Embed Size (px)
Citation preview

Opening dynamics of 8-oxoguanine in DNAAlicia E. Everya,b and Irina M. Russua*
8-oxoguanine is a major lesion of genomic DNA that results from oxidation of guanine by reactive oxygen species.The repair of this lesion is initiated by 8-oxoguanine glycosylases, which excise the damaged base by “flipping” itoutside the DNA double helix. The molecular mechanisms involved in the specific recognition of the damagedbase by the enzyme are not yet fully understood. Several models have proposed that, in DNA, the base pair between8-oxoguanine and cytosine may possess altered dynamic properties that could help the enzyme locate the lesionand could favor the selective extra-helical flipping of the damaged base. To test this proposal, we have characterizedthe spontaneous opening of the base pair between 8-oxoguanine and cytosine in a DNA double helix using NMRspectroscopy and proton exchange. The results show that the rate of spontaneous opening of 8-oxoguanine andthe lifetime of the base in the extra-helical state are the same as those of a canonical guanine-cytosine base pair,in the same base sequence context. This finding suggests that the opening dynamics of 8-oxoguanine, when pairedwith cytosine in DNA, does not play a significant role in the recognition of the lesion by glycosylases. Copyright ©2013 John Wiley & Sons, Ltd.Supporting information may be found in the online version of this paper
Keywords: 8-oxoguanine; base-pair opening; proton exchange; NMR
The genomes of all organisms are under continuous attack byendogenous and exogenous agents. Products of the metabolism,environmental chemicals, and UV and ionizing radiation caninduce chemical modifications of DNA bases, such as oxidation,deamination, and alkylation. These lesions alter the informationstored in the genome and thus, play key roles in aging andcarcinogenesis (Hamilton et al., 2001; Friedberg et al., 2005).A major oxidative lesion of the genome is 8-oxoguanine(i.e., 7,8-dihydro-8-oxoguanine; henceforth, abbreviated G*,Figure 1) in which guanine is oxidized at the C8 position by reac-tive oxygen species such as hydroxyl radical, superoxide, andhydrogen peroxide. The lesion is repaired by 8-oxoguanine DNAglycosylases (e.g., bacterial MutM and human OGG1), whichexcise the damaged base by cleaving the C10-N glycosidic bond.Extensive biochemical and crystallographic investigations
have demonstrated that 8-oxoguanine glycosylases performtheir function by extruding the damaged base from the interiorof the DNA double helix and placing it in a G*-specificpocket of the enzyme (Fromme et al., 2004). This “extra-helicalbase-flipping”mechanism is employed by a variety of DNA repairand modification enzymes including DNA methylases and mostother DNA glycosylases (David et al., 2007; Friedman and Stivers,2010). A question of great current interest is the mechanism bywhich the damaged base is initially selected for extra-helicalflipping. Recognition of the damaged base represents anexceedingly difficult task because the number of G* lesions inthe genome is much smaller than the number of undamagedguanines (i.e., on the average, one G* lesion per million ofundamaged guanines) (Fromme and Verdine, 2004). The diffi-culty is compounded by the fact that G* differs from undamagedG by only two atoms (i.e., the oxo group at C8 and the hydrogenatom at N7), and its base pair with cytosine is isomorphous tothe Watson–Crick GC base pair (Figure 1). Furthermore, the pres-ence of the lesion does not induce significant perturbations inneighboring base pairs and in the overall geometry of the DNAdouble helix (Oda et al., 1991; Lipscomb et al., 1995; Plum et al.,
1995; Crenshaw et al., 2011). This paucity of structural signatureshas led to the proposal that the damaged DNA site may havealtered dynamic properties that could serve as signals for itsrecognition by the enzyme. In these models, the enzyme takesadvantage of the spontaneous opening of DNA base pairs, whichis driven by thermal energy (Priyakumar and MacKerell, 2006). Atambient temperatures, all DNA base pairs open and closespontaneously, one base pair at a time, and at rates determinedby their chemical nature (e.g., AT vs. GC) and by their neigh-boring bases (Gueron and Leroy, 1995; Russu, 2004). The openstate generated by these motions could already be on thebase-flipping pathway of glycosylases (Friedman and Stivers,2010). The opening motions could also contribute to the specificrecognition of the lesion. For example, the glycosylase coulddistinguish the damaged site if the G*C base pair opened moreoften than a canonical base pair. The search for the damaged sitewould also be aided if the G*C base pair spent a longer time inthe extra-helical state than a canonical base pair. Alterations inthe opening dynamics of the G*C base pair have been suggestedby molecular dynamics simulations, which show that the ener-getic barrier for extrusion of G* from a G*C base pair is signifi-cantly reduced relative to that for extrusion of an undamagedG from a GC base pair (Cheng et al., 2005). Experimentalevidence for the existence of such alterations in the kinetics ofopening of the G*C base pair is currently lacking. To address this
* Correspondence to: IrinaM. Russu, Department of Chemistry,Wesleyan University,Middletown, CT 06459, USA.E-mail: [email protected]
a A. E. Every, I. M. RussuDepartment of Chemistry and Molecular Biophysics Program, WesleyanUniversity, Middletown, CT 06459, USA
b A. E. EveryDepartment of Molecular and Cell Biology, University of Connecticut, Storrs,CT 06250, USA
Research Article
Received: 28 August 2012, Revised: 18 December 2012, Accepted: 22 December 2012, Published online in Wiley Online Library
(wileyonlinelibrary.com) DOI: 10.1002/jmr.2262
J. Mol. Recognit. 2013; 26: 175–180 Copyright © 2013 John Wiley & Sons, Ltd.
175
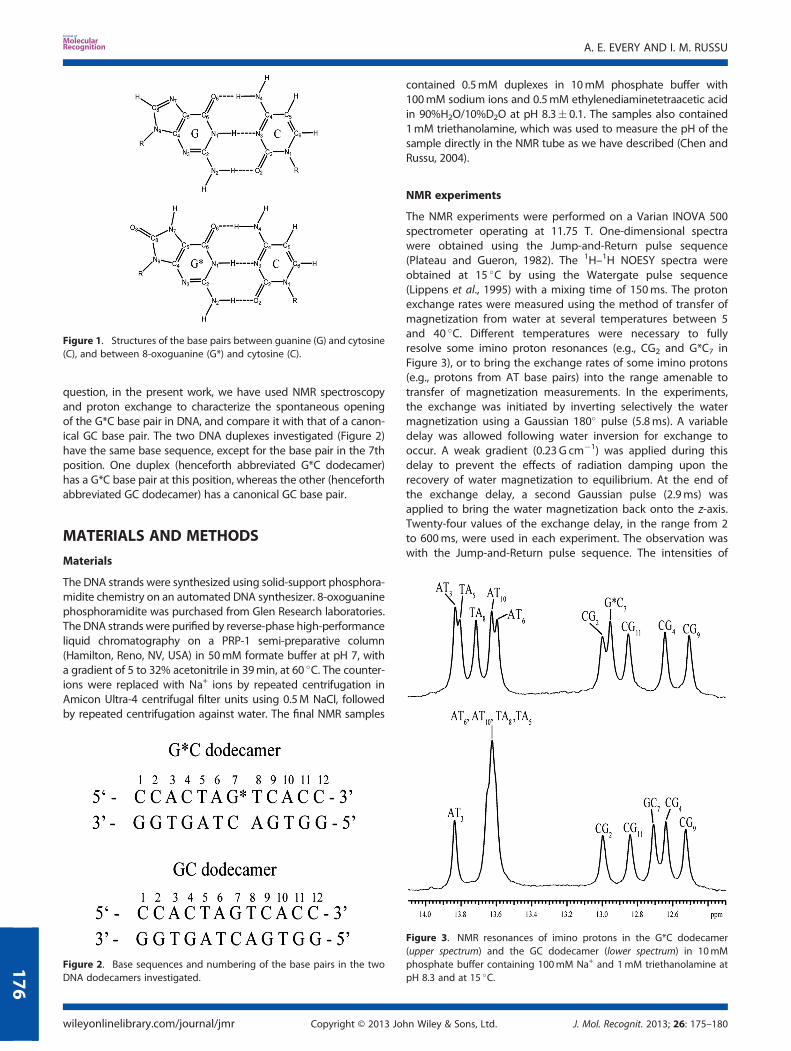
question, in the present work, we have used NMR spectroscopyand proton exchange to characterize the spontaneous openingof the G*C base pair in DNA, and compare it with that of a canon-ical GC base pair. The two DNA duplexes investigated (Figure 2)have the same base sequence, except for the base pair in the 7thposition. One duplex (henceforth abbreviated G*C dodecamer)has a G*C base pair at this position, whereas the other (henceforthabbreviated GC dodecamer) has a canonical GC base pair.
MATERIALS AND METHODS
Materials
The DNA strands were synthesized using solid-support phosphora-midite chemistry on an automated DNA synthesizer. 8-oxoguaninephosphoramidite was purchased from Glen Research laboratories.The DNA strands were purified by reverse-phase high-performanceliquid chromatography on a PRP-1 semi-preparative column(Hamilton, Reno, NV, USA) in 50mM formate buffer at pH 7, witha gradient of 5 to 32% acetonitrile in 39min, at 60 �C. The counter-ions were replaced with Na+ ions by repeated centrifugation inAmicon Ultra-4 centrifugal filter units using 0.5M NaCl, followedby repeated centrifugation against water. The final NMR samples
contained 0.5mM duplexes in 10mM phosphate buffer with100mM sodium ions and 0.5mM ethylenediaminetetraacetic acidin 90%H2O/10%D2O at pH 8.3� 0.1. The samples also contained1mM triethanolamine, which was used to measure the pH of thesample directly in the NMR tube as we have described (Chen andRussu, 2004).
NMR experiments
The NMR experiments were performed on a Varian INOVA 500spectrometer operating at 11.75 T. One-dimensional spectrawere obtained using the Jump-and-Return pulse sequence(Plateau and Gueron, 1982). The 1H–1H NOESY spectra wereobtained at 15 �C by using the Watergate pulse sequence(Lippens et al., 1995) with a mixing time of 150ms. The protonexchange rates were measured using the method of transfer ofmagnetization from water at several temperatures between 5and 40 �C. Different temperatures were necessary to fullyresolve some imino proton resonances (e.g., CG2 and G*C7 inFigure 3), or to bring the exchange rates of some imino protons(e.g., protons from AT base pairs) into the range amenable totransfer of magnetization measurements. In the experiments,the exchange was initiated by inverting selectively the watermagnetization using a Gaussian 180� pulse (5.8ms). A variabledelay was allowed following water inversion for exchange tooccur. A weak gradient (0.23G cm�1) was applied during thisdelay to prevent the effects of radiation damping upon therecovery of water magnetization to equilibrium. At the end ofthe exchange delay, a second Gaussian pulse (2.9ms) wasapplied to bring the water magnetization back onto the z-axis.Twenty-four values of the exchange delay, in the range from 2to 600ms, were used in each experiment. The observation waswith the Jump-and-Return pulse sequence. The intensities of
Figure 2. Base sequences and numbering of the base pairs in the twoDNA dodecamers investigated.
Figure 3. NMR resonances of imino protons in the G*C dodecamer(upper spectrum) and the GC dodecamer (lower spectrum) in 10mMphosphate buffer containing 100mM Na+ and 1mM triethanolamine atpH 8.3 and at 15 �C.
Figure 1. Structures of the base pairs between guanine (G) and cytosine(C), and between 8-oxoguanine (G*) and cytosine (C).
A. E. EVERY AND I. M. RUSSU
wileyonlinelibrary.com/journal/jmr Copyright © 2013 John Wiley & Sons, Ltd. J. Mol. Recognit. 2013; 26: 175–180
176
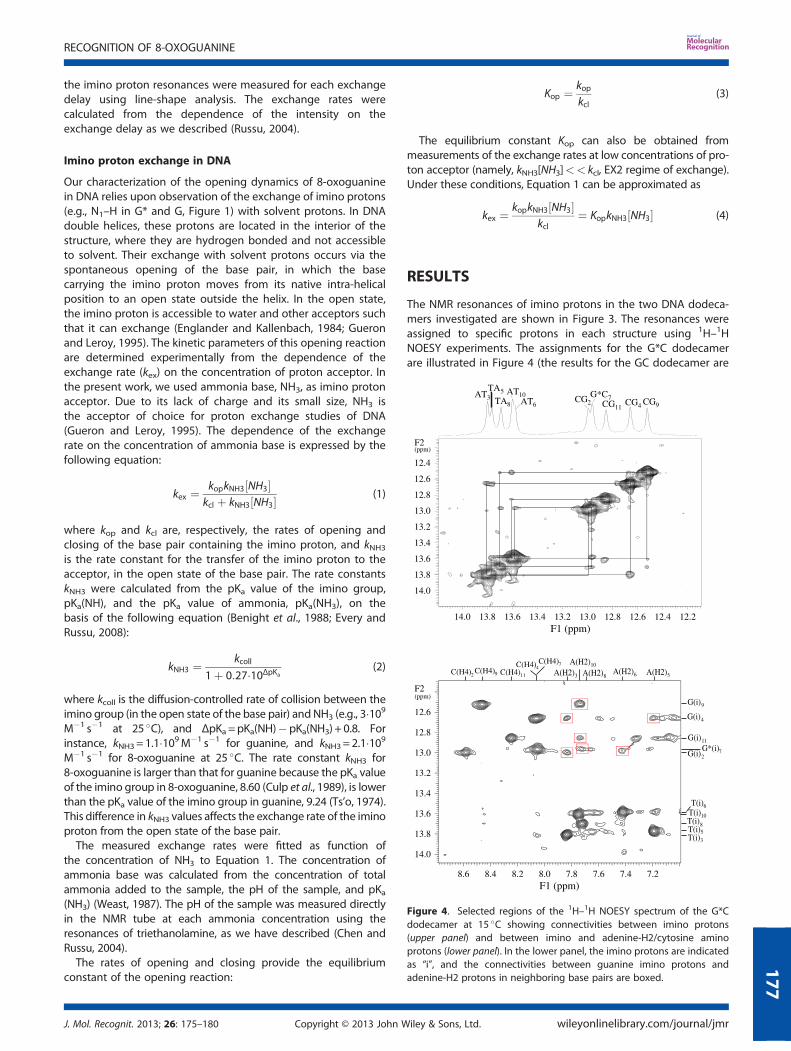
the imino proton resonances were measured for each exchangedelay using line-shape analysis. The exchange rates werecalculated from the dependence of the intensity on theexchange delay as we described (Russu, 2004).
Imino proton exchange in DNA
Our characterization of the opening dynamics of 8-oxoguaninein DNA relies upon observation of the exchange of imino protons(e.g., N1–H in G* and G, Figure 1) with solvent protons. In DNAdouble helices, these protons are located in the interior of thestructure, where they are hydrogen bonded and not accessibleto solvent. Their exchange with solvent protons occurs via thespontaneous opening of the base pair, in which the basecarrying the imino proton moves from its native intra-helicalposition to an open state outside the helix. In the open state,the imino proton is accessible to water and other acceptors suchthat it can exchange (Englander and Kallenbach, 1984; Gueronand Leroy, 1995). The kinetic parameters of this opening reactionare determined experimentally from the dependence of theexchange rate (kex) on the concentration of proton acceptor. Inthe present work, we used ammonia base, NH3, as imino protonacceptor. Due to its lack of charge and its small size, NH3 isthe acceptor of choice for proton exchange studies of DNA(Gueron and Leroy, 1995). The dependence of the exchangerate on the concentration of ammonia base is expressed by thefollowing equation:
kex ¼ kopkNH3 NH3½ �kcl þ kNH3 NH3½ � (1)
where kop and kcl are, respectively, the rates of opening andclosing of the base pair containing the imino proton, and kNH3is the rate constant for the transfer of the imino proton to theacceptor, in the open state of the base pair. The rate constantskNH3 were calculated from the pKa value of the imino group,pKa(NH), and the pKa value of ammonia, pKa(NH3), on thebasis of the following equation (Benight et al., 1988; Every andRussu, 2008):
kNH3 ¼ kcoll1þ 0:27�10ΔpKa (2)
where kcoll is the diffusion-controlled rate of collision between theimino group (in the open state of the base pair) and NH3 (e.g., 3�109M�1 s�1 at 25 �C), and ΔpKa =pKa(NH)� pKa(NH3) + 0.8. Forinstance, kNH3= 1.1�109M�1 s�1 for guanine, and kNH3= 2.1�109M�1 s�1 for 8-oxoguanine at 25 �C. The rate constant kNH3 for8-oxoguanine is larger than that for guanine because the pKa valueof the imino group in 8-oxoguanine, 8.60 (Culp et al., 1989), is lowerthan the pKa value of the imino group in guanine, 9.24 (Ts’o, 1974).This difference in kNH3 values affects the exchange rate of the iminoproton from the open state of the base pair.The measured exchange rates were fitted as function of
the concentration of NH3 to Equation 1. The concentration ofammonia base was calculated from the concentration of totalammonia added to the sample, the pH of the sample, and pKa(NH3) (Weast, 1987). The pH of the sample was measured directlyin the NMR tube at each ammonia concentration using theresonances of triethanolamine, as we have described (Chen andRussu, 2004).The rates of opening and closing provide the equilibrium
constant of the opening reaction:
Kop ¼ kopkcl
(3)
The equilibrium constant Kop can also be obtained frommeasurements of the exchange rates at low concentrations of pro-ton acceptor (namely, kNH3[NH3]<< kcl, EX2 regime of exchange).Under these conditions, Equation 1 can be approximated as
kex ¼ kopkNH3 NH3½ �kcl
¼ KopkNH3 NH3½ � (4)
RESULTS
The NMR resonances of imino protons in the two DNA dodeca-mers investigated are shown in Figure 3. The resonances wereassigned to specific protons in each structure using 1H–1HNOESY experiments. The assignments for the G*C dodecamerare illustrated in Figure 4 (the results for the GC dodecamer are
F1 (ppm)12.212.412.612.813.013.213.413.613.814.0
F2(ppm)
12.4
12.6
12.8
13.0
13.2
13.4
13.6
13.8
14.0
CG2 CG11CG4 CG9
G*C7AT6
AT3AT10
TA8
TA5
F1 (ppm)7.27.47.67.88.08.28.48.6
F2(ppm)
12.6
12.8
13.0
13.2
13.4
13.6
13.8
14.0
A(H2)5
A(H2)10
A(H2)3 A(H2)8A(H2)6
G(i)2
G(i)11
G*(i)7
G(i)4
G(i)9
T(i)3
T(i)10T(i)8
T(i)6
T(i)5
C(H4)7C(H4)4C(H4)9C(H4)2 C(H4)11
Figure 4. Selected regions of the 1H–1H NOESY spectrum of the G*Cdodecamer at 15 �C showing connectivities between imino protons(upper panel) and between imino and adenine-H2/cytosine aminoprotons (lower panel). In the lower panel, the imino protons are indicatedas “i”, and the connectivities between guanine imino protons andadenine-H2 protons in neighboring base pairs are boxed.
RECOGNITION OF 8-OXOGUANINE
J. Mol. Recognit. 2013; 26: 175–180 Copyright © 2013 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
177
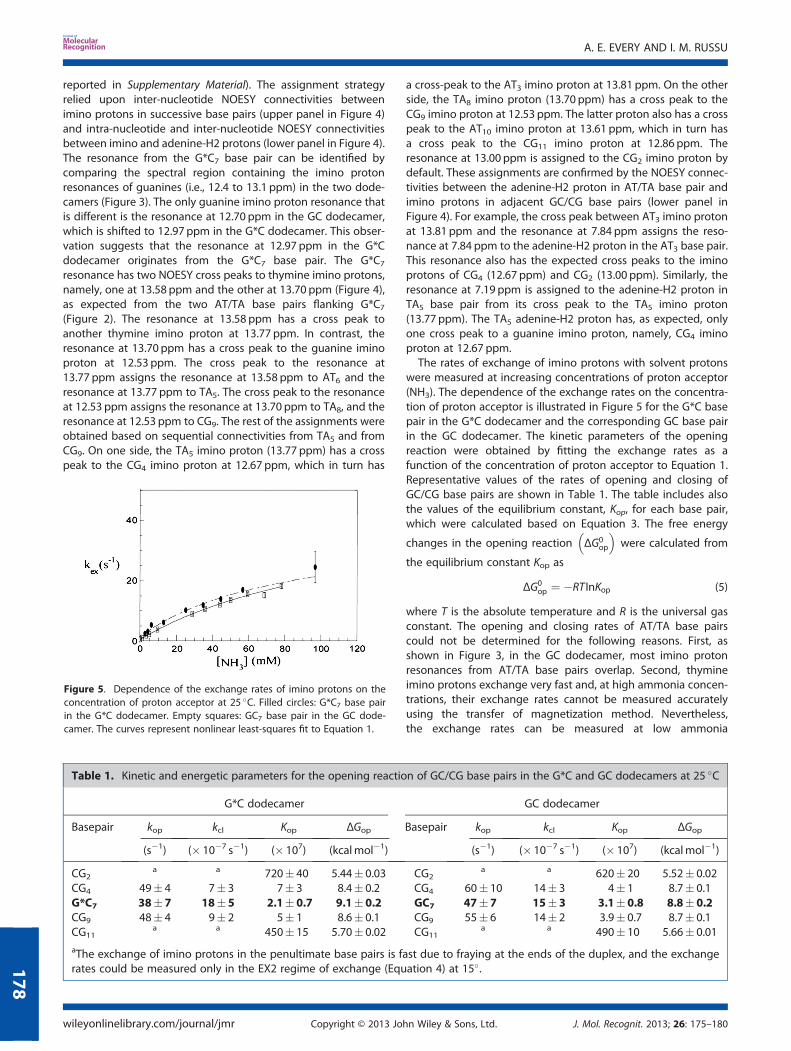
reported in Supplementary Material). The assignment strategyrelied upon inter-nucleotide NOESY connectivities betweenimino protons in successive base pairs (upper panel in Figure 4)and intra-nucleotide and inter-nucleotide NOESY connectivitiesbetween imino and adenine-H2 protons (lower panel in Figure 4).The resonance from the G*C7 base pair can be identified bycomparing the spectral region containing the imino protonresonances of guanines (i.e., 12.4 to 13.1 ppm) in the two dode-camers (Figure 3). The only guanine imino proton resonance thatis different is the resonance at 12.70 ppm in the GC dodecamer,which is shifted to 12.97 ppm in the G*C dodecamer. This obser-vation suggests that the resonance at 12.97 ppm in the G*Cdodecamer originates from the G*C7 base pair. The G*C7resonance has two NOESY cross peaks to thymine imino protons,namely, one at 13.58 ppm and the other at 13.70 ppm (Figure 4),as expected from the two AT/TA base pairs flanking G*C7(Figure 2). The resonance at 13.58 ppm has a cross peak toanother thymine imino proton at 13.77 ppm. In contrast, theresonance at 13.70 ppm has a cross peak to the guanine iminoproton at 12.53 ppm. The cross peak to the resonance at13.77 ppm assigns the resonance at 13.58 ppm to AT6 and theresonance at 13.77 ppm to TA5. The cross peak to the resonanceat 12.53 ppm assigns the resonance at 13.70 ppm to TA8, and theresonance at 12.53 ppm to CG9. The rest of the assignments wereobtained based on sequential connectivities from TA5 and fromCG9. On one side, the TA5 imino proton (13.77 ppm) has a crosspeak to the CG4 imino proton at 12.67 ppm, which in turn has
a cross-peak to the AT3 imino proton at 13.81 ppm. On the otherside, the TA8 imino proton (13.70 ppm) has a cross peak to theCG9 imino proton at 12.53 ppm. The latter proton also has a crosspeak to the AT10 imino proton at 13.61 ppm, which in turn hasa cross peak to the CG11 imino proton at 12.86 ppm. Theresonance at 13.00 ppm is assigned to the CG2 imino proton bydefault. These assignments are confirmed by the NOESY connec-tivities between the adenine-H2 proton in AT/TA base pair andimino protons in adjacent GC/CG base pairs (lower panel inFigure 4). For example, the cross peak between AT3 imino protonat 13.81 ppm and the resonance at 7.84 ppm assigns the reso-nance at 7.84 ppm to the adenine-H2 proton in the AT3 base pair.This resonance also has the expected cross peaks to the iminoprotons of CG4 (12.67 ppm) and CG2 (13.00 ppm). Similarly, theresonance at 7.19 ppm is assigned to the adenine-H2 proton inTA5 base pair from its cross peak to the TA5 imino proton(13.77 ppm). The TA5 adenine-H2 proton has, as expected, onlyone cross peak to a guanine imino proton, namely, CG4 iminoproton at 12.67 ppm.The rates of exchange of imino protons with solvent protons
were measured at increasing concentrations of proton acceptor(NH3). The dependence of the exchange rates on the concentra-tion of proton acceptor is illustrated in Figure 5 for the G*C basepair in the G*C dodecamer and the corresponding GC base pairin the GC dodecamer. The kinetic parameters of the openingreaction were obtained by fitting the exchange rates as afunction of the concentration of proton acceptor to Equation 1.Representative values of the rates of opening and closing ofGC/CG base pairs are shown in Table 1. The table includes alsothe values of the equilibrium constant, Kop, for each base pair,which were calculated based on Equation 3. The free energy
changes in the opening reaction ΔG0op
� �were calculated from
the equilibrium constant Kop as
ΔG0op ¼ �RT InKop (5)
where T is the absolute temperature and R is the universal gasconstant. The opening and closing rates of AT/TA base pairscould not be determined for the following reasons. First, asshown in Figure 3, in the GC dodecamer, most imino protonresonances from AT/TA base pairs overlap. Second, thymineimino protons exchange very fast and, at high ammonia concen-trations, their exchange rates cannot be measured accuratelyusing the transfer of magnetization method. Nevertheless,the exchange rates can be measured at low ammonia
Figure 5. Dependence of the exchange rates of imino protons on theconcentration of proton acceptor at 25 �C. Filled circles: G*C7 base pairin the G*C dodecamer. Empty squares: GC7 base pair in the GC dode-camer. The curves represent nonlinear least-squares fit to Equation 1.
Table 1. Kinetic and energetic parameters for the opening reaction of GC/CG base pairs in the G*C and GC dodecamers at 25 �C
G*C dodecamer GC dodecamer
Basepair kop kcl Kop ΔGop Basepair kop kcl Kop ΔGop
(s�1) (� 10�7 s�1) (� 107) (kcalmol�1) (s�1) (� 10�7 s�1) (� 107) (kcalmol�1)
CG2a a 720� 40 5.44� 0.03 CG2
a a 620� 20 5.52� 0.02CG4 49� 4 7� 3 7� 3 8.4� 0.2 CG4 60� 10 14� 3 4� 1 8.7� 0.1G*C7 38� 7 18� 5 2.1� 0.7 9.1�0.2 GC7 47�7 15� 3 3.1�0.8 8.8� 0.2CG9 48� 4 9� 2 5� 1 8.6� 0.1 CG9 55� 6 14� 2 3.9� 0.7 8.7� 0.1CG11
a a 450� 15 5.70� 0.02 CG11a a 490� 10 5.66� 0.01
aThe exchange of imino protons in the penultimate base pairs is fast due to fraying at the ends of the duplex, and the exchangerates could be measured only in the EX2 regime of exchange (Equation 4) at 15�.
A. E. EVERY AND I. M. RUSSU
wileyonlinelibrary.com/journal/jmr Copyright © 2013 John Wiley & Sons, Ltd. J. Mol. Recognit. 2013; 26: 175–180
178

concentrations, that is, in the EX2 regime of exchange (Equation4) allowing determination of the opening equilibrium constantsKop. In the G*C dodecamer, the Kop values that we measuredfor AT/TA base pairs at 15 �C are: (93� 2)� 10�7 for AT3;(114� 2)� 10�7 for TA5; (68� 2)� 10�7 for AT6; (60� 1)� 10�7
for TA8; and (134� 2)� 10�7 for AT10. All these values are inthe range previously found by our laboratory for AT/TA basepairs in a variety of non-damaged DNA duplexes (Chen andRussu, 2004; Coman and Russu, 2005a, 2005b).
DISCUSSION
The main results obtained in the present work pertain to thekinetics of opening and closing of the G*C base pair. As shownin Table 1, the rates of opening and closing of the G*C base pairare the same as those of a canonical GC base pair in the samebase sequence context. These findings provide several insightsinto the mechanisms by which the G* base is located and iden-tified by glycosylases. The “extra-helical capture” mechanismfor recognition of G* proposes that the damaged base mayreside outside the DNA helix longer than a native base, thusgiving the enzyme enough time to capture it (Verdine andBruner, 1997). The lifetime of a base pair in the open, extra-helical state is related to the rate of base-pair closing as
t extra-helicalð Þ ¼ 1kcl
(6)
On the basis of our results for the G*C base pair (Table 1), theaverage lifetime of 8-oxoguanine in the open, extra-helical stateis ~5 ns. This lifetime is comparable with that of guanine in thecanonical GC base pair in the same position of the GC duplex.Hence, the enzyme cannot distinguish the damaged base froma native undamaged base simply based on the lifetime of thebases outside the helix. The opening and closing rates of theG*C base pair also help define the role of spontaneous base-pairopening in the enzyme’s search for the damaged base. Singlemolecule studies of human OGG1 have shown that the enzymeslides along the DNA helix with a diffusion constant of 5� 106
(base pair)2/s (Blainey et al., 2006). This value predicts that theenzyme slides over a mean distance of 440 base pairs duringits mean lifetime in the DNA-bound state (i.e., 25ms),corresponding to a rate of ~1.8� 104 base pair/s. The chancethat the enzyme finds the G*C base pair in its extra-helical stateduring this time is exceedingly small because the rate at whichthe base pair opens (e.g., 38 s�1, Table 1) is much lower thanthe rate of diffusion of the enzyme along the DNA. Furthermore,because of the high rate of closing (18� 107 s�1, Table 1), thelifetime of the G*C base pair in the extra-helical state (~5 ns) istoo short to allow for a kinetically efficient encounter betweenthe enzyme and the open base pair. These observationstherefore suggest that the spontaneous opening of the G*C basepair in DNA does not occur on a timescale compatible withone-dimensional search of the enzyme for the lesion site alongthe DNA.An alternative mechanism for the recognition of the G*C base
pair by glycosylases proposes that the lesion site is selectedbecause of the lower energetic cost of moving the damagedbase from its intra-helical to its extra-helical state. (Plum et al.,1995; Verdine and Bruner, 1997; Banerjee et al., 2006; Davidet al., 2007) The intra-helical stability of the G*C base pair in
the DNA double helix can be defined based on the equilibrium
constant (Kop) or the free energy change ΔG0op
� �of its opening
reaction (Equations 3 and 5). The opening equilibrium constantfor the G*C base pair is ~2�10�7 (Table 1). This value correspondsto a free energy cost for opening the G*C base pair of ~9 kcalmol�1 (at 25 �C). For the canonical GC base pair in the sameposition of the GC dodecamer helix, the opening free energy,hence the stability, is, within experimental errors, the same asthat for the G*C base pair. The implication of these findings isthat the stability of the G*C base pair in the DNA double helixdoes not represent a feature of the lesion site that can be recog-nized by glycosylases.
The stability of the G*C base pair in a DNA duplex structure hasbeen recently investigated by Núñez and co-workers using NMRspectroscopy and imino proton exchange (Crenshaw et al., 2011).Because of the choice of proton acceptor (i.e., glycine) and theexperimental conditions used (pH 7.5 at 8 �C), the exchange rateswere measured only at low proton acceptor concentrations,namely, ≤1.5mM. This range corresponds to the EX2 regime ofexchange (Equation 4 and Figure 5). As a result, Núñez andco-workers were unable to determine the rates of opening andclosing of the G*C base pair, and compare them to thecorresponding rates of an undamaged GC base pair. Nevertheless,analysis of their exchange data in the EX2 regime allowed determi-nation of the opening equilibrium constants, Kop. Like the presentwork, their results showed that the Kop value for the G*C base pairis the same as that for a canonical GC base pair.
The NMR results obtained in our work also provide new insightinto the long-range effects of the G*C base pair upon theundamaged base pairs in DNA. As shown in Figure 3, most iminoproton resonances of the undamaged bases in the G*C dodeca-mer occur at the same spectral positions as the correspondingresonances of the GC dodecamer. Small changes are observedfor the resonances from TA8 and TA5 base pairs: in the G*C dode-camer, these resonances shift by 0.11 and 0.16 ppm, respectively,from their positions in the GC dodecamer. A change in the chem-ical shift of the TA8 resonance may be expected because of theproximity of this base pair to the damaged base. The change inthe TA5 resonance suggests that the G*C base pair induces smallchanges in the local conformation of canonical base pairs, whichare removed from it. Such conformational changes are, however,sufficiently small that they do not affect the opening kinetics andthe stability of distant base pairs (Table 1). For example, for theCG4 and CG9 base pairs, which are 3 and 2 positions removedfrom the damaged base, the opening and closing rates in theG*C dodecamer are, within experimental errors, the same asthose in the GC dodecamer. Similarly, for the AT3 base pair, theopening equilibrium constant is (93� 2)� 10�7 in the G*Cdodecamer and (86� 3)� 10�7 in the GC dodecamer (at 15 �C).Moreover, as discussed in the Results section, the values of theopening equilibrium constant that we measured in the G*Cdodecamer are all in the range previously found by our labora-tory in a variety of other non-damaged DNA duplexes (Chenand Russu, 2004; Coman and Russu, 2005a, 2005b). These results,therefore, indicate that the long-range effects of the G*C basepair, if any, are very small.
In summary, the present work has shown that, in DNA, therates of spontaneous opening and closing, and the stability ofthe G*C base pair are indistinguishable from those of a canonicalGC base pair, in the same base sequence context. Therefore,recognition of the G*C base pair does not rely upon its intrinsic
RECOGNITION OF 8-OXOGUANINE
J. Mol. Recognit. 2013; 26: 175–180 Copyright © 2013 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
179

dynamic and energetic properties. The opening properties of theG*C base pair could, however, be altered by the binding of theglycosylase to the DNA. For example, molecular dynamics simu-lations predict that the bending of the DNA helix induced by the
enzyme (Qi et al., 2009) enhances the opening propensity of theG*C base pair such that its extrusion from the helix is facilitated.Experimental definition of these changes awaits the protonexchange study of damaged DNA in complex with the glycosylase.
REFERENCES
Banerjee A, Santos WL, Verdine GL. 2006. Structure of a DNA glycosylasesearching for lesions. Science 311: 1153–1157.
Benight AS, Schurr JM, Flynn PF, Reid BR, Wemmer DE. 1988. Melting of aself-complementary DNA minicircle. Comparison of optical meltingtheory with exchange broadening of the nuclear magneticresonance spectrum. J Mol Biol 200: 377–399.
Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. 2006. A base-excision DNA-repair protein finds intrahelical lesion bases by fastsliding in contact with DNA. Proc Natl Acad Sci USA 103: 5752–5757.
Chen C, Russu IM. 2004. Sequence-dependence of the energetics ofopening of AT base pairs in DNA. Biophys J 87: 2545–2551.
Cheng X, Kelso C, Hornak V, de los Santos C, Grollman AP, Simmerling C.2005. Dynamic behavior of DNA bases containing 8-oxoguanine.J Am Chem Soc 127: 13906–13918.
Coman D, Russu IM. 2005a. A nuclear magnetic resonance investigationof the energetics of basepair opening pathways in DNA. BiophysJ 89: 3285–3292.
Coman D, Russu IM. 2005b. Base-pair opening in three DNA unwindingelements. J Biol Chem 280: 20216–20221.
Crenshaw CM, Wade JE, Arthanari H, Frueh D, Lane BF, Nunez ME. 2011.Hidden in plain sight: subtle effects of the 8-oxoguanine lesion onthe structure, dynamics, and thermodynamics of a 15-base pairoligodeoxynucleotide duplex. Biochemistry 50: 8463–8477.
Culp SJ, Cho BP, Kadlubar FF, Evans FE. 1989. Structural and conforma-tional analyses of 8-hydroxy-20-deoxyguanosine. Chem Res Toxicol2: 416–422.
David SS, O’Shea VL, Kundu S. 2007. Base-excision repair of oxidative DNAdamage. Nature 447: 941–950.
Englander SW, Kallenbach NR. 1984. Hydrogen exchange and structuraldynamics of proteins and nucleic acids. Q Rev Biophys 16: 521–655.
Every AE, Russu IM. 2008. Influence of magnesium ions on spontaneousopening of DNA base pairs. J Phys Chem B 112: 7689–7695.
Friedberg EC, Walker GC, Sieda W, Wood RD, Schultz RA, Ellenberger T.2005. DNA repair and mutagenesis, 2nd edn. ASM Press:Washington, D. C.
Friedman JI, Stivers JT. 2010. Detection of damaged DNA bases by DNAglycosylase enzymes. Biochemistry 49: 4957–4967.
Fromme JC, Verdine GL. 2004. Base excision repair. Adv Prot Chem 69: 1–41.
Fromme JC, Banerjee A, Verdine GL. 2004. DNA glycosylase recognitionand catalysis. Curr Opin Struct Biol 14: 43–49.
Gueron M, Leroy J-L. 1995. Studies of base pair kinetics by NMR measure-ment of proton exchange. Methods Enzymol 261: 383–413.
Hamilton ML, Remmen HV, Drake JA, Yang H, Guo ZM, Kewitt K,Walter CA, Richardson A. 2001. Does oxidative damage to DNAincrease with age? Proc Natl Acad Sci USA 98: 10469–10474.
Lippens G, Dhalluin C, Wieruszeski J-M. 1995. Use of a water flip-back pulsein the homonuclear NOESY experiment. J Biomol NMR 5: 327–331.
Lipscomb LA, Peek ME, Morningstar ML, Verghis SM, Miller EM, Rich A,Essigmann JM, Williams LD. 1995. X-ray structure of a DNA decamercontaining 7,8-dihydro-8-oxoguanine. Proc Natl Acad Sci USA 92:719–723.
Oda Y, Uesugi S, Ikehara M, Nishimura S, Kawase Y, Ishikawa H, Inoue H,Ohtsuka E. 1991. NMR studies of a DNA containing 8-hydroxydeoxy-guanosine. Nucleic Acids Res 19: 1407–1412.
Plateau P, Gueron M. 1982. Exchangeable proton NMR without base-linedistortion, using new strong-pulse sequences. J Am Chem Soc104: 7310–7311.
Plum GE, Grollman AP, Johnson F, Breslauer KJ. 1995. Influence of theoxidatively damaged adduct 8-oxodeoxyguanosine on the confor-mation, energetics, and thermodynamic stability of a DNA duplex.Biochemistry 34: 16148–16160.
Priyakumar UD, MacKerell AD Jr. 2006. Computational approaches forinvestigating base flipping in oligonucleotides. Chem Rev 106:489–505.
Qi Y, Spong MC, Nam K, Banerjee A, Jiralerspong S, Karplus M, Verdine GL.2009. Encounter and extrusion of an intrahelical lesion by a DNArepair enzyme. Nature 462: 762–768.
Russu IM. 2004. Probing site-specific energetics in proteins and nucleicacids by hydrogen exchange and NMR spectroscopy. MethodsEnzymol 379: 152–175.
Ts’o POP. 1974. Basic principles in nucleic acid chemistry, vol. I. AcademicPress: New York.
Verdine GL, Bruner SD. 1997. How do DNA repair proteins locatedamaged bases in the genome? Chemistry & Biology 4: 329–334.
Weast RC. 1987. CRC handbook of chemistry and physics, 67th edn. CRCPress: Boca Raton, FL.
A. E. EVERY AND I. M. RUSSU
wileyonlinelibrary.com/journal/jmr Copyright © 2013 John Wiley & Sons, Ltd. J. Mol. Recognit. 2013; 26: 175–180
180





























