Embed Size (px)
Citation preview

Applied Catalysis A: General 245 (2003) 363–376
Optimization of cyclohexene epoxidation with dilutehydrogen peroxide and silica-supported
titanium catalysts
José M. Fraile∗, José I. Garcıa, José A. Mayoral, Eugenio VispeDepartamento de Qu´ımica Orgánica, Instituto de Ciencia de Materiales de Aragón, Facultad de Ciencias,
Universidad de Zaragoza—C.S.I.C., E-50009 Zaragoza, Spain
Received 25 October 2002; received in revised form 6 December 2002; accepted 6 December 2002
Abstract
The results of activity and selectivity in the epoxidation of cyclohexene with dilute hydrogen peroxide can be improvedby careful optimization of the catalyst properties and the reaction conditions. The type of silica support, the silanizationwith long hydrocarbon chains and mainly the slow addition of hydrogen peroxide are important factors. This slow additionnot only noticeably improves the selectivity in terms of epoxidation products but also leads to a better recoverability of thecatalyst, showing the role of the radical pathway in deactivation and titanium leaching. In the best case the catalyst can bereused at least three times with very low titanium leaching, and an overall productivity of 228 mmol epoxidation products permmol Ti.© 2002 Elsevier Science B.V. All rights reserved.
Keywords:Heterogeneous titanium catalysts; Epoxidation; Hydrogen peroxide; Recoverability
1. Introduction
Dilute hydrogen peroxide is one of the most con-venient oxidants due to its ease of handling, highcontent of active oxygen and absence of byproducts[1]. However, in the epoxidation of alkenes, severalside reactions can take place, such as oxidation inthe allylic positions, ring-opening of the epoxide byhydrolysis or solvolysis, epoxide rearrangement oreven total breakdown of the C=C double bond. Cy-clohexene is one of the most difficult cases given thatthe first two problems, namely allylic oxidation and
∗ Corresponding author. Tel.:+34-976-76-22-72;fax: +34-976-76-20-77.E-mail address:[email protected] (J.M. Fraile).
epoxide ring-opening, occur extensively. Followingthe success found on using TS-1 for the oxidation ofdifferent organic compounds with hydrogen peroxide[2], titanium catalysts have been widely studied inan attempt to overcome the size limitations imposedby the microporosity of TS-1[3]. However, neitherclosely related Ti-zeolites[4] nor mesoporous crys-talline Ti-silicas [5] have attained the high activityand selectivity of TS-1.
Many attempts have been made to design more ef-ficient catalysts to improve the performance in epoxi-dation with hydrogen peroxide, either by imparting ahydrophobic character to the catalyst surface[6–9] or,more recently, by addition of ammonium salts[10].On the other hand, we demonstrated the possibility ofusing silica-supported catalysts with dilute hydrogen
0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved.doi:10.1016/S0926-860X(02)00643-9
364 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
peroxide[11], under milder conditions than those usedwith other related solids[12,13], with a positive roleof titanium dispersion[14].
Another important problem is that in the scarcecases where catalyst recovery has been studied, ex-tensive leaching has been observed both with sup-ported and framework titanium catalysts[6]. EvenTS-1 suffers from this problem when allylic alco-hols are epoxidized[15]. Titanium leaching has alsobeen observed in the case of silica-supported catalysts[11,14].
In contrast to the high degree of activity in the areaof catalyst design, very little attention has been paidto the role of the reaction conditions in this process. Avariety of solvents, reaction temperatures and reagentsratios have been used. Although in most papers theaddition of hydrogen peroxide in one lot is described,some authors have reported a slow addition[13,16–18]without any further explanation about the role of thisexperimental condition. Only in one case[12] the re-sults with the two types of addition are compared. Inthat paper 70% hydrogen peroxide was used becauseof the effect of water on the catalyst deactivation, andthe improvement in activity and selectivity with theslow addition of the oxidant was ascribed to the loweramount of water present in the earlier stage of the re-action.
In this paper we describe the important effect of theslow addition of H2O2 on the epoxidation of cyclo-hexene with silica-supported catalysts, paying specialattention to the stability and recovery of the catalyst.
Table 1Analysis of the catalysts
Catalyst Ti (mmol/g) C (mmol/g)
Fresha Used-Ab Used-Bc Freshd Used-Ab Used-Bc
SiO2-Ti(OiPr) 0.24 (1.07) 0.13 0.17 0.85 4.12 2.71Aerosil-Ti(OiPr) 0.10 (0.80) 0.07 0.09 1.16 3.84 1.97SiO2(TMS)-Ti(OiPr) 0.24 (0.39) 0.17 0.21 4.16 (3.25) 7.02 4.66SiO2(C18)-Ti(OiPr) 0.23 (0.58) 0.16 0.20 12.77 (12.23) 12.89 12.64SiO2(TMS)-Ti(OiPr) (0.1)e 0.09 (0.39) – 0.10 3.31 (3.25) – 3.90SiO2(TMS)-Ti(DD) (0.1)e 0.10 (0.39) – 0.11 4.92 (3.25) – 4.58SiO2-Ti(TTS) 0.11 – 0.14 3.57 – 3.17
a In parenthesis the maximum possible loading of titanium.b After three runs with H2O2 added in one lot.c After three runs with slow addition of H2O2.d In parenthesis the carbon content of the support.e In parenthesis the expected titanium loading.
The silica-supported titanium catalysts have been fur-ther optimized under these new reaction conditions.
2. Experimental
2.1. Preparation of the catalysts
All the silica-based solids were dried at 140◦C un-der vacuum for 12 h prior to any treatment or use ascatalysts. SiO2(TMS) and SiO2(C18) were preparedby treatment of silica (Merck 60) (5 g) with hexam-ethyldisilazane and dimethyloctadecylsilyl chloride(11 mmol), respectively, in anhydrous toluene (15 ml)under reflux under an Ar atmosphere for 1 h. Thesolids were filtered off, washed with ethanol, water,ethanol and ether and dried under vacuum.
Titanium catalysts were prepared by treatment ofthe silica support (5 g) with Ti(OiPr)4 (1.5 mmol) inanhydrous toluene (20 ml) under reflux under an Aratmosphere for 48 h. Some of the samples were pre-pared with half the amount of titanium (seeTable 1).Water was carefully excluded to prevent the forma-tion of titania particles. The maximum possible chargeof titanium was obtained by treatment of the differ-ent supports with an excess of Ti(OiPr)4 (2.5 mmol/gsupport) under the same conditions.
SiO2(TMS)-Ti(DD) was prepared by treatmentof SiO2(TMS)-Ti(OiPr) (1 g) with decane-1,2-diol(0.45 mmol) in anhydrous toluene (20 ml) under refluxfor 6 h under an Ar atmosphere. A portion (15 ml) of
J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 365
the solution was distilled, the remaining suspensionwas cooled to room temperature and the solid wasfiltered off, washed with toluene and dichloromethaneand finally dried under vacuum.
SiO2-Ti(TTS) was prepared by treatment of Merck60 silica (1 g) with 0.3 mmol tris(isooctadecanoate)-isopropoxytitanium (KR TTS from Kenrich Petro-chemicals Inc., USA) in anhydrous toluene (10 ml)under reflux for 2 h.
2.2. Characterization of the catalysts
Titanium analyses were carried out by plasmaemission spectroscopy on a Perkin-Elmer Plasma 40emission spectrometer. Carbon analyses were carriedout using a Perkin-Elmer 2400 elemental analyzer.Infrared spectra were recorded on a Mattson GenesisSeries FTIR spectrometer and a Nicolet Avatar 360FTIR spectrometer. Spectra of the organic part weretaken from self-supported wafers treated under vac-uum (<10−5 Torr) at 140◦C in a cell equipped withNaCl windows. Spectra of the silica part were takenfrom wafers diluted with KBr treated in the same way.DR-UV spectra were recorded on a Unicam UV-4spectrometer equipped with a SpectralonTM RSA-UC-40 Labsphere integrating sphere. Thermogravimetricanalysis was carried out on a SDT 2960 Simultane-ous TGA–DTA apparatus (TA Instruments). Sampleswere heated under nitrogen flow (130 cm3/min) from
Table 2Comparison of the results obtained in the epoxidation of cyclohexene with dilute hydrogen peroxide depending on the silica support andthe type of additiona
Entry Catalyst Addition Run Yield (%)b Contributiondirectc
Epoxide/diol
Epoxide Diol Enol Chhp
1 Merck silica At once 1 4.2 1.7 17.2 25.8 0 –2 Slowd 1 0.0 1.2 5.3 10.1 0 –3 SiO2-Ti(OiPr) At once 1 31.4 28.4 36.0 9.6 21 53/474 3 31.9 9.2 35.1 24.0 5 78/225 Aerosil-Ti(OiPr) At once 1 44.3 12.4 26.5 9.2 30 78/226 2 12.5 9.4 17.0 30.5 5 57/437 SiO2-Ti(OiPr) Slowd 1 21.8 29.3 7.8 9.2 56 43/578 3 20.4 28.9 10.3 16.8 42 41/59
a Reaction conditions: 50 mmol cyclohexene, 2.5 mmol H2O2 (30%), 5 ml tert-butanol, 200 mg catalyst, 80◦C, 24 h.b Determined by GC and calculated with respect to the maximum. Epoxide: cyclohexene oxide; diol:trans-cyclohexane-1,2-diol; enol:
cyclohex-2-en-1-ol; chhp: cyclohex-2-enyl hydroperoxide.c Estimation of the contribution of the direct non-radical epoxidation with hydrogen peroxide to the total productive conversion of
hydrogen peroxide (see text).d Addition in 6 h; total reaction time 11 h (24 h in the case of silica).
100 to 500◦C (10◦C/min) and then under air flowfrom 500 to 650◦C (10◦C/min). Step-scanned X-raydiffraction patterns were collected at room tempera-ture from 3◦ in 2θ up to 60◦, using a D-max Rigakusystem with rotating anode.
2.3. Catalytic tests
All the catalysts were dried at 140◦C under vacuumfor 12 h prior to use. All the reagents and solvents(synthesis grade 99%) were used as received. The cat-alyst (200 mg) was added to a solution of cyclohexene(5 or 2.5 ml, 50 or 25 mmol) intert-butanol (4 ml).The reaction mixture was heated at 80◦C and H2O2(0.28 ml, 30%, 2.5 mmol) diluted withtert-butanol(1 ml) was slowly added (6 or 2.5 h, seeTables 2–4)using a syringe pump. Ethylene glycol dimethyl ether(1 ml, internal standard) was added and the reactionmixture was stirred at 80◦C for an additional periodof time. The catalyst was filtered off, washed withdichloromethane (5× 5 ml), dried under vacuum andreused under the same conditions. The yields weredetermined by GC[11] and the total conversion ofH2O2 was confirmed by iodometric titration of thefinal solution.
Some of the catalysts were also tested under thesame conditions with the addition of H2O2 and theinternal standard in one lot at the beginning of thereaction.
366 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
Table 3Comparison of the results obtained in the epoxidation of cyclohexene with dilute hydrogen peroxide and silanized catalystsa
Entry Catalyst Addition Run Yield (%)b Contributiondirectc
Epoxide/diol
Epoxide Diol Enol Chhp
1 SiO2(TMS)-Ti(OiPr) At once 1 14.5 17.2 25.5 4.8 9 46/542 3 5.2 5.7 9.4 14.4 3 48/523 Slowd 1 34.5 44.4 8.1 6.4 71 44/564 3 13.5 29.9 10.4 23.0 33 31/696 SiO2(C18) Slowd 1 0.0 2.0 8.1 12.0 0 –7 SiO2(C18)-Ti(OiPr) At once 1 12.6 25.3 23.2 2.1 23 33/678 3 10.5 5.3 12.7 4.1 8 66/349 Slowd 1 23.2 39.4 2.7 1.6 87 37/63
10 2 15.6 40.3 3.6 8.1 69 28/7211 3 18.2 34.2 5.9 10.5 59 35/65
a Reaction conditions: 25 mmol cyclohexene, 2.5 mmol H2O2 (30%), 5 ml tert-butanol, 200 mg catalyst, 80◦C, 24 h.b Determined by GC and calculated with respect to the maximum. Epoxide: cyclohexene oxide; diol:trans-cyclohexane-1,2-diol; enol:
cyclohex-2-en-1-ol; chhp: cyclohex-2-enyl hydroperoxide.c Estimation of the contribution of the direct non-radical epoxidation with hydrogen peroxide to the total productive conversion of
hydrogen peroxide (see text).d Addition in 6 h; total reaction time 11 h.
Table 4Comparison of the results obtained in the epoxidation of cyclohexene with dilute hydrogen peroxide and catalysts modified in theenvironment close to titaniuma
Entry Catalyst Addition Run Yield (%)b Contributiondirectc
Epoxide/diol
Epoxide Diol Enol Chhp
1 SiO2(TMS)-Ti(OiPr) (0.1) Slowd 1 32.5 31.8 6.3 13.6 59 51/492 3 12.6 21.4 6.9 26.3 29 37/633 SiO2(TMS)-Ti(DD) (0.1) Slowd 1 29.2 19.1 8.2 14.3 47 60/404 3 13.8 8.8 10.1 27.7 14 61/395 SiO2-Ti(TTS) Slowd 1 22.7 33.5 7.3 9.0 60 40/606 3 22.6 27.5 9.5 11.9 49 45/557 Aerosil-Ti(OiPr) Slowe 1 36.1 38.8 8.3 5.0 71 48/528 3 33.9 22.5 9.4 17.5 47 60/40
a Reaction conditions: 25 mmol cyclohexene, 2.5 mmol H2O2 (30%), 5 ml tert-butanol, 200 mg catalyst, 80◦C, 24 h.b Determined by GC and calculated with respect to the maximum. Epoxide: cyclohexene oxide; diol:trans-cyclohexane-1,2-diol; enol:
cyclohex-2-en-1-ol; chhp: cyclohex-2-enyl hydroperoxide.c Estimation of the contribution of the direct non-radical epoxidation with hydrogen peroxide to the total productive conversion of
hydrogen peroxide (see text).d Addition in 2.5 h; total reaction time 7.5 h.e Addition in 6 h; total reaction time 11 h.
3. Results and discussion
3.1. Preparation of the catalysts and effectof the type of support
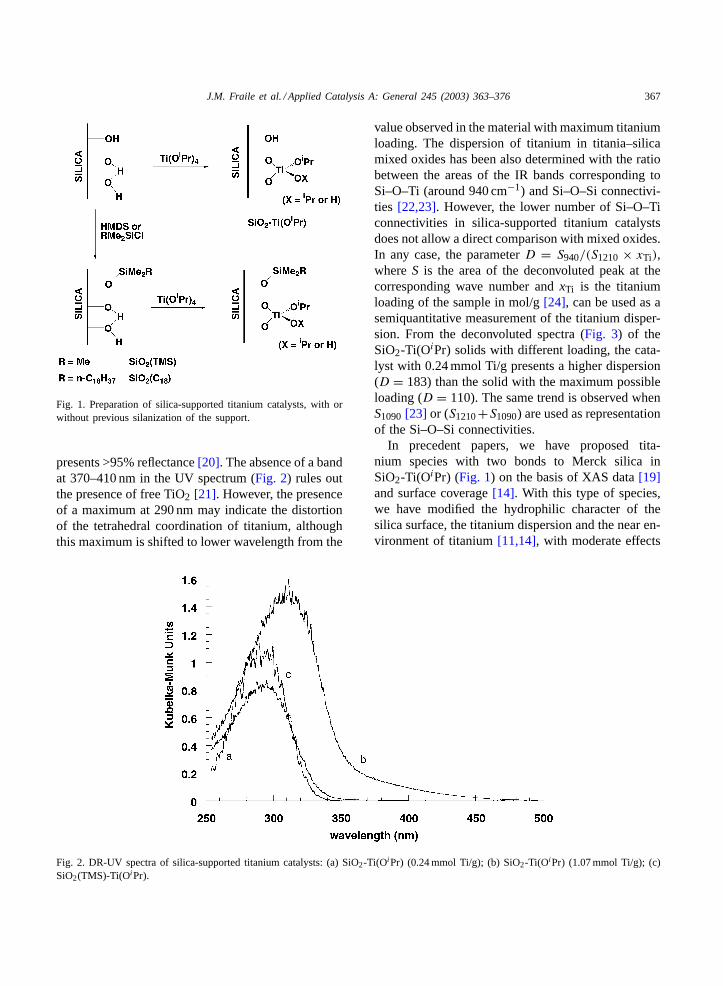
The SiO2-Ti(OiPr) catalyst was prepared by treat-ment of silica (Merck 60) with Ti(OiPr)4 (Fig. 1) togive a material with low titanium loading (Table 1). In
previous work, a solid prepared in the same way withthe maximum loading was characterized by EXAFSand XANES [19]. Large particle TiO2 was not de-tected on that solid and spectra fitted well with tetra-hedral titanium centers. The XRD diffraction patternof the low-loaded SiO2-Ti(OiPr) catalyst did not showany crystalline phase. DR-UV spectra were recordedin the range of 250–600 nm, where SpectralonTM
J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 367
Fig. 1. Preparation of silica-supported titanium catalysts, with orwithout previous silanization of the support.
presents >95% reflectance[20]. The absence of a bandat 370–410 nm in the UV spectrum (Fig. 2) rules outthe presence of free TiO2 [21]. However, the presenceof a maximum at 290 nm may indicate the distortionof the tetrahedral coordination of titanium, althoughthis maximum is shifted to lower wavelength from the
Fig. 2. DR-UV spectra of silica-supported titanium catalysts: (a) SiO2-Ti(OiPr) (0.24 mmol Ti/g); (b) SiO2-Ti(OiPr) (1.07 mmol Ti/g); (c)SiO2(TMS)-Ti(OiPr).
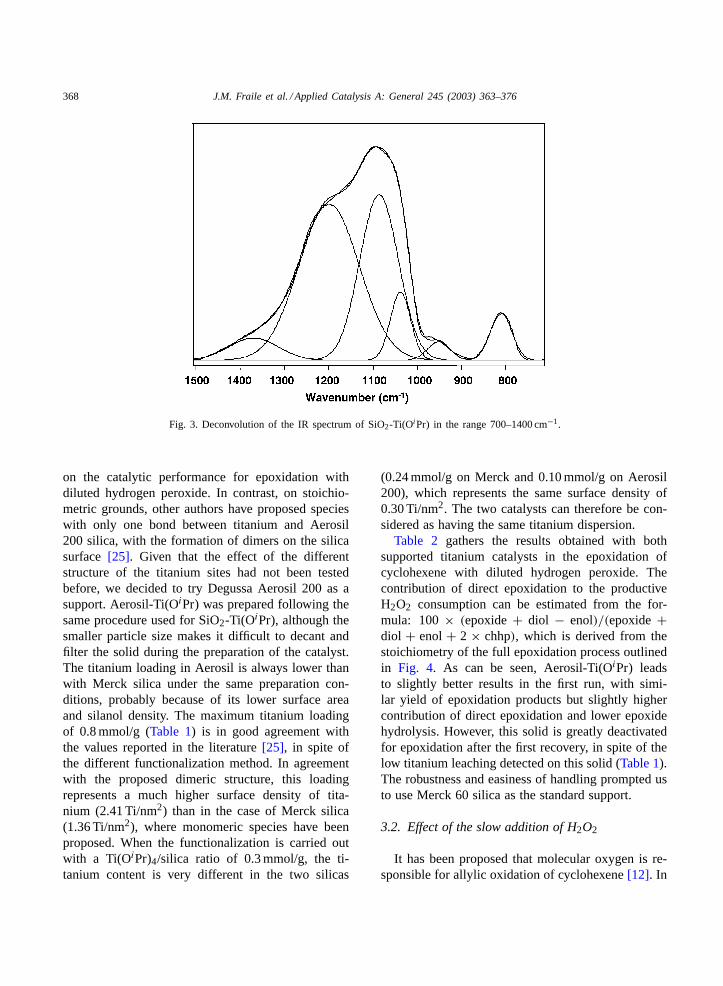
value observed in the material with maximum titaniumloading. The dispersion of titanium in titania–silicamixed oxides has been also determined with the ratiobetween the areas of the IR bands corresponding toSi–O–Ti (around 940 cm−1) and Si–O–Si connectivi-ties [22,23]. However, the lower number of Si–O–Ticonnectivities in silica-supported titanium catalystsdoes not allow a direct comparison with mixed oxides.In any case, the parameterD = S940/(S1210 × xTi),whereS is the area of the deconvoluted peak at thecorresponding wave number andxTi is the titaniumloading of the sample in mol/g[24], can be used as asemiquantitative measurement of the titanium disper-sion. From the deconvoluted spectra (Fig. 3) of theSiO2-Ti(OiPr) solids with different loading, the cata-lyst with 0.24 mmol Ti/g presents a higher dispersion(D = 183) than the solid with the maximum possibleloading (D = 110). The same trend is observed whenS1090 [23] or (S1210+S1090) are used as representationof the Si–O–Si connectivities.
In precedent papers, we have proposed tita-nium species with two bonds to Merck silica inSiO2-Ti(OiPr) (Fig. 1) on the basis of XAS data[19]and surface coverage[14]. With this type of species,we have modified the hydrophilic character of thesilica surface, the titanium dispersion and the near en-vironment of titanium[11,14], with moderate effects
368 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
Fig. 3. Deconvolution of the IR spectrum of SiO2-Ti(OiPr) in the range 700–1400 cm−1.
on the catalytic performance for epoxidation withdiluted hydrogen peroxide. In contrast, on stoichio-metric grounds, other authors have proposed specieswith only one bond between titanium and Aerosil200 silica, with the formation of dimers on the silicasurface[25]. Given that the effect of the differentstructure of the titanium sites had not been testedbefore, we decided to try Degussa Aerosil 200 as asupport. Aerosil-Ti(OiPr) was prepared following thesame procedure used for SiO2-Ti(OiPr), although thesmaller particle size makes it difficult to decant andfilter the solid during the preparation of the catalyst.The titanium loading in Aerosil is always lower thanwith Merck silica under the same preparation con-ditions, probably because of its lower surface areaand silanol density. The maximum titanium loadingof 0.8 mmol/g (Table 1) is in good agreement withthe values reported in the literature[25], in spite ofthe different functionalization method. In agreementwith the proposed dimeric structure, this loadingrepresents a much higher surface density of tita-nium (2.41 Ti/nm2) than in the case of Merck silica(1.36 Ti/nm2), where monomeric species have beenproposed. When the functionalization is carried outwith a Ti(OiPr)4/silica ratio of 0.3 mmol/g, the ti-tanium content is very different in the two silicas
(0.24 mmol/g on Merck and 0.10 mmol/g on Aerosil200), which represents the same surface density of0.30 Ti/nm2. The two catalysts can therefore be con-sidered as having the same titanium dispersion.
Table 2 gathers the results obtained with bothsupported titanium catalysts in the epoxidation ofcyclohexene with diluted hydrogen peroxide. Thecontribution of direct epoxidation to the productiveH2O2 consumption can be estimated from the for-mula: 100× (epoxide+ diol − enol)/(epoxide+diol + enol+ 2 × chhp), which is derived from thestoichiometry of the full epoxidation process outlinedin Fig. 4. As can be seen, Aerosil-Ti(OiPr) leadsto slightly better results in the first run, with simi-lar yield of epoxidation products but slightly highercontribution of direct epoxidation and lower epoxidehydrolysis. However, this solid is greatly deactivatedfor epoxidation after the first recovery, in spite of thelow titanium leaching detected on this solid (Table 1).The robustness and easiness of handling prompted usto use Merck 60 silica as the standard support.
3.2. Effect of the slow addition of H2O2
It has been proposed that molecular oxygen is re-sponsible for allylic oxidation of cyclohexene[12]. In

J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 369
Fig. 4. Reaction pathways in the epoxidation of cyclohexene with dilute hydrogen peroxide.
our experience it appears that the use of an inert at-mosphere does not significantly modify the selectivityof the reaction[14], hence the origin of the molecularoxygen must lie in the self-decomposition of hydrogenperoxide (Eq. (1)). This reaction is highly exother-mic and is greatly accelerated by a slight increasein temperature. Moreover, the presence of titaniumdoubles the decomposition rate, as tested by iodomet-ric titration at short reaction times in the absence ofcyclohexene. Given that this side reaction is secondorder in H2O2, whereas the direct epoxidation is firstorder, a drastic decrease in the H2O2 concentrationin the reaction medium would be expected to slowdown the decomposition rate, with a subsequent lowerconcentration of molecular oxygen in the solutionand a less important effect on the direct non-radicalepoxidation:
2H2O2 → 2H2O + O2 (1)
Thus, the positive role of the slow addition of hydro-gen peroxide may be more related to the lower H2O2decomposition than to the lower water content in thereaction mixture[12]. Then, we decided to test the ef-fect of this important parameter on the epoxidation ofcyclohexene with our silica-supported titanium cata-lysts.
First we studied the effect with the catalyst SiO2-Ti(OiPr) by addition of diluted hydrogen peroxidewith part of thetert-butanol solvent at the slower rateof our syringe pump (6 h). Control experiments werecarried out with non-functionalized silica. The resultsobtained in this reaction are shown inTable 2along
with those obtained with the addition in one lot atthe beginning of the reaction. It can be seen that,with silica, the yields were considerably lower in thecase of slow addition of H2O2, a fact that confirmsthe much lower concentration of molecular oxygen inthe reaction solution. The use of the Si-Ti(OiPr) cat-alyst led to yields in epoxidation products (epoxideand diol) that were slightly lower in the slow additionreaction (51% versus 60%), but the amount of prod-ucts arising from allylic oxidation was much smaller;36% yield of the enol versus 7.8% with slow ad-dition. The contribution of direct epoxidation to theproductive H2O2 consumption increased considerably(from 21 to 56%) upon slow addition of the perox-ide, showing the really important role played by thedecomposition of H2O2 to O2 (Eq. (1)) in the radicalpathway. In contrast, the rate of epoxide hydrolysis issimilar under both sets of conditions, in spite of theconcomitant slower addition of water to the reactionmedium.
Another relevant difference between these reactionsconcerns the recovery of the catalysts used under thetwo sets of experimental conditions. The catalyst usedunder normal conditions cannot be efficiently recov-ered due to a significant loss of activity for directepoxidation (only 5% contribution in the third run).However, the yield in epoxidation products is almostthe same in the third run with the catalyst used underslow addition conditions, with only a slight increasein the amount of products arising from allylic oxida-tion. In this case the results represent 42% contributionof direct epoxidation. This lower level of deactivation

370 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
is accompanied by the conservation of the high ac-tivity for epoxide hydrolysis in the recycled catalyst.Another important effect of slow addition is a signif-icantly lower degree of titanium leaching (Table 1).This is the first time that the slow addition of H2O2has been recognized as the origin of an enhancementof the catalyst stability.
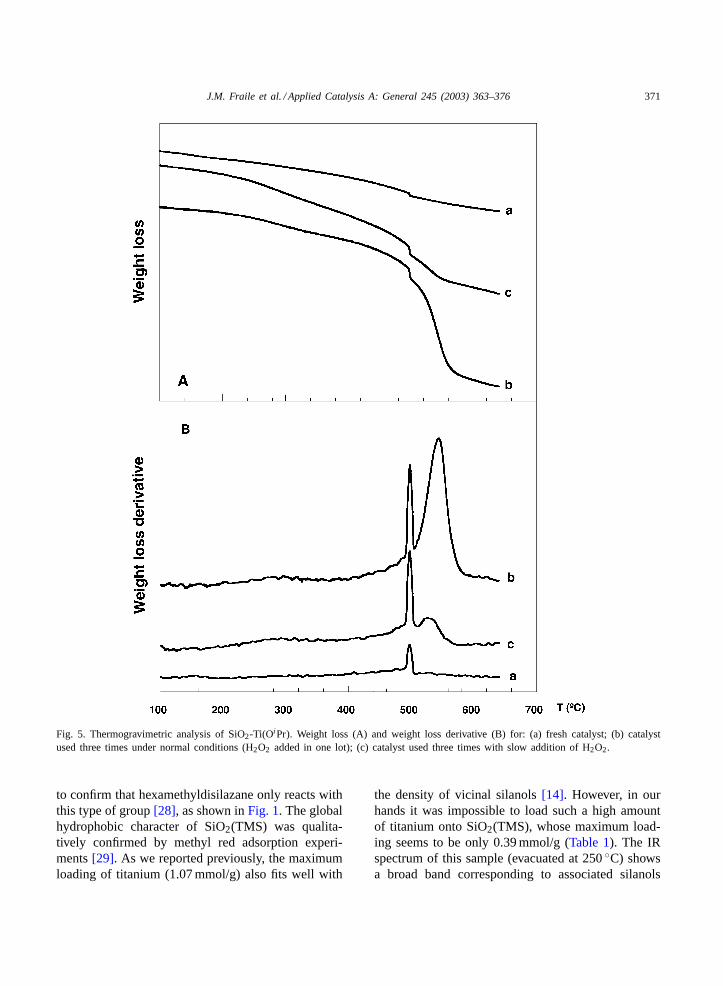
This observation is in agreement with the role ofpolyoxygenated products in the leaching of titaniumfrom TS-1 [15]. In our case, these byproducts mightbe the result of the epoxidation of 2-cyclohexenoland subsequent epoxide hydrolysis. The low concen-tration and the complexation to titanium would ex-plain that this type of byproduct is not detected inthe reaction mixture. The suppression of the radicalpathway would account for the lower titanium leach-ing in the case of slow addition of H2O2. Additionalevidence for the suppression of the radical mecha-nism is the lower carbon content in the recovered cat-alyst (Table 1) on employing slow H2O2 addition.In fact, the carbon content in the fresh catalyst isonly 0.83 mmol/g (1.15iPr/Ti) and this increases upto 2.7 mmol/g after three runs with slow addition—clearly lower than the 4.1 mmol/g obtained after threeruns under normal conditions. Moreover, this carboncontent cannot be eliminated by desorption in nitrogenflow up to 500◦C, as shown by the thermogravimet-ric analysis (Fig. 5). Whereas most of weight loss isdue to desorption in the solid used under slow additionconditions, the solid used under normal conditions re-quires combustion at high temperature, demonstratingthe strong adsorption or the non-volatile character ofthe organic deposits. This result indicates that poly-oxygenated byproducts are not only capable of leach-ing some of the titanium but also of poisoning theremaining sites, leading to a further reduction in cat-alytic activity.
The slow addition of hydrogen peroxide also givesrise to a positive effect on the epoxidation of an alkenethat does not suffer from selectivity problems, e.g. cy-clooctene. In this case the decomposition of hydro-gen peroxide leads only to a reduction in the yieldof epoxide (26% under normal conditions), which in-creased to 59% on using the slow addition technique.These results are in good agreement with the con-tribution of the direct epoxidation, given that in thecase of cyclooctene the radical pathway leads only tonon-productive H2O2 decomposition.
3.3. Effect of the hydrophobic characterof the support
After optimization of the reaction conditions, we at-tempted to optimize the catalyst. It has been proposedthat the good results obtained with TS-1 in epoxida-tion reactions with hydrogen peroxide are due to thehydrophobic nature of the surface, as shown by ad-sorption measurements[26]. Indeed, with other tita-nium catalysts the introduction of hydrophobic groupson the silica-like surface has a positive effect[8]. Inour case, the silanization of the silica with trimethylsi-lyl groups (Fig. 1) did not give rise to any effecton the catalytic performance under normal conditions[14]. However, the dramatic improvement in resultsobtained with the slow addition of hydrogen perox-ide prompted us to test the new conditions with thishydrophobic catalyst SiO2(TMS)-Ti(OiPr). As can beseen inFig. 2, the DR-UV spectrum of this catalyst isnearly superposable to that of SiO2-Ti(OiPr), in agree-ment with the identical titanium loading of both solids(Table 1).
The test of SiO2(TMS)-Ti(OiPr) was carried outwith only 10 equivalents of alkene, in an attempt toapproach more closely the conditions of a practicalapplication[1]. The results obtained are gathered inTable 3. As occurred with the hydrophilic catalyst, theslow addition of hydrogen peroxide is highly benefi-cial for the selectivity of the epoxidation in that thecontribution of the direct pathway to the H2O2 con-sumption is 71%, compared with 9% obtained undernormal conditions. It is remarkable that, in the case ofthe hydrophobic catalyst, even the total yield of epox-idation products increases on using the new reactionconditions. The lack of any effect of the hydrophobiccharacter on the epoxide hydrolysis is not in agree-ment with the results obtained with Ti-MCM-41[8],probably because of the minor role of the silica sur-face on the hydrolysis process.
In the case of zeolites and MCM-41, it seems clearthat the change in character of the micro- or meso-pores from hydrophilic to hydrophobic is significantenough to control the access of water to the catalyticsites placed inside the pores. However, in the caseof amorphous macroporous silica the situation maybe different. The number (Table 1) of trimethylsilylgroups present (1.1 mmol/g) fits well with the densityof isolated silanols in Merck 60 silica[27] and seems
J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 371
Fig. 5. Thermogravimetric analysis of SiO2-Ti(OiPr). Weight loss (A) and weight loss derivative (B) for: (a) fresh catalyst; (b) catalystused three times under normal conditions (H2O2 added in one lot); (c) catalyst used three times with slow addition of H2O2.
to confirm that hexamethyldisilazane only reacts withthis type of group[28], as shown inFig. 1. The globalhydrophobic character of SiO2(TMS) was qualita-tively confirmed by methyl red adsorption experi-ments[29]. As we reported previously, the maximumloading of titanium (1.07 mmol/g) also fits well with
the density of vicinal silanols[14]. However, in ourhands it was impossible to load such a high amountof titanium onto SiO2(TMS), whose maximum load-ing seems to be only 0.39 mmol/g (Table 1). The IRspectrum of this sample (evacuated at 250◦C) showsa broad band corresponding to associated silanols
372 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
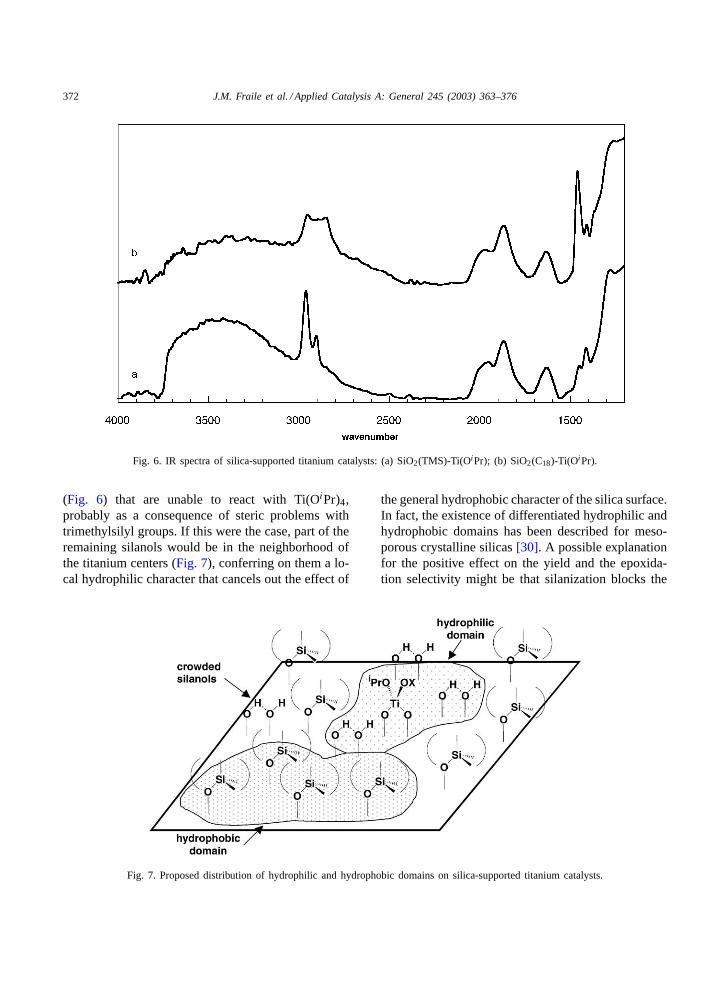
Fig. 6. IR spectra of silica-supported titanium catalysts: (a) SiO2(TMS)-Ti(OiPr); (b) SiO2(C18)-Ti(OiPr).
(Fig. 6) that are unable to react with Ti(OiPr)4,probably as a consequence of steric problems withtrimethylsilyl groups. If this were the case, part of theremaining silanols would be in the neighborhood ofthe titanium centers (Fig. 7), conferring on them a lo-cal hydrophilic character that cancels out the effect of
Fig. 7. Proposed distribution of hydrophilic and hydrophobic domains on silica-supported titanium catalysts.
the general hydrophobic character of the silica surface.In fact, the existence of differentiated hydrophilic andhydrophobic domains has been described for meso-porous crystalline silicas[30]. A possible explanationfor the positive effect on the yield and the epoxida-tion selectivity might be that silanization blocks the

J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 373
access of H2O2 to impurity centers present in the sil-ica support[31], which are responsible for part of itsdecomposition.
In contrast, the behavior of the catalyst after re-covery is not as good as expected and a significantdecrease both in yield and contribution of the directepoxidation (only 33%) are observed in the third run.Neither the amount of adsorbed byproducts (only0.5 mmol C/g) nor the titanium leaching (Table 1)accounts for this loss of selectivity.
With these results in mind, we tried to further in-crease the hydrophobic character of the silica by usinga silane with longer hydrocarbon chains. With this aim,SiO2(C18) was prepared by treatment of silica withdimethyloctadecylsilyl chloride. In this case the degreeof silanization (Table 1) was lower than with the TMSgroup (0.61 mmol silane/g) probably because of stericproblems. The large number of methylene groups leadsto a broad band in the 2960–2850 cm−1 region ofthe IR spectrum (Fig. 6), together with a significantincrease in the intensity of the band at 1460 cm−1.As a consequence of the lower silane functionaliza-tion, the maximum titanium loading (0.58 mmol/g) ishigher than in the case of SiO2(TMS). Both types ofsilanization give rise to similar values for the max-imum available silanol groups at around 1.8 mmolOH/g, taking into account the grafting of the titaniumspecies through two bonds. With regard to epoxidation(Table 3), the yield of epoxidation products is lowerthan that obtained with SiO2(TMS), but the contri-bution of the direct epoxidation shows a significantincrease, reaching the highest value (87%) of all thecatalysts described in this paper. As far as catalyst re-covery is concerned, the solid with C18 groups is alsomore stable with regard to both yield and selectivity(59% contribution of direct epoxidation in the thirdrun). Once again the high degree of epoxide hydroly-sis is remarkable, in spite of the highly hydrophobiccharacter of the silane used in this case. It is notewor-thy that the total yield of epoxidation products afterthree runs is around 100 mmol/mmol Ti, which com-pares well with results obtained with more sophisti-cated mesoporous crystalline supports.
One very significant aspect for the hydrophobic cat-alysts is the low level of titanium leaching after threeruns, with a total H2O2/Ti ratio of around 180. Fromthe conclusions obtained for TS-1[15], titanium leach-ing may be due to the presence of highly oxygenated
byproducts that form stable complexes with titaniumand favor the breakdown of the Si–O–Ti bonds. Thehigher stability of the silica-supported titanium cata-lysts under these new reaction conditions correlateswell with the smaller amounts of byproducts gener-ated in the reaction.
3.4. Modification of dispersion and immediateenvironment around titanium
Titanium dispersion has a positive effect on activ-ity and selectivity, as demonstrated by our previousresults [14]. Other authors have also indicated thatthis factor is responsible for the promising results ob-tained with solids prepared by ion-beam implantation[32]. Given this information we tried to improve theresults with SiO2(TMS)-Ti(OiPr) by increasing thetitanium dispersion to values around 0.1 mmol Ti/g.Unfortunately, the low titanium loading led to verysmall bands in both DR-UV and IR spectra, makingit difficult to draw any conclusion about dispersion.Some tests showed that the results were not modifiedif the addition of H2O2 was carried out over only 2.5 h,with an additional reaction time of 5 h. All the resultsin Table 4were obtained under such conditions. Ascan be seen, the results associated with a higher dis-persion level are comparable to those obtained withhigher titanium loading, although a slightly loweryield (64% versus 79%) and contribution of the di-rect epoxidation (59% versus 71%) is obtained in theformer case. This trend is also reflected in the resultswith the recovered catalyst. However, it is worth not-ing that the total productivity of this catalyst afterthree runs is 175 mmol of epoxidation products permmol Ti, with no detectable leaching of titanium.
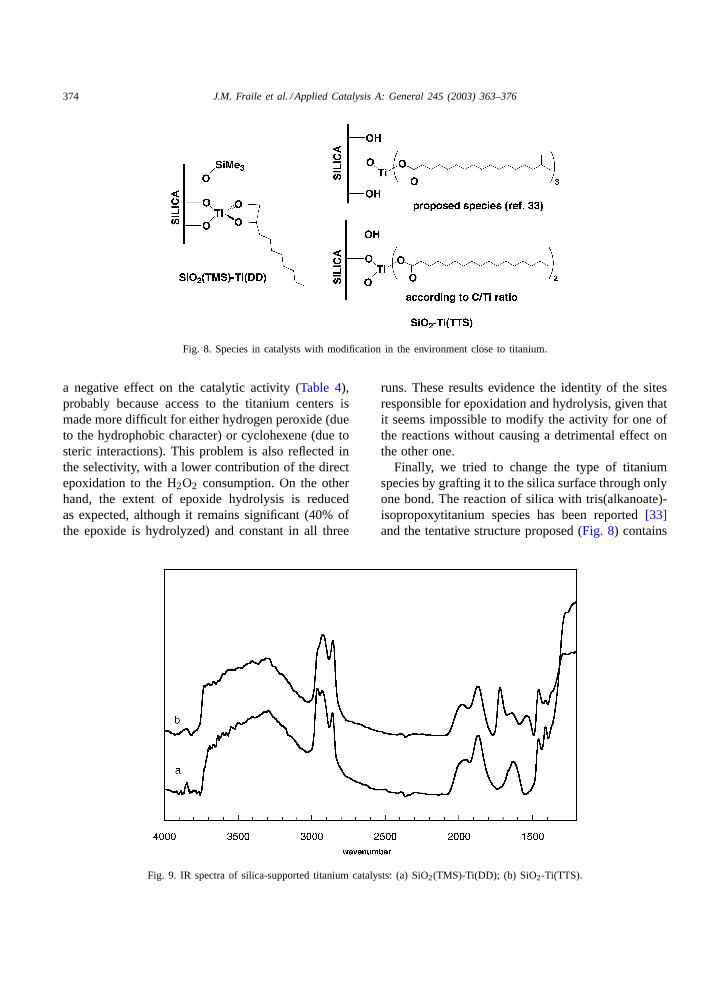
As mentioned above, the silanization of the sil-ica surface does not have an important influence onthe epoxide hydrolysis. We then tried to increasethe hydrophobic character of the immediate envi-ronment around titanium through substitution of theisopropoxy groups[11,14] by a hydrophobic diol,namely decane-1,2-diol, by distillation of the iso-propanol/toluene azeotrope. The C/Ti ratio (Table 1)is consistent with the almost exclusive presence ofthe chelate species shown inFig. 8. The substitutionis also evidenced in the IR spectrum (Fig. 9) by theintense bands at 2928 and 2859 cm−1, which aretypical of methylene groups. This substitution has
374 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
Fig. 8. Species in catalysts with modification in the environment close to titanium.
a negative effect on the catalytic activity (Table 4),probably because access to the titanium centers ismade more difficult for either hydrogen peroxide (dueto the hydrophobic character) or cyclohexene (due tosteric interactions). This problem is also reflected inthe selectivity, with a lower contribution of the directepoxidation to the H2O2 consumption. On the otherhand, the extent of epoxide hydrolysis is reducedas expected, although it remains significant (40% ofthe epoxide is hydrolyzed) and constant in all three
Fig. 9. IR spectra of silica-supported titanium catalysts: (a) SiO2(TMS)-Ti(DD); (b) SiO2-Ti(TTS).
runs. These results evidence the identity of the sitesresponsible for epoxidation and hydrolysis, given thatit seems impossible to modify the activity for one ofthe reactions without causing a detrimental effect onthe other one.
Finally, we tried to change the type of titaniumspecies by grafting it to the silica surface through onlyone bond. The reaction of silica with tris(alkanoate)-isopropoxytitanium species has been reported[33]and the tentative structure proposed (Fig. 8) contains

J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376 375
the titanium species with only one bond to the sil-ica surface and the three long hydrophobic alkanoatemoieties maintained. This species would be highlyinteresting for epoxidation, due to its presumablyhighly hydrophobic character, and we prepared thecorresponding catalyst SiO2-Ti(TTS) with the isooc-tadecanoate derivative (KR TTS). However, the C/Tiratio (Table 1) is not consistent with the species pro-posed but is consistent with titanium grafted throughtwo bonds to silica and bearing two isooctadecanoategroups (Fig. 8). The presence of the alkenoate moietiesis clearly shown in the IR spectrum (Fig. 9) by theintense C–H(st) bands in the 2950–2840 cm−1 regionand the carbonyl band at 1720 cm−1. In any case, thissolid shows only slightly higher activity and selectivity(Table 4) compared to the analogous SiO2-Ti(OiPr),although it is difficult to ascribe this effect to ei-ther the presence of TTS or to the higher titaniumdispersion.
Given the disappointing results obtained with thetris(alkanoate) approach, we decided to try DegussaAerosil 200, in spite of the problems detected in theepoxidation with addition of the oxidant in one lot. Theresults in epoxidation (Table 4) are somewhat surpris-ing. Aerosil-Ti(OiPr) is highly active, with 75% yieldin epoxidation products—a figure that represents aproductivity of 94 mmol/mmol Ti—much higher thanthe 57% obtained with one-lot addition (Table 2). It isalso a selective catalyst, with the highest contributionof the direct epoxidation to H2O2 consumption of allthe non-silanized catalysts. Most surprising, however,is the stability of the catalyst as it shows very lowleaching (Table 1) and still moderate activity and se-lectivity even in the third run, despite the fact that thereis only one silica–titanium bond. The overall produc-tivity in epoxidation products is 228 mmol/mmol Ti,with only 69 mmol allylic oxidation products/mmolTi. This promising result reveals that the titaniumspecies on Aerosil-Ti(OiPr) is more active than thaton Merck silica, although also more sensitive to de-activation. The slow addition of hydrogen peroxide isthen greatly positive for this type of catalyst. In anattempt to increase the hydrophobic character of thissupport, Aerosil 200 was treated with hexamethyld-isilazane to obtain Aerosil(TMS). However, this solidwas very difficult to handle, because it forms a thickgel in toluene which is difficult to separate by filtra-tion or centrifugation. In view of this, we are currently
working in the preparation of a silica support with thesurface properties of Aerosil 200 but the practical ad-vantages of Merck 60.
4. Conclusions
This work demonstrates the important role of oxy-gen produced by the decomposition of hydrogen per-oxide in the allylic oxidation side reaction. The slowaddition of the oxidant not only improves the yield but,more importantly, the selectivity to epoxidation prod-ucts through the direct epoxidation mechanism and forthe first time a positive effect on the catalyst stabil-ity is reported. Silica-supported titanium species arethen efficient catalysts for epoxidation with dilute hy-drogen peroxide under mild conditions. The silaniza-tion of the silica surface and titanium dispersion havepositive effects on yield and selectivity but not on theepoxide hydrolysis. The hydrophilic environment oftitanium may be responsible for this lack of effect,but its modification with hydrophobic diol or alka-noate produces a significant decrease in activity to-gether with a small reduction in the level of hydrolysis.The textural properties of the silica support seem tobe crucial for the efficiency of the supported titaniumspecies. Aerosil, which has a lower surface area andsilanol density, seems to be a better support and leadsto a highly active and selective titanium catalyst. Thisfinding opens the way to further developments in thisdirection.
Acknowledgements
This work was made possible by the generous fi-nancial support of the C.I.C.Y.T. (Project PPQ2002-04012). Kenrich Petrochemicals Inc. is gratefullyacknowledged for the generous gift of KR TTS.
References
[1] W.R. Sanderson, Pure Appl. Chem. 72 (2000) 1289.[2] (a) M.G. Clerici, G. Bellusi, U. Romano, J. Catal. 129 (1991)
159;(b) M.G. Clerici, P. Ingallina, J. Catal. 140 (1993) 71.
[3] (a) R. Murugavel, H.W. Roesky, Angew. Chem. Int. Ed. Engl.36 (1997) 477;

376 J.M. Fraile et al. / Applied Catalysis A: General 245 (2003) 363–376
(b) I.W.C.E. Arends, R.A. Sheldon, M. Wallau, U. Schuchardt,Angew. Chem. Int. Ed. Engl. 36 (1997) 1145;(c) R.A. Sheldon, M. Wallau, I.W.C.E. Arends, U. Schuchardt,Acc. Chem. Res. 31 (1998) 485.
[4] (a) A. Corma, P. Esteve, A. Martınez, S. Valencia, J. Catal.152 (1995) 18;(b) N. Jappar, Q. Xia, T. Tatsumi, J. Catal. 180 (1998) 132.
[5] (a) T. Blasco, A. Corma, M.T. Navarro, J. Pérez-Pariente, J.Catal. 156 (1995) 65;M.J. Dıaz-Cabañas, L.A. Villaescusa, M.A. Camblor, Chem.Commun. (2000) 761.
[6] L.Y. Chen, G.K. Chuah, S. Jaenicke, Catal. Lett. 50 (1998)107.
[7] T. Blasco, M.A. Camblor, A. Corma, P. Esteve, A. Martınez,C. Prieto, S. Valencia, Chem. Commun. (1996) 2367.
[8] (a) T. Tatsumi, K.A. Koyano, N. Igarashi, Chem. Commun.(1998) 325;(b) A. Corma, M. Domine, J.A. Gaona, J.L. Jordá, M.T.Navarro, F. Rey, J. Pérez-Pariente, J. Tsuji, B. McCulloch,L.T. Nemeth, Chem. Commun. (1998) 2211;(c) M.B. D’Amore, S. Schwarz, Chem. Commun. (1999) 121;(d) J. Bu, H.-K. Rhee, Catal. Lett. 65 (2000) 141;(e) J. Bu, H.-K. Rhee, Catal. Lett. 66 (2000) 245.
[9] (a) S. Klein, W.F. Maier, Angew. Chem. Int. Ed. Engl. 35(1996) 2230;(b) H. Kochkar, F. Figueras, J. Catal. 171 (1997) 420.
[10] (a) Y. Goa, P. Wu, T. Tatsumi, Chem. Commun. (2001) 1714;(b) M. Ogura, S. Nakata, E. Kikuchi, M. Matsukata, J. Catal.199 (2001) 41.
[11] J.M. Fraile, J.I. Garcıa, J.A. Mayoral, E. Vispe, J. Catal. 189(2000) 40.
[12] E. Jorda, A. Tuel, R. Teissier, J. Kervennal, J. Catal. 175(1998) 93.
[13] (a) S.A. Holmes, F. Quignard, A. Choplin, R. Teissier, J.Kervennal, J. Catal. 176 (1998) 173;(b) S.A. Holmes, F. Quignard, A. Choplin, R. Teissier, J.Kervennal, J. Catal. 176 (1998) 182.
[14] J.M. Fraile, J.I. Garcıa, J.A. Mayoral, E. Vispe, J. Catal. 204(2001) 146.
[15] (a) L. Davies, P. McMorn, D. Bethell, P.C. Bulman Page, F.King, F.E. Hancock, G.J. Hutchings, Chem. Commun. (2000)1807;(b) L. Davies, P. McMorn, D. Bethell, P.C. Bulman Page,F. King, F.E. Hancock, G.J. Hutchings, Phys. Chem. Chem.Phys. 3 (2001) 632;
(c) L.J. Davies, P. McMorn, D. Bethell, P.C. Bulman Page,F. King, F.E. Hancock, G.J. Hutchings, J. Catal. 198 (2001)319.
[16] S.B. Kumar, S.P. Mirajkar, G.C.G. Pais, P. Kumar, R. Kumar,J. Catal. 156 (1995) 163.
[17] M.C. Capel-Sanchez, J.M. Campos-Martin, J.L.G. Fierro,M.P. de Frutos, A. Padilla Polo, Chem. Commun. (2000) 855.
[18] A. Corma, M.J. Dıaz-Cabañas, M.E. Domine, F. Rey, Chem.Commun. (2000) 1725.
[19] J.M. Fraile, J. Garcıa, J.A. Mayoral, M.G. Proietti, M.C.Sánchez, J. Phys. Chem. 100 (1996) 19484.
[20] A Guide to Reflectance Coatings and Materials,http://labsphere.com/.
[21] F. Geobaldo, S. Bordiga, A. Zecchina, E. Giamello, G.Leofanti, G. Petrini, Catal. Lett. 16 (1992) 109.
[22] C.A. Müller, M. Maciejewski, T. Mallat, A. Baiker, J. Catal.184 (1999) 280.
[23] M.A. Holland, D.M. Pickup, G. Mountjoy, E.S.C. Tsang, W.Wallidge, R.J. Newport, M.E. Smith, J. Mater. Chem. 10(2000) 2495.
[24] D.C.M. Dutoit, M. Schneider, A. Baiker, J. Catal. 153 (1995)165.
[25] A.O. Bouh, G.L. Rice, S.L. Scott, J. Am. Chem. Soc. 121(1999) 7201.
[26] G. Langhendries, D.E. De Vos, G.V. Baron, P.A. Jacobs, J.Catal. 187 (1999) 453.
[27] (a) C.G. Armistead, A.J. Tyler, F.H. Hambleton, S.A. Mitchell,J.A. Hockey, J. Phys. Chem. 73 (1969) 3947;(b) K. Schrijnemakers, P. Van Der Voort, E.F. Vansant, Phys.Chem. Chem. Phys. 1 (1999) 2569.
[28] (a) J.P. Blitz, C.C. Meverden, R.E. Diebel III, Langmuir 14(1998) 1122;(b) B.H. Wouters, T. Chen, M. Dewilde, P.J. Grobet, Micro-porous Mesoporous Mater. 44–45 (2001) 453.
[29] (a) I. Shapiro, I.M. Kolthoff, J. Am. Chem. Soc. 72 (1950)776;(b) A. Heckel, D. Seebach, Chem. Eur. J. 8 (2002) 560.
[30] D. Brunel, A. Cauvel, F. Di Renzo, F. Fajula, B. Fubini, B.Onida, E. Garrone, New J. Chem. 24 (2000) 807.
[31] C. Beck, T. Mallat, A. Baiker, Catal. Lett. 75 (2001) 131.[32] (a) Q. Yang, C. Li, S. Yuan, J. Li, P. Ying, Q. Xin, W. Shi,
J. Catal. 183 (1999) 128;(b) Q. Yang, C. Li, S. Wang, J. Lu, P. Ying, Q. Xin, W. Shi,Stud. Surf. Sci. Catal. 130 (2000) 221.
[33] A. Voelkel, T. Grzeskowiak, J. Mater. Chem. 11 (2001) 1288.





























