Embed Size (px)
Citation preview

Office for ResearchResearch Compliance
Research Compliance Officer - Tanta Myles

Research Compliance
• Animal Subjects (IACUC)• Human Subjects (IRB)• Export Control• Conflict of Interest• Scientific Misconduct

Research Compliance
• Animal Subjects (IACUC)• Human Subjects (IRB)• Export Control• Conflict of Interest• Scientific Misconduct

Human SubjectsInstitutional Review Board (IRB)
Purpose:
•Review research proposals, projects, and activities that involve human subjects; •determine for each activity or proposed activity if human subjects will be placed at risk; •if risk is involved, determine if the importance of the knowledge to be gained warrants allowing the subject to accept the risks; •oversee protection of rights and welfare of subjects.

IRB Basics
• Applies to funded and unfunded research
• Submission of a protocol and approval by the IRB is mandatory for any faculty, staff or student who will interact with a human being for purposes of conducting research
• Research may not begin before approval from the IRB is given

• Annual Training (Required)• Centralized IRB submission• All protocols must be submitted to
the Office For Research (OR) -152 Rose Administration.
New Policy and Procedure
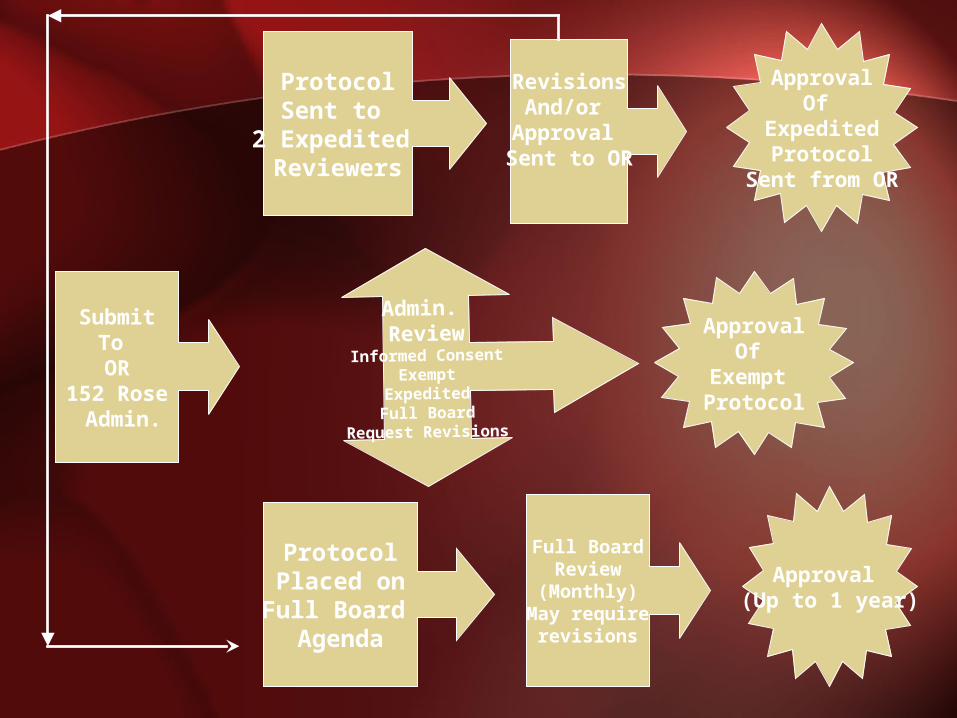
SubmitTo OR
152 Rose Admin.
Admin. Review
Informed ConsentExempt
ExpeditedFull Board
Request Revisions
ApprovalOf
Exempt Protocol
ProtocolSent to
2 Expedited Reviewers
ProtocolPlaced on
Full Board Agenda
Full BoardReview
(Monthly)May require
revisions
Approval (Up to 1 year)
RevisionsAnd/or
Approval Sent to OR
ApprovalOf
ExpeditedProtocol
Sent from OR

IRB Membership
• Shall have at least five members, • No IRB may consist entirely of members of one
profession.• Shall include at least one member whose
primary concerns are in scientific areas.• At least one member whose primary concerns
are in nonscientific areas.• At least one member who is not otherwise
affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution.
• No IRB may have a member participate in the IRB's initial or continuing review of any project in which the member has a conflicting interest, except to provide information requested by the IRB.

Medical IRB Membership• Marianne Woods PhD, JD, Chairperson • John Higginbotham, PHD, MPH , Vice Chair
• Phillip Bishop, EdD
• Karen Burgess, MD
• William Owings, MD
• Sharol Jacobson, PhD, RN, FAAN
• Olivia Kendrick, Dr. P.H., RD
• Jan Claisson
• Tom Stem
• Carpantato Myles
• Bishop Earnest Palmer (Community Member)
• Vacant (Faculty Senate Representative)
• Mike Spearing, JD (Non-Voting Liaison Members)

Non-Medical IRB Membership• Marianne Woods , Chairperson - Associate Vice President for Research
• Tom Stem, Vice Chairperson - Director, Research and Consulting Lab
• Stuart Usdan - Health Sciences, CHES
• K. Michele Kacmar - Durr-Fillauer Chair of Business Ethics and Professor of Management
• Jennifer Lav - Associate Professor, School of Law
• Sharol Jacobson - Associate Dean for Research and Practice
• Edward Merrill - Associate Professor, Psychology
• James Leeper - Chair and Professor, Psychiatry and Behavioral Medicine
• Lucinda Roff - Professor, School of Social Work
• Thomas Ward - Senior Research Fellow, Center for Creative Media
• Valeria Rudolph-Rivers (Community Member) • Hoyt Winslett, Jr. (Community Member)
• John B. Vincent (Faculty Senate Representative)
• Mike Spearing (Non-Voting Liaison Members) • Keith McDowell (Non-Voting Liaison Members)

Rules and Regulations
• Office for Human Research Protections (OHRP) The OHRP is a division of the U.S. Department
of Health and Human Services. The homepage contains news, information, and links relevant to the IRB policies and procedures, including an IRB member guidebook.
• Food and Drug Administration (FDA)FDA's Guidance on Protection of Human Subjects. The FDA's information sheets regarding the projection of human participants for IRBs and investigators; includes a guide on informed consent.

Rules and Regulations
• Title 45 Code of Federal Regulation (CFR) Part 46, Protection of Human Subjects, DHHS
• Title 21, CFR, Parts 50, 54, 56, and 312, FDA,
• U.S. Food and Drug Administration Information Sheets for the “Guidance for Institutional Review Boards and Clinical Investigators.”

OHRP Faults University of Washington
The Office of Human Research Protections (OHRP) has faulted institutional review boards (IRB) at the University of Washington for failing to aggressively regulate research trials under their review, according to the Tacoma News Tribune. OHRP issued a determination letter to the institution that criticized IRBs for failing to delay studies until objections over consent or monitoring were satisfied. Further, the IRBs did not properly follow procedures for approving research involving prisoners and children, the report noted. It faulted the university for allowing IRB-shopping in the case of one researcher who was not satisfied with the first review he received on a proposal and questioned whether IRB members have adequate knowledge of human subjects regulations and adequate authority to perform their functions. The university has said that it is working with OHRP to resolve deficiencies in its human subjects research program.

What is research?
A systematic investigation designed to develop or contribute to generalizable knowledge.
45 CFR 46.102(d)

A Human Subject is defined as:A living individual about whom an
investigator obtains data through intervention or interaction or obtains identifiable private information.
45 CFR 46.102(d)

Categories of Review
• The type of review your project needs rests solely with the IRB.
– Exempt Review – Expedited Review– Full Board Review

Categories of Human Research
• Exempt Research:– Educational testing– Surveys without individual’s
identification– Use of existing data, documents, and – Records with no identifiers– No links to subjects
• The IRB makes the determination that a study is exempt not the individual investigator

Categories of Human Research
Expedited Research• IRB review performed by two
designees.• Criteria for IRB review and approval are
the same for Expedited and Full review.• Study may either be approved or
referred to the full IRB for review.• The review is reported to full Board.• The study is subject to continuing
review at least annually.

Categories of Human Research
Full IRB Review•Conducted at a convened meeting
•Must have a majority of members present including at least one member who is a non-scientist
•IRB actions
»Approve
»Approve with revisions
»Table
»Disapprove
»Resubmit with revisions
•Investigator notified in writing of actions and comments

IRB Review
To approve research the IRB must determine that:– Risk to subjects are minimized– Risks to subjects are reasonable in relation to
subject benefit– Subject selection is equitable– Informed consent obtained– Provisions are adequate for monitoring safety– Provisions to protect subject privacy and data
confidentiality are adequate– When subjects are likely vulnerable to
coercion or undue influence additional safeguards to protect subject rights and welfare have been included

IRB Review and Approval
• Approval is for up to one year, may be less depending on level of risk
• All approved research is subject to continuing review• Required documentation for Continuing Review
» number of subjects accrued» description of any adverse events or unanticipated
problems» summary of any recent literature» copy of the current informed consent

IRB Review and Approval
• ANY change in the protocol, consent form or other study documents must be reviewed and approved by the IRB before they are implemented
• ANY unanticipated problems, adverse events or protocol deviations must be promptly reported to the IRB

Advertising for Research Subjects
• IRB review and approval for ads in paper, TV, radio, flyers, videos, internet postings• Compensation, if any, may be mentioned• Required:
» Lay language» Purpose and eligibility criteria» Mention research» Not coercive» Contact information

Informed consent is a process of
information exchange that
takes place between the
prospective subject and the
investigator, before, during
and sometimes after the study.
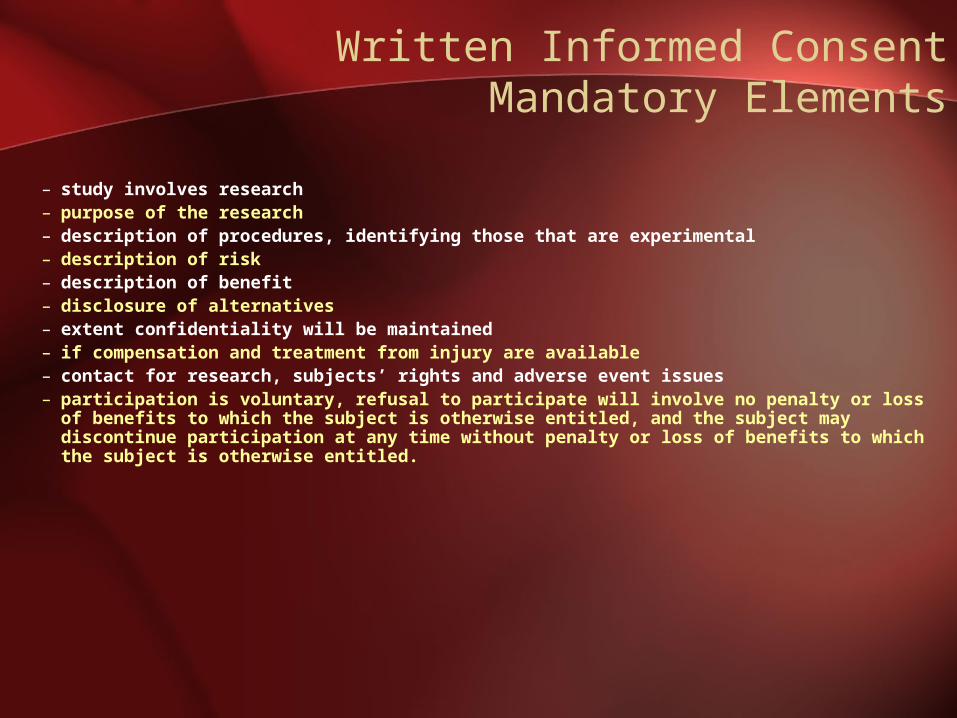
Written Informed ConsentMandatory Elements
– study involves research– purpose of the research– description of procedures, identifying those that are experimental– description of risk– description of benefit– disclosure of alternatives– extent confidentiality will be maintained– if compensation and treatment from injury are available– contact for research, subjects’ rights and adverse event issues– participation is voluntary, refusal to participate will involve no penalty or loss of
benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled.

When May IRB Waive Consent Process?
• Minimal risk • Rights and welfare not
adversely affected• Not practical without waiver• Provide information after
participation• Procedures do not require
written consent outside of research

New Policy and Procedure
• Annual Training (Required)– http://osp.ua.edu/irbtraining.html– Medical or Non-Medical
• Centralized IRB submission
All protocols must be submitted to the Office For Research (OR) -152 Rose Administration or [email protected].

Current Concerns
• Updating of IRB website to become more user friendly.
• Revision of IRB Policies and Procedures
• Revision of Forms• IRB Accreditation• Implementing eProtocol software
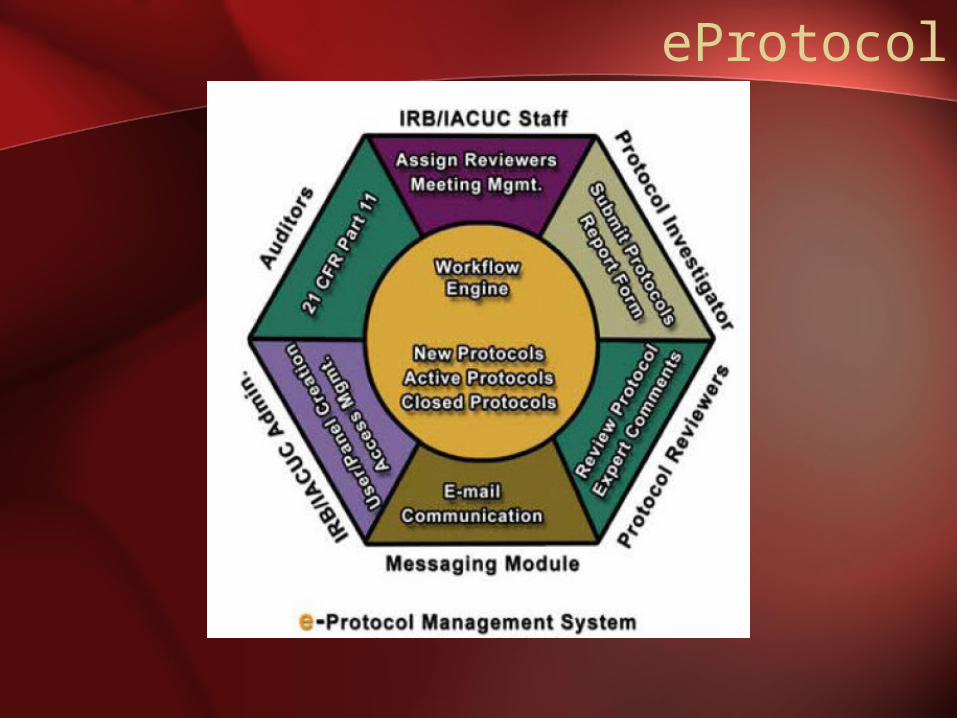
eProtocol

• Investigators have the ability to create and submit the protocol online utilizing such features as: templates to Create a Protocol,Protocol Updates, Save Protocols, Check for Completeness, Clone Submitted or Approved protocols, and Submit Renewals or Revisions.
• Dashboard based interface to access latestevents/status of protocols
• Automatic recommendation of the mandatory and suggested guidelines for the protocol.
• View and Print Approval Letters
• Online View/Response to comments by Reviewers
eProtocol protocol submission

Contact Information
• Tanta Myles – Research Compliance Officer205.348.5152 or [email protected]
• Ed Shirley – Research Compliance Specialist205.348.5152 or [email protected]
• Jeanelle Graham – Research Compliance Specialist205.348.5152 or 205.348.6457
• http://osp.ua.edu/Research_compliance.html

Office for ResearchResearch Compliance
Research Compliance Officer - Tanta Myles




![Systemic Thinking Approach to Teaching and learning Chemistry [STATLC] By Dr. Kamel A.M Professor of physiological psychology Tanta University Tanta –](https://img.pdfslide.us/doc/110x75/56649efe5503460f94c137ae/systemic-thinking-approach-to-teaching-and-learning-chemistry-statlc-by-dr.jpg)
























