Embed Size (px)
Citation preview

J Oral Med Oral Surg 2019;25:34© The authors, 2019https://doi.org/10.1051/mbcb/2019015
https://www.jomos.org
Educational Article
Oral and skin manifestations of tuberous sclerosis complexLafont Jacinthe1,*, Catherine Jean-Hughes1,2, Lejeune Mathilde1, Ordioni Ugo1,3,Lan Romain1,2, Campana Fabrice1,4
1 Aix Marseille Univ, APHM, La Timone, Service d’Odontologie, 264 rue Saint Pierre, 13385 Marseille Cedex 5, France2 UMR 7268 ADES, Aix-Marseille/EFS/CNRS, faculté de médecine-secteur nord, Boulevard Pierre-Dramard, 13344 Marseille Cedex 15,France
3 Centre Massilien de la Face, 24 avenue du Prado, 13006 Marseille, France4 UMR_S910. Centre de génétique médicale de Marseille, Aix Marseille Univ, Campus Timone, 27 Bd J Moulin, 13385 Marseille Cedex 5,France
(Received: 6 June 2018, accepted: 29 May 2019)
Keywords:tuberous sclerosis /oral manifestations /skin manifestations
* Correspondence: Jacinth
This is an Open Access article dun
Abstract -- Tuberous sclerosis complex is a genetic disease characterized by multisystemic hamartomas with variableand non-specific clinical manifestations. The disease is associated with mutations of genes encoding the proteinshamartin and tuberin. The hamartin/tuberin complex plays an anti-tumor function by inhibiting mammalian target ofrapamycin. The diagnostic criteria for the disease were reviewed at a consensus conference in 2012. Evidence ofmutations of tuberous sclerosis complex 1 or 2 genes has become a clinical and independent diagnostic criterion.Among the clinical criteria used, two oral criteria include the presence of three or more enamel pits and the presenceof two or more oral fibromas. Several dermatological criteria are included within these criteria and are of interest inour specialty when these are localized at the cephalic extremity.
Introduction
Tuberous sclerosis (Bourneville disease) or tuberoussclerosis of the brain is a genetic disorder characterized bymultisystemic hamartomas with variable and non-specificclinical manifestations. The diagnostic criteria for the diseasewere updated in a consensus conference in 2012 [1] The mostsignificant development was the inclusion of a genetic criterionallowing the diagnostician to make diagnosis independently.Among the clinical diagnostic criteria, two oral criteria wereincluded as minor criteria. The presence of three or moreenamel pits was introduced as a diagnostic criterion during thisrevision, and the presence of gingival fibromas was replaced bythe presence of two or more oral fibromas. Moreover, severaldermatological criteria were included within the major andminor diagnostic criteria, which are of interest in our specialty,particularly when they are localized around the cephalicextremity.
The aim of this article was to emphasize the manifestationsand management of oral and dermatological localizations oftuberous sclerosis.
istributed under the terms of the Creative Commons Arestricted use, distribution, and reproduction in any
Definition, epidemiology, and genetics
The first description of the disease was made by vonRecklinghausen in 1862 [1], and in 1880 Bourneville gave itthe name tuberous sclerosis. Its prevalence has been estimatedat 1 case per 10,000 to 25,000 individuals [2,3]. Thisprevalence is probably underestimated due to undiagnosedcases [4]. Tuberous sclerosis belongs to the group of classicalphacomatoses [2] with neurofibromatosis types 1 and 2,Sturge–Weber–Krabbe syndrome, Von Hippel–Lindau disease,and various neuroectodermal dysembryoplasias. It is anautosomal dominant genetic disease with almost completepenetrance; however, two-thirds of individuals develop thedisease following pathogenic de novo variation [2,3]. Thedisease is associated with a pathogenic mutation of two genes.However, despite the advances in diagnostic techniques, nomutation is detected in 15%–20% of the cases, not excludingdiagnosis [1,4]. In 31% of the patients, a mutation is identifiedin the tuberous sclerosis complex 1 gene (TSC1) located onchromosome 9 (9q34), and in 69% of the patients, a mutationis identified the TSC2 gene located on chromosome 16(16p13.3) [4,5]. The genes TSC1 and TSC2 respectively encodehamartin and tuberin [4] that combine to form a hamartin–tuberin complex. Mutation of either of the proteins renders thecomplex inactive [4]. This complex shows anti-tumor activity
ttribution License (http://creativecommons.org/licenses/by/4.0), which permitsmedium, provided the original work is properly cited.
1

Table I. Diagnostic criteria for tuberous sclerosis (Bourneville disease) according to the 2012 consensus conference. Red: oralcriteria; blue: dermatological criteria.
J Oral Med Oral Surg 2019;25:34 L. Jacinthe et al.
by inhibiting the activity of mammalian target rapamycintarget (mTOR) protein [4], which is a regulatory kinase for cellproliferation and growth [4].
Diagnosis
Tuberous sclerosis has a predominantly neurocutaneousexpression characterized by multisystemic hamartomas associ-ated with neuropsychiatric manifestations such as mentalretardation and epilepsy [2]. Many symptoms associated withtuberous sclerosis are not pathognomonic, which poses
2
diagnostic difficulties. The Washington International Consen-sus Conference in 2012 [1,5] modified the diagnostic criteriafor this disease.
A genetic diagnostic criterion was introduced during thisrevision [1,5]. The presence of pathogenic mutations of the TSC1or TSC2 genes allows for a definitive diagnosis of tuberoussclerosis, independent of the associated clinical manifestations.
The second diagnostic criterion is clinical [1]. Clinicalmanifestations are grouped into 11 major and 6 minor criteria(Tab. I). Diagnosis is considered definitive in the presence oftwo major clinical criteria or one major clinical criterion and
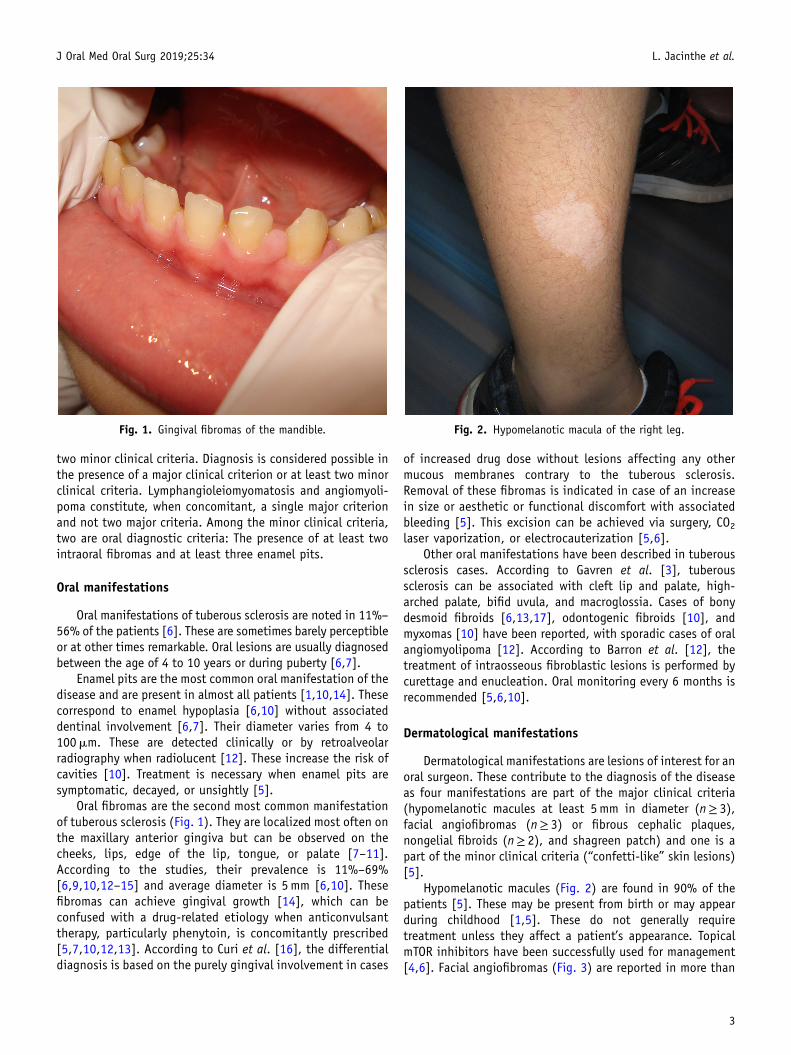
Fig. 1. Gingival fibromas of the mandible. Fig. 2. Hypomelanotic macula of the right leg.
J Oral Med Oral Surg 2019;25:34 L. Jacinthe et al.
two minor clinical criteria. Diagnosis is considered possible inthe presence of a major clinical criterion or at least two minorclinical criteria. Lymphangioleiomyomatosis and angiomyoli-poma constitute, when concomitant, a single major criterionand not two major criteria. Among the minor clinical criteria,two are oral diagnostic criteria: The presence of at least twointraoral fibromas and at least three enamel pits.
Oral manifestations
Oral manifestations of tuberous sclerosis are noted in 11%–56% of the patients [6]. These are sometimes barely perceptibleor at other times remarkable. Oral lesions are usually diagnosedbetween the age of 4 to 10 years or during puberty [6,7].
Enamel pits are the most common oral manifestation of thedisease and are present in almost all patients [1,10,14]. Thesecorrespond to enamel hypoplasia [6,10] without associateddentinal involvement [6,7]. Their diameter varies from 4 to100mm. These are detected clinically or by retroalveolarradiography when radiolucent [12]. These increase the risk ofcavities [10]. Treatment is necessary when enamel pits aresymptomatic, decayed, or unsightly [5].
Oral fibromas are the second most common manifestationof tuberous sclerosis (Fig. 1). They are localized most often onthe maxillary anterior gingiva but can be observed on thecheeks, lips, edge of the lip, tongue, or palate [7–11].According to the studies, their prevalence is 11%–69%[6,9,10,12–15] and average diameter is 5mm [6,10]. Thesefibromas can achieve gingival growth [14], which can beconfused with a drug-related etiology when anticonvulsanttherapy, particularly phenytoin, is concomitantly prescribed[5,7,10,12,13]. According to Curi et al. [16], the differentialdiagnosis is based on the purely gingival involvement in cases
of increased drug dose without lesions affecting any othermucous membranes contrary to the tuberous sclerosis.Removal of these fibromas is indicated in case of an increasein size or aesthetic or functional discomfort with associatedbleeding [5]. This excision can be achieved via surgery, CO2laser vaporization, or electrocauterization [5,6].
Other oral manifestations have been described in tuberoussclerosis cases. According to Gavren et al. [3], tuberoussclerosis can be associated with cleft lip and palate, high-arched palate, bifid uvula, and macroglossia. Cases of bonydesmoid fibroids [6,13,17], odontogenic fibroids [10], andmyxomas [10] have been reported, with sporadic cases of oralangiomyolipoma [12]. According to Barron et al. [12], thetreatment of intraosseous fibroblastic lesions is performed bycurettage and enucleation. Oral monitoring every 6 months isrecommended [5,6,10].
Dermatological manifestations
Dermatological manifestations are lesions of interest for anoral surgeon. These contribute to the diagnosis of the diseaseas four manifestations are part of the major clinical criteria(hypomelanotic macules at least 5mm in diameter (n≥ 3),facial angiofibromas (n≥ 3) or fibrous cephalic plaques,nongelial fibroids (n≥ 2), and shagreen patch) and one is apart of the minor clinical criteria (“confetti-like” skin lesions)[5].
Hypomelanotic macules (Fig. 2) are found in 90% of thepatients [5]. These may be present from birth or may appearduring childhood [1,5]. These do not generally requiretreatment unless they affect a patient’s appearance. TopicalmTOR inhibitors have been successfully used for management[4,6]. Facial angiofibromas (Fig. 3) are reported in more than
3
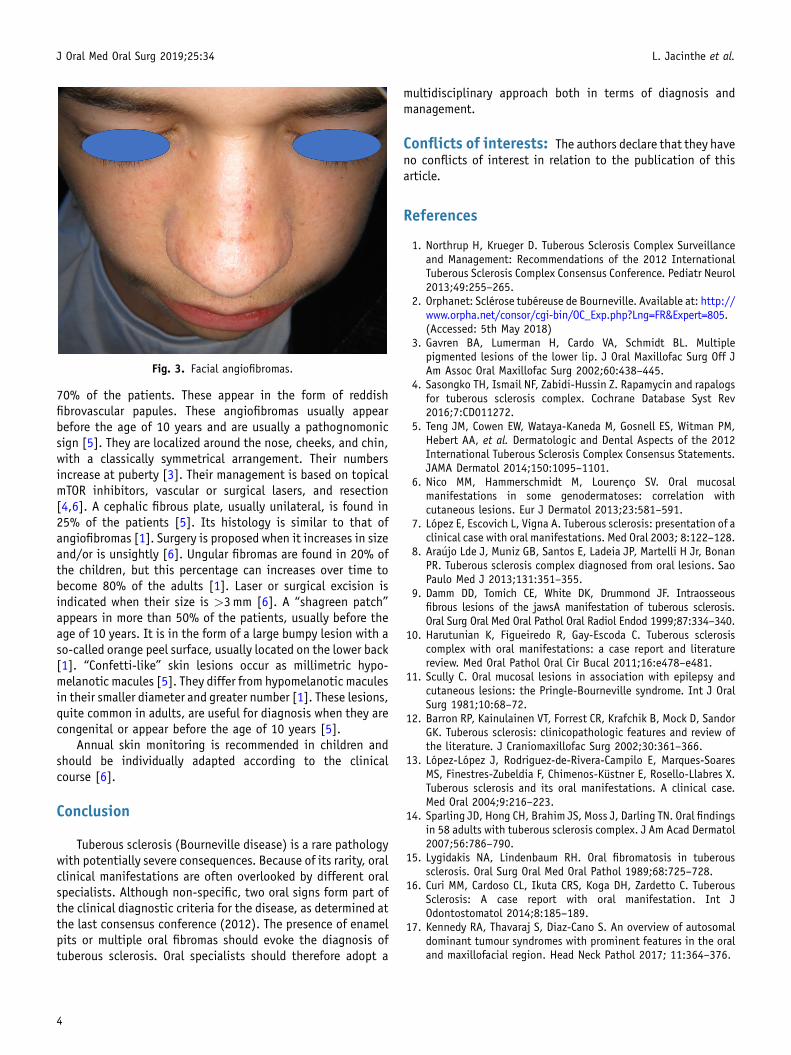
Fig. 3. Facial angiofibromas.
J Oral Med Oral Surg 2019;25:34 L. Jacinthe et al.
70% of the patients. These appear in the form of reddishfibrovascular papules. These angiofibromas usually appearbefore the age of 10 years and are usually a pathognomonicsign [5]. They are localized around the nose, cheeks, and chin,with a classically symmetrical arrangement. Their numbersincrease at puberty [3]. Their management is based on topicalmTOR inhibitors, vascular or surgical lasers, and resection[4,6]. A cephalic fibrous plate, usually unilateral, is found in25% of the patients [5]. Its histology is similar to that ofangiofibromas [1]. Surgery is proposed when it increases in sizeand/or is unsightly [6]. Ungular fibromas are found in 20% ofthe children, but this percentage can increases over time tobecome 80% of the adults [1]. Laser or surgical excision isindicated when their size is >3mm [6]. A “shagreen patch”appears in more than 50% of the patients, usually before theage of 10 years. It is in the form of a large bumpy lesion with aso-called orange peel surface, usually located on the lower back[1]. “Confetti-like” skin lesions occur as millimetric hypo-melanotic macules [5]. They differ from hypomelanotic maculesin their smaller diameter and greater number [1]. These lesions,quite common in adults, are useful for diagnosis when they arecongenital or appear before the age of 10 years [5].
Annual skin monitoring is recommended in children andshould be individually adapted according to the clinicalcourse [6].
Conclusion
Tuberous sclerosis (Bourneville disease) is a rare pathologywith potentially severe consequences. Because of its rarity, oralclinical manifestations are often overlooked by different oralspecialists. Although non-specific, two oral signs form part ofthe clinical diagnostic criteria for the disease, as determined atthe last consensus conference (2012). The presence of enamelpits or multiple oral fibromas should evoke the diagnosis oftuberous sclerosis. Oral specialists should therefore adopt a
4
multidisciplinary approach both in terms of diagnosis andmanagement.
Conflicts of interests: The authors declare that they haveno conflicts of interest in relation to the publication of thisarticle.
References
1. Northrup H, Krueger D. Tuberous Sclerosis Complex Surveillanceand Management: Recommendations of the 2012 InternationalTuberous Sclerosis Complex Consensus Conference. Pediatr Neurol2013;49:255–265.
2. Orphanet: Sclérose tubéreuse de Bourneville. Available at: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=805.(Accessed: 5th May 2018)
3. Gavren BA, Lumerman H, Cardo VA, Schmidt BL. Multiplepigmented lesions of the lower lip. J Oral Maxillofac Surg Off JAm Assoc Oral Maxillofac Surg 2002;60:438–445.
4. Sasongko TH, Ismail NF, Zabidi-Hussin Z. Rapamycin and rapalogsfor tuberous sclerosis complex. Cochrane Database Syst Rev2016;7:CD011272.
5. Teng JM, Cowen EW, Wataya-Kaneda M, Gosnell ES, Witman PM,Hebert AA, et al. Dermatologic and Dental Aspects of the 2012International Tuberous Sclerosis Complex Consensus Statements.JAMA Dermatol 2014;150:1095–1101.
6. Nico MM, Hammerschmidt M, Lourenço SV. Oral mucosalmanifestations in some genodermatoses: correlation withcutaneous lesions. Eur J Dermatol 2013;23:581–591.
7. López E, Escovich L, Vigna A. Tuberous sclerosis: presentation of aclinical case with oral manifestations. Med Oral 2003; 8:122–128.
8. Ara�ujo Lde J, Muniz GB, Santos E, Ladeia JP, Martelli H Jr, BonanPR. Tuberous sclerosis complex diagnosed from oral lesions. SaoPaulo Med J 2013;131:351–355.
9. Damm DD, Tomich CE, White DK, Drummond JF. Intraosseousfibrous lesions of the jawsA manifestation of tuberous sclerosis.Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;87:334–340.
10. Harutunian K, Figueiredo R, Gay-Escoda C. Tuberous sclerosiscomplex with oral manifestations: a case report and literaturereview. Med Oral Pathol Oral Cir Bucal 2011;16:e478–e481.
11. Scully C. Oral mucosal lesions in association with epilepsy andcutaneous lesions: the Pringle-Bourneville syndrome. Int J OralSurg 1981;10:68–72.
12. Barron RP, Kainulainen VT, Forrest CR, Krafchik B, Mock D, SandorGK. Tuberous sclerosis: clinicopathologic features and review ofthe literature. J Craniomaxillofac Surg 2002;30:361–366.
13. López-López J, Rodriguez-de-Rivera-Campilo E, Marques-SoaresMS, Finestres-Zubeldia F, Chimenos-Küstner E, Rosello-Llabres X.Tuberous sclerosis and its oral manifestations. A clinical case.Med Oral 2004;9:216–223.
14. Sparling JD, Hong CH, Brahim JS, Moss J, Darling TN. Oral findingsin 58 adults with tuberous sclerosis complex. J Am Acad Dermatol2007;56:786–790.
15. Lygidakis NA, Lindenbaum RH. Oral fibromatosis in tuberoussclerosis. Oral Surg Oral Med Oral Pathol 1989;68:725–728.
16. Curi MM, Cardoso CL, Ikuta CRS, Koga DH, Zardetto C. TuberousSclerosis: A case report with oral manifestation. Int JOdontostomatol 2014;8:185–189.
17. Kennedy RA, Thavaraj S, Diaz-Cano S. An overview of autosomaldominant tumour syndromes with prominent features in the oraland maxillofacial region. Head Neck Pathol 2017; 11:364–376.





























