidation or otherNOVA).Painaisecompariwns properties of the channel (Cotella et al.,posthatest).E,Normalized 2012; Wu et al., 2013). ~~Ie refer to theseares, n =18), and Tg-C73A
mice simply as Tg-C73A and Tg-WT. To9.3, and 5A mV for non 1'g, ~zrect expression of hI~GNR 1 mainly incortex and hippocampus, we subclonedthe channel in the thy1.2 cassette, whichrepresents the standard for expression in
these regions of the mouse brain (Aigner et a1.,1995). Transgenic
mice were constructed using pronuclear injection. We obtained 3Tg-WT founders and 4 Tg-C73A founders. All transgenic ani-
mals heterozygous in hKCNBI were normal in size and weight(32.4 -~- 1.0, 32.1 -} 0.6, and 32.6 ± 1.5 g, respectively, for
3-month-old non-Tg, Tg-WT, and Tg-C73A mice of either sex;
N = 30 mice/group) and did not e~ibit any apparent phenotype.In contrast, while homozygous Tg-C73A were normal, hon~ozy-
gous Tg-WT developed more slowly and exhibited high mortal-
ity rates after LFP injury (58 vs 12% of homozygous Tg-C73A). In what follows, we present results obtained with line
#51 (7'g-WT) and line #18 (Tg-C73A), both heterozygous inhKCNBI (mortality rates after LFP injury, 13 and 9%, respec-tively) ,
Transgenic KCNBl channels are expressed in cortex
and hippocampusTo confirm that hKCNBI was expressed in the cortex and hip-
pocampus, coronal frozen sections cut from the brains of
3-month-old animals of either sex were fixed and stained with anantibody against the HA tag (anti-HA) that recognizes only trans-
genic hKCNB 1 and, separately, with an antibody against KCNB1
(anti-KCNB1) that detects both mouse (mKCNB 1) and human
KCNB1 (hKCNBl) through a conserved C-terminal e~itope(HMLPGGGAHGSTRDQSI). As expected, hKCNBI was de-tected only in the corte~c and hippocampus oftransgenic sections,
whereas total KCNB i was detected also in nontransgenic sections(Fig. 2A). Furthermore, we did not detect appreciable KCNB1
11088 • J. Neurosci.,October26, 2016 ~ 36(43):11084-11096
expression in other areas of the brain. A B injuredStaining with either KCNB1 or HA anti-body revealed typical cluster distribution _ oo ~iP
of KCNB1 channels in the plasma mem- H2O2 + + ,~ c ~1
brave of both cortical (Fig. 2B) and hip- ~~T - - + ~av co ,boo
pocampal neurons (Fig. 2C; Rhodes et al.,1995; Lim et al., 2000; O'Connell et al., 200 ~„~ ~ 1
2006, ZO10; Sarmiere et al., 2008; Fox et kDaal., 2013). 100
Transgenic hKCNB 1 channels Ccontribute to outward K+ current inhippocampal neuronsTo assess the relative amounts of endoge- gnous and transgenic KCNB1 protein and ~consequently to have a rough measure of athe level of overexpression of the latter, ~~half brains were lysed, immnnvblotted, oand analyzed by densitometry. Results oftour cxperimcnts with thc anti-KCNB1antibody indicated that the amounts oftransgenic channels were comparable inthe two transgenic lines and were abouthalfthe aniounts of eudogeuous chancels figure4. ThelevelofKC(Fig.3A).Whensimilarexperimentswere thebrainsofinjurednon-Tgperformed with the anti-HA antibody, HzOZ or2Q mMredudng ag
hKCNB 1 was not detected in non-Tg uninjured brains of the Indic
brains, as effected, and its levels were KCNBt o~dation (oxidation r
similar in the two transgenic lines, in ~eHAtag(anti-HA,N=3
agreement with the results of the experi- ~9nificant except Tg-Wf v
menu with anti-KCNB1 (Fig. 3B). To ~iledStudent'stten).D,~iometry and detected with an
assess the contribution of transgenic ~~~tesq.N=3experiKCNB1 channels to total neuronal cur-rent, we recorded somatic whole-cell cur-rents in primary hippoca~upal neurons obtained fromnontransgenic and transgenic embryos. Voltage jumps rangingfrom —80 to +80 mV (from an holding voltage of —80 mV)
evoked robust outward currents that reversed around —40 mV(Fig. 3C,D; data are shown without subtraction ofleak currents).
Steady-state current amplitudes at +80 mV were comparable inTg-WT and Tg-C73A transgenic neurons and were roughly---40%larger than in non-Tg neurons, consistent with biochem-ical results (Fig. 3A) and with previous studies that showed thatC73A KCNB1 channels conduct like WT KCNBI channels (Co-tella et al., 2012). Normalized macroscopic conductances (Fig.3E~ fitted to a Boltzmann function exhibited half-activation
(V112) values around 0 mV in good agreement with the results ofothers (Frazzini et al., 2016). The V1~2 values of transgenic con-ductances were slighfly more positive than those of nontrans-
gznic conductances (V,~Z = 1.8 ± 0.3, 8.7 ± 2.1, and 5.7 ± 1.1mV for non-Tg, Tg-W'I', and Tg-C73A, respectively), but thesedifferences were not statistically significant.
Overall, these data indicate that the transgenic channels areexpressed in neurons ofthe cortex and hippocampus and that thecys73 to ala replacement in KCNB1 does not affect the channels'ftmction~l ~tttribt~tes ortheir ~tbilityto cluster in the pl~sm~ mem-brane. We cannot rule out that the resolution of our analysis mayhave missed micro differences in cluster distributions of the
transgenic channels. However, since clustering affects KCNB1conduction (O'Connell et al., 2006, 2010; Fox et al., 2013), con-sidering that there were no differences in total outward K + cur-rents expressed in Tg-WT and Tg-C73A hippocampal neurons,
this possibility seems unlikely.
D1.0 anti-HA
0 0.~ ,~
0 0.5 0.~ ,~~ ~
o ~ o
0.0 -~r—r~~~ ~o ~~ 1
Yu et al. • Oxidation of KCN81 in TBI
un-injured
~~ ~ L~Pc~c Sao tau
anti-HA (CHO cells)
1.0
# t
0.5
0.0'1~ 1~ O'•
WT:C73A cDNA ratio
NBt oxidation islow in theTg-t73A brain.A, Representative Western blots oftotal KCN61 channels inmicein theabsencein thesamplebufferofdenaturantsorreducing agentsorin thepresenceof1.0 mM
ent DTT. B, RepresentativeWestem blots oftotal KCN61 channeh in the injured (quantification in Q orated genotypes in the absence ofdenaturants or redudng agents. C, Densitometric quanHficaHon ofatio) using an antibody against total KCN61(anti-KCN61,N = 5 experiments) or an antibody againstexperiments). In experimentswithanti-KtNB1, p = 0.005, with pairwisee comparisonsall statis6caltyerws non-Tg (one-way ANOVA/fukeys post hoc test). In experiments with anti-HA, p = 0.011(two-daHonratios ofwild-rypeand 03A hKCNB1 channels expressed in CHO cells at the indicated stoichi-ti-HA. p = 0.00012, with pairwisecompariwns all statistkaltysignificant (one-way ANOVA/Tuke~smenu.*p < 0.05; **p <.0.01.
Oxidation of KCNB 1 is negligible in Tg-C73A miceThe LFP injury enabled us to expose live animals to conditions of
oaudative stress in a reproducible fashion. However, this ap-
control DTDP
3.0Q~ 2.5~'y 2.0a~~ 1.5
~ 1.0c'x~ 0.5Q
~~
c~ 'rV7
figare5. Tg-C73Ahippocampalneuronsareresistanttooxidant-inducedapoptosis.Repre-senWtiveimages ofcultured primary hippo<ampal neurons of the indicted genotypes in con-trol (no oxidant, bright liyht) orexpased to 50 µM OTDP for 5 min and staineQwith MnexNi-V6hafteroxidatlon (fluorescentlight).QuantiWtiveassessmentofMnexin-Vfluorescence(pro-portional to the number of cells undergoing apaptosis, a~bihary unite) 6 h after oxldafion isshown on the right Scale bars:200 µm. p c 0.0001, with pairwisecompariwnsall statisticaltysignificantexceptnon-Tg verwsTg-Wf (one-way ANOVA/fukey's postha test). Neuronswereobtained horn N = 2 non-Tg, 4 Tg-VdT, and 4 Tg-C73A embryos. *p < 0.05.
anti-KCNB11.0 r----I
,r
0.5
0.0y~ .~A ,bP
~~ Cp ~~ 1

Yu etal. • Oxidation of KCN61 in TBI J. Neurosci., 0~tober 26, 2016.36(43):1108 4-1 1 096.11089
A 100 B 100 C 100 ~ 50
"~ pre-trial.. -~• * * * y ~ 40
pre-trial := * ~- L
co
U 50 U 50 U 50 sham o Z~
~ ,iccep ,+~-+ U
J J —~1 G
injured J 10
0 0 0 0
c °' ~~.~3P 10 20 10 20 ~°' ~~ ~3P
~° ~~ o~ Uay post injury Day post injury ro .~~
E F G50 50
* injured ,~
.o
30 * * ~ 30 sham ~.a * a ~
~ 20 ~ 20 °'U U ~''
~ ~Q y ~0
J J Eh--
2 3 4 5 6 7 2 3 4 5 6 7
Day post injury Day post injury
40 **--,--,
~~~~~~
oc~~ ~n~PL
~~
m.,0.6
E
~ 0.4
~aN
E0.2
C
d
2 3 4 5 6 7
Day post injury
Figure6. KCNB1oxidationnegadvelyaffecubehavioraloutcome.A,Latenrytofallfromtherotadngrodforthelndicatedgroupsofmice2dbeforesurgery.Differencesbetweenmeansarenotstatisticalty significant (one-way ANOVA). N =16,16, and 16 for non-Tg, Tg-Wf, and Tg-C73A groups, respectively. 8, latenry to fall from the rotaHngrodfor inJurednon-Tg (circles), Tg-Wf(squares), and Tg-C73A (Mangles) mke. p <— 0.014 for longitudinal statlstical comparisons at each individual dpi (one-way ANOVA). Pairvvise comparisons are all stadsticalty significant exceptTg-Wf versus non-Tg at 1 dpl, Tg-Vllf versus non-Tg at 21 dpi, and non-Tg versus Tg-03A at 21 dpi. (Tukey's postha test). In three experimenu, N = 7, 7, and 8 mice, respectively, for non-Tg,Tg-WT, and Tg-Q3A. C, Latenry to fall from the rotating rod for non-Tg (drcles), Tg-WT (square), and Tg-C73A (triangles) shams. Differences between means are not statisticalty signficant
(one-way ANOVA). Inthreeexperiments, N = 7, 7, and 8 mice, respectNdy, for non-Tg, Tg-WT, and Tg-C73A. D, latency to reach the platform of the indicated genotypes 4 d before surgery.
Differences between means are not statistically significant (one-way ANOVA). N = 21,19, and 17, respectivety, for non-Tg, Tg-WT, and Tg-C73A mice. f, Latenry to reach the platform of injured
non-Tg (circles), Tg-WT (squares), and Tg-O3A (fiangles) mice. p < 0.0007 for longitudinal stathtical comparisons at each individual dpi (on~way ANOVA). Pairwise comparimns are all
statistically significant exceptTg-Nlf versus non-Tg at 2 dpi, non-Tg verws Tg-C73A at 3 dpi, and non-Tg versus Tg-C73A at4 dpi (Tukey's posthoc test). In six experiments, N =12,10, and 9 mice,
respectively, for non-ig, Tg-WT, and Tg-C73A. F, Latenry to reach the platform ofnon-Tg (circles), Tg-IN~ (squares), and Tg-L73A (triangles) shams. The response ofinjured Tg-C73A mice (dotted
line) isshown forcompariwn. Differencesbetween means are notstatistically significant(one-way ANOUA),Insixexperiments, N =1, 7, and 7 mice, respectively, for non-Tg,Tg-WT, and Tg-C73A.
G, Conwlidated memory retention test forthe indicated groups of mice. For LFP-injured mice, p = 0.0001, with pairwise comparisons all sCatlsticalty significant (one-way ANOVA/Tukey's posthoc
test). For sham mice, differences between means are not statistically significant (one-way ANOYA). In six e~cperiments, N =12,10, and 9 mice, respectively, for non-Tg, Tg-Nlf, and Tg-(73A, and
N = 7, 7, and 7 mice for their respective shams. H, Mean swimming speeds of injured and sham mice. Symbols are as in the otherpanels.Differences between means are not statisticaly significant
(one-way ANOVA). Speedswerecakulatedusing EthoVision XT software.ln slxexperiments, N =12,10, and 9 mice, respectivety,fornon-Tg,Tg-WT, andTg-C73A, andN = 7, 7, and 7 micefortheir
respective shams. *p < 0.05; ~"`p < 0.01.
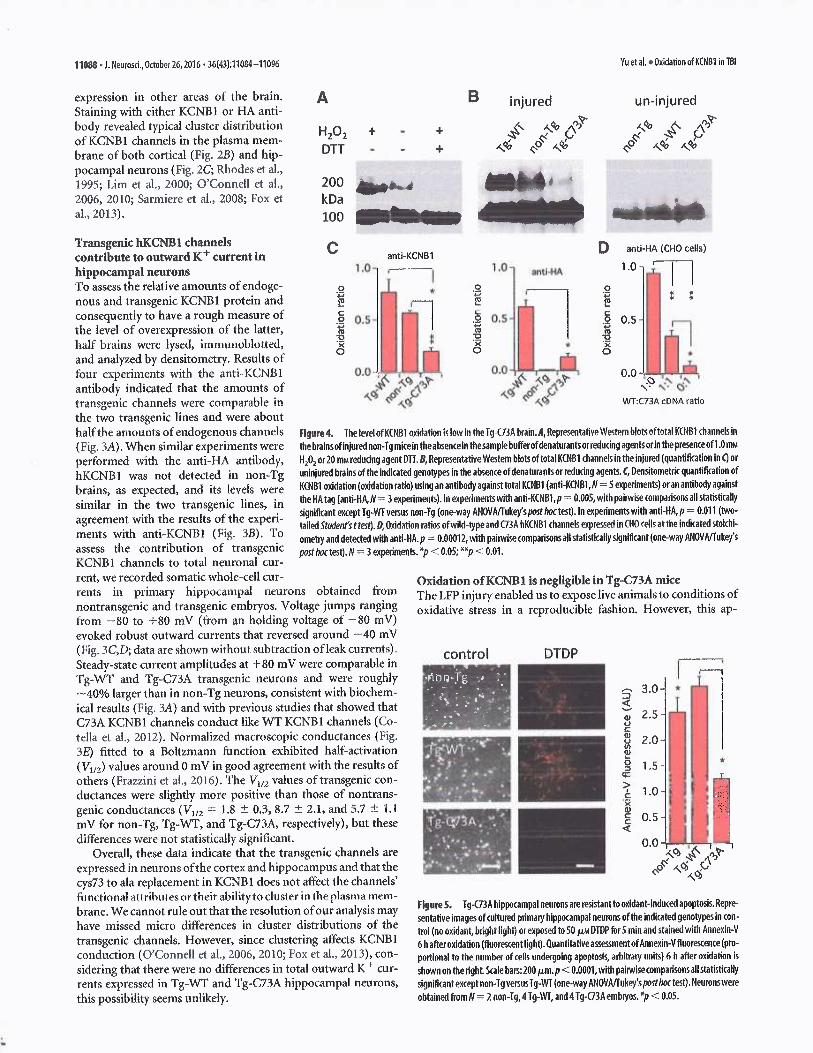
proach was valid only if KCNB1 oxidation remained low in theinjured Tg-C73A brain. To answer this crucial question we as-sessedthe eactent of KCNB 1 oxidation in the brains of the variousgenotypes using Western blot analysis as done previously (Co-tella et a(., 2012). Oxidized KCNB1 channels in the mouse brainor heterologously expressed in mammalian cells form oligomersheld together Uy disulfide bridges that run with multiple molec-ularmasses ranging from 170 to 500 kDa and that are suppressedby reducing agents DTT and (3-mercaptoethanol (Cotella et al.,2012). Tndeeci, a fraction oftotal KCNR1 channels from lysates ofinjured non-Tg brains were run as oligomers with a molecularmass of --200 kDa in the absence of reducing or denaturingagents (Fig. 4A). This fraction of oligomers was enriched by~--40% following exposure to 1.0 mtv~ hydrogen peroxide (H102)and was abolished by 20 mNt D'TT. KCNB 1 oligomers were pres-ent in lysates of injured Tg-WT and non-Tg brains and in verylow amounts in Tg-C73A lysates~ but not in uninjured brains
(Fig. 4A). Densitometric analysis indicated that the ratio betweenthe oligomeric and monomeric bands (oxidation ratio; Cotella etal., 2012), which gives a measure ofthe level of KCNB 1 oxidation,was moderately increased in Tg-WT mice compared to non-Tgmice (359'o increase; Fig. 4C), and most importantly, this ratiowas significantly decreased in the brains of Tg-C73A compared tonon-Tg mice (70% decrease). Similar oJcidation ratios were ob-tained asing the anti-HA antibody, e:rcept that no protein wasdetected in lysates of non-Tg brains, as expected (Fig. 4C). Thelow level of KCNB1 oxidation in the Tg-C73A brain c.nuld liedue to the formation of heteromeric channels composed ofendogenous and transgenic subunits, as the mouse and humanchannel share 97% amino acid sequence identity (CLUSTALWalignment, https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_clustalw.html). To test this idea, we ex-pressed WT/C73A heterometric channels in Chinese hamster
ovary cells and oxidized them with 1.0 r:i;~~ hydrogen peroxide

11090 • J. Neurosci., October 26, 2016.36(43):11084-11096
as done previously {Cotella et ai., 2012).In cells transfected with an equal ratio ofwild-type and C73A cDNA, the oxida-tion ratio was roughly one-third that ofcells expressing the wild-type channelalone (Fig. 4D), a fraction comparable#o that detected in injtrrecj Tg-C73Abrains. The lack of oligomerization pre-vents C73A mutant channels heterolo-gously expressed in mammalian cells toinduce apoptosis in response to an oxi-dative challenge (Wu et al., 2013). SinceKCNB1 oligomerization was low in Tg-C73A brains, their neurons should. beresistant to o~udant-induced apoptosis.Therefore, we challenged cultures of pri-maryhippocampal neurons with DTDP, apotent oxidant, and assessed apoptosis.Representative pictures of cells in controlor oxidized and stained with apoptoticmarker Annexin-V are shown in Figure 5.Thus, in agreement with previous studies(Redman et al., 2007), non-Tg neuronswere susceptible to DTDF-induced apo-ptosis. However, while Tg-WT neuronsexhibited levels of apoptosis comparableto those of non-Tg cells, Tg-C73A neu-rons were significantly less susceptible.Together, these data indicated that oxida-tion of KCNB1 and the toxic effects asso-ciated with it are low in the brains of Tg-C73A mice.
non-T T -WT T -C73A
'c
N
WU
Et
Tg-C73A mice perform better thannan-Tg and Tg-WT mice on the rotarodTo determine the impact of NCNB 1 chan- ~nel o~dation on the function ofthe brain, v ~ ~we tested the animals on the acceleratingrotarod device (rotarod) and the MWM,which respectively provide sensitive mea-sures of the function of the motor and ~ ~A ~ ,gP ~Asensory cortex and the hippocampus co~~~~~coc.~(Smith et al., 1995; Laurer and McIntosh,1999), the areas of the brain most affected injuredby the injury in our LFP model (the injury
F~ure8. Typical lesionsoccurs at the motor and sensory cortex, antibody per hippocampalwhich is overlying the hippocampus; Al- ~gni&antexceptshamnon-der et al., 2011). Two days before under- ~g~~ions(3brains,2fieldgoing surgery, the mice were exposed to genotypesatfdpi.p<o.0the rotarod paradigm. This procedure sham non-TgversusshamTserved the double purpose of allowing the (2-3 brains, 2 fieldsofviewanimals to acclimatize to thenewprotocol oftheindicatedgenotypesaand allowing us to identify impaired ani- shamTg-WT,non-Tgversusmall or detect differences due to genotype angle mean was calculatedand normalize data to baseline response.Hc7wever, ~tll mice behaved similarly dtrring pretesting before in-jury,indicating that genotype did not affect baseline motor func-tion (Fig. 6A). Tn contrast, mice subjected to the LFP injury had ashorter latency to fall relative to their shams, and most impor-tantly, Tg-C73A injured mice could remain longer on the rotat-ing cylinder than Tg-WT or non-Tg mice (Fig. 6B). There werenn significant differences between the latencies ofthe shams (Fig.6C), strengthening the notion that in the absence of events that
~~3P~~
sham
injured sham
-a 80 GFAPa~
~ o so 21 dpi.~40 **
N
v~ ZO
~ o
~s` 3 ~` ~~ , • co'~AA~,~co A~~ P
injured shamFigure 7. Inflammatan is decreased in the cortex ofTg-C73A mke following the LFP injury. Representative imagesofcoronalcortical sections from the brains of the indicated genotypes stained with GFAP antibody and the mean number of cells positive forGfAP per section at the indicaked dpi. Scale bar, 200 µm. At both 7 and 21 dpi, p ~ O.Od01, with painaise comparisotn allstatistically signlfiwnt except sham non-Tg versus sham Tg-Wf (on~way ANWMukey's posthoc test). Ead~ single mean wascakulated hom 18 sections from 3 brains (6 fields of view/section). **p < 0.01.
A **
20 GFAP
g **
zoFLJC
0.~N tOdv
0
o ~0 5~~ oc g~~~Pc .~0 0 ~o;
injured sham
C **
c 6 I i"F'I I; ~ caspase-37( 1t
~4~11~1
Yu et al. • Oxidation of KCNBi in TBI
**
2
T'o ,. ;co .~ ~ A ~,~c ~ A 1~P
injured sham
ofTBl are ameliorated inthe hippocampi ofTg-C73A mice.A, Mean number of cells positive for GFAPsection of the indicated genotypes at 7 dpi. p < 0.0001 with pairwise compariwns all statisticallyTgversussham Tg-WT (one-wayANOVIUTukey'sposthoctest). Each single meanwascalculatedfroms ofview/section).8, Mean numberofcdls positive for fUC per hippocampal section ofthe indicated001 with pairwise comparisons all statistically significant except sham non-Tg versus sham Tg-WT andg-C73A (one-way ANOVMukey's posthoctest). Each single mean was calwlated from 8 —12 sections/section). (, Mean numberof cells positive for activated caspase-3 antibody per hippacampal seRiont 7 dpi. p < 0.0001 with pairwlse comparisons all statisticalty significantexceptshamnon-Tg versussham Tg-C73A, and sham Tg-VYT versus sham Tg-C73A (one-way ANOVA/fukey's post ha test). Eachfrom 18 sections (3 brains, 2 fieMs ofview/section). **p < 0.01.
trigger the release of ROS, the transgenic mice are normal atbaseline. In all groups of mice, latencies reached a peak aroundthe first week after injury and then either remained stable or, as inthe case of Tg-C73A, moderately decreased. This trend probablyreflects the fact that the animals were getting acquainted to thedevice (in fact, latencies were shorter presurgery, probably be-cause the animals experienced the device for the first time; Fig.6A). We and others have observed this behavior in previous stud-

Yu et al. • O~dadon of KCN61 in i6~
Ta51e 1. Cortical sectlons stainings
Injured
J. Neurosci., October 2b, 201b • 35(43):19084-i109b ~ it091
Shamnon-Tg Tg-Wf Tg-C73A non-Tg Tg-WT Tg-C73AFUC 72.4 ± 33 (9) 885 -* 2.T (9) 482 ± 15 (11) 27.0 ± 2S (1z) 335 -* 1.1(12} 20.1 ± 0.9 (8)
Caspase-3 24.4 ± 0.6 (18) 30.6 ± 0.9 (18) 18S ± 0.6 (18) 5.7 ± 0.2 (18) 6.7 ± 03 (18) 4.6 ± 03 (18)Vehkle pautinib Vehicle DasatinibGfAP 74.6 ± 2.4 (18) 37.0 -* 1.7 (}8) 19.6 ± 0.6 (18) 15.0 ± 0.7 (18)FUC 61.9 ± 1.6 (12) 31.8 ± 1.2 (12) T7.9 t 0.9 (18) 12.7 -±- 0.9 (18)Mean num6ero(ceNspercorticalsectbn, positiveforthe indicated markersat 1dpi. The numberofsectbns (maximumnurrberofsedwns/brain,6) isIndicated inparentheses. ForFUC,toprow,p <0.0001,exceptforshamnon-Tgversussham Tg-WT and sham non-Tgversus sham Tg-Q3A (not rtatis6caNy signifiaM). forcaspase-3,p <0.0001, except forsham non-Tg versus sham Tg-WT, shamnort-Tgversm sham Tg-03A, and shamTg-Wiversus shamTg~L73A (notstatistkalty sgnifrcant). ForGFAP, p < 0.0001,except forsham vehideversus shamdasatinib (nMstatlsticaflysignificant). ForFUC,bottom mw, px:0.0001, and all painvaecomparisons are statisticaly significant (one-wayANOVA/Tukey iposthoctest).
ies (Cernak et al., 2014; Chen et al., 2014; Rowe et al., 2014; Alderet al., Z01 G), and since it was independent ofthe injury, we did notinvestigate this matter further.
Tg-C73A mice perform better than non-Tg and Tg-WT micein the Morris water mazeSpatial learning and memory were assessed in the MWM usingindependent groups of mice. As observed in the rotarod test, allmice behaved similarly in the presurgery trial using a visible plat-form, and therefore baseline adjustments were not performed(Fig. 6D). During the training period after surgery (Pig. 6E~ andin the test of memory consolidation that was administered theday after the last training (Fig. 6G), injured. Tg-C73A mice per-formed significantly better than non-Tg and Tg-WT', with thelatter being the most cognitively impaired. Indeed, injured Tg-C73A animals did not e~ibit any appreciable loss of cognitiveliznction in the MWM (Fig. 6F, compaze injured mice andshams). To exclude (or quantify) that impaired motor functioncould have affected the ability of the animals to reach the piat-form, wemeasured swimming speeds, which were comparable inthe various groups of nice, suggesting that the improved outcome in the Tg-C73A mice was not due to increased activity(Fig. bH}.
Typical histological lesions of TBI are reduced in cortex andhippocampus of Tg-C73A miceinflammation, disniption of axonal transport followed by axonalswelling, and finally degeneration and neuronal death are threehallmark lesions of TBI that affect many processes including mo-tor function, memory, and. spatial learning (Smith et al., 1991;Miyazaki et x1.,1992; Hamm et x1.,1993; Hicks et a1.,1993;Dixonet x1.,1999; Royo et al., 2003; Johnson et al.> 2013; DeKosky et al.,2013). Therefore, we compared the extent ofthese lesions in cor-onal sections of the region of injury throughout the cortex andhippocampus that we stained against glial fibrillary acidic protein(cortex, Fig. 7; hippocampus, Fig. 8A), which, being a marker forglia (astrocytes) and since gliosis accompanies inflammation,provides indirect evidence for inflammation (Jacque et al., 1978;Vos et al., 2010); Fluoro-Jade C (FLJC, hippocampus, Fig. SB;cortex, Table 1), a markerfor neurodegeneration (Schmued et al.,1997); and activated caspase-3 (hippocampus> Fig. 8C; cortex,Table 1), a marker fcr neuronal apoptnsis (cas~ase-3 stainingcolocalizes with NeuN but not GFAP; Beer et al., 2000; Clark etal., 2000; McEwen and Springer, 2005; Alder et al., 2016). Repre-sentativeimages of GFAP staining in cortical sections of the var-ious genotypes at 7 and 21 dpi along with quantitative analysesare shown in Figure 7. Thus, consistent with the course of atraumatic event, GFAP staining revealed the onset of an inflam-
m~CII~k' ~rp~~~~, remitting over time that was high in T~~WT and
A80
x60
.,.. s
~ 40
'~' Z~ injured
to 20
B80
v <~~j.
60 r0
c~' 40~ shamJ
zo -r--,--~T10 20
Day post injury Day post injuryCv 50
4Q * Injured
a 30a x~° 20 ~ r
toU r-r-r-r-r-i
2 3 4 5 6 7Day post injury
E
Dv 50
40 sham
0 30a r
20
~o
o ~-r-,--r-~2 3 4 5 6 7Day post injury
F~ 40
v
~ 20
~ o
Jer ayaO
0.6E
~ d.4a
0.2N
~ 0.0 ~r~2 3 4 5 6 7Day post injury
Figure 9. Dasatlnib improves behavioral outcome in lFP-injured mice. A, latenry ro fallfrom the rotating rod of non-Tg injured mice injected intraperitonealty da1y with 23 mgJkgdasafinib (diamonds) or vehicle (drdes). p s 0.039 for statistical comparisons at each individ-ua!dpi (one-tailed Students t test). !n five experiments, N = 8 and 8 animals for vehick~ anddasatlnib-injected mke, respectively. 8, Latenry to fall from the rotating rod of sham non-Tgmice injected with dasatlnib (diamonds) orvehicle (circles). Differencesbetween meansare notstatistkaltysignificant(one-tailed Student'sttest),Infiveexperiments,N= 8and7animalsforvehicle-and dasatinib-injected mice, respectively. C, latenry to ready the platform of injurednon-Tg mice injected with dasaUn~b (d'~amonds) a vehicle (circles). Where indicatcd,p < 0.045 faste6riicalcompariwnsatead~ inckvidual dpi (one-4ikdStudenYsttest),In fivee~erimenhs,N = 9and9 animalsforvehide-anQclasadnil~injectedmice, respectin~dy. D, Latwxytoreachthe p6itfamofsham non-Tg mke treated with dasatinib (diamonds) orvehkle(drdes).Thedotted the indkatesinjured micetr~eated with dasatlnib. Differences between meansarenotstaHsdcally signiflcant(one-tailedStudents ttest). Infiveexperiment, N = 8 and 8animals forvehidrand dasatlnib-injectedmke,respectivdy.E,Coriwkdatedmemory retentlontestfatheindkatedgroupsofmke.p = 0.025(one~tai7ed Students t tesQ. In five expetinteiits, N - 9 and 9 animate fur veAide- and dasatin~rInf ected mkerespectively, and N = 8and 8 forthdrres~ectiveshams.f, Mean swimming speedsofinjuredEfiUed~+m6ols) andsham(houowrymbds)micetreatedwi~hdasatinib(diamonds}orvehicle{drcies?. ~►ences between meaosarenotstatlsdcaly~gniflcant(one-wayANOVA),In fiveexper-iments,N = 9 and 9animakforvehide-and dasatinitrinjected mke, res~ectivety, and N = 8 and 8rnrm~irr s~m~ *a ~ o.~: ~*~ ~ a,ot,

11092.1. Neurosci., October 2b, 2016.36(43):11084-11096 Yu et al, • Oxidation of KC~IB1 in TBI
low in Tg-C73A sections. Results of im- 7 dpi 14 dpi 21 dpi vehiclemunostaining in hippocampal sections at7 dpi are summarized in Figure 8 and in
.~`s~~ 20cortical sections in Table 1. In all cases,damage was maxima( in Tg-WT and min-
~ ~ oimal in Tg-C73A brains underscoring the ~ ~ oexistence of a causative relationship be- ~
tween KNCB1 oacidation, tissue destruc- ~ 0 20 dasatinibtion, and behavioral deficit following the
LFP injury. Thus, where KCNB1 oxida- ~ ~otion was exacerbated, neuronal damage v pwas increased and the outcome of TBI was
severe, whereas where KCNB1 oxidationwas suppressed, the outcome of TBI,
4 20along with tissue damage, were improved.We further notice that inflammation,
~ to shamveh.neurodegeneration, and apoptosis were
~ omoderately more pronounced in the ~sham brains of Tg-WT compared to ~
4 zonon-Tg and Tg-C73A micc. This may in-
~ sham das.dicate moderate oxidation in the shams,
~ ~ ~probably due to the surgery, or, alterna- ~ ptively, the existence of KCNBI-
,~aQ'1,,aQ~ti,~aQ~independent mechanisms that contriUuteto tissue damage.Weconcludethato~da- Figure l0. ApoptosisisdecreasedinthecortexofmicetreatedwithdasaNnib.Exemplarimagesofcorticalsectionsstainedwithtion of KCNB1 channels resulting from anandbodyagainstactivatedcaspase-3andmeannumberofcelkpositiveforanti-caspase-3persectionattheindicateddpi.5cakTBI is an event that contributes toward bar, 200 µm. p < 0.0001 for longitudinal statistical anatyses at each individual dpi with pairwise comparisons all statisticaltytissue damage and impacts behavioral ~9~~~antexceptshamvehicleversusshamdasatlmbat7dpi,shamvehideversusshamdasati~batl4dpi,andinjureddasaNniboutcomes. versus sham vehicle at 21 dpi (one-way ANOVA/fukey's post hottest). Each single mean was calculated from 12 sedans (2 brains)at 7 dpi or from 18 sections (3 brains) at 14 and 21 dpi (6 fields ofview/section). *"p < 0.01.Mice treated with dasatinib performbetter than vehicle mice in the rotarodPrevious in vitro sCudies showedthat o~cidation ofhKCNBl chan-nels favors the activation of Src tyrosine kinases, which in turninitiate an apoptoric cascade (Fig. l; Wu et al., 2013). Therefore,we sought to determine whether phazmacological strategies thatact to inhibit the activities of Src kinases could improve neuronallesions and behavioral outcome of LFP-injured animals. To thisend we used dasat'rnib, an FDA-approved Src kinases inhibitorthat is blood—brain barrier permeable and pharmacologically ac-tive in the brain (Porkka et al., 2008; Agarwal et al., 2012). Miceinjected with dasatinib (25 mg/kg daily starting 2 h after surgery;Luo et al.> 2006; Hasi~na and Aggarwal, 2012; Katsumata et al.,2012) or vehicle and their respective shams were subjected tothe rotarod and the MWM protocols, and the results of theseexperiments are illustrated in Figure 9. Thus, dasatinib signif-icantly incrcascd latency to fall at all days after injury rclativcto vehicle-treated mice (Fig. 9A) and had no effect on shamanimals (Fig. 9B).
Mice treated with dasatinib performed better than vehiclemice in the Morris water mazeDuring the 6 d training period (Figs. 9C,D) and in the memoryretention test the following day (Fig. 9E), injured mice in-jectedwith dasatinib performed significantly better than vehi-cle mice. In fact, there was no significant difference in thebehavioral responses of injured mice treated with the drug andthe shams (Fig. 9D). Mean swimming speeds, displayed inFigure 9F, were similar in all groups, excluding that differ-ences in the fitness of the animals might have impacted theoutcome of the tests.V UII.V lll~. Vl llll, l4JtJ.
Histological lesions of TBI are reduced in the brains of micetreated with dasatinibCortical and hippocampal sections were stained with anti-caspase-3 (Fig. 10, Table 1) and GFAP and FLJC (Fig. l 1, TaUte 1),As expected, dasatinib treatment resulted in a marked decrease ofthe number of cells positive for the various markers compared tovehicle in both cortical (Fig. 10, Table 1) and hippocampal sec-tions (Fig. 11). The number of positive cells to any marker wasmoderately higher in sham animals injected with vehicle com-pared to those injected with dasatinib (albeit these differenceswere generally not statistically significant). The protective effectof the drug in sham animals may reflect baseline oxidation ofKCNB1 channels or, alternatively, inflammatory processes asso-ciatedwith the surgery. We conclude that inhibition of the activ-ity of Srctyrosine kinases significandyameliorates typical lesionsof TBI, leading to improved motor function and spatial memoryduring the critical phase of the LFP injury.
Oxidation of KCNB 1 chamiels is associated with Srckinases activityTo determinewhetheroxidation afKCNBI channelswas respon-siblefor the activation ofthe Src kinases following the LFP injuryand therefore link the effect of dasatinib on recovery post TBI toKCNB1 oxidation, we biochemically assessed the fraction of ac-tivated Src kinases in the brains of our mice using an antiphns-phorylated Src family antibody that detects phosphorylationstatus of tyr416, a residue conserved in all members of the Srckinase family (Konig et al., 2008). Representative immunoblotsof total and activated (phosphorylated) Src proteins in the brainsof mice injected with vehicle or dasatinib and separately in thebrains ofnon-Tg, Tg-W'T, and Tg-C73A animals, along with den-sitometric analyses, are illustrated ;n Fi~~t,r~ ~ y. S~~ ~~t4~FiaJy rwi~aJ1LV111~. LA.II. ({11[31 J~.J~ [l.l1. lAlUJll (1 ~.1~ 11

Yu et al..Oxidation of KCNBt in TBI
is-~I~1 ts-~I 1 ,.
f+V
aU S
,er~~O~a~\~ra~ ~ra~ ae
5 h
'~
°v' s
[~7
er~~\G agcy Jec. day.J O,~y yratr yrpc~
CO
4U
N
v 2
0
Jr``\ 5~c`v ,er F aye Oa a ~~~~ hca
A non-Tg B
.~~~ a'rc F' C, ~~ v,~ ply g'c,~ '`~' '`~ ``°~ J~~
pSrc g7ml`e'L"+~Av~!+Hd9Guktf:-~y'l+at..+"~:~,:Ntd, c' .r-eM.l.~vk .tiz~diia
kDaSrc - 57
---1.0 i.o ~ t.o
N N .s:" ~0 0
0.5 L 0.5 ~ ~ 0.5n I- -o0 o p'd. o.
0.0 0.0 0.0.fie .c~ ,~(~ '~P '~a'Je'c Oaf'` ,~o; ~~-G~ coy Je
Figure 12. Thea<UvityafSrckinasesisneyligiMeinthebrainsafTy-C73Amice.A,Representa4and phosphorylated Src (pSrc) in the brains of injured non-Tg mice injected with vehicle ordasatinibnon-Tg mice, and inthe txains ofinjured Tg-WT, Tg-C73A, and non-Tg mice. Animals were killed aimmunopredpitatedwith anantibodythat recognrzestotal Srcand immunoblottedwith anantiphorylated at tyr416. Densitometric quantifications are expressed as the fractions of activated Srnormalized tototal5rc).p = 4.0017 forvehide/dasatlnib (one-tailed Student's hest; N = 3 expertTg-YVT, and Tg-C73A with painvise comparisons all statlsticallysignificant exceptTg-WT versus non-posthoc test; N = 3 experiments). 8, Representative Western blot showing KCN61 oligomeia in tmice injected with vehicle or dasatinib and densitometric quan4flca6on of three experiments. Aniimmunoblotted wfth anfi-KCN61 antlbody. p = 0.00045 with painvise comparisons all stadsticadasatinib versus sham vehicle and sham vehkleversus sham dasatinib (one-way ANOVA/Tukey's pn0.01.
significantly inhibited in animals subjected to dasatinib therapy,confirming that dasatinib acts upon the Src pathway (Fig. 12A).Most importantly, the fraction of activated Src kinases was de-creased in the brains of Tg-C73A animals and increased in thebrains of Tg-WT animals compared to control (Fig. 12A). Previ-ousstudies have indicated that Src kinases act to induce oxidative
J. Neuroxi., October 2G, 2016 ~ 36(43):11 08 4-11096.11093
stress, possibly via a pathway that destabi-lizes mitochondria (Wu et al., 2013). Thisimplies that KCNB1 oxidation should below in mice treated with dasatinib. In-deed, the amounts of KCNB1 oligomerswere significantly decreased in the brainsofmicetreated with the cin~gcampared tovehicle, and in fact, their amounts werecomparable to those of sham mice (Fig.12B).
Together these data indicate that in thebrains where oxidation of KCNB1 wasprevented, the activity of Src kinases wasnegligible, and vice versa, in the brainswhere Src activity was inhibited, oJcida-tion of KCNB 1 channels was also low. Weconclude that there is a causative relation-ship between oxidation of ICCNBI chan-nels and Src tyrosine kinases activities.
DiscussionTo determine the role that oxidation ofKCNB 1 K + channels has for the fianctionof the brain, we developed a transgenic
~ ~c~ ~~ Ooh mouse expressing a KCNB1 mutant~~o'~ ~F ~F (C73A) resistant to redox and pursued a
~a g'~ y'~ pharmacological approach that directlyimpinges on the apoptotic pathway acti-
~ #► 200 vated in response to oxidation of thekDa channel. We found that decreasing the
~M~ 100 amount of oxidizable ICCNBI channelsby genetic means is strongly protectivein a mouse model of moderate TBI, acondition characterized by high oxidative
** stress. Thus, neuronal damage induced bythe LFP injury was markedly reduced inTg-C73A mice, and consequently, theanimals exhibited improved behavioraloutcome after TBI. Moreover, the detri-mental effects of the neuroto~c pathwayactivated in responseto KCNB1 oxidation
,coe ,~,o~ Jer' Qay' could be neutralized by dasatinib, which
aya ~~ a~ ameliorated the devastating effects of TBIo ~,r yr during both the primary and secondaryinjury processes. Furthermore, Src kinaseveWestemMotshowingtotal activities were significantly depressed in,inthebrainsofvehiclesham the brains of Tg-C73A mice, and, vicet7d~i,andbrainlysateswere versa, the drug was associated with smallbody that recogn~es Src phos- amounts of oxidized channels. Overall,c kinases (phosphorylated Src these findings demonstrate that oxidationments);p = 0.011 for non-Tg,
Tg,(one-wayANOVA/fukeys of KCNB1 channels represents a mecha-hebrainsofinjuredandsham nism of cognitive and functional impair-mahwere kitled at 7 dpi and ment in verteUrates and further validate aity significant except injured molecular model for its toxicity that
sthatest).'~p<0.05;**p< emerged from in vitro evidence. Despite alarge volume of reseazch, no medicationhas Veen proven effective for the treat-ment of TBI in humans. Protein kinases
are one of the most investigated drug targets by the pharmaceu-tical industry, but the development ofkinase-based therapies forbrain diseases remains a challenge (Chico et al., 2009). Thus thesefindings not only underscore the pathological nature of oxidationof KCNB1 channels in TBI, but they may further suggest a new
Figure 11. Typical lesions of TBI are ameliorated in the hippocampi of mice Veated with dasa6nib. A, Mean number of celkpositive for GFAP per hippocampal section of the indicated groups of mice at 7 dpi. Each single mean was wkulated from 18sections (3 brains, 2 fields of view/section). B, Mean namber of cells positive ro FUC per hippocampal section of the indicatedgroups of mice at 7 dpf. Ead~ single mean was calculated from 12-18 sections (2-3 brains, 2 fields of viewlsedion). C, Meannumber ofcellspositive toanti-caspase-3 per hippocampal section of the indicated groups of mice at 7 dpi. Each single mean wascalculated from 12 sedans (2 brains, 2 fieMs of viewlsection). In A—C, p < 0.0 1 with pairwise comparisons all statisticallysigniflcant except sham vehide versus sham dasatinib (one-way ANOVA/fukey's posthoc test). **p < O.Oi.

1T~94 t 1. fle~itusci., Giinb~ 26, 20~6.35f#3):t 154-1;Q"n'r
etiera~~~zri~ a~pruaeh fir a wn~iiei~n chat affects ntiili~~Yi~worldwide.
~3r~ ~~r~;.r~t ~sr€~blsst~ cvi~~~ trans~es~i~, a~~ra~~.hes is the ~~Ee~_tial side effects of protein overexpression. The mice used in thisi~ae~e~t~~~i.a~~ *~z~•e n~.r~a~~l.4aa :~a~ ~~i~ ~u~i~lat~ s~~v~}a~}3~~. ~r-rnally, did not: present any tissue damage, did nat show any ap-paret~t ~~heno~v~e, xncl, limi#~ci t~ the rn~nitive ~hilitiec ~esesseein this stady, they were normal. 'Thus, it appears that a moderateKGNB 1 fain of function is well taleratied in the brain even thoughwe cannot cornpleYely exclude that the transgenic mice may havedeveloped some sork of cognitive decg~n~~ensati~n that went un-detected in our investigations. These findings also argue againstthe idea that augmented KCNB 1 current is proapaptotic, as sug-gested by some in vitro studies (for review, see Sesti et al., 201:4).Moreover, since the N terminus and the C terminus of the chan-nel physical#y interact during the channeYs activation (Ju et at.,2003; I~obi•insky ~t al., 2006) and oligomexization probably linkstote trvv termini ta~et.her tihrough tlistx~fiit i~riciges, it is possii~Iethat the antiapoptotic effect of some KCNB 1 inhibitors (I,itY et a1.,2013; Zhou et al., 2016) stems fr~tn their atiilit}= to prc~ent oti-gomerization rather than conduction, or, alternatively, as in thecase e.f he~ie ~ ~~~e~aa~-1 in a=a all ry ~rzadel~ ~f Al;~lari~raer's ~iis-ease,through interfering with ICCNB 1 regalatory pathways (Het-tiarachchi et al., 2014 : In contrast, the 11f c,;~s of the LFP injurywere markedly aggravated in Tg-WT mice compared to non-Tginice> suggesting that the increased amount of oxidized KCNB1channels in the former is proportional to the extent of the neu-ranal damage and behavioral deficits. 5peca et al. (2014) haveshown that in KCNB 7 KO mice the absence of KCNB i currentcauses hyperexcitability, which in turn correlates with cognitiveimpairment and sasceptibitityto seizure. Oxidized KCNB~ chan-nels da not conduct current (Cotella et aL, 2U1L; Frazzini et' al.,2016}, an~i this may complain zhe cognitive impairment of LFi~-injured mice. On the other hand, the potent effect of dasatinibw~ul~ ax~ue that ~teui-unaf da~~~~~e is tic underlying Cause, but itmust be considered that inhibition of Src tyrosine kinases resultsin lo~~~r levels of o~dz~ed---mod ~~eref~r€ npncondueting---KCNB1 channels. Thus, it is likely that decreased current anda~+optotic stimuli both c.~ntril~ute to the neuronal and Uehavi~raldeficits observed in injured mice, and future investigations willdissect the individual contribution of each to TBI.
Oxidation of a K+ channel as a mechanism of neuronal vul-nerability was initially demonstrated in Caenorha6ditis elegans(Gal and Sesti, 2009). The channel that undergoes oxidation inthe worm, KVS-1, is a homolog.of KCNBI (Rojas et al., 2008),and in both channels the effects of oxidation are mediated byconserved cysteine residues, cys113 in KVS-1 and cys73 inNCNB I. I`he #inclings reported here that show that oxidation ofK ' channels contribute to cognitive impairment underscore thehigh degree of ~atiservation of this niechanis~~7 of neuronal vu~-nerabilityand filrther broaden its potential relevance. For exam-~S4e, it ~s well esla'uiis~~c~ chat tie agi~~g hi~~a~,a}npt~s of rodentsdevelops hyperexcitability (Landfeld et al., ].986; Barnes et al.,1987; Barnes, 1994; Papath~odoropoulos and Kostopoulos,1996), and it may not be coincidental that KCNB1 undergoesoxidation in the brains of naturally wing mice (~~tella et al..,2012). Moreover, KCNB1 channels are oxidized in hippocampalnearons oFthe 3x-Tg-AD mouse where they promote hyperex-citability (Frazzini et al., 2016), while inhibition of Src kinasesattenuates tnicrogliosis in BV2 marine cells incubated with~3-amyloid aligomers (Dhar~van and Combs, 2012). In summary,the evidence presented here would argue that oxidation ofKCNB1 channels is a mechanism that contributes to cognitive
Yu e; al..~x~dati-~i QiKEld61 in TS!
deficit iti nutntai a~ir~~ as vvefi in ~1lzheirnet's disease—albeit todifferent e~:tentsr— and by analogry, that dasatinib may represent aY~ti~ ~c~d~l €~€ t~~ra~ae~tic t~te~~ve~Atzofs its ~euraue~e~ratiyedisease.
ReferencesA~rwal S, lvtitta~atli 12K> Zellmer DM> Gallarda TL, Danelson R, Seiler C,
Decker SA, Santacruz KS, Pokorny JL, Sarkaria jN, Elmquist WF, OhlfestJR (2012) Active efflux of Dasatinib from the brlin lirniYs efficacya}~ainst n~urinz gti~bh3toma: biva~ inlpiii~ti~n~ for the ~-tini~i use ~fmolecularly targeted agents. Mol Cancer Ther 11:2183-2192. Cross RefMedline
Aigner L, Arber S, Kapfharnmer JP, Latix T, Schneider C, Botteri F, BrennerHR, Caroni P (X995) Overe~ression of the neural growth-associatedprotein vAi>-~#3 induces nerve sprouting in the aclutt nervous system oftransgenic mice. CeIl 83:269-278. Crossltef Meclline
Alder J, Fujioka W, t.ifshitz J, Crockett I>P, Thakker-Varia S (201 l) Lateralfluid percussion: model of traumatic brain injury in mice. J Vis Exp 54:e3063. Medlixze
Alder ), Fujioka W, Uiarratana A, Wissocki J,'i'hakkar k, Vuong Y, Yatel B,Chakraborty T, Elsabeh R, Parikh A, Girn HS, Crockett D, Thakker-VariaS (2~16j Genetic aatd plta~5nacological itate~ve~itin~ of c3xe g75~TTRpathway alters morphological and beliaviourat recovery following trau-matic brain injury in mice. Brain Inj 30:48-65. CrossRef~Meclline
$arnes C;A (1994} Normat aging: regionally specific changes in hippocam-palsynaptic transmission. Trends Nearosci 17:13-18. CrossRef Mediine
T+<ernec C.A., R~Zc~ G, A4cltiaugl~tLzn ~L (ly$"✓) incre~tssc$ e~eciratc~sis; mt~}`lingin aged rat }uippocampus: 1 possible mechanism for cellular excitlbllitychanges. J Corny Neuro1259:549-558. CrossRef Medline
Beer R, Franz G, Srinivasan A, Hayes TtL, Pike BR Newcomb )K, Zhao X,Schmutzhard E, Poewe W, Kampfl A (2000) Ternportl profile and cellseal~type da~tritr~ateaaa of actavated caspass-~ £~itQa~ e~eriancntat tr~u-matic brain injury. J Neurochenn 75:1264-1273. Meclline
Cai 5Q, Sesti F (2009) O~cidation of a potassiwm channel causes progressivesensory function loss during aging. Nat Neurosci 12:b11-617. CrossRefM.edline
Ger~~k i, Sh'iF~g I~, ~avids~3i 3> Fla~~t~~a~ti S (2010 A car=~3 a~~~~cse. s~cn~lei ofpenetrating brain injury. Front Neurol 5:209. Med(ine
Ghent,NTaoH,YangKH,AbeIT,Mu~neyDF (2014) Amadifiedcontrolledcortical impact technique to model mild traumatic brain injury mechan-ics in mice. Front Neuro15:100. Nleciline
Chico I,K, Va~~ Eit3ik LJ, f'Va~te~sajn I3Iv1 (2ETa3} Tar~tix~g ~nutes~a liinascs i~zcentr<1( nervous system disorders. Nat Rev 17rug Discov 8:892-909.CrassRef Medline
Chou JL, 5henoy DV, Thomas N, Choudhary PK, Laferia FM, Goodman SR,Breen GA (2011) Early dysregulation of rile mitochondria) proteome ina n~ause model of zh4zheinrer's disease. J Prat~ntics ,"4:~4Cib-4i9.CrossKef Medtine
Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon GE, Seidberg NA,Melick J, Laeffert JE, Nathaniel PD, Jin KL, Graham SH (2000)Caspase-3 mediated neuronal death after traumatic brain injury in rats.J Neuroshem 74J4t?-753. Medline
Corps KN, Roth T'L, McGavern DB (2015) Inflammation and neuroprotec-tion in Xraumatic brain injury. j:AMA .Neurol 72:355--362. CrossRefMedline
Cotella U, Hernandez-Enz'iquez B, Wu R, Li R, Pan Z, LeveIlle J, Link CD,Ud~o 5, Mesh F (2 12} Toxic role of ic+ chlnnel oacidaYion in mamma-lianbrain. J Neurosci 32:4133-4144. CrossRef IvTedline
DeKasky 3T, Rlennew K, Ikanc~movic MLA, Candy S (2A13) tict~te andchronic trautnaric encephalopathies: pathogenesis and biomarkers. NatRev Neuro19:192-200. CrossRef Iv[edline
Dhawan G, CornUs GK ("1012) Inhibirion of Src Icinase activity attenuatesamyloid associated microgliosis in a murine model ofAlzheimer's disease.T Neuxain4amnn~tLou 9:I1~. Medline
Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, Wolfson BM,Marion DW (1999) Amantadine improves wafer maze performancewithout afYecCing motorbehavior fof(owingtraumatic brain injury in rats.Restor Neurol Neurosci 14:285-294. Medline
L2~meniccana d~2, He;nnpsteaci PAL, Cl~;asa A4V (W(~47) PrQ-~C~£ secrEted~y as-trocytes promotes motor neuron cell death. Mol Cell Neurosci 34:271-279. CrossRef Medline
Du J, Hank LL, Phillips-Tansey E, Russell )T, McBain CT (2000) Frequency-

Yuetal:.OxidationrofK(N61 in TBF
dependent ~~egulation of rat 1~lppocam~al somato=denriritle e ecitabilityby the K+ channel subunit Kv2.1. J Physio1522: i9-31. C~rossRef Medline
FRx PD, Lo~#us RI, Ta~n~C~E~ ~Iit~ (2013) Regullt~on afKv2.~ K(+) conduc-tance Uycell surface channel density. J Neurosci 33:1259-1270. CrossRefMedline
rrazzir~i V, ~ita~`luetf S, Somba iVi, Ndval'i~a' R, IvlotaliiCo' f;, Mari~~id' IvllA,Sensi SL (2016) tlltered Kv2.1 funcrioning promotes increased excit-
akzilitK in h~~~Qcam~a~ ncux4ns of an P,lzheimgr's di~seasc maas~ m4dei.Cell Death Dis 7:e2100. CrossRef Nledline
Hamm I2J, Lyeth BG, Jenkins LW, O'Dell DM, Pike BR (1993) Selectivecogmirive impairment following traumatic brain injury in rats. Behav
Brain Res 59:169-173. CrossRef MedlineEIasima N, Aggarwal ~~ (2012} Caner=1iMked Farg~ts mocle~lHted Uy cur-.
cumin. Int J Biochem Mol Bio13:328-351. MledlineHettiarachclii N, Dallas Ivt, AI-Owais M, Griffiths H, Hooper N, Scragg J,
Boyle J, Peers C (2014) Heme oxygenase-1 protects against Alzheimer'sarnyloid-beta(1-42)-induced toxicity via carbon mono~tide producrion.Cell Deadt Dis S:e1569. CrossRef Medl ne
Hicks RR, Smith DH, Lowenstein DH, Saint M1rie R, McIntosh TK (1993)Mild experimental brain injury in the. rat induces cognitive deficits asso-ciated with regional neuronal loss in the hippocampus. J Neurotrauma10:405-414. CrossRef Medline
Hiebert JB, Shen Q, Thimcnesch AR, Pierce JD (2015) Tinumatic br<iui in-jury and mitochondrial dysfunction. Am J Med Sci 350:132-13$.crossxef Ntealine
Hille B (2001) Ionic channels of excitable membranes, Ed 3. Sunderland,NTt1: Sinauer.
Jacque CM, Vinner C, Kujas M, Raoul M, Racadot J, Baumann NA (1978)Determination of filial fibrillary acidic protein (GFAP) in human Uraintumor~5. J Neurol Sci 35:147-155. Crosslte£Medline
Johnson VE, Stewart W, Smifh DH (2013) A~conal pathology in traumaricbrain injury. F~tp Neurol 246:35-43. Meciline
Ju M, Stevens L, Leadbitter E, Wray ll (2003) The Roles of N- and
C-terminal determinants in the activation of the Kv2.1 potassium chan-nel. J Biol Chem 278:12769-12778. CrossRef Medline
Katsumata R, Ishigaki S, Katsuno M, Kawai K, Sone J, Huang Z, Adachi H,Tanaka F, Ur~no P, Sobue G (2012) c-Abl inliiUition delays motor neu-ron degeneration in the G93A mouse, an animal model of amyotrophiclateral sclerosis. PI.oS One 7:e46185. CrvssRef Medline
Icobxlnst y E, Stevens 1, Kazmi Y, Wray D, Soid~tov NM (2006) Moleculazrearrangements of the KVL1 potassium channel termini associated with
valta~C gating, J Brol Chem 287:19233-19240. CrossRef MedlmeRonig H, Copland M, Chu S, Jove R, Holyoake TL, Bhati~l R (2008) Effects
of dasatinib on S&C kinase activity and downstream intraceAular signal-ing inprimitive chronic myelogenous lcukeania liematopoietic cells. Can-cerRes 68:9624-9633. CrossRef Medline
Landfield PW, Pider TA, Applegate MD (1986) Theeffects of high_MgL-F-to-Ca2+ratios on frequencypotentiation in hippocampaislices ofyoungand aged rats. J Neurophysiol.56:797-811. Mecllirae
Laurer HL, Mcintosh TK (1999} Expernnental models of brain trauma.Carr Opin Newro112:715-721. GrossRef Tvtedline
~,ifshita J (2008) Fluid percussion injury model. In: Animal models of acute
neurological injiuies (Chen J, Xu ZC, Xu XM, Zhang )H, eds), pp 369-384. New York: Humana.
Lim ST, Antonucci DE, Scannevin RH, Trimmer JS (2000) A novel targetingsignal for proximal clustering of the Kv2.1 K+ channel in hippocampaineurons. 2Jeuron 2.538S-397: CrossRef Medline
Liu H, Liu J, Liang S, Xiong H (2013) Plasma gelsolin protects HIV-1 gp120-inducecl neuronal. injury via voltage-gates(. K channel Kv2.L Moll CellNeurosci 57:73-82. Medline
Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney D~, Davis J,NicMlliazr f35, Conte V, L.:iurer HL, Stein S, 5tocchetci N, Mctiztasli'1'K(2005) Temporal window of vulnerability to reperitive eacperiment~llronei~ssive bra9n injiery. Ns t;ros~trgery 56:364-3'74; disctLssic~n 364-374,CrossRef IVtedline
Luo FR, Yang Z, Camuso A, Smykla R, McGlinchey K, Falter K, Flefleh C,Castaneda S, Inigo I, Kan D, Wen ML, Kramer R, Blackwood-Chirchir A,Lee FY (2006) Dasatinib (BMS-354825) pharmacokinerics and pharma-endgry3am+_e.l~ian~xrkera_sn.;~r~ianal..maclPic ~xre.~lcf n}zt+_t+_v~1 c~~+ica_I ea~n-sure. Clip Cancer Res 12:7180-9186. Cross.Ref Medl9ne
M.asel BE, DeWitt DS~2010) Traumatic brain injury: a disuse process, not
an event. j Neurotrauma 27:1529-1540. CrassRef Mediine
J: Neurosci:, OetobeF26; 2076 ~ 3643):11084-71096•• tY@95
IvF~Ewcn Mi., Syringer JL (2005} A ma~yin~ study of easpase-3lhvariojrfollowing acute spinal cord contusion its rats. J Histochem Cytochem
53:8Q9-819. CrossRef MedlineMcIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faders AL
(1989) Traumatic brain injury in the rat: characterization of a lateralfluid=percussion model. Neazoscience 28:233-244: CrossRef Medline
McManus MJ, MurphyNiP, Franklin JL (2011) The mitochondria-targeted
~r~tiu~cidant MitoQ prevznts less of s~aaYial tnetnoiy retenrion :trjc~ t~trlyneuropathology in a transgenic mouse model of Alzheimer's disease.J Neurosci 31:15703-15715. Cz~ossRef Medline
Miyazaki S, Katayama Y, Lyeth BG, Jenkins LW, DeWitt DS, Goldberg SJ,Newlon PG, I-Tayes RL (1992) Enduring suppression of hippocampaltong-tet~m yotentiation following trawnatic brain injury in rat. ~inrn Res585:335-339. CrossRef Medlrne
MuzakoshiH,'I'rimmerJS (1994) IdentificationoftheKv2.~TC+channel asa major component of the delayed rectifier K+ current in rat hippoclm-
pal neurons. J Nearosci 19:1728-1735. MediineO'C;onnell KM, Rolig AS, Whitesell )D, Tamkun MM (2006) Kv2.1 yotas-
sium channels are retained witihin dynamic cell surface microdomains
that are defined by ~ }~erlmeter fence. J Neut~~sci 2f:9609-9C,18. C:xossRefNiediine
O'Connell KM, Loftus R, Tamkun MM (2010) Loc~tli~ltion-dependent ac-
tivity ofthe Kv2.i dehyed-rectifier ~C+ channel. Proc Natl Acid Sci U S A107:12351-12356. CrossRef Medtine
Oddu S, Caccamo A, Shepherd JD, Murphy MP, Uolde TE; Kayed R, Mether-
~te R, Tvlattson MP, Akbari Y, LaFerla PM (2003) Triple-transgenic
model ofAlzhenner's.disease with plaques andtangles: intrncellularAGetaand synaptic dysfunction. Neuron 39:409-421. CrossRef Medline
Papafheodaropoulos C, Kostopoulos G (1996} Age-related changes in ex-citabilityand recurrent inhibition in the rat CA1 hippocampal region. EurJ Neurosci 8:510-520. CrossRef Meclline
Patel R, Sesti F (2016) Qxid~tian of ion channels ici the lging nervous sys-tem. Brain Res 1639:174-185. CrossRef Med(ine
Porkka K, Koskenvasa P, Lunddn T, Rimpii~inen 1, Mustjoki S, Smykla R,Wild R, Luo R, Arnan M, $rethon B, Eccersley L, Hjorth-Hansen H,
HSglund M, IClamova H, Knutsen H, Parikh S, Raffouu E, Gruber F,Brito-Babapul2e ~, F~anibret H; et al. (2008) Dasatinib crosses theblood-brain Uarrier and is an efficient therapy for central nervous systemPhiladelphia chromosome-positive leukemia. Blood. 112:1005-1012.CrossRef Medline
Redman PT, He K, I-Iartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitln
ES, Aizenman E (2007) Apuptotic surge of potassium currents is medi-ated Uy p38 phosphorylaYion of Kv2.1. Proc Natl Acad Sci U S A 1.04:356&-3573. CrpssRef Medline
Rhodes Kj, Keflbaugh SA, Barrezueta NX, Lopez KL, Trimmer JS (1995)Association and colocalization of K+ channel alpha- and Ueta-suUunityolypeytides in rat brain. J Neurosci 15:5,360-53'71. Medline
Rojas P, Garst-Orozco J, Baban B, de Santiago-Castillo JA, CovarruUias M,5aikoffL (2008) Gumulativeacrivationofvoltage-dcpendentKVS-lyo-tassium channels. J Neurosci 28:757-765. CrassRef Ntedtins
Roth. TL, Nayak D, Atanasijevic T, Kozetsky AP, Latour LL, McGavern DB
(2014) Tthnscranial amelioration of inflammation and cell death afterbntinvnjury. Nature 505:223-228. CrossRef Medtine
Ltuwe 12K, Harxison jI, O'Hara BF, Lifsliitz J (2014) Recovery of neurolo~-
ical function despite immediate sleep disruption fallowing diffuse braininjury in the mouse: clinical relevasTee to rnedicallyunfrested concussion.
Sleep 37:743-752. MedlineRoyo NC, Schouten JW, Pu1p CT, Shimizu S, M:u~klund N, Graham DI,
McIntosh TK (2003) From cell death to neuronal regenerarion: buildinga new brain after traumatic brain injury. J Neuropathol E~cy Neuro162:801-$lt. CrossRefMedline
Sarmiere PD, Weigle CM, Tamkan MTVI (2008) The Kv2.1 K-I- channel tar-gets to the anon inirial segment of hippocarnpal and cortical neurons inculture and in situ. BMC Neurusci 9:112. CrossRef Mediine
Schrnued LC, AlUertson C, Slikker WJ Jr (1997) Fluoro-Jade: a novel fluo-rvcilimmefor tliesensitive and reliable histocliemical localization of i~eu-ron~l degeneration. Brain Res 751:37-46. Cx~ossRef Medline
Sensi SL,. Ra,~~~a~elli IG, Pra2zini ~.', Mascetra N (2U08) Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse
model of tV2heimer's disease. Fop Gerontol 43:488-492. CrossRe£Medline

19096.1. Neurosci., October 26, 2016 ~ 36(43):11084-11096
Sesti F, Wu X, Liu S (2014} Oxidation of KCNBI IC(+)channels in centralnervous system and beyond. World J Biot Chem 5:85-92. Mext(ine
Shen Q, HieUert JB, Hartwell J, Thimmesch AR, Pierce JD (epub ahead ofprint Ivfay 31, 2016) Systematic review of traumaric brain injury and theimpact of antioxidant therapy on clinical outcomes. Worldviews EvidBased Nurs. doi: 10.11 i llwvn.12167. lvtedtine
Smith DH, Okryama K, Thomas MJ, Clausen B, McIntosh TK (1991) Evalua-tion of memory dysfunction following experimental brain injury using theMorris water maze. J Nenrori~autna 8:259-269. Crossi2ef Mediuie
Smith DH, Soarer HD, Pierce JS, Perlman KG, Slatman KE, Heaney DF,Dixon GE, McIntosh TK (1995) A model of parasagittal controlled cor-tical impact in the mouse: cognitive and histopathologic effects. J Neu-rotrauml 12:169-178. CrossRef Medline
Smith IF, Hitt B, Green KN, Oddo S, I,aFerla FM (2005) Bnhanced caffeine-induced Cat+ release in the 3xTg-AD mouse model of Alzheimer's dis-ease. J Neurochem 94:1711-1718. CrossRef redline
Speca DJ, Ogata G, Mandikian D, Bishop H[, Wiler SW, Eum K, Wenzel HJ,Doesq ET, Matt L, Campi KI„ Golub MS, Nerbonne JM, Hell JW, TrainorBC, Sack JT, Schwartzkroin PA, Trimmer JS (2014) Deletion of theKv2.1 delayed rectifier potassium channel leads to neuronal and behav-ioral hyperexcitability.Genes $rain Behav 13:394-408. CrossRef Medline
Thakker-Norio S, Alder J, Crozzer RA, Plummer MR, Black IB (2001) Ra63A
Yu et al. • Oxidation of KCNBI in TBI
is Y•egaired for braui-derived neurotruphic factar-uiduced synapCic ylas-ticity: transcriptional analysis at the poyulation and single-cell levels.J Neurosci 21:6782-6790. Medline
Thompson HJ, Lifshicz J, Marklund N, Grady MS, Graham AI, Hovda DA,McIntosh TK (2005) Lateral fluid percussion brain injury: a 15-yearreview and evaluation. J Neurotrauma 22:42-75. Cx•ossRef Medline
Vos PE, Jacobs B, Andriessen TM, Lamers KJ, Sorm GF, Beems T, EdwardsM, Rosmalen CF, Vissers JL (2010) GFAP and S100B are biomarkers oftraumatic brain injury: an observational cohort study. Neurology 75:1786-1793. CrossRefMedtine
Wu X, Hernandez-Enriquez B, $anas M, Xu R, Sesri F (2013) Ivtolecu(arMechanisms Underlying the Apoptotic Effect of KCNBI K+ ChannelQ~darion. J $iol Chem 2&&:4128-4134. CrossRef Medline
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mito-chondrial bioenergetic deficit precedes Alzheimer's pathology in femalemouse model of AlzAeimer's disease. Proc 1Vat1 Acad Sci U 5 A 106:14670-14675. CrassRef Mecfline
7,hou'T'T, Quan LL, Chen LP, Du T, Sun KX, Zhang JC, Yu L, Li Y, Wan P,Ct►en LL, Jiang BH, Hu LH, Chen J, Shen X (2016) SP6616 as a newKv2.1 channel inhibitor efficiently promotes beG1-cell survival involvingboth PKG/Erkl/2 and CaM/PI3KlAkt signlling pathways. Cell Death Dis7:e221G. CrossRef Medline





























