Embed Size (px)
Citation preview

OF1 | CANCER DISCOVERY march 2016 www.aacrjournals.org
ReseaRch aRticle
Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120Shih-Hsun Chen1, Youyan Zhang1, Robert D. Van Horn1, Tinggui Yin1, Sean Buchanan1, Vipin Yadav1, Igor Mochalkin2, Swee Seong Wong3, Yong Gang Yue3, Lysiane Huber1, Ilaria Conti4, James R. Henry2, James J. Starling1, Gregory D. Plowman1, and Sheng-Bin Peng1
1Oncology Research, Eli Lilly and Company, Indianapolis, Indiana. 2Discovery Chemistry Research and Technologies, Eli Lilly and Company, Indianapolis, Indiana. 3Tailored Therapeutics, Eli Lilly and Company, Indianapolis, Indiana. 4Oncology Business Unit, Eli Lilly and Company, Indianapolis, Indiana.Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
abstRact We have identified previously undiscovered BRAF in-frame deletions near the αC-helix region of the kinase domain in pancreatic, lung, ovarian, and thyroid cancers.
These deletions are mutually exclusive with KRAS mutations and occur in 4.21% of KRAS wild-type pancreatic cancer. siRNA knockdown in cells harboring BRAF deletions showed that the MAPK activity and cell growth are BRAF dependent. Structurally, the BRAF deletions are predicted to shorten the β3/αC-helix loop and hinder its flexibility by locking the helix in the active αC-helix-in conformation that favors dimer formation. Expression of L485-P490–deleted BRAF is able to transform NIH/3T3 cells in a BRAF dimer–dependent manner. BRAF homodimer is confirmed to be the dominant RAF dimer by proximity ligation assays in BRAF deletion cells, which are resistant to the BRAF inhibitor vemurafenib and sensitive to LY3009120, a RAF dimer inhibitor. In tumor models with BRAF deletions, LY3009120 has shown tumor growth regression, whereas vemurafenib is inactive.
SIGNIFICANCE: This study discovered oncogenic BRAF deletions with a distinct activation mechanism dependent on the BRAF dimer formation in tumor cells. LY3009120 is active against these cells and represents a potential treatment option for patients with cancer with these BRAF deletions, or other atypical BRAF mutations where BRAF functions as a dimer. Cancer Discovery; 6(3); 1–16. ©2016 AACR.
Corresponding Author: Sheng-Bin Peng, Lilly Research Laboratories, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN 46285. Phone: 317-433-4549; Fax: 317-276-1414; E-mail: [email protected]: 10.1158/2159-8290.CD-15-0896©2016 American Association for Cancer Research.
iNtRODUctiONSomatic mutations in the BRAF gene were discovered
in 2002 in melanoma, where they behave as potent onco-genes and activate downstream MAPK signaling and cancer cell growth (1–5). BRAF mutations have subsequently been found in many other tumor types, including thyroid, ovarian,
colorectal, and non–small cell lung cancers, as well as in hairy cell leukemia and Langerhans cell histiocytosis (1, 6–9). In melanoma, the V600E hotspot mutation is particularly preva-lent, but mutations affecting side chains other than valine 600 (non-V600 or atypical) have been described either adja-cent to the activation segment (e.g., L597, G596, F595, and
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF2
E586) or within the glycine-rich GXGXXG motif (e.g., G464, G466, and G469) of the kinase domain (1, 10). In patients with melanoma, atypical BRAF mutations were detected in 37 out of 499 (7.4%) patient specimens (11). In colorectal cancer samples with BRAF mutations, it has been reported that approximately 50% of them are V600E, and the remain-ing 50% are atypical mutations, i.e., D594V and T599I (12). BRAF mutations are found in 6.7% of lung adenocarcinomas, and 80% of these are atypical BRAF mutations (13). In addi-tion to missense mutations, oncogenic BRAF fusions were also identified in cancers of the thyroid and prostate, mela-noma, and astrocytomas (14–19). These fusions either encode protein partners that contribute coiled-coil (CC) or zinc-fin-ger dimerization motifs to produce constitutively activated BRAF dimers, or remove at least the first eight exons of BRAF that are known to promote BRAF dimerization.
Recent mechanistic studies suggest that BRAFV600E protein functions as a monomer, whereas most of the atypical BRAF-mutant proteins function as dimers (5, 20–23). The ectopic expression of BRAFV600E induces constitutive activation of downstream ERK1/2 signaling, including negative feedback regulation of RTKs and RAS. Among BRAFV600E-expressing cells, BRAF monomer remains the primary driver of MEK1/2 and ERK1/2 signaling with minimal contribution from RTKs and RAS, thus making it an attractive anticancer target
(4, 24). This led to the identification and FDA approval of the BRAF-selective inhibitors vemurafenib and dabrafenib. Both inhibitors showed antitumor activities in BRAF-mutant xeno-graft models (25–27), and significant clinical benefit among patients with BRAF-mutant melanoma (28–30). However, these first-generation BRAF drugs are not effective inhibitors of dimeric forms of RAF, including RAS-activated RAF dimers, many atypical BRAF mutants, BRAF splice forms, and BRAF fusions. Indeed, vemurafenib and dabrafenib have been shown to induce the dimerization of RAF proteins and pro-mote paradoxical pathway activation of the MAPK pathway in BRAF wild-type (WT) cells (31–33). The paradoxical activa-tion is thought to explain the promotion of tumor growth and metastasis observed with BRAF inhibitors in BRAF WT preclinical models (33, 34). Clinically, these compounds pro-mote skin side effects including keratoacanthomas and squa-mous cell carcinomas (29, 30). We have recently developed LY3009120, a pan-RAF and RAF dimer inhibitor currently in clinical studies. LY3009120 is able to effectively inhibit active RAF dimers with minimal paradoxical activation (35, 36). In this study, we have discovered novel BRAF aberrant variants, which have in-frame deletions within or adjacent to residues L485-P490 of the αC-helix region in patient samples and cell lines of pancreatic, lung, and ovarian cancers. Further analy-ses revealed that the L485-P490–deleted BRAF is an activating
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF3 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
BRAF mutation, functions as a BRAF homodimer, and is able to transform the cells. Tumor cells with these BRAF deletions are resistant to the BRAF selective inhibitor vemurafenib but sensitive to the RAF dimer inhibitor LY3009120 in vitro and in vivo. LY3009120 represents a potential opportunity for treat-ment of patients with cancer with BRAF deletions or other atypical BRAF mutations where BRAF is activated as a dimer.
ResUltsIdentification and Confirmation of BRAF In-Frame Deletions within or Adjacent to Residues L485-P490 of the aC-Helix Region in Cancer Cell Lines and Patient Samples
In an unbiased screen of a large panel of tumor cell lines for their sensitivity to MAPK pathway inhibitors, we observed that BxPC-3 cells were very sensitive to the RAF dimer inhibi-tor LY3009120, but not sensitive to the BRAF-selective inhibi-tors vemurafenib or dabrafenib (36). BxPC-3 cells, derived from a pancreatic adenocarcinoma, are unusual for not having a KRAS mutation. As part of our Lilly internal genomics effort, we have genetically characterized the tumor cell line panel by whole exome sequencing (WES). When we carefully evaluated these data for mutations in RAS pathway genes in BxPC-3 cells, including small in-frame deletions which are easy to be ignored by routine data analysis, we discovered that BxPC-3 cells have a 5-amino acid deletion near the αC-helix region of the BRAF kinase domain (V487-P492>A). One other cell line, NCI-H2405, also stood out in our tumor cell line profiling for being sensitive to LY3009120, but insensitive to BRAF-selective inhibitors, despite having no well-described mutations in RAS pathway genes (36). Close inspection of WES data revealed an in-frame deletion in H2405 affecting the same region of the BRAF gene in BxPC-3 cells (Table S1). Further searching of cell line databases identified one additional cell line, OV-90, an ovarian adenocarcinoma with a similar BRAF N486-P490 deletion, and sensitive to LY3009120, but not vemurafenib. These in-frame deletions were further verified and con-firmed by Sanger sequencing (i.e., BxPC-3; Supplementary Fig. S1A–S1C).
These results hinted that the small in-frame deletions might explain the high sensitivity of these cells to LY3009120 and therefore would indicate a potential subset of patients who could benefit from this drug. To evaluate if these in-frame deletions occur in patients with cancer, we analyzed the publicly available databases from The Cancer Genome Atlas (TCGA) and The International Cancer Genome Consortium (ICGC). Indeed, several variations of these in-frame dele-tions were discovered in pancreatic cancer patient samples (T488-Q493>K, N486-Q490 deletion) and thyroid carcinoma samples (P490-Q494, T488-P492, N486-P490 deletion) from TCGA studies. Two pancreatic cancer patient samples harbor-ing in-frame deletion variants (T488-Q493>K and N486-P940 deletion) were also found in ICGC studies (Supplementary Table S1). Further analysis revealed that these BRAF in-frame deletions are mutually exclusive from RAS and BRAFV600 muta-tions. In all above reported cases, no missense mutation in KRAS (G12, G13, Q61), NRAS (Q61), or BRAF (V600) was found. This is especially relevant in pancreatic cancers that have a high prevalence of KRAS mutation. Overall, the rate of
BRAF deletion in patients with KRAS WT pancreatic cancer is 4.21% based on TCGA and ICGC studies (Fig. 1A). For thyroid carcinomas, 3 out of 506 patients (0.59%) were identified to have the BRAF in-frame deletion. These incidence rates are likely underestimated due to the technical challenge in detect-ing a deletion and the limitations of sequencing technologies.
BRAF In-Frame Deletions Activate MAPK Signaling in Tumor Cells and Ectopically Expressed Cells
To evaluate if these in-frame BRAF deletions activate down-stream signaling, we conducted RAF isoform–specific knock-down with siRNA in tumor cell lines H2405, BxPC-3, and OV-90. As demonstrated in Fig. 1B with H2405 cells, siRNA knockdown of BRAF alone, or combinations of BRAF and other RAF isoforms, showed significant decreases in phospho-MEK and ERK. However, knockdown of ARAF or CRAF alone, or their combination, had minimal effects on phospho-MEK or ERK levels. Similar siRNA knockdown results were observed in BxPC-3 (Fig. 1C) and OV-90 (Fig. 1D) cells, although the degree of phospho-MEK and ERK inhibition was different among these cells, likely due to differences in BRAF knock-down efficiency. These results suggest that BRAF is a major isoform to activate MAPK signaling in these tumor cells. To further confirm pathway activation, we transfected a represent-ative BRAF L485-P490 deletion (∆BRAF) with or without the RAF dimer–deficient mutation (BRAF R509H) into HEK293 cells. As shown in Fig. 1E, ∆BRAF caused significant elevation of phospho-MEK and ERK. Interestingly, the R509H mutation showed significantly reduced activation, suggesting that the BRAF deletion may function as a RAF dimer. In addition to activating MAPK signaling, BRAF in-frame deletions appear to be important for tumor cell proliferation. siRNA knockdown of BRAF alone showed significant inhibition of cell prolifera-tion in H2405 (Fig. 1F), BxPC-3 (Fig. 1G), and OV-90 (Fig. 1H) cells. However, knockdown of either ARAF or CRAF had no statistically significant effect on cell proliferation as compared with controls. Overall, the MAPK activation and cell prolifera-tion data suggest that the identified BRAF deletions are activat-ing alterations and potentially oncogenic.
BRAF Deletions Transform Cells in a BRAF Dimer–Dependent Manner
To validate if BRAF deletions possess oncogenic transforma-tion activities, mouse NIH/3T3 cells were stably transfected with ∆BRAF and then grown in soft-agar culture. In three independent studies, ectopic expression of ∆BRAF was able to transform NIH/3T3 cells and promote colony formation in soft agar (Fig. 2A and Supplementary Fig. S2), whereas expression of WT BRAF revealed no transformation activity (Supplemen-tary Fig. S2). As a positive control, the anchorage-independent growth of NIH/3T3 cells was also observed with the expression of BRAFV600E. Again, ∆BRAF with a R509H mutation did not support anchorage-independent growth in soft-agar culture (Fig. 2A and B and Supplementary Fig. S2), indicating that ∆BRAF-promoted anchorage-independent growth is depend-ent on BRAF dimerization. Additionally, the ectopic expression of ∆BRAF in NIH/3T3 cells elevated MAPK signaling as evalu-ated by phospho-MEK and ERK levels, whereas a concomitant R509H mutation reduced phospho-MEK and ERK (Fig. 2C), consistent with the observation in HEK293 cells (Fig. 1E).
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF4
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
Figure 1. Prevalence of BRAF deletions in thyroid and pancreatic patient samples and BRAF dependency of MAPK activation in tumor cells and HEK293 cells harboring BRAF deletions. A, prevalence of BRAF deletions in thyroid and pancreatic patient samples. PACA-AU, pancreatic cancer Australia; PACA-CA, pancreatic cancer Canada. B–D, knockdown of BRAF, but not ARAF or CRAF, inhibits MEK and ERK phosphorylation (p) in BRAF deletion cells. H2405, BxPC-3, and OV-90 cells with endogenous expression of BRAF deletions were transfected with control scramble siRNA or ARAF, BRAF, or CRAF siRNA either individually or in combination, as indicated. Cell lysates were analyzed for MEK and ERK phosphorylation by Western blot analysis. E, ectopic expres-sion of BRAF deletion activates MAPK signaling in HEK293 cells. MEK and ERK phosphorylation in HEK293 cells transfected with pcDNA3.1 vector control or FLAG-tagged BRAF L485-P490 deletion (∆BRAF) with or without the RAF dimer disrupting mutation (BRAFR509H). F–H, knockdown of BRAF, not ARAF or CRAF, inhibits cell proliferation in BRAF deletion cells. Cell viability (mean ± SEM, relative to control) of H2405, BxPC-3, and OV-90 transfected with control, ARAF, BRAF, or CRAF siRNA were analyzed by the CellTiter-Glo assay (*, P < 0.05; **, P < 0.01, one-tailed t test).
A
B
E F G H
C D
Cancer type
Thyroid
Pancreas
Pancreas
Pancreas
Pancreas
TCGA
TCGA
ICGC PACA-AU
ICGC PACA-CA
TCGA + ICGC
Study
506
131
392
112
635
H2405
HEK293 H2405 BxPC-3
BxPC-3
Con
trol
Con
trol
FLAG
120** *
*
100
Rel
ativ
e lu
min
esce
nce
Rel
ativ
e lu
min
esce
nce
Rel
ativ
e lu
min
esce
nce
80
60
40
20
0Control ARAF BRAF CRAF
120
100
80
60
40
20
0Control ARAF BRAF CRAF
120
100
80
60
40
20
0Control ARAF BRAF CRAF
pMEK
pERK
ERK
∆BR
AF
∆RA
FB
R50
9H
AR
AF
BR
AF
CR
AF
A &
BR
AF
A &
CR
AF
B &
CR
AF
A &
B &
CR
AF
Con
trol
AR
AF
BR
AF
CR
AF
A &
BR
AF
A &
CR
AF
B &
CR
AF
A &
B &
CR
AF
Con
trol
AR
AF
BR
AF
CR
AF
A &
BR
AF
A &
CR
AF
B &
CR
AF
A &
B &
CR
AF
siRNA
ARAF
BRAF
Samplecounts
3
2
1
1
4
BRAFdeletion cases
0.59%
1.53%
0.26%
0.89%
0.63%
Overallprevalence
38
43
14
95
Sample count(KRAS WT)
5.25%
2.33%
7.14%
4.21%
Prevalencein KRAS WT
OV-90
CRAF
pMEK
pERK
β-Actin
OV-90
Overall, these results strongly suggest that ∆BRAF is an activat-ing and oncogenic alteration, because the anchorage-independ-ent growth is one of the hallmarks of cell transformation.
To further evaluate the role of the in-frame BRAF deletions in the transformation of tumor cells, H2405 and OV-90 were transfected with ARAF, BRAF, or CRAF siRNA and grown in soft-agar culture. On the one hand, as demonstrated in Fig. 2D–F,
knockdown of BRAF resulted in a significant decrease in colony formation. On the other hand, knockdown of either ARAF or CRAF had minimal effect compared with the control. This suggests that BRAF plays the most important role among RAF isoforms in maintaining transformation activities in these tumor cells. It is also noteworthy that knockdown of KRAS showed minimal effects on transformation activities (Fig. 2G–I)
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF5 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
or MAPK signaling (Fig. 2J) in these BRAF deletion–expressing tumor cells, suggesting that these BRAF deletions function as activating mutations in a KRAS-independent manner.
BRAF In-Frame Deletions Mainly Function as BRAF Homodimers
To understand if BRAF deletions mainly function as BRAF homodimers or BRAF/CRAF heterodimers, we developed in situ proximity ligation assays (PLA) as described previously
(37, 38). As a control for BRAF homodimers, we transfected A375 cells with a construct encoding the p61BRAFV600E splice variant. Consistent with previous observations that p61BRAFV600E mainly functions as a BRAF homodimer (5), the ectopic expression of p61BRAFV600E exhibits a strong in situ PLA signal for BRAF homodimers but minimal detect-able BRAF/CRAF heterodimer signal (Fig. 3A). HeLa cells treated with EGF to induce BRAF/CRAF heterodimers served as a positive control for the ability of our PLA system to
Figure 2. ∆BRAF is an oncogenic alteration and transforms NIH/3T3 cells in a BRAF-dimer dependent manner. A, ∆BRAF is able to transform NIH/3T3 cells in a BRAF dimer–dependent manner. NIH/3T3 cells stably expressing FLAG-tagged ∆BRAF with or without R509H mutation, MYC-tagged BRAFV600E, or empty vector were seeded in soft-agar plates for colony formation. Colonies were examined by light microscopy after 3 weeks, and representative images from at least three independent studies were taken. B, quantification of colonies in soft-agar growth of A. The numbers of colonies are expressed as mean ± SEM from three experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001, relative to control, one-tailed t test). C, ∆BRAF activates MAPK signaling in a BRAF dimer–dependent manner in NIH/3T3 cells. Cell lysates from NIH/3T3 expressing the indicated BRAF proteins were examined for MEK and ERK phosphorylation. D, BRAF, but not ARAF or CRAF, is important for cell proliferation in tumor cells with BRAF deletions. H2405 and OV-90 cells were seeded in soft-agar plates 48 hours after transfection with indicated control, ARAF, BRAF, or CRAF siRNA. Representative images from at least two independent studies were taken after 3 weeks. E and F, quantification of colonies in soft-agar growth of D. G, KRAS is not important in tumor cells harboring BRAF dele-tions. H2405 and OV-90 cells were seeded in soft agar 48 hours after transfection with control or KRAS siRNA. Representative images from at least two independent studies were taken after 3 weeks. H and I, quantification of colonies in soft-agar growth of G. J, KRAS-independent MAPK activation in tumor cells with BRAF deletions. Cell lysates from H2405 and OV-90 with KRAS knockdown were analyzed for MEK and ERK phosphorylation.
A B C
D
G H I J
E F
Control ∆BRAF ∆BRAF R509H BRAF V600E
0
60
50
40
30
Num
ber
of H
2405
col
onie
s
Num
ber
of H
2405
col
onie
s
Num
ber
of O
V-9
0 co
loni
es
Num
ber
of O
V-9
0 co
loni
es
20
10
0
60 n.s.n.s.
H2405
− + − ++ − + −
OV-90
KRAS siRNACtrl siRNA
KRAS
pMEK
pERK
β-Actin
50
40
30
20
10
0
60
50
40
30
20
10
0Control KRAS Control KRAS
60
50
40
30
20
10
0Control ARAF BRAF CRAF Control ARAF BRAF CRAF
Control ∆BRAF ∆BRAFR509H
BRAFV600E
BRAF
Con
trol
∆BR
AF
∆BR
AF
R50
9H
BR
AF
V60
0E
10
20
30
Num
ber
of c
olon
ies
40
**
***
**
***
50
Control
Control KRAS
H24
05O
V-9
0
H24
05O
V-9
0
ARAF BRAF CRAF
MYC
FLAG
pMEK
pERK
β-Actin
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF6
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
detect this dimeric species (38). As revealed in Supplementary Fig. S3A, treatment of HeLa cells by EGF induced a clear PLA signal for BRAF/CRAF heterodimer. With PLAs for BRAF homodimers and BRAF/CRAF heterodimers established, we examined the status of RAF dimers in tumor cells. As
shown in Fig. 3A, H2405, BxPC-3, and OV-90 cells harboring BRAF in-frame deletions all showed clear evidence for BRAF homodimers but not for BRAF/CRAF heterodimers, suggest-ing that the BRAF homodimer is the major RAF dimer in these cells (Fig. 3A and B).
Figure 3. BRAF in-frame deletions mainly function as BRAF homodimers. A, detection of BRAF/CRAF heterodimers and BRAF homodimers in H2405, BxPC-3, OV-90, and A375 cells ectopically expressing p61BRAFV600E using in situ PLA. B, quantification of in situ PLA signals of A (mean ± SEM). The number of PLA signals per cell with at least 1,000 cells for all reactions in triplicate was quantified and analyzed by Cellomics ArrayScan VTI Reader and HCS software (**, P < 0.01; ***, P < 0.001, one-tailed t test). C, detection of BRAF/CRAF heterodimers and BRAF homodimers in HEK293 cells ectopically expressing BRAFE586K, BRAF L485-P490 deletion (∆BRAF) with or without R509H, or empty vector (control) using PLA. PLA signals (red spots) were examined under a confocal microscope and representative images were shown from at least two or three independent experiments. D, quantification of in situ PLA signals of C (mean ± SEM) in HEK293 expressing the indicated BRAF proteins. E, detection of BRAF homodimer by IP and Western blot analysis. HEK293 cells stably expressing FLAG-tagged ∆BRAF or ∆BRAF R509H protein were transfected with vectors encoding MYC-tagged ∆BRAF followed by IP using anti-FLAG or anti-MYC antibody–conjugated beads. The input and IP-prepared proteins were subjected to Western blot analysis with anti-FLAG and anti-MYC antibodies. F, CRAF S338 phosphorylation and MAPK activation in ∆BRAF-transfected HEK293 and NIH/3T3 cells.
A
C
E F
D
BA375 p61V600E
Control
BR
AF
/CR
AF
hete
rodi
mer
sB
RA
F/C
RA
Fhe
tero
dim
ers
H2405 BxPC-3 OV-90
12BRAF/CRAFBRAF/BRAF
BRAF/CRAFBRAF/BRAF
10
8*** ***
***
*** **
***
Sig
nals
per
cel
lS
igna
ls p
er c
ell
6
4
2
0
25
20
15
10
5
0Control BRAF
E586K∆BRAF
∆BRAFFLAG
Input
∆BRAF-MYC
FLAG
HEK293 NIH/3T3
Con
trol
∆BR
AF
Con
trol
∆BR
AF
MYC
FLAG
pCRAF S338
CRAF
BRAF
pMEK
pERK
ERK
β-Actin
MYC
FLAG
MYC
β-Actin
− + − +
IP: FLAG
IP: MYC
∆BRAFR509H
A375p61V600E
H2405
BR
AF
/BR
AF
hom
odim
ers
***
***
BxPC-3 OV-90
BRAF E586K ∆BRAF ∆BRAF R509H
BR
AF
/BR
AF
hom
odim
ers
∆BRAF R509HFLAG
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF7 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
The existence of BRAF homodimers in tumor cells, and the demonstration that Arg509 is critical for signaling of these deletion mutants, indicated that the mutations may activate downstream signaling by promoting the forma-tion of dimers just like many other oncogenic BRAF altera-tions. To test this idea, we transfected ∆BRAF into HEK293 cells to detect in situ dimerization using PLA. As a positive control, ectopic expression of BRAFE586K, which has been shown to promote RAF dimerization (22, 38, 39), exhibited a strong PLA signal for the BRAF homodimer (Fig. 3C). In contrast, BRAF homodimers were minimal in cells express-ing CRAFE478K, a CRAF dimer–promoting mutation, serving as a negative control (Supplementary Fig. S3B). Similar to BRAFE586K, the ectopic expression of the ∆BRAF predomi-nantly promoted BRAF homodimers, while a low level of BRAF/CRAF heterodimer signal was detected. The homodi-mer signal was substantially reduced by R509H BRAF dimer– deficient mutation (Fig. 3C and D). Further, we cotransfected two constructs encoding the same ∆BRAF with different tags, FLAG or MYC, into HEK293 cells. As shown in Fig. 3E, immunoprecipitation (IP) with a FLAG antibody was able to pull down the MYC-tagged BRAF protein. Similarly, IP with a MYC antibody was able to pull down the FLAG-tagged BRAF protein, confirming the formation of BRAF homodimers. Consistent with the PLA results, BRAF R509H mutation reduced the BRAF dimer formation in these IP studies (Fig. 3E). All together, the PLA and IP studies show that ∆BRAF promotes homodimer formation in cells, providing an expla-nation for oncogenicity of this new class of BRAF mutations.
The low-level PLA signal for BRAF/CRAF heterodimers may be an artifact of the system used but could, conceivably, instead reflect a cryptic role of CRAF in the signaling path-way activated by ∆BRAF. To determine whether CRAF may be active in BRAF deletion–mediated MAPK activation, we transfected ∆BRAF into HEK293 and NIH/3T3 cells. ∆BRAF activated phospho-MEK and ERK in both cell lines (Fig. 3F). However, CRAF protein was not activated in either cell line based on phosphorylation of serine 338, suggesting that CRAF is not functionally engaged in ∆BRAF-mediated MAPK activation. In three tumor cell lines with BRAF deletions and A375 cells with a BRAFV600E mutation, the endogenous phospho-CRAF levels are generally low with the exception of BxPC-3 cells, whereas phospho-CRAF activities are generally higher in KRAS-mutant tumor cells (Supplementary Fig. S4).
BRAF Deletions Shorten the b3/aC-Helix Loop and Hinder Its Flexibility by Locking the Helix in the Active aC-Helix-in Conformation That Favors BRAF Dimerization
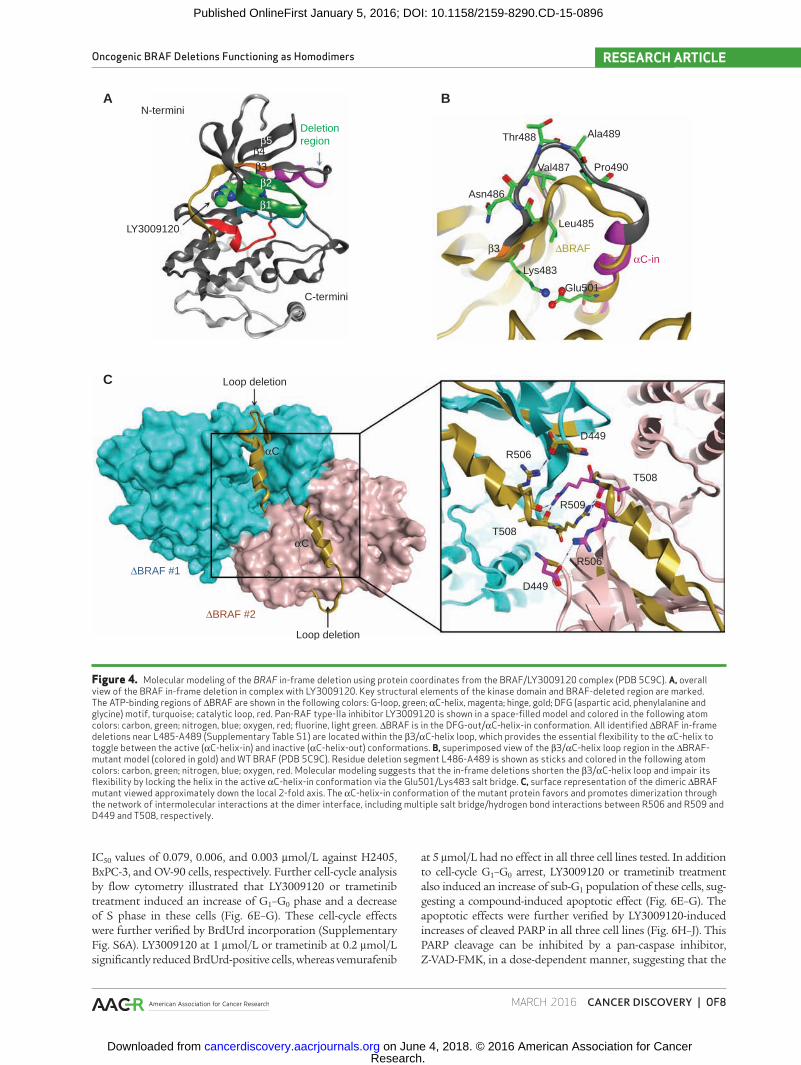
To explain how BRAF in-frame deletions promote protein dimerization and activation of the MAPK signaling pathway, we located structural regions corresponding to the identified deletions in the crystal structure of the BRAF kinase domain in complex with LY3009120 (PDB 5C9C). Based on the struc-tural information, all in-frame deletions listed in Supple-mentary Table S1 were mapped to the β3/αC-helix loop, an essential structural region responsible for a conformational movement of the αC-helix between the αC-helix-in and αC-helix-out conformations (Fig. 4A). Molecular modeling sug-gests that the in-frame deletions shorten the β3/αC-helix loop
and hinder the flexibility of αC-helix by locking it in the αC-helix-in conformation via the Glu501/Lys483 salt bridge (Fig. 4B). Although this αC-helix-in conformation accommodates binding of type IIa inhibitors (e.g., LY3009120), it disfavors the type IIb binders (e.g., vemurafenib) that require the αC-helix-out binding conformation. Meanwhile, the αC-helix-in con-formation of BRAF promotes protein dimerization through the network of intermolecular interactions at the dimer sur-face, including multiple salt bridge/hydrogen bond interac-tions between Arg506 and Arg509, and Asp449 and Thr508, respectively (Fig. 4C). To summarize, the active αC-helix-in conformation stabilized by a shortened β3/αC-helix loop region in the BRAF in-frame deletion mutants favors BRAF dimerization, leading to MAPK pathway activation. This is consistent with previous reports suggesting that the αC-helix-in conformation of the RAF proteins promotes dimer formation (36, 40).
BRAF Deletion–Mediated MAPK Activation Is Sensitive to LY3009120, a RAF Dimer Inhibitor, but Resistant to Vemurafenib
Next, we investigated the sensitivity of the BRAF deletion–mediated MAPK activation to MAPK pathway inhibitors. As revealed in Fig. 5A, vemurafenib fails to reduce phospho-MEK or ERK, at concentrations below 10 μmol/L in all three cell lines—H2405, BxPC-3, and OV-90—that harbor BRAF deletions. Similarly, treatment with another BRAF-selective inhibitor, dabrafenib, revealed minimal effects on MEK and ERK phosphorylation in H2405 and BxPC-3 and modest inhibition in OV-90 (Supplementary Fig. S5A). In contrast, LY3009120 demonstrated potent and dose-dependent inhibi-tion of phospho-MEK and ERK with significant inhibition observed at 0.01 μmol/L in all three cell lines (Fig. 5B). Similar to LY3009120, the MEK inhibitor trametinib is potent and active at inhibiting the phospho-MEK and ERK activities in these cells (Fig. 5C). These results further solidify the notion that the BRAF deletions function as RAF dimers. The sen-sitivities of BRAF-deleted cells to LY3009120 are similar to BRAFV600E-mutant A375 cells and are considerably more sensi-tive than KRAS-mutant HCT116 cells based on phospho-ERK inhibition (Fig. 5D and E) and cell proliferation (Supple-mentary Fig. S5B and S5C). To further verify these results, we compared the inhibitory activities of vemurafenib and LY3009120 in HEK293 cells transfected with ∆BRAF. Con-sistent with results obtained in H2405, BxPC-3, and OV-90 cells, LY3009120 exhibited dose-dependent inhibition of phospho-MEK and ERK in HEK293 cells (Fig. 5F). However, vemurafenib showed no inhibitory activity (Fig. 5G).
Growth of Tumor Cells Harboring BRAF Deletion Is Sensitive to LY3009120, but Resistant to Vemurafenib In Vitro
We then evaluated the in vitro growth inhibitory activities of MAPK pathway inhibitors to tumor cells harboring BRAF dele-tions. As shown in Fig. 6A–D, LY3009120 demonstrated a con-centration-dependent cell growth inhibition with IC50 values of 0.04, 0.087, and 0.007 μmol/L against H2405, BxPC-3, and OV-90 cells, respectively. However, vemurafenib had minimal activity inhibiting the cell growth of these cells. Again, the MEK inhibitor trametinib showed potent cell growth inhibition with
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896
march 2016 CANCER DISCOVERY | OF8
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
Figure 4. Molecular modeling of the BRAF in-frame deletion using protein coordinates from the BRAF/LY3009120 complex (PDB 5C9C). A, overall view of the BRAF in-frame deletion in complex with LY3009120. Key structural elements of the kinase domain and BRAF-deleted region are marked. The ATP-binding regions of ∆BRAF are shown in the following colors: G-loop, green; αC-helix, magenta; hinge, gold; DFG (aspartic acid, phenylalanine and glycine) motif, turquoise; catalytic loop, red. Pan-RAF type-IIa inhibitor LY3009120 is shown in a space-filled model and colored in the following atom colors: carbon, green; nitrogen, blue; oxygen, red; fluorine, light green. ∆BRAF is in the DFG-out/αC-helix-in conformation. All identified ∆BRAF in-frame deletions near L485-A489 (Supplementary Table S1) are located within the β3/αC-helix loop, which provides the essential flexibility to the αC-helix to toggle between the active (αC-helix-in) and inactive (αC-helix-out) conformations. B, superimposed view of the β3/αC-helix loop region in the ∆BRAF-mutant model (colored in gold) and WT BRAF (PDB 5C9C). Residue deletion segment L486-A489 is shown as sticks and colored in the following atom colors: carbon, green; nitrogen, blue; oxygen, red. Molecular modeling suggests that the in-frame deletions shorten the β3/αC-helix loop and impair its flexibility by locking the helix in the active αC-helix-in conformation via the Glu501/Lys483 salt bridge. C, surface representation of the dimeric ∆BRAF mutant viewed approximately down the local 2-fold axis. The αC-helix-in conformation of the mutant protein favors and promotes dimerization through the network of intermolecular interactions at the dimer interface, including multiple salt bridge/hydrogen bond interactions between R506 and R509 and D449 and T508, respectively.
A
C
BN-termini
C-termini
∆BRAF #1
∆BRAF #2
Loop deletion
LY3009120
β5
β2
β1
β4
αC
αC
Loop deletion
D449
Thr488 Ala489
Pro490Val487
Asn486
Leu485
Lys483
Glu501
R506
R509
T508
D449
R506
T508
β3
β3αC-in
∆BRAF
Deletionregion
IC50 values of 0.079, 0.006, and 0.003 μmol/L against H2405, BxPC-3, and OV-90 cells, respectively. Further cell-cycle analysis by flow cytometry illustrated that LY3009120 or trametinib treatment induced an increase of G1–G0 phase and a decrease of S phase in these cells (Fig. 6E–G). These cell-cycle effects were further verified by BrdUrd incorporation (Supplementary Fig. S6A). LY3009120 at 1 μmol/L or trametinib at 0.2 μmol/L significantly reduced BrdUrd-positive cells, whereas vemurafenib
at 5 μmol/L had no effect in all three cell lines tested. In addition to cell-cycle G1–G0 arrest, LY3009120 or trametinib treatment also induced an increase of sub-G1 population of these cells, sug-gesting a compound-induced apoptotic effect (Fig. 6E–G). The apoptotic effects were further verified by LY3009120-induced increases of cleaved PARP in all three cell lines (Fig. 6H–J). This PARP cleavage can be inhibited by a pan-caspase inhibitor, Z-VAD-FMK, in a dose-dependent manner, suggesting that the
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF9 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
Figure 5. BRAF deletion–mediated MAPK activation is sensitive to LY3009120, a RAF dimer inhibitor, and trametinib, but resistant to vemurafenib, a BRAF monomer inhibitor. A–C, phospho-MEK and ERK levels of H2405, BxPC-3, and OV-90 cells treated with vemurafenib, LY3009120, or trametinib. Cells were treated at indicated concentrations for 2 hours, and cell lysates were analyzed for MEK and ERK phosphorylation by Western blotting. D and E, phospho-MEK and phospho-ERK inhibition of BRAFV600E-mutant A375 and KRASG13D-mutant HCT116 cells by LY3009120. F–G, phospho-MEK and phospho-ERK inhibition by LY3009120 and vemurafenib in ∆BRAF-transfected HEK293 cells. HEK293 cells stably expressing ∆BRAF were treated with LY3009120 or vemurafenib at indicated concentrations for 2 hours. Cell lysates were analyzed for MEK and ERK phosphorylation by Western blotting.
A
B
C
D E
F G
H2405 BxPC-3V
emur
afen
ibLY
3009
120
LY30
0912
0
Tram
etin
ib
LY30
0912
0
LY30
0912
0
Vem
uraf
enib
LY30
0912
0
Tram
etin
ib
Tram
etin
ib
Vem
uraf
enib
Vem
uraf
enib
0 .01 .08 .4 2 10 0 .01 .08 .4 2 10 0 .01 .08 .4 2 10
0 .01 .08 .4 2 10 0 .01 .08 .4 2 10 0 .01 .08 .4 2 10
0 .01 .08
A375 (BRAFV600E)
HEK293 ∆BRAF HEK293 ∆BRAF
0 .01 .03 0.1 0.3 1 3 10 0 .01 .03 0.1 0.3 1 3 10
HCT116 (KRASG13D)
0 0.00
30.
010.
030.
1
0.3
1 3 0 0.00
030.
001
0.00
30.
010.
030.
1
0.3
1 3 10
.4 2 10 0 .01 .08 .4 2 10 0 .01 .08 .4 2 10
µmol/L
pMEK
pERK
β-Actin
µmol/L
pMEK
pERK
β-Actin
µmol/L
pMEK
pERK
β-Actin
µmol/L
pMEK
pERK
β-Actin
µmol/L
pMEK
pERK
FLAG
BRAF
β-Actin
µmol/L
pMEK
pERK
FLAG
BRAF
β-Actin
OV-90
LY30
0912
0
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF10
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
Figure 6. Growth of tumor cells harboring BRAF deletion is sensitive to LY3009120, but resistant to vemurafenib in vitro. A–C, antiproliferation activi-ties of vemurafenib, LY3009120, and trametinib in H2405, BxPC-3, or OV-90 cells assessed by the CellTiter-Glo assay. The cells were treated for 72 hours with different inhibitors at indicated concentrations. D, antiproliferation IC50 of vemurafenib (Vem), LY3009120 (LY), or trametinib (Tra) in H2405, BxPC-3, and OV-90 cells. IC50 was calculated via sigmoidal dose-response curve using GraphPad Prism 4 software. E–G, cell-cycle analysis by flow cytom-etry of H2405, BxPC-3, or OV-90 cells treated with vemurafenib (5 μmol/L), LY3009120 (1 μmol/L), or trametinib (0.2 μmol/L). Cells were subjected to PI staining at 72 hours after treatment. Dead cells are indicated as debris. Representative histograms are shown from three independent experiments. H–J, apoptosis analysis of H2405, BxPC-3, and OV-90 cells treated with vemurafenib (5 μmol/L) or LY3009120 (1 μmol/L) for 2, 24, and 48 hours, respectively. Cell lysates were analyzed for MEK and ERK phosphorylation and cleaved PARP (cPARP) induction by Western blotting.
A
E
F
G
H I J
B C D
120H2405
DMSO
%G1 = 54.64
%G2–M = 14.47%S = 30.90
%Debris = 9.98
%G1 = 47.62
%G2–M = 11.79%S = 40.60
%Debris = 13.77
%G1 = 43.30
%G2–M = 28.97%S = 27.74
%Debris = 15.86
%G1 = 54.80
%G2–M = 35.58%S = 9.61
%Debris = 61.01
%G1 = 43.17
%G2–M = 32.12%S = 24.70
%Debris = 18.93
%G1 = 57.80
%G2–M = 32.48%S = 9.72
%Debris = 54.76
%G1 = 63.47
%G2–M = 12.07%S = 24.47
%Debris = 43.94
%G1 = 45.10
%G2–M = 11.77%S = 43.13
%Debris = 11.22
%G1 = 73.67
%G2–M = 12.07%S = 14.26
%Debris = 26.86
%G1 = 70.84
%G2–M = 12.32%S = 16.84
%Debris = 43.13
%G1 = 60.88
%G2–M = 12.89%S = 26.23
%Debris = 9.77
%G1 = 71.47
%G2–M = 12.09%S = 16.44
%Debris = 34.17
LY3009120 Vemurafenib Trametinib
BxPC-3 OV-90IC50
(nmol/L)
H2405
BxPC-3
OV-90
Vem
>10,000
>10,000
4,033.0
LY
40.0
87.4
7.1
Tra
79.1
6.1
3.1
100
80
Rel
ativ
e lu
min
esce
nce
60
40
20
120
100
80
Rel
ativ
e lu
min
esce
nce
60
20
40
0
120
100VemurafenibLY3009120
Trametinib80
Rel
ativ
e lu
min
esce
nce
60
20
40
0−1 0
00
4080
120
160
070
140
210
280
0 50 100 150PI
200 250
0 50 100 150PI
200 250
0 50 100 150PI
H2405
DMSO LY30091202 24 48 2 24 48
Vemurafenib
BxPC-3 OV-90
DMSO LY30091202 24 48 2 24 48
Vemurafenib DMSO LY30091202 24 48 2 24 48 h
pERK
pMEK
cPARP
β-Actin
Vemurafenib
200 250
060
120
180
240
0 50 100 150PI
200 250
100
200
300
400
Num
ber
Num
ber
Num
ber
Num
ber
080
160
240
320
400
0 50 100 150PI
200 250 0 50 100 150PI
200 250
Num
ber
080
160
240
320
400
Num
ber
070
140
210
280
0 50 100 150PI
200 250 0 50 100 150PI
200 250
Num
ber
050
100
150
200
Num
ber
0 50 100 150PI
200 250
080
160
240
320
400
Num
ber
H24
05B
xPC
-3O
V-9
0
500
600
0 50 100 150PI
200 250
100
020
030
040
0
Num
ber 50
060
0
0 50 100 150PI
200 250
020
040
0
Num
ber
600
0 50 100 150PI
200 250
010
020
030
0
Num
ber 40
050
0
1 2Compound: Log (nmol/L) Compound: Log (nmol/L)
3 4 −1 0 1 2 3 4 5Compound: Log (nmol/L)
−1 0 1 2 3 4 5
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF11 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
apoptosis is caspase dependent (Supplementary Fig. S6B). In contrast to LY3009120, vemurafenib treatment had minimal effects on cell-cycle G1–G0 arrest or apoptosis of these tumor cells (Fig. 6E–J). Similar to vemurafenib, dabrafenib treatment did not induce apoptosis of H2405 and BxPC-3 cells based on cPARP (Supplementary Fig. S6C).
To extend the analysis of LY3009120 against tumor cells har-boring atypical BRAF mutations where BRAF proteins mostly function as dimers, we tested 14 additional cell lines, including 2 PDX cell lines, BXF 1218L and RXL 1183L, as shown in Sup-plementary Table S2. LY3009120 is active against the majority of these tumor cell lines in vitro. Among them, 12 of 14 cell lines exhibited absolute IC50 values from 0.045 to 0.58 μmol/L LY3009120, whereas vemurafenib was generally inactive.
Xenograft Tumors Harboring BRAF Deletions Are Sensitive to LY3009120, but Resistant to Vemurafenib In Vivo
We then attempted to develop rat xenograft models with H2405, BxPC-3, and OV-90 cells. Both H2405 and BxPC-3 cells were able to grow tumors consistently, whereas OV-90 cells failed to grow tumors in nude rats. To assess the in vivo sensitivity to MAPK pathway inhibitors, we treated the xeno-graft tumors with LY3009120 or vemurafenib as described. As demonstrated in Fig. 7A and Supplementary Fig. S7A, in the H2405 xenograft model, treatment of LY3009120 at 15 or 30 mg/kg achieved almost complete tumor growth regression, whereas vemurafenib treatment at 20 mg/kg had no antitumor growth activity despite achieving significant single-agent activity in melanoma BRAFV600E-mutant models (36, 41). Similarly, in the BxPC-3 xenograft model, LY3009120 at 15 or 30 mg/kg demonstrated significant tumor growth inhibition and partial regression, whereas vemurafenib had no antitumor effect (Fig. 7B; Supplementary Fig. S7B). Western blot analysis of the tumor lysates from these studies revealed that LY3009120 significantly inhibited phospho-MEK and phospho-ERK, whereas vemurafenib did not (Fig. 7C and D). Further analysis revealed that treatment of LY3009120 at 15 or 30 mg/kg inhibited downstream phospho-MEK and ERK by approximately 70% and 60%, respectively, in the H2405 model (Fig. 7E), and significant inhibition of phospho-MEK (61% at 15 mg/kg; 71% at 30 mg/kg) and phospho-ERK (66% at 15 mg/kg; 75% at 30 mg/kg) by LY3009120 was also observed in the BxPC-3 model (Fig. 7F). Based on the inhibi-tion of tumor growth and downstream signaling, LY3009120 treatment at 15 mg/kg achieved nearly maximum effect. In both studies, LY3009120 appeared to be well tolerated at 15 and 30 mg/kg with no significant body weight loss (Supple-mentary Fig. S7C and S7D). Overall, the results from these in vivo studies are completely consistent with in vitro observa-tions. Xenograft tumors with a BRAF deletion are sensitive to RAF dimer inhibitor LY3009120 and resistant to the BRAF monomer inhibitor vemurafenib. In both studies, the tumor growth inhibition induced by LY3009120 was correlated with phospho-MEK and ERK inhibition within the tumors.
DiscUssiONBRAF inhibitors vemurafenib and dabrafenib are active in
BRAFV600-mutant melanoma. However, these inhibitors are
less active in cells expressing WT BRAF and paradoxically activate downstream RAF–MEK–ERK signaling and promote tumor growth in cells with activating RAS mutations (33, 34). Consequently, these inhibitors should be used with caution in patients whose tumors harbor a RAS mutation. Recent studies have revealed that these BRAF-selective inhibitors promote BRAF and CRAF dimerization, an essential step in the paradoxical pathway activation (31–33). It has now become more evident that vemurafenib and dabrafenib pri-marily bind one of the two protomers of the asymmetric RAF dimers and thereby fail to effectively inhibit downstream signaling (36). LY3009120 is a pan-RAF inhibitor that binds both protomers of RAF dimers and effectively inhibits down-stream signaling (35, 36). Due to their distinct mechanisms of action, LY3009120, but not vemurafenib, is active against tumor cells with RAF in-frame deletions identified in this study and many other atypical BRAF mutations where BRAF functions as dimers (Table S2). Our data provide additional evidence that LY3009120 is a RAF dimer inhibitor.
In this study, we have discovered novel oncogenic BRAF in-frame deletions with a distinct activating mechanism dependent on BRAF dimer formation in human cancers. In addition to cell lines, the BRAF deletions were also identi-fied in patients with pancreatic cancer or thyroid carcinoma with overall prevalence of 0.63% and 0.59%, respectively, and 4.2% frequency occurring in KRAS WT pancreatic cancer. The prevalence of BRAF deletions is likely underestimated because the current sequencing technologies and analytic tools are mainly designed for identification of point mutations and less favorable for identification of small in-frame deletions. In addition to BRAF in-frame deletions, other atypical BRAF mutations and BRAF fusions where BRAF functions as dimers also occur in many cancer types, including melanoma, lung, colorectal, and pancreatic cancers. In lung adenocarcinoma, the overall BRAF mutation frequency is approximately 6.7%, and 80% of these are atypical (13). In colorectal cancer, BRAF mutations are present in about 10% of patients, with 50% atypical (12, 13). Although the frequencies of these BRAF alterations in lung, colon, and pancreatic cancers are low, the disease-related mortality of these cancers irrespective of their mutational subtype remains high: 158,000 and 50,000 deaths per year in the United States in lung cancer and colon can-cer, respectively, which suggests a clear unmet medical need (42, 43). LY3009120 may have the potential for treatment of this unique patient population.
We found that BRAF deletions are mutually exclusive with RAS mutations, suggesting that BRAF deletions are potential oncogenes. Indeed, we have confirmed that they are activating and oncogenic alterations. In three cancer cell lines, BxPC-3, H2405, and OV-90, harboring BRAF deletions, siRNA knockdown of BRAF, but not CRAF or ARAF, signifi-cantly reduced phospho-MEK and ERK levels, and ectopic expression of ∆BRAF enhanced phospho-MEK and ERK acti-vation in HEK293 and NIH/3T3 cells. Knockdown of BRAF by siRNA or inhibition by LY3009120 inhibited proliferation of tumor cells harboring BRAF deletions. Ectopic expression of ∆BRAF is able to transform NIH/3T3 cells and promotes anchorage-independent growth that is comparable with that caused by the BRAFV600E mutation. Finally, xenograft models developed with tumor cells harboring a BRAF deletion are
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF12
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
Figure 7. Xenograft tumors harboring BRAF deletion are sensitive to LY3009120, but resistant to vemurafenib. A and B, antitumor activities of LY3009120 (LY) and vemurafenib (Vem) in H2405 (A) and BxPC-3 (B) models. Xenografts were treated with vemurafenib (20 mg/kg), LY3009120 (15 or 30 mg/kg), or vehicle twice daily for 3 to 4 weeks (8 animals per treatment group) and tumor volumes (mean ± SEM) were measured every 3 to 5 days. C and D, inhibition of phospho-MEK and phospho-ERK in tumor lysates. H2405 and BxPC-3 tumors were lysed following the completion of the treatment and analyzed with Western blotting for MEK and ERK phosphorylation. E and F, densitometric analysis (mean ± SEM, relative to vehicle groups) of the levels of phospho-MEK and phospho-ERK in H2405 and BxPC-3 tumors after normalized to total MEK and ERK using ImageJ software (***, P < 0.001; ****, P < 0.0001, one-tailed t test).
A B
C
E F
D
018 21 24 27 30
Day after tumor implant
Vehicle
H2405 H2405 BxPC-3 BxPC-3
0
**** ****
*** ***
Vehicl
e
LY 1
5 m
g/kg
LY 3
0 m
g/kg
Vem 2
0 m
g/kg
Vehicl
e
LY 1
5 m
g/kg
LY 3
0 m
g/kg
Vem 2
0 m
g/kg
20
40
60
80
Rel
ativ
e de
nsito
met
ry(p
ME
K/M
EK
)
100
120
0
20
40
60
80
Rel
ativ
e de
nsito
met
ry(p
ER
K/E
RK
)
100
120
******** ****
****
Vehicl
e
LY 1
5 m
g/kg
LY 3
0 m
g/kg
Vem 2
0 m
g/kg
0
20
40
60
80
Rel
ativ
e de
nsito
met
ry(p
ME
K/M
EK
)
100
120
Vehicl
e
LY 1
5 m
g/kg
LY 3
0 m
g/kg
Vem 2
0 m
g/kg
0
20
40
60
80
Rel
ativ
e de
nsito
met
ry(p
ER
K/E
RK
)100
120
15 mg/kgLY LY
33 36 39 42
1,000
2,000
3,000
4,000
Tum
or v
olum
e (m
m3 ) 5,000
6,000 Vehicle
H2405 BxPC-3
Vem (20 mg/kg, BID)
LY (15 mg/kg, BID)
LY (30 mg/kg, BID)
018 21 24 3027 33 36
Day after tumor implant
39 42 4845 51
200
400
600
800
Tum
or v
olum
e (m
m3 ) 1,000
1,200 Vehicle
Vem (20 mg/kg, BID)
LY (15 mg/kg, BID)
LY (30 mg/kg, BID)
30 mg/kg Vem
pMEK
pERK
MEK
ERK
β-Actin
Vehicle 15 mg/kgLY LY
30 mg/kg Vem
pMEK
pERK
MEK
ERK
β-Actin
highly sensitive to inhibition by the RAF dimer inhibitor LY3009120, and the tumor growth inhibition is associated with downregulation of phospho-MEK and ERK. Overall, these data support the conclusion that the novel BRAF dele-tions are activating and oncogenic alterations.
We found that MAPK activation by BRAF deletions is depend-ent on homodimerization. Ectopic expression of ∆BRAF acti-vated phospho-MEK and ERK in HEK293 and NIH/3T3 cells,
and BRAF dimer-deficient mutation R509H significantly reduced MEK and ERK activation, suggesting that BRAF-engaged dimer is important for pathway activation. Similarly in soft-agar culture, BRAF deletion with a dimer-deficient R509H substitution failed to transform NIH/3T3 cells. In situ PLA demonstrated that the BRAF homodimer is the major RAF dimer formed in tumor cells or transfected HEK293 cells harboring these BRAF in-frame deletions, and co-IP analysis
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF13 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
revealed that the BRAF deletion is able to form BRAF homodi-mers. Structural analysis revealed that these BRAF deletions shorten the β3/αC-helix loop and hinder its flexibility by lock-ing the helix in the active αC-helix-in conformation that favors BRAF dimerization. Finally, tumor cells with these BRAF dele-tions are sensitive to the RAF dimer inhibitor LY3009120 but resistant to the BRAF monomer inhibitor vemurafenib in vitro and in vivo. As indirect evidence, transfection of ∆BRAF into HEK293 or NIH/3T3 cells activated the MAPK activity in a CRAF-independent manner. All together, these data suggest that these BRAF deletions function as BRAF homodimers.
In the original description of BRAF mutations in cancer, BRAFV600E was only 1 of the 14 BRAF alterations identified in cell lines and primary tumor samples (1). Since then, nearly 300 distinct missense mutations have been found in tumor samples and cancer cell lines (44). These missense mutations encompass over 100 of the 766 BRAF amino acids, but most of the mutations occur in the activation loop (A-loop) near V600, or in the phosphate-binding loop (P-loop) at residues 464–469 (10). According to a model based on crystal structures of the BRAF kinase domain, both loops interact with each other via hydrophobic interactions (2). Disruption of this interaction by V600 mutations results in a conformational change within the kinase domain and full activation of BRAF. Many non-V600 oncogenic variants of BRAF have been shown to activate signaling by promoting the formation of active dimers, and our data suggest that BRAF deletions also promote dimer formation. Structural analysis suggests an explanation for this ability and points to a novel mechanism of dimer promo-tion. The BRAF deletions described here all have a 5-amino acid deletion just outside the P-loop, within the β3/αC-helix loop, a region at the interface critical for RAF dimerization. The β3/αC-helix loop provides flexibility to the αC-helix, allowing movement between the active (αC-helix-in) and inac-tive (αC-helix-out) conformation. The 5-amino acid deletion shortens the β3/αC-helix loop, impairing its flexibility and fixing the helix in the active αC-helix-in conformation. The αC-helix-in conformation of the BRAF proteins favors and promotes BRAF dimerization as described (36, 40). Consistent with this model, tumor cells with BRAF deletions are sensitive to the RAF dimer inhibitor LY3009120, but resistant to the BRAF monomer inhibitor vemurafenib in vitro and in vivo. In principle, the structural studies do not preclude the possible stabilization of BRAF heterodimers with ARAF or CRAF. However, we have not found evidence for a substantial contri-bution of heterodimers. For example, transfection of ∆BRAF into HEK293 or NIH/3T3 cells activated the MAPK activity in a CRAF-independent manner. The activation mechanism proposed here is consistent with recent findings showing that N-terminally truncated BRAF proteins, and many other atypi-cal BRAF mutations, function as BRAF homodimers (5, 23).
The activating effect of the BRAF deletion described here highlights a region of the kinase domain that could play an important role in the normal control of RAF activity. In addi-tion to BRAF, similar 5-amino acid deletions near the αC-helix domain of a protein kinase were also identified in other tar-gets, such as EGFR and HER2. In EGFR, the exon 19 deletions including E746-A750 were characterized in non–small cell lung cancer (45, 46). For HER2, another EGFR family mem-ber, the L755-T799 deletion was recently identified in breast
cancer (47). All these deletions have been found to be activat-ing mutations. Therefore, the activating mechanism of BRAF deletions identified in this study might represent a common mechanism for activating other protein kinases.
MethODsCell Culture, Antibodies, and Reagents
BxPC-3, H2405, OV-90, A375, HCT116, NIH/3T3, and HEK-293 cells were obtained from the ATCC from 2010 to 2013 and stored within a central cell bank that performs cell line characterizations. All these cells were passaged for less than 2 months, after which new cul-tures were initiated from vials of frozen cells. Characterization of the cell lines was done by a third-party vendor (RADIL), which included profiling by PCR for contamination by various microorganisms of bacterial and viral origin. As a result, no contamination was detected. The samples were also verified to be of human origin without mam-malian interspecies contamination. The alleles for 9 different genetic markers were used to determine that the banked cells matched the genetic profile that has been previously reported. H2405 cells were grown in ACL-4 medium (ATCC), whereas NIH/3T3, HEK-293, and A375 cells were maintained in DMEM supplemented with 10% FBS (Invitrogen). HCT116 cells were cultured in McCoy’s 5A with 10% FBS (Invitrogen), and BxPC-3 cells were cultured in RPMI with 10% FBS. OV-90 cells were grown in a 1:1 mixture of MCDB 105 medium containing a final concentration of 1.5 g/L sodium bicarbonate and Medium 199 containing a final concentration of 2.2 g/L sodium bicarbonate with 15% FBS (Thermo Scientific). The BRAF-selective inhibitor vemurafenib, the pan-RAF inhibitor LY3009120, and the MEK inhibitor trametinib were synthesized by Eli Lilly and Com-pany. All siRNAs were obtained from Dharmacon (ON-TARGETplus siRNA). siRNA transfections were performed using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manu-facturer’s instructions. All plasmids were created using standard cloning methods with pcDNA3.1 (Invitrogen) as a vector. All plasmid transfections were carried out using FuGENEHD transfection rea-gents (Promega) as per the manufacturer’s instructions.
Deletion Detection from Sequencing AnalysisBRAF deletion calls on cancer cell lines were aggregated by search-
ing through the repositories COSMIC v71 and Sanger Institute’s Cancer Cell Line Project, and exome sequencing variant calls from Broad Institute’s Cancer Cell Line Encyclopedia (CCLE), as well as internally generated exome sequencing data. Internal exome data were prepared using Agilent Sure-Select 38 Mbp all-exon capture library sequenced on the Illumina HiSeq 2000 platform, generat-ing approximately 80× paired-end reads. Variant calling on internal exome and CCLE exome data were performed using the BWA-mem aligner v0.7.4 (mapped to GRCh37) and called with GATK lite v2.3, Freebayes v0.9.10, and Samtools mpileup v0.1.19. TCGA mutation data (MAF files) were downloaded from the Broad Institute’s GDAC firehose (2014_10_17 release). RNA-sequencing data (fastq files) from TCGA were downloaded from http://cghub.ucsc.edu under controlled access in accordance with the data-user agreement, and mapped to human genome GRCh37 using the GSNAP (2013-11–27) aligner. BRAF deletions based on RNA-sequencing data from TCGA and CCLE were identified by searching for reads with at least 5 base deletions in the BRAF genomic region, and analysis was done to determine the consequence of the change if it resulted in an in-frame deletion. For TCGA pancreatic and thyroid cancer samples, we further confirmed the deletion identified by searching through the mapped reads from the whole exome sequencing data where there were cases in which the deletion was not reported from the mutation data downloaded from Broad’s firehose. The ICGC data (release 17) was accessible from http://dcc.icgc.org.
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF14
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
Preparation of Cell Lysates, Western Blot Analysis, and Cell Proliferation Assay
Cell lysate preparation, Western blot analysis, and cell proliferation assay were performed as described previously (41, 48).
Transfections and CoimmunoprecipitationA375 cells transfected with BRAFV600E with deleted amino acids
169–380 (A375 p61V600E) and HEK293 transfected with BRAF E586K, CRAF E478K, ∆BRAF, and ∆BRAF R509H were generated using pcDNA3.1 vectors under G418-containing medium selection and evaluated for BRAF and FLAG or MYC-tagged protein expression as described previously (36). For coimmunoprecipitation, HEK293 cells stably expressing FLAG-tagged ∆BRAF or ∆BRAF R509H pro-teins were transfected with pcDNA3.1 vector encoding MYC-tagged ∆BRAF followed by immunoprecipitation using anti-FLAG (Sigma) or anti-MYC magnetic beads (Cell Signaling Technologies). The IP-prepared proteins were next subjected to Western blot analysis as previously described (36).
Colony Transformation Assay in Soft-Agar CultureFor ectopic expression, NIH/3T3 cells transfected with ∆BRAF,
∆BRAF with R509H, BRAFV600E, or BRAF WT constructs or parental vector (pcDNA3.1) were selected in G418-containing medium for 2 weeks. The transfected cells (3 × 104) in growth media with 0.3% agar were plated on top of 0.5% agar medium in 6-well tissue culture plates. Formation of spherical colonies was evaluated after 3 weeks under a microscope. For target protein knockdown with siRNAs, the transformation assay for H2405 and OV-90 cells was performed with the same procedure at 48 hours after transfection.
In Situ Proximity Ligation AssayPLA was conducted and validated according to the manufactur-
er’s instructions (Olink Bioscience) as previously described (37, 38). Briefly, cells grown on glass slides or 96-well plates were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100 before being incubated with 1% BSA-blocking solution overnight at 4°C. For detection of BRAF homodimers, monoclonal BRAF antibodies were first conjugated to PLUS and MINUS PLA oligonucleotides using the Duolink II Probemaker system (37). For detection of BRAF and CRAF heterodimers, the primary antibodies were directly used and followed by incubation with PLUS and MINUS oligonucleo-tide-conjugated PLA probes (38). The bound proximity probes were then visualized as red spots with Duolink In Situ Detection Rea-gents Orange (Olink Bioscience) and detected under a confocal fluo-rescent microscope. The nuclear staining with Hoechst 33342 was used to delineate the cells, and the quantification of the number of in situ PLA signals per cell was further analyzed by Cellomics ArrayScan VTI Reader and HCS software (Thermo Scientific). At least 1,000 cells were analyzed in each 96-well plate for all reactions in triplicate.
Cell-Cycle AnalysisCell-cycle analysis was performed as described previously (36, 41).
Cells treated with DMSO or inhibitors for 72 hours were collected and fixed in 70% ethanol for 30 minutes at −20°C. After being washed with PBS, fixed cells were stained with propidium iodide/Triton X-100 staining solution and incubated for 30 minutes at room tem-perature. Fixed cells were then subjected to flow cytometric analysis on a Beckman Coulter FC 500 Cytomics flow cytometer. Data were analyzed with ModFit LT 3.0 (Verity House Software).
BrdUrd Incorporation AssayTumor cell lines H2405, BxPC-3, and OV-90 were grown in 6-well
plates, and the growth medium was changed and the cells were treated with DMSO or inhibitors the following day. One hour prior
to the end of the 72 hours of treatment time, cell medium was spiked with 10 μmol/L BrdUrd. After 1 hour of incubation with BrdUrd, cells were harvested and fixed in 70% EtOH at −20°C. Cells were then washed with PBS/BSA, incubated with 2N HCl/FBS for 20 minutes, and treated with sodium borate. To determine the amount of BrdUrd incorporation, cells were stained with isotype control or FITC-conjugated anti-BrdUrd antibody (BD Pharmingen) for 30 minutes, washed with PBS/BSA, and incubated in propidium iodide (Life Technologies) for 30 minutes before reading on a flow cytometer. Data were analyzed with FlowJo software.
In Vivo Xenograft StudiesIn vivo studies were performed in accordance with the American
Association for Laboratory Animal Care institutional guidelines. All the experimental protocols were approved by The Eli Lilly and Company Animal Care and Use Committee. Briefly, 5 × 106 to 10 × 106 tumor cells in a 1:1 Matrigel mix (0.2 mL total volume) were injected subcutaneously into the right hind flank of female NIH nude rats (Taconic Biosciences). After tumors reached a desired size of approximately 300 mm3, animals were randomized into groups of 8 for efficacy studies. Drugs (LY3009120 or vemurafenib) were administered orally (gavage) in 0.6-mL volume of vehicle with the dose schedules described in each study. Tumor growth and body weight were monitored over time to evaluate efficacy and signs of toxicity as described (41).
Disclosure of Potential Conflicts of InterestY.G. Yue is Director, Computational Biology, at Boehringer Ingel-
heim. No potential conflicts of interest were disclosed by the other authors.
Authors’ ContributionsConception and design: S.-H. Chen, S. Buchanan, V. Yadav, J.R. Henry, J.J. Starling, G.D. Plowman, S.-B. PengDevelopment of methodology: S.-H. Chen, Y. Zhang, R.D. Van Horn, T. Yin, S.-B. PengAcquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.-H. Chen, Y. Zhang, R.D. Van Horn, T. Yin, L. HuberAnalysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.-H. Chen, Y. Zhang, S. Buchanan, I. Mochalkin, S.S. Wong, Y.G. Yue, S.-B. PengWriting, review, and/or revision of the manuscript: S.-H. Chen, V. Yadav, I. Mochalkin, S.S. Wong, I. Conti, J.R. Henry, G.D. Plowman, S.-B. PengAdministrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S.-H. Chen, R.D. Van Horn, T. Yin, S.S. Wong, L. Huber, G.D. PlowmanStudy supervision: S.-B. Peng
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received July 27, 2015; revised December 28, 2015; accepted December 30, 2015; published OnlineFirst January 5, 2016.
REFERENCES 1. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al.
Mutations of the BRAF gene in human cancer. Nature 2002;417: 949–54.
2. Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855–67.
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

OF15 | CANCER DISCOVERY march 2016 www.aacrjournals.org
Chen et al.RESEARCH ARTICLE
3. Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melano-mas. Cancer Cell 2012;22:668–82.
4. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401–9.
5. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aber-rantly spliced BRAF(V600E). Nature 2011;480:387–90.
6. Tiacci E, Schiavoni G, Martelli MP, Boveri E, Pacini R, Tabarrini A, et al. Constant activation of the RAF-MEK-ERK pathway as a diag-nostic and therapeutic target in hairy cell leukemia. Haematologica 2013;98:635–9.
7. Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919–23.
8. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 2004;5:875–85.
9. Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy. Pharmacol Ther 2014;142:176–82.
10. Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004;6:313–9.
11. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, et al. Frequency and spectrum of BRAF mutations in a retrospec-tive, single-institution study of 1112 cases of melanoma. J Mol Diagn 2013;15:220–6.
12. Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, et al. Simi-larity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res 2002;62:6451–5.
13. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495–501.
14. Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, Lyle PL, et al. BRAF fusions define a distinct molecular subset of melano-mas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013;19:6696–702.
15. Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010;16: 793–8.
16. Lee NV, Lira ME, Pavlicek A, Ye J, Buckman D, Bagrodia S, et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells through [corrected] MAPK activation. PLoS One 2012;7:e39653.
17. Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest 2005;115:94–101.
18. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673–7.
19. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846.
20. Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, et al. The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev 2012;26:1945–58.
21. Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, et al. Functional analysis of mutations within the kinase activa-tion segment of B-Raf in human colorectal tumors. Cancer Res 2003;63:8132–7.
22. Roring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J 2012;31:2629–47.
23. Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that deter-mine their sensitivity to pharmacologic inhibition. Cancer Cell 2015; 28:370–83.
24. Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res 2004;64:2338–42.
25. Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res 2010;70:5518–27.
26. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010;467:596–9.
27. King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One 2013;8:e67583.
28. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809–19.
29. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
30. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
31. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140:209–21.
32. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibi-tors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
33. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
34. Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal 2014;7:ra30.
35. Henry JR, Kaufman MD, Peng SB, Ahn YM, Caldwell TM, Vogeti L, et al. Discovery of 1-(3,3-Dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a Pan-RAF inhibitor with minimal paradoxical acti-vation and activity against BRAF or RAS mutant tumor cells. J Med Chem 2015;58:4165–79.
36. Peng SB, Henry JR, Kaufman M, Lu WP, Smith B, Vogeti S, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 2015;28:384–98.
37. Fichter CD, Timme S, Braun JA, Gudernatsch V, Schopflin A, Bogatyreva L, et al. EGFR, HER2 and HER3 dimerization patterns guide targeted inhibition in two histotypes of esophageal cancer. Int J Cancer 2014;135:1517–30.
38. Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 2013;49:751–8.
39. Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009;461:542–5.
40. Wang X, Kim J. Conformation-specific effects of Raf kinase inhibi-tors. J Med Chem 2012;55:7332–41.
41. Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregula-tion. Mol Cancer Ther 2014;13:2253–63.
42. Holderfield M, Deuker MM, McCormick F, McMahon M. Target-ing RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014;14:455–67.
43. U.S. Cancer Statistics Working Group. United States Cancer Statis-tics: 1999–2012 Incidence and Mortality Web-based Report. Atlanta:
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

march 2016 CANCER DISCOVERY | OF16
Oncogenic BRAF Deletions Functioning as Homodimers RESEARCH ARTICLE
U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; c2015. Avail-able from: www.cdc.gov/uscs
44. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–50.
45. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung can-cer to gefitinib. N Engl J Med 2004;350:2129–39.
46. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefi-tinib therapy. Science 2004;304:1497–500.
47. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification nega-tive breast cancer. Cancer Discov 2013;3:224–37.
48. Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF recep-tor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem 2012;287:28087–98.
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896

Published OnlineFirst January 5, 2016.Cancer Discov Shih-Hsun Chen, Youyan Zhang, Robert D. Van Horn, et al. Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120
Deletions That Function as Homodimers andBRAFOncogenic
Updated version
10.1158/2159-8290.CD-15-0896doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerdiscovery.aacrjournals.org/content/suppl/2016/01/05/2159-8290.CD-15-0896.DC1
Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerdiscovery.aacrjournals.org/content/early/2016/02/19/2159-8290.CD-15-0896To request permission to re-use all or part of this article, use this link
Research. on June 4, 2018. © 2016 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Published OnlineFirst January 5, 2016; DOI: 10.1158/2159-8290.CD-15-0896