Embed Size (px)
Citation preview

OCULAR MANIFESTATIONS O F SUBACUTE NECROTIZING ENCEPHALOMYELOPATHY (LEIGH'S DISEASE)
RUFUS O. HOWARD, M.D., AND DANIEL M. ALBERT, M.D.
New Haven, Connecticut
In 1951, Leigh1 described an infant who was apparently normal at birth, but developed a central nervous system disorder characterized by somnolence, blindness, deafness, and spasticity of limbs at the age of six months. The baby's condition deteriorated and he died six weeks later. Neuropathologic examination revealed bilateral, focal, sub-acute necrotic lesions extending from the thalamus to the pons. Leigh noted "a striking similarity between the tissue reaction here and that found in the Wernicke syndrome." Several similar reports have been added to the literature, and this disease has been termed subacute necrotizing encephalomye-lopathy ( S N E ) , or Leigh's disease. The eye findings have received little attention.
In the present report, the ocular manifestations of Leigh's disease in three children from one family are reported and the ocular histopathologic findings from one of the children are described. In addition, the ocular abnormalities in affected patients reported in the literature are summarized.
CASE REPORTS
Case 1—This boy was first seen at the age of 15 months for evaluation of weakness and lethargy with acute onset. During the first 12 months of life the boy had been entirely normal, but after this age his physical and mental development did not progress. A review of the family history revealed that two sisters had died of SNE, while an older brother and twin sister were normal (Fig. 1). At the time of the initial examination the patient could not support his head, he slumped forward when placed in a sitting position, and was unable to walk. He had bilateral blepharoptosis and horizontal jerk
From the Department of Ophthalmology, Yale University School of Medicine, New Haven, Connecticut. This work was supported in part by Public Health Service Research Grants EY002S3 and EY00002 from the National Eye Institute.
Reprint requests to Rufus O. Howard, M.D., Department of Ophthalmology, Yale University School of Medicine, New Haven, Connecticut 06510.
nystagmus, but his visual acuity appeared grossly normal and his optic nerves and fundi appeared normal. A diagnosis of SNE was made on the basis of the physical findings and his family history. He was treated with 1-2 g of oral thiamine daily. He gradually gained weight, was able to pull himself to a sitting position, and walked with assistance. His blepharoptosis remained but his nystagmus decreased.
At 21 months of age, the patient suddenly developed severe weakness and lethargy, and was admitted to Yale-New Haven Hospital. On examination, he showed generalized hypotonia with inability to support his head. He had an intention tremor and diminished tendon reflexes. Periodically, he had sobbing respirations. Positive eye findings included bilateral blepharoptosis, horizontal jerk nystagmus, and esotropia due to bilateral lateral rectus palsy. The ophthalmoscopic examination appeared normal.
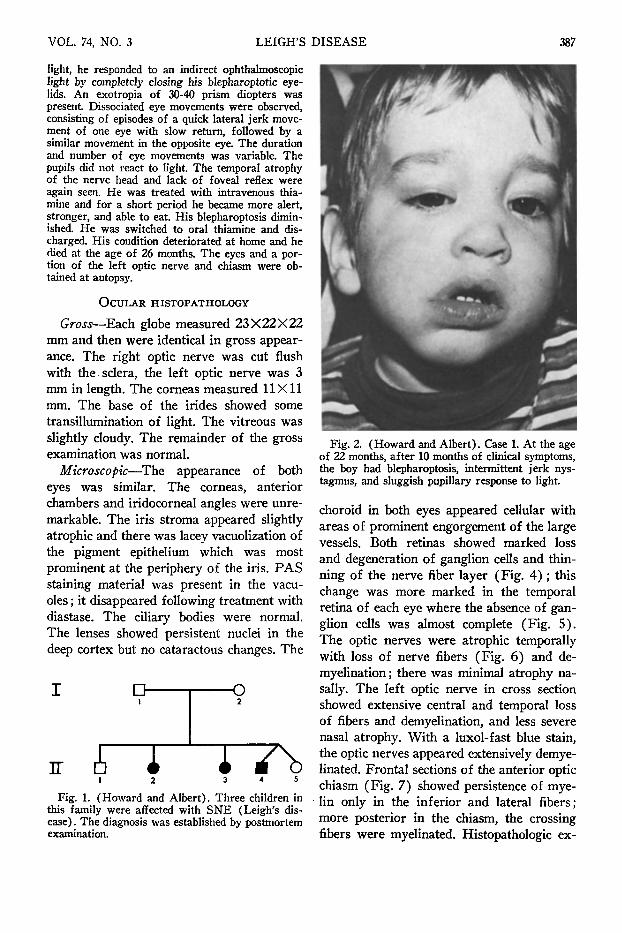
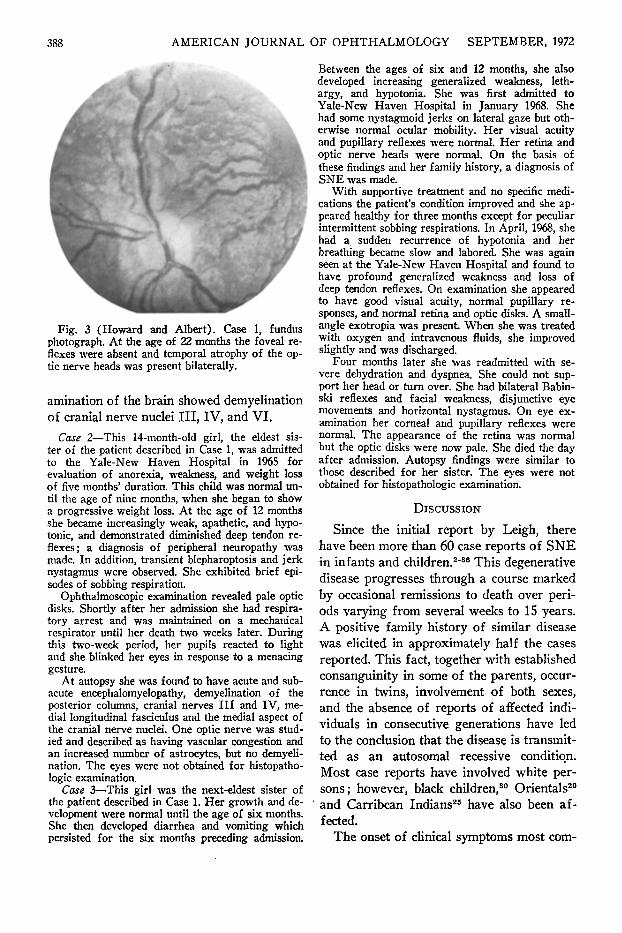
The morning after admission, the child developed respiratory arrest and was treated with endotra-cheal intubation, mechanical ventilation, and intravenous thiamine propyl disulfide. He began breathing spontaneously 48 hours later and continued to improve over the following days and was discharged from the hospital. An eye examination at the age of 22 months (Fig. 2) again revealed bilateral blepharoptosis, intermittent left jerk nystagmus, and sluggish pupillary response to light. Extraocular motility was normal, with no evidence of estropia; corneal sensation and tear production as measured by the Shirmer test were normal. Ophthalmoscopic examination showed absence of the foveal reflexes and temporal atrophy of the optic nerve heads (Fig. 3). The choroidal blood vessels were prominent. The patient was able to see and pick up objects as small as 1 to 2 mm. The pupils showed an unusual sensitivity to the dilating drops (1% tropicamide [My-driacyl] and 5% homatropine) and remained dilated five days following instillation of two drops of each drug. Soon after, when his pupils were dilated with two drops of 1% tropicamide alone, they remained dilated for about 36 hours.
For six weeks following the initiation of thiamine therapy the patient showed steady improvement with regard to his alertness and general muscle tone. His subsequent course, however, was characterized by progressive deterioration punctuated by temporary remissions. These periods of improvement seemed to correspond to transient elevations of the thiamine levels in the blood and spinal fluid. By the age of 25 months he was lethargic, malnourished, had marked ataxia of head, limbs, and trunk, and was unable to sit or stand. He was readmitted to Yale-New Haven Hospital. On physical examination his deep tendon reflexes were diminished or absent. Although he could not fixate on or follow a
386
VOL. 74, NO. 3 LEIGH'S DISEASE 387
light, he responded to an indirect ophthalmoscopic light by completely closing his blepharoptotic eyelids. An exotropia of 30-40 prism diopters was present. Dissociated eye movements were observed, consisting of episodes of a quick lateral jerk movement of one eye with slow return, followed by a similar movement in the opposite eye. The duration and number of eye movements was variable. The pupils did not react to light. The temporal atrophy of the nerve head and lack of foveal reflex were again seen. He was treated with intravenous thia-mine and for a short period he became more alert, stronger, and able to eat. His blepharoptosis diminished. He was switched to oral thiamine and discharged. His condition deteriorated at home and he died at the age of 26 months. The eyes and a portion of the left optic nerve and chiasm were obtained at autopsy.
OCULAR HISTOPATHOLOGY
Gross—Each globe measured 23X22X22 mm and then were identical in gross appearance. The right optic nerve was cut flush with the sciera, the left optic nerve was 3 mm in length. The corneas measured 11X11 mm. The base of the irides showed some transillumination of light. The vitreous was slightly cloudy. The remainder of the gross examination was normal.
Microscopic—The appearance of both eyes was similar. The corneas, anterior chambers and iridocorneal angles were unremarkable. The iris stroma appeared slightly atrophie and there was lacey vacuolization of the pigment epithelium which was most prominent at the periphery of the iris. PAS staining material was present in the vacuoles ; it disappeared following treatment with diastase. The ciliary bodies were normal. The lenses showed persistent nuclei in the deep cortex but no cataractous changes. The
I D 1 O 1 2
π A l W^b I 2 3 4 5
Fig. 1. (Howard and Albert). Three children in this family were aiïected with SNE (Leigh's disease) . The diagnosis was established by postmortem examination.
Fig. 2. (Howard and Albert). Case 1. At the age of 22 months, after 10 months of clinical symptoms, the boy had blepharoptosis, intermittent jerk nystagmus, and sluggish pupillary response to light.
choroid in both eyes appeared cellular with areas of prominent engorgement of the large vessels. Both retinas showed marked loss and degeneration of ganglion cells and thinning of the nerve fiber layer (Fig. 4) ; this change was more marked in the temporal retina of each eye where the absence of ganglion cells was almost complete (Fig. 5). The optic nerves were atrophie temporally with loss of nerve fibers (Fig. 6) and de-myelination; there was minimal atrophy nasally. The left optic nerve in cross section showed extensive central and temporal loss of fibers and demyelination, and less severe nasal atrophy. With a luxol-fast blue stain, the optic nerves appeared extensively demye-linated. Frontal sections of the anterior optic chiasm (Fig. 7) showed persistence of mye-lin only in the inferior and lateral fibers; more posterior in the chiasm, the crossing fibers were myelinated. Histopathologic ex-
388 AMERICAN JOURNAL OF OPHTHALMOLOGY SEPTEMBER, 1972
Fig. 3 (Howard and Albert). Case 1, fundus photograph. At the age of 22 months the foveal reflexes were absent and temporal atrophy of the optic nerve heads was present bilaterally.
amination of the brain showed demyelination of cranial nerve nuclei I I I , IV, and V I .
Case 2—This 14-month-old girl, the eldest sister of the patient described in Case 1, was admitted to the Yale-New Haven Hospital in 1965 for evaluation of anorexia, weakness, and weight loss of five months' duration. This child was normal until the age of nine months, when she began to show a progressive weight loss. At the age of 12 months she became increasingly weak, apathetic, and hypo-tonic, and demonstrated diminished deep tendon reflexes; a diagnosis of peripheral neuropathy was made. In addition, transient blepharoptosis and jerk nystagmus were observed. She exhibited brief episodes of sobbing respiration.
Ophthalmoscopic examination revealed pale optic disks. Shortly after her admission she had respiratory arrest and was maintained on a mechanical respirator until her death two weeks later. During this two-week period, her pupils reacted to light and she blinked her eyes in response to a menacing gesture.
At autopsy she was found to have acute and sub-acute encephalomyelopathy, demyelination of the posterior columns, cranial nerves III and IV, medial longitudinal fasciculus and the medial aspect of the cranial nerve nuclei. One optic nerve was studied and described as having vascular congestion and an increased number of astrocytes, but no demyelination. The eyes were not obtained for histopatho-logic examination.
Case 3—This girl was the next-eldest sister of the patient described in Case 1. Her growth and development were normal until the age of six months. She then developed diarrhea and vomiting which persisted for the six months preceding admission.
Between the ages of six and 12 months, she also developed increasing generalized weakness, lethargy, and hypotonia. She was first admitted to Yale-New Haven Hospital in January 1968. She had some nystagmoid jerks on lateral gaze but otherwise normal ocular mobility. Her visual acuity and pupillary reflexes were normal. Her retina and optic nerve heads were normal. On the basis of these findings and her family history, a diagnosis of SNE was made.
With supportive treatment and no specific medications the patient's condition improved and she appeared healthy for three months except for peculiar intermittent sobbing respirations. In April, 1968, she had a sudden recurrence of hypotonia and her breathing became slow and labored. She was again seen at the Yale-New Haven Hospital and found to have profound generalized weakness and loss of deep tendon reflexes. On examination she appeared to have good visual acuity, normal pupillary responses, and normal retina and optic disks. A small-angle exotropia was present When she was treated with oxygen and intravenous fluids, she improved slightly and was discharged.
Four months later she was readmitted with severe dehydration and dyspnea. She could not support her head or turn over. She had bilateral Babin-ski reflexes and facial weakness, disjunctive eye movements and horizontal nystagmus. On eye examination her corneal and pupillary reflexes were normal. The appearance of the retina was normal but the optic disks were now pale. She died the day after admission. Autopsy findings were similar to those described for her sister. The eyes were not obtained for histopathologic examination.
DISCUSSION
Since the initial report by Leigh, there have been more than 60 case reports of S N E in infants and children.2"36 This degenerative disease progresses through a course marked by occasional remissions to death over periods varying from several weeks to 15 years. A positive family history of similar disease was elicited in approximately half the cases reported. This fact, together with established consanguinity in some of the parents, occurrence in twins, involvement of both sexes, and the absence of reports of affected individuals in consecutive generations have led to the conclusion that the disease is transmitted as an autosomal recessive condition. Most case reports have involved white persons; however, black children,30 Orientals2 0
' and Carribean Indians2 5 have also been affected.
The onset of clinical symptoms most com-
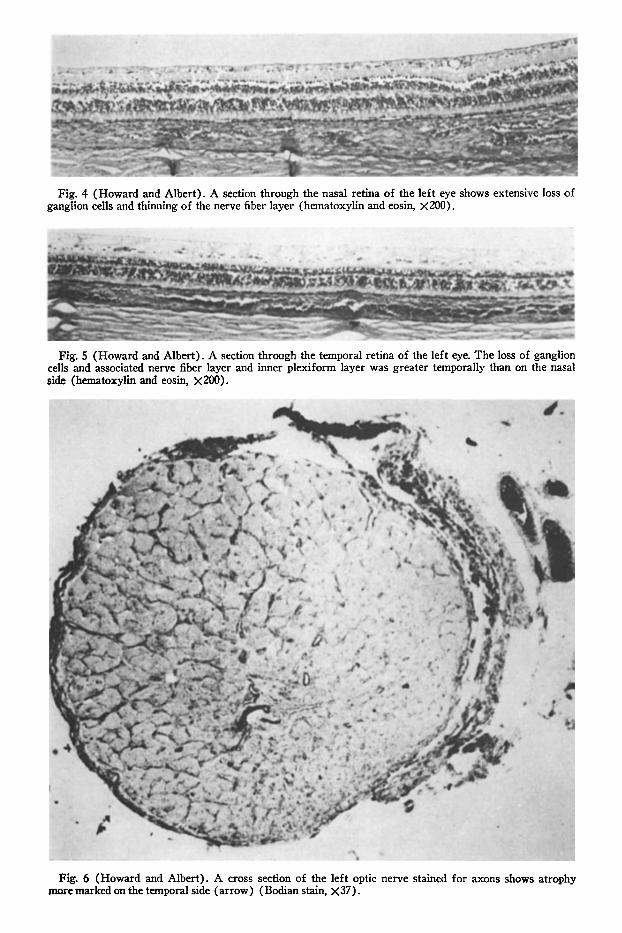
Fig. 4 (Howard and Albert). A section through the nasal retina of the left eye shows extensive loss of ganglion cells and thinning of the nerve fiber layer (hematoxylin and eosin, X200).
Fig. 5 (Howard and Albert). A section through the temporal retina of the left eye. The loss of ganglion cells and associated nerve fiber layer and inner plexiform layer was greater temporally than on the nasal side (hematoxylin and eosin, X200).
Fig. 6 (Howard and Albert). A cross section of the left optic nerve stained for axons shows atrophy more marked on the temporal side (arrow) (Bodian stain, X37).

390 AMERICAN JOURNAL OF OPHTHALMOLOGY SEPTEMBER, 1972
Fig. 7 (Howard and Albert). A frontal section through the anterior portion of the optic chiasm showing extensive demyeliniation (Luxol-fast blue, X28).
monly occurred between the ages of two months and six years. The clinical manifestations of SNE are so variable and nonspecific that in the absence of a history of this disease in siblings the correct diagnosis has seldom been made prior to autopsy. Its onset is generally insidious, with initial symptoms variously described as feeding difficulty, failure to thrive, generalized weakness, and hypotonia. Developmental regression and respiratory difficulties including a peculiar sobbing respiration may appear early. Cerebrospinal fluid protein is often normal, but may be elevated. Biopsy of the cerebrum,25 peripheral nerves,80 or rectum was performed in some cases but has not been helpful in establishing a correct diagnosis. In most patients, death resulted from respiratory failure.
The clinical ocular manifestations of SNE are due to changes in the central nervous system and cranial nerves. Although there have been few detailed clinical or histopatho-logic ocular evaluations of affected individuals, at least one abnormal ocular finding was mentioned in the description of 61 previously reported patients (Table 1). Abnormal eye movements, including nystagmus, disjunctive eye movements, and bizarre rolling eye movements, were the most frequently described ocular signs. Two patients with nystagmus subsequently developed complete external ophthalmoplegia. Strabismus was present in 19 case reports: 11 of these pa
tients had exotropia, six had esotropia, and the deviation was not specified in two. In some reports, strabismus seemed to result from ocular muscle palsy.
TABLE 1 OCULAR FINDINGS IN 65 PATIENTS WITH SNE
Findings No. Cases
Blepharoptosis Abnormal eye movements
Nystagmus Ophthalmoplegia Other
Strabismus Exotropia Esotropia Not specified
Parinaud's syndrome, facial palsy, neuroparalytic keratitis
Pupils Dilated Miotic Diminished light response
Estimated visual acuity Diminished No light perception Cortical blindness
Field defect (central scotoma) Fundus (absent foveal reflex)
Normal fluorescein angi-ography
Optic nerve abnormality (color, atrophy, blurring)
Normal optic nerve Facial nerve palsy
21!»
J72,3,14,16,19,21,2S,30,33,36 g3,7,15(f), 19,25,30,33»
122.6,8,11,21,27-29,36»
104.M?.20,21»
62.".»* 221,26
1»
415,21,24 51,8,11,15,31
151,2,5,6,7,8,21,24,25, 28,31,33»
g3,8,19,25,28,31,32* 51,5,6,19
Is
l3
335»
235
lgl-3,5,8,16,17,22,27,28, 31,32,35,36»
144-6,12,19,26,27,30,33* 33,26»
* This report.

VOL. 74, NO. 3 LEIGH'S DISEASE 391
Visual acuity was estimated to be either partially or completely diminished in 14 patients. One patient was considered to have cortical blindness, although his pupils were described as reacting poorly to light. One 15-year-old boy had Parinaud's syndrome with a peripheral facial palsy and neuroparalytic keratitis.
Pupillary abnormalities have included dilated pupils, miotic pupils, and diminished or absent pupillary response to light. The prolonged pupillary dilatation in our patient (Case 1) could be related to impairment of the third cranial nerve, as evidenced by de-myelination of his third cranial nerve found at autopsy.
Apart from pallor of the disk, no retinal vascular abnormality or macular or focal retinal lesions have been previously reported. The foveal reflex was absent in one of the patients described in the present report (Case 1). The optic nerves have been atrophie or shown primary atrophy in 17 patients. Most of the reports in the literature do not specify whether optic atrophy and other ocular abnormalities were unilateral or bilateral.
On histopathologic examination varying degrees of demyelination of the optic nerves, optic tracts, and chiasms has been observed, and partial or complete necrosis of the cranial nerve nuclei, particularly III , IV, V, VI, VIII, IX, X and XII . The optic nerves were stated to have appeared normal histo-logically in three reports. The finding of symmetrical necrosis with vascular proliferation, predominantly in the basal ganglia, brain stem, and spinal cord is a constant finding. Significantly, in S NE the mamillary bodies are usually unaffected; there have been few exceptions.9'22 Since autopsy findings in SNE resemble those in Wernicke's encephalopathy, the possibility of a defect in thiamine metabolism has been investigated. In patients with SNE, blood and brain levels of thiamine are normal, as are the thiamine pyrophosphate dependent enzymes,27·28·34
transketolase, pyruvic dehydrogenase, and a-
ketoglutarate. The isolation of a factor that inhibits the reversible conversion of thiamine pyrophosphate to thiamine triphosphate34
appears to provide the best index of a defect in thiamine metabolism at this time. This inhibitory factor was present in the urine, CSF, and serum of the patients in Cases 1 and 3. Other individuals with SNE have been shown to have this inhibitory factor.37
In four affected patients, cerebral tissue has been analyzed and thiamine triphosphate has been absent, while the thiamine level of cerebral tissue has been normal.
The significance of isolated reports of muscle phosphofructokinase deficiency,27
cystathioninuria,32 and liver pyruvate decar-boxylate deficiency29 is uncertain at this time. The elevation of blood pyruvate and lactate levels in affected children described in the literature did not occur in the patients described here. Previous studies have failed to demonstrate a viral etiology,12 or abnormalities in brain lipids or in the trace metals in the brain.30
The treatment of SNE has not been successful in the past. Lipoic acid has been used to counter the elevation of blood pyruvate and lactate which occurs in some SNE patients, and which is thought to result from a defect in pyruvate decarboxylation. While brief clinical improvement has been reported, the ultimate course of the disease was not altered. Oral and parenteral thiamine in low doses has not been effective. Treatment with high doses of oral or parenteral thiamine and thiamine propyl disulfide has been associated with temporary clinical improvement in some cases. The dramatic reversal of respiratory failure in response to such treatment described in Case 1 apparently has not been previously reported.
SUMMARY
Subacute necrotizing encephalomyelopa-thy, or Leigh's disease, occurred in three children in one family. Each child developed normally for the first few months of life, then physical growth and development ar-

392 AMERICAN JOURNAL OF OPHTHALMOLOGY SEPTEMBER, 1972
rested. They became weak and ocular neurologic abnormalities appeared, including ble-pharoptosis, nystagmus, strabismus secondary to ocular muscle palsy, and optic atrophy. Each child expired after a brief illness. On histologie examination of the eyes of one child, vacuolation of the iris pigment epithelium was present. In the retina, there was loss of ganglion cells, and the nerve fiber layer was thinned. The optic nerves and chiasm showed demyelination.
ACKNOWLEDGMENT
We thank J. H. Pincus, M.D., who allowed us to examine his patient during life, helped us obtain the eyes for histopathologic examination, and reviewed the manuscript. The histopathology slides were reviewed at the Armed Forces Institute of Pathology. The AFIP Accession number is 13S4569.
REFERENCES
1. Leigh, D. : Subacute necrotizing encephalo-myelopathy in an infant. J. Neurol. Neurosurg. Psychiat. 14:216, 1951.
2. Feigan, I., and Wolf, A. : A disease in infants resembling chronic Wernicke's encephalopathy. J. Pediat. 45:243, 1954.
3. Garcin, R., Grüner, J., and Godlewski, S. : Spongiose disséminée de l'encéphale évoluant clini-quement par poussées chez un enfant malgache. Rev. Neurol. 95 :273, 1956.
4. Christensen, E., Melchior, J., and Plum, P. Combined lesions of basal ganglia, medulla oblon-gata, and spinal cord in a 10-year-old boy. Acta Paediat. 45 :396, 1956.
5. Richter, R. : Infantile subacute necrotizing encephalopathy with predilection for the brain stem. J. Neuropath. Exp. Neurol. 16:281, 1957.
6. Wohlwill, F., and Paine, R.: Progressive de-myelinating leukoencephalopathy. Neurology 8:285, 1958.
7. Ule, G. : Über eine der Wernickeschen Pseu-doencephalitis entsprechende Encephalopathie bei Kindern. Virchow's Arch. Path. Anat. 332:204, 1959.
8. Ford, F. : Diseases of the Nervous System in Infancy, Childhood, and Adolescence, 5th ed. New York, McGraw-Hill, 1966, p. 303.
9. Tuthill, C. R. : Der morphologische Wernicke-Komplex in frühem Kindesalter. Arch. Psych. Neurol. 200:520, 1960.
10. Crome, L. : Neuropathological changes in diseases caused by inborn errors of metabolism. In Holt, K. S., and Milner, J. (eds.) : Neurometabolic Disorders of Childhood. London, E. and S. Livingstone, 1964, p. 37.
11. Poser, G, and Bogaert, L. : Van Leuco-et-po-lio-encephalopathies symétriques nécrosantes. Rev. Neurol. 103:3, 1960.
12. Reye, R. D. K. : Subacute necrotizing ence-phalomyelopathy. J. Path. Bacteriol. 79:165, 1960.
13. Tom, M. I., and Rewcastle, N. B. : Infantile subacute necrotizing encephalopathy. Neurology 12 : 624, 1962.
14. Christensen, E., Melchior, J. C, and Plum, P. : Infantile chronic necrotizing encephalopathy. Acta Paediat. 52:304, 1963.
15. Aronson, S. M., and Okazaki, H. : Clinical neuropathological conference. Dis. Nerv. Syst. 24: 630, 1963.
16. Tuthill, C. R., and Henn, R. : Wernicke-Syn-drom in Kindesalter ohne Anzeichen von Mangelernahrung. Arch. Psych. Neurol. 205:116, 1964.
17. Peterson, H. C, and Alvord, E. C. : Necrotizing encephalopathy with predilection for the brainstem. Tr. Am. Neurol. Assoc. 89:104, 1964.
18. Tariska, I., and Haraszti, A. : Subacut nekro-tizalo enkephalopathia 10 eves fiuban. Gyermekg-yogyaszat 15:129, 1964.
19. Bargeton-Farkas, E., Cochard, A. M., Bris-saud, H. E., Robain, O., and LeBalle, J. C. : Encephalopathie Infantile Familiale avec Necrose Bilaterale et Symétrique des Corps Stries. J. Neurol. Sei. 1:429, 1964.
20. Namike, H. : Subacute necrotizing encephalo-myelopathy. Arch. Neurol. 12:98, 1965.
21. Ebels, E. J., Blokzijl, E. J., and Troelstra, J. A. : A Wernicke-like encephalomyelopathy in children (Leigh), an inborn error of metabolism? Hel-vet. Paediat. Acta 20:310, 1965.
22. Worsley, H. E., Brookfield, R. W., Elwood, J. S., Noble, R. L., and Taylor, W. H. : Lactic aci-dosis with necrotizing encephalopathy in two sibs. Arch. Dis. Child. 40:492, 1965.
23. Thieffry, S., Farkas-Bargeton, E., Martin, C, and Lyon, G. : Encéphalite Nécrosante Subaique de L'enfant. Rev. Neurol. 113: 105, 1965.
24. Lewis, A. J. : Infantile subacute necrotizing encephalopathy. Canad. M. A. J. 93:878, 1965.
25. Lakke, J. P. W. F., Ebels, E. J., and ten Thye, O. J. : Infantile necrotizing encephalomyelopathy (Leigh). Arch. Neurol. 16:227, 1967.
26. Robinson, F., Solitare, G. B., Lamarche, J. B., and Levy, L. L. : Necrotizing encephalomyelopathy of childhood. Neurology 17:472, 1967.
27. Clayton, B. E., Dobbs, R. H., and Patrick, A. D. : Leigh's subacute necrotizing encephalopathy : Clinical and biochemical study, with special reference to therapy with lipoate. Arch. Dis. Child. 42: 467, 1967.
28. Greenhouse, A. H., and Schneck, S. A. : Sub-acute necrotizing encephalornyelopathy. Neurology 18:1,1968.
29. Hommes, F. A., Polman, H. A., and Reerink, J. D. : Leigh's encephalomyelopathy : An inborn error of gluconeogenesis. Arch. Dis. Child. 42:423, 1968.
30. Kamoshita, S., Aguilar, M. J., and Landing, B. H. : Infantile subacute necrotizing encephalomyelopathy. Am. J. Dis. Child. 116:120, 1968.
31. Richter, R. B. : Infantile subacute necrotizing encephalopathy (Leigh's disease). Neurology 18: 1125, 1968.
VOL. 74, NO. 3 LEIGH'S DISEASE 393
32. Weil, M. L., Shaw, K. N. F., and Menkes, J. : Cystathionuria accompanying necrotizing encepha-lomyelopathy of childhood. Neurology 18:301, 1968.
33. Hardman, J. M., Allen, L. W., Baughman, F. A., and Waterman, D. F. : Subacute necrotizing en-cephalopathy in late adolescence. Arch. Neurol. 18 : 478, 1968.
34. Pincus, J. H., Itokawa, Y., and Cooper, J. R. : Enzyme-inhibiting factor in subacute necrotizing encephalomyelopathy. Neurology 19:841, 1969.
35. Grover, W. D., Green, W. R., and Pileggi, A. J. : Ocular findings in subacute necrotizing encepha-lomyelitis. Am. J. Ophth. 70 :S99, 1970.
36. Borit, A. : Leigh's necrotizing encephalomyelopathy (neuro-ophthalmological abnormalities). Arch. Ophth. 85:439, 1971.
37. Cooper, J. R., Pincus, J. H., Itokawa, Y., and Piros, K. : Experience with phosphoryl transferase inhibition in subacute necrotizing encephalomyelopathy myelopathy. New Eng. J. Med. 283:793, 1970.
O P H T H A L M I C M I N I A T U R E
A new ophthalmoscope. H . Knapp
Tr. Am. Ophth. Soc. 2:107,1873-75





























