Embed Size (px)
Citation preview

Novel Oncolytic Adenovirus Selectively Targets Tumor-AssociatedPolo-Like Kinase1and Tumor Cell ViabilityJianfeng Zhou, Qinglei Gao, Gang Chen, Xiaoyuan Huang, Yunping Lu, Kanyan Li, Daxing Xie,Liang Zhuang, Jingniu Deng, and DingMa
Abstract Purpose: Polo-like kinase 1 (plk1) is a serine/threonine protein kinase essential for multiplemitotic processes. Previous observations have validated plk1as a promising therapeutic target.Despite being conceptually attractive, the potency and specificity of current plk1-based therapiesremain limited.We sought to develop a novel plk1-targeting strategy by constructing an oncolyticadenovirus to selectively silence plk1in tumor cells.Experimental Design: Two artificial features were engineered into one wild-type adenovirustype 5 (wt-Adv5) genome to generate a new oncolytic adenovirus (M1). First, M1contains a27-bp deletion in E1A region, which confers potent, oncolytic efficacy. Second, M1is armedwitha fragment of antisenseplk1cDNAthat substitutes the E3 regionencoding 6.7K andgp19K. In thisdesign, tumor-selective replication of M1would activate the native adenovirus E3 promoters toexpress the antisense plk1 cDNA preferentially in tumor cells and silence tumor-associated plk1protein.Results: By virtue of combining oncolysis with plk1targeting, M1exhibited potent antitumoralefficacy in vitro and in vivo. Systemic administration of M1plus cisplatin induced complete tumorregression in 80% of orthotopic hepatic carcinoma model mice that were otherwise resistant tocisplatin and disseminatedmetastases.Conclusions: Coupling plk1targeting with oncolysis had shown superior antitumor efficacy.Present findings would benefit the development of novel oncolytic adenoviruses generally appli-cable to awide range of molecule-based therapeutics.
Polo-like kinase 1 (plk1) is a serine/threonine protein kinaseessential for multiple mitotic processes, including functionalmaturation of centrosomes and establishment of the bipolarspindle (1). Elevated expression of plk1 is common in manytypes of cancer, and is correlated with poor clinical prognosis(2, 3). Targeting plk1 by antisense oligonucleotide, smallinterfering RNAs, or small molecule chemicals induced mitoticarrest, apoptosis, and tumor regression, which validated plk1 asa promising therapeutic target in devising novel molecule-based anticancer therapies (3, 4). Despite being conceptuallyattractive, there are some limitations to plk1-targeting strate-gies. Plk1 is also expressed at the highest levels in normaltissues with actively proliferating cell populations (5); thus,
depletion of plk1 without tumor selectivity could potentiallycause aberrant mitoses and contribute to de novo tumorformation (6). In addition, insufficient and nonselectivedelivery of small interfering RNAs or oligonucleotide to targetshas greatly reduced clinical potential of such therapies.Development of novel potent strategies to selectively silenceplk1 in tumor cells is thus of fundamental importance.
The use of adenovirus mutants that preferentially replicatein and lyse tumor cells, known as oncolytic adenoviruses,represents a promising new platform for the treatment ofcancer (7). For engineering oncolytic adenovirus, smalldeletions in E1B or E1A encoding region are made toattenuate viral replication and cytolysis in normal tissues butnot in tumor cells (7, 8). The most studied oncolytic adeno-virus, thus far named onyx-015 (also known as dl1520 orCI-1042), is an adenovirus with the E1B 55-kDa gene deleted.Onyx-015 has shown encouraging clinical outcome in clinicaltrials (9). More recently, investigators have identified thatdl922-947 or D24, an adenovirus type 5 (Adv5) mutant, con-tains a 24-bp deletion in the E1A protein conserved region-2(CR2) known to be necessary for retinoblastoma (Rb) proteinbinding and showed much more improved antitumoralefficacy than onyx-015 whereas retaining tumor selectivity(10, 11). Other attempts by incorporating a therapeutic trans-gene gene into onyx-015 have augmented the antitumoralefficacy (12, 13). To optimize the insertion of a therapeutictransgene into the oncolytic adenovirus, Hawkins et al. haverecently developed a gene delivery system by replacing E3
Cancer Therapy: Preclinical
Authors’Affiliation: Cancer Biology Research Center, TongJi Hospital, TongJiMedical College, Huazhong University of Science and Technology,WuHan, Hubei,P.R. ChinaReceived 5/18/05; revised 8/3/05; accepted 9/7/05 .Grant support:China State Key Basic Research Program 2002CB513100 and theNational Science Foundation of China grant 30271472.The costs of publication of this article were defrayed in part by the payment of pagecharges.This article must therefore be hereby marked advertisement in accordancewith18 U.S.C. Section1734 solely to indicate this fact.Note: J. Zhou and Q. Gao contributed equally to this work.Requests for reprints: Ding Ma, Cancer Biology Research Center, TongJiHospital, 1095 Jie-Fang Avenue,WuHan, HuBei, 430030, P.R. China. Phone: 86-27-8366-2475; Fax: 86-27-8366-2680; E-mail: [email protected].
F2005 American Association for Cancer Research.doi:10.1158/1078-0432.CCR-05-1085
www.aacrjournals.org Clin Cancer Res 2005;11(23) December1, 20058431
corrected 10/23/19;
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from

6.7K/gp19K of wt-Adv5 with transgenes (14). In this system,the transgene can be efficiently expressed by native E3 promo-ters at a level superior to that by human cytomegalovirusmajor immediate-early promoter/enhancer (hCMV promoter).More importantly, expression of the transgene is dependenton active viral DNA replication.These pioneering studies cited above have laid the conceptual
groundwork for us to develop a novel oncolytic adenovirus forselective targeting of tumor-associated plk1 and tumor cells. Wehypothesize that an adenovirus mutant of this kind could begenerated by replacing 6.7K/gp19K in an E1A-modified Adv5with an antisense plk1 cDNA. In this mutant, a small deletionin E1A CR2 region is expected to differentiate tumor fromnormal cells and to confer tumor-selective replication. Conse-quently, tumor-permissive replication of the mutant wouldactivate the native adenovirus E3 promoters to drive expressionof the antisense plk1 cDNA preferentially in tumor cells andsilence tumor-associated plk1 protein.We report here the construction and characterization of M1,
a novel E1A CR2-deleted Adv5 with a fragment of antisenseplk1 cDNA inserted into the deleted 6.7K/gp19K region. Byvirtue of targeting both plk1 protein and tumor cells, M1showed potent antitumor efficacy superior to its parent E1ACR2 adenovirus mutant in vivo and in vitro. Most strikingly,systemic administration of M1 plus low dose of cisplatininduced complete tumor regression in 80% of orthotopichepatic carcinoma model mice that was otherwise resistant tocisplatin and disseminated metastases.
Materials andMethods
Viruses and cells. Adv5/dE1A with deletion of amino acids 121 to129 in E1A CR2 was constructed in this laboratory following describedelsewhere (15). M1 was driven from Adv5/dE1A through replacement ofthe 6.7K/gp19K open reading frame in the E3 region by a fragment ofreverse plk1 cDNA (bases 960-161). The virus mutant was constructed byhomologous recombination in 293 cells (American Type CultureCollection, Rockville, MD; ref. 16). The replication-deficient adenovirusvector Adv-TK, containing a herpes simplex virus-thymidine kinase(HSV-TK) gene under control of a Rous sarcoma virus long terminalrepeat promoter in the region of the excised E1 adenoviral genes, wasconstructed in this laboratory following our previous description (17).Adv-TK has an intact E3 region and was used as a control vehicle.Wt-Adv5 was obtained from American Type Culture Collection. Thefollowing tumor cell lines varying in p53 and Rb status and tissue oforigin were obtained from American Type Culture Collection: breastcarcinoma (MCF-7, p53, and Rb wild type; MDA-MB-231, p53 mutant,and Rb wild type), cervical carcinoma (HeLa, p53 inactivated by humanpapillomavirus E6 and mutant Rb), lung carcinoma (A549, p53 wildtype, and Rb mutant), hepatocellular carcinoma (HepG2, p53 wild type,and Rb mutant). Ovarian carcinoma cell line A2780 with wild-type p53and mutant Rb was obtained from the China Center for Type CultureCollection (Shanghai, P.R. China). Lung-derived primary humanmicrovascular endothelial cells (MEVC) and human normal hepatocyteswere purchased from Cambrex Bio Science Rockland, Inc. (Rockland,ME) and cultured following the manufacturer’s instructions and ourprevious description (18).
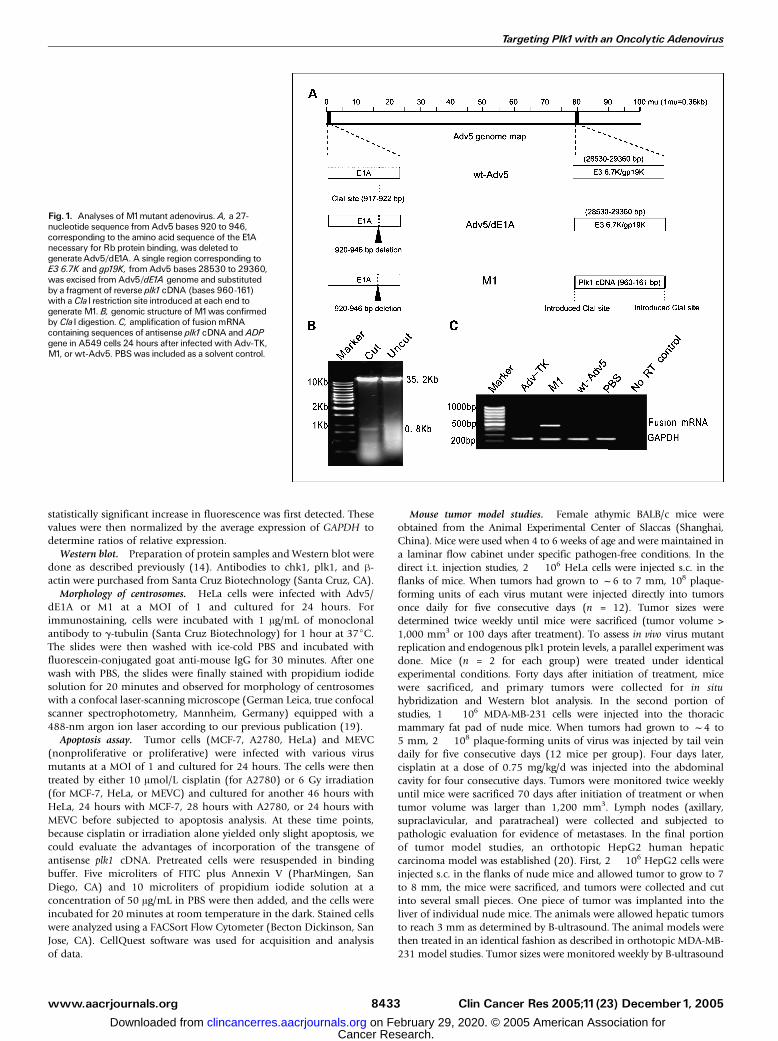
Verification of M1 by ClaI restriction mapping. 293 cells wereinfected with M1 at a multiplicity of infection (MOI) of 10. After 3 daysof culture, 293 cells were collected for isolation of viral genome DNA.Two micrograms of virus DNA were digested with ClaI and separatedon a 0.8 % agarose gel.
Detection of fusion mRNA containing sequences of antisense plk1 cDNAand ADP gene. A549 cells were infected with Adv-TK, M1, or wt-Adv5
at a MOI of 5. After 24 hours of incubation, A549 cells were collectedfor extraction of total RNA. Two micrograms of DNase I– treated totalRNA were used for reverse transcriptase reaction. For amplification offusion transcripts containing sequences of antisense plk1 cDNA andADP gene, 5V-end sense primer from antisense plk1 cDNA (5V-TCTCG-AAGCACTTGGCAAAG-3V) and 3V-end antisense primer complimentaryto the coding region of ADP (5V-GGGTGTAGCACAATGATGGG-3V) wereused. Five microliters of reverse transcriptase mixture were amplifiedusing PCR. glyceraldehyde-3-phosphate dehydrogenase (GAPDH) waschosen as an endogenous marker to check the integrity of cDNA.A 5V-end sense primer (5V-ACGGATTTGGTCGTATTGGG-3V) and 3V-endantisense primer (5V-TGATTTTGGAGGGATGTCGC-3V) were used toamplify a 230-bp-long sequence in GAPDH mRNA.
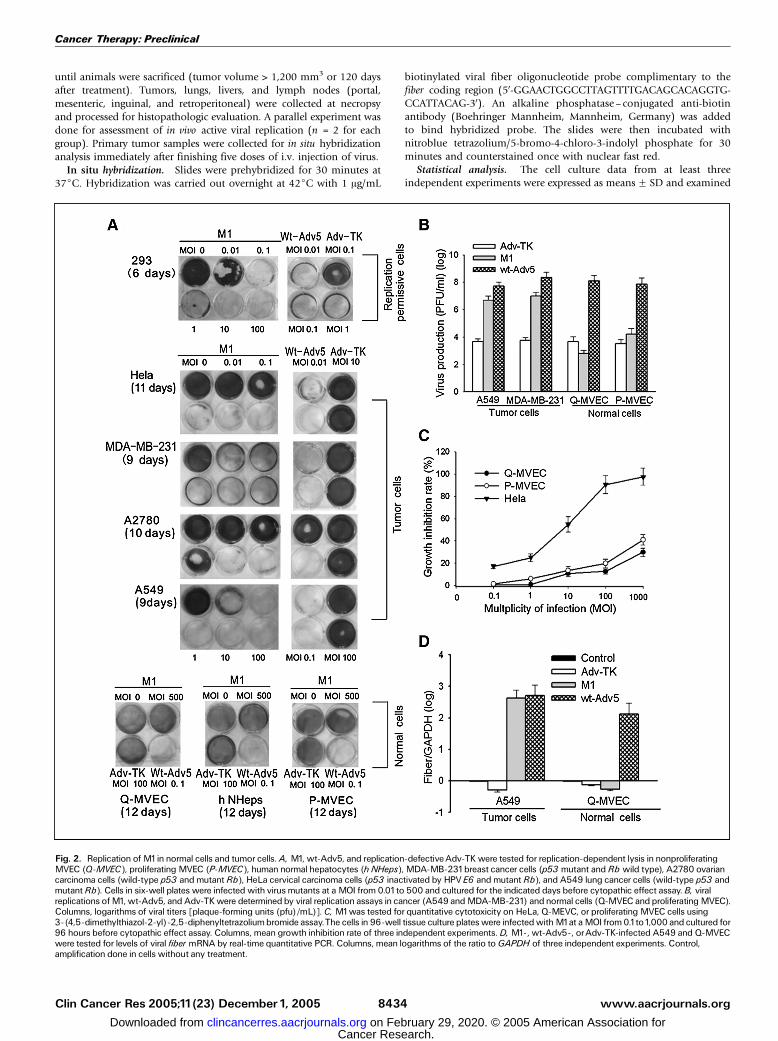
In vitro cytopathic effects and replication assays. Seventy percentconfluent cells were infected with Adv-TK, M1, or wt-Adv5 for 90minutes at a range of MOI from 0.01 to 500 and then switched tocomplete culture medium. Nonproliferating MEVC (Q-MEVC) wasprepared by growing MEVC to complete confluence and maintained inthis state for 2 days before initiation of infection. Cytopathic effectassays were done on the day on which complete cytopathic effect forwt-Adv5 was achieved at a MOI of 0.1. For cytopathic effect assays, theculture plates were stained with crystal violet solution (Sigma ChemicalCo., St. Louis, MO). For viral replication assays, cells were infected withM1, Adv-TK, or wt-Adv5 at a MOI of 5. Cells were cultured for another48 hours and scraped into culture supernatant. Cell lysates wereprepared by three cycles of freezing and thawing and titered on 293cells according to TCID50 method following instructions in the AdEasyapplication manual (Quantum Biotechnologies, Montreal, Quebec,Canada) and are presented as plaque forming units. Results are themeans of titers for quadruplicate samples.
Quantitative cytotoxicity assay. The cytopathic effect and replication
assays showed qualitative comparison of the cytotoxicity effects of the
viruses studied. These results were further confirmed by quantitative
cytotoxicity assay using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-
tetrazolium bromide method. For 3-(4,5-dimethylthiazol-2-yl)-2,5-
diphenyltetrazolium bromide assay, The HeLa, Q-MEVC, or P- MEVC
cells were plated in 96-well tissue culture plates at a density of 5 � 104/
mL. The HeLa or P-MEVC cells were grown to 70 % confluence, infected
with M1 for 90 minutes at a MOI of 0.1 to 1,000, and then switched to
complete culture medium for culture in a humidified atmosphere with
5% CO2 at 37jC for 96 hours. To prepare Q-MEVC cells, MEVC were
made proliferation-free following growth to complete confluence and
maintained in this state for at least 2 days before initiation of infection.
After incubation, the cells were stained with 3-(4,5-dimethylthiazol-2-
yl)-2,5-diphenyltetrazolium bromide reagent at a concentration of
5 mg/mL (Sigma Chemical). The absorbance was determined at a
wavelength of 570 nm. Results represented the means of growth
inhibition rates from three independent experiments. Growth inhibi-
tion rate was calculated following formula: growth suppression rate
(%) = (1 � A570 of experimental wells) / A570 of mock control wells � 100%.Real-time reverse transcription-PCR. Cells were infected with
Adv-TK, wt-Adv5, or M1 at a MOI of 5. After 24 hours of culture,total RNA was extracted, and 2 Ag RNA was treated with DNase I(Life Technologies, Grand Island, NY) before single-strand cDNAsynthesis. To amplify gene-specific transcripts, 5V-end sense primer(5V-AAATGTGGCAGTCAAATAC-3V), 3V-end antisense primer (5V-ATA-GGTTAGGCATAAATCC-3V), and FITC-labeled primer (5V-TGGC-TCCAATATCTGGAACA-3V) for fiber gene; 5V-end sense primer (5V-GAAATGGACGGAATTATTACAG-3V), 3V-end antisense primer(5V-TTTCTGACGCTTGGTTGG-3V), and FITC-labeled primer (5V-ACCTACGACAGTAATACCACCGGAC-3V) for E3 14.7K; and 5V-endsense primer (5V-ACGGATTTGGTCGTATTGGG-3V), 3V-end antisenseprimer (5V-TGATTTTGGAGGGATGTCGC-3V), and FITC labeled primer(5V-TGACTGCTATAACTCGATGA-3V) for GAPDH gene were used.Following the PCR reaction, a melting curve assay was done todetermine the purity of the amplified product. Threshold cycle value(CT) is given for each sample and indicates the cycle at which a
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(23) December1, 2005 8432
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
statistically significant increase in fluorescence was first detected. Thesevalues were then normalized by the average expression of GAPDH todetermine ratios of relative expression.
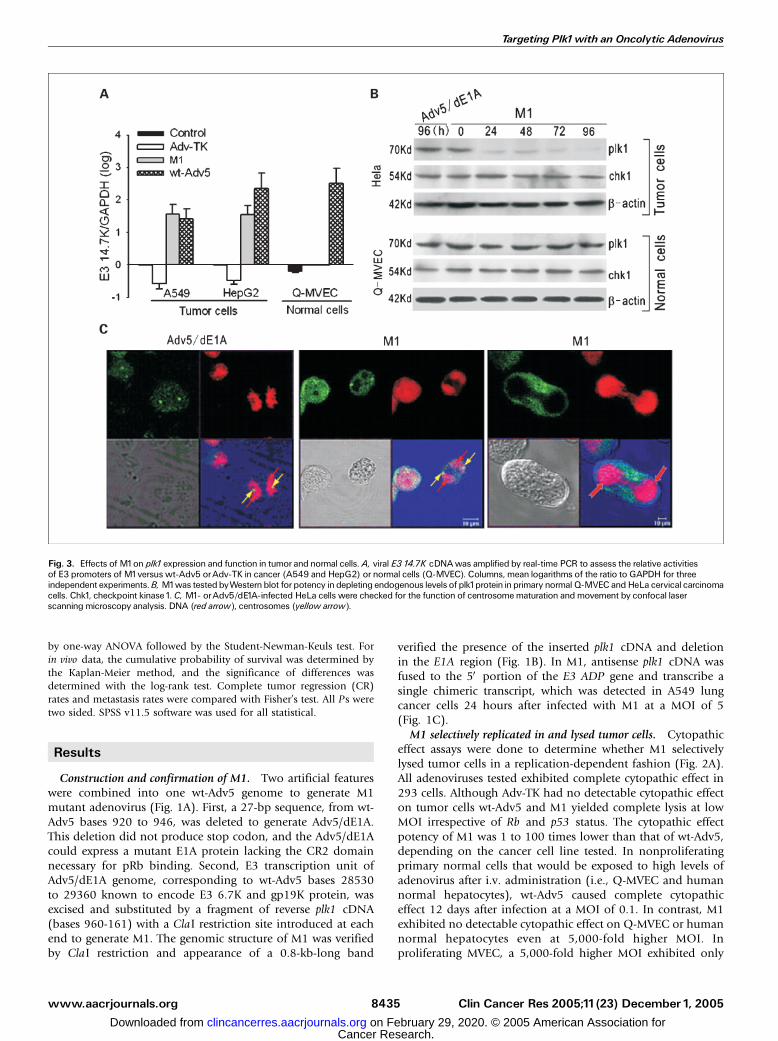
Western blot. Preparation of protein samples and Western blot weredone as described previously (14). Antibodies to chk1, plk1, and h-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Morphology of centrosomes. HeLa cells were infected with Adv5/dE1A or M1 at a MOI of 1 and cultured for 24 hours. For
immunostaining, cells were incubated with 1 Ag/mL of monoclonalantibody to g-tubulin (Santa Cruz Biotechnology) for 1 hour at 37jC.The slides were then washed with ice-cold PBS and incubated withfluorescein-conjugated goat anti-mouse IgG for 30 minutes. After one
wash with PBS, the slides were finally stained with propidium iodidesolution for 20 minutes and observed for morphology of centrosomeswith a confocal laser-scanning microscope (German Leica, true confocalscanner spectrophotometry, Mannheim, Germany) equipped with a
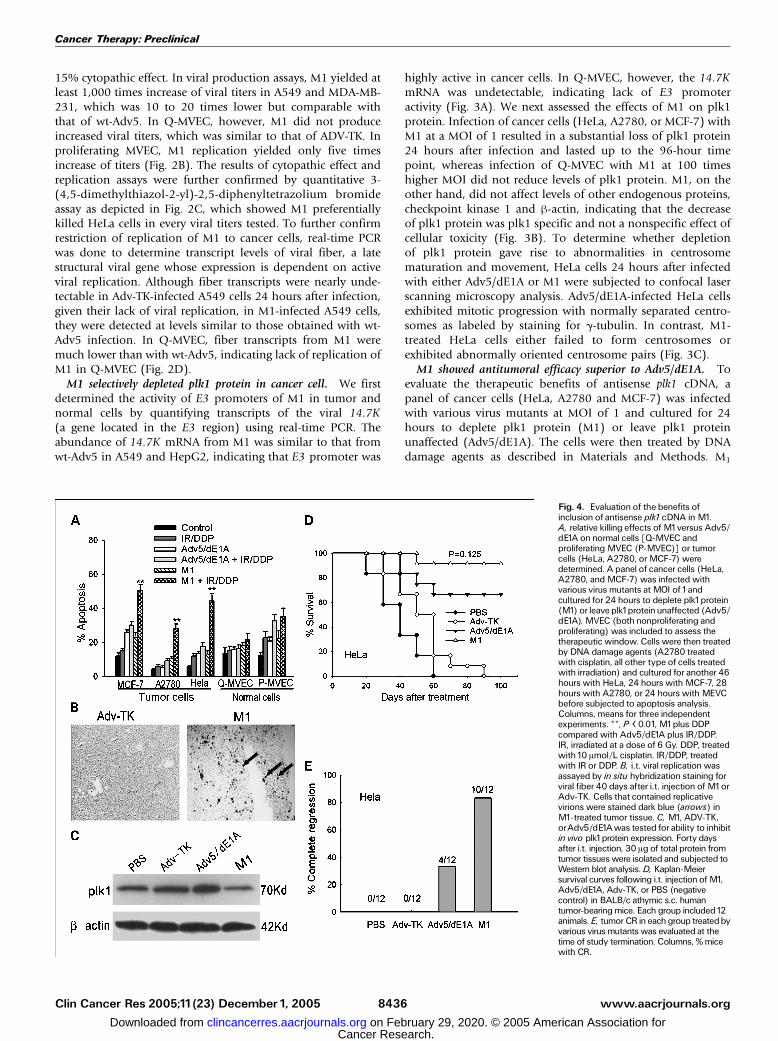
488-nm argon ion laser according to our previous publication (19).Apoptosis assay. Tumor cells (MCF-7, A2780, HeLa) and MEVC
(nonproliferative or proliferative) were infected with various virus
mutants at a MOI of 1 and cultured for 24 hours. The cells were then
treated by either 10 Amol/L cisplatin (for A2780) or 6 Gy irradiation
(for MCF-7, HeLa, or MEVC) and cultured for another 46 hours with
HeLa, 24 hours with MCF-7, 28 hours with A2780, or 24 hours with
MEVC before subjected to apoptosis analysis. At these time points,
because cisplatin or irradiation alone yielded only slight apoptosis, we
could evaluate the advantages of incorporation of the transgene of
antisense plk1 cDNA. Pretreated cells were resuspended in binding
buffer. Five microliters of FITC plus Annexin V (PharMingen, San
Diego, CA) and 10 microliters of propidium iodide solution at a
concentration of 50 Ag/mL in PBS were then added, and the cells were
incubated for 20 minutes at room temperature in the dark. Stained cells
were analyzed using a FACSort Flow Cytometer (Becton Dickinson, San
Jose, CA). CellQuest software was used for acquisition and analysis
of data.
Mouse tumor model studies. Female athymic BALB/c mice wereobtained from the Animal Experimental Center of Slaccas (Shanghai,
China). Mice were used when 4 to 6 weeks of age and were maintained in
a laminar flow cabinet under specific pathogen-free conditions. In the
direct i.t. injection studies, 2 � 106 HeLa cells were injected s.c. in theflanks of mice. When tumors had grown to f6 to 7 mm, 108 plaque-
forming units of each virus mutant were injected directly into tumors
once daily for five consecutive days (n = 12). Tumor sizes weredetermined twice weekly until mice were sacrificed (tumor volume >
1,000 mm3 or 100 days after treatment). To assess in vivo virus mutant
replication and endogenous plk1 protein levels, a parallel experiment wasdone. Mice (n = 2 for each group) were treated under identical
experimental conditions. Forty days after initiation of treatment, mice
were sacrificed, and primary tumors were collected for in situ
hybridization and Western blot analysis. In the second portion ofstudies, 1 � 106 MDA-MB-231 cells were injected into the thoracic
mammary fat pad of nude mice. When tumors had grown to f4 to
5 mm, 2 � 108 plaque-forming units of virus was injected by tail veindaily for five consecutive days (12 mice per group). Four days later,
cisplatin at a dose of 0.75 mg/kg/d was injected into the abdominal
cavity for four consecutive days. Tumors were monitored twice weeklyuntil mice were sacrificed 70 days after initiation of treatment or when
tumor volume was larger than 1,200 mm3. Lymph nodes (axillary,
supraclavicular, and paratracheal) were collected and subjected topathologic evaluation for evidence of metastases. In the final portion
of tumor model studies, an orthotopic HepG2 human hepatic
carcinoma model was established (20). First, 2 � 106 HepG2 cells were
injected s.c. in the flanks of nude mice and allowed tumor to grow to 7to 8 mm, the mice were sacrificed, and tumors were collected and cut
into several small pieces. One piece of tumor was implanted into the
liver of individual nude mice. The animals were allowed hepatic tumorsto reach 3 mm as determined by B-ultrasound. The animal models were
then treated in an identical fashion as described in orthotopic MDA-MB-
231 model studies. Tumor sizes were monitored weekly by B-ultrasound
Fig. 1. Analyses ofM1mutant adenovirus.A, a 27-nucleotide sequence from Adv5 bases 920 to 946,corresponding to the amino acid sequence of the E1Anecessary for Rb protein binding, was deleted togenerateAdv5/dE1A. A single region corresponding toE3 6.7K and gp19K, from Adv5 bases 28530 to 29360,was excised from Adv5/dE1A genome and substitutedby a fragment of reverse plk1cDNA (bases 960-161)with a ClaI restriction site introduced at each end togenerate M1. B, genomic structure of M1was confirmedby ClaI digestion. C, amplification of fusionmRNAcontaining sequences of antisense plk1cDNA and ADPgene in A549 cells 24 hours after infected with Adv-TK,M1, or wt-Adv5. PBSwas included as a solvent control.
Targeting Plk1with an Oncolytic Adenovirus
www.aacrjournals.org Clin Cancer Res 2005;11(23) December1, 20058433
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
until animals were sacrificed (tumor volume > 1,200 mm3 or 120 days
after treatment). Tumors, lungs, livers, and lymph nodes (portal,
mesenteric, inguinal, and retroperitoneal) were collected at necropsyand processed for histopathologic evaluation. A parallel experiment was
done for assessment of in vivo active viral replication (n = 2 for each
group). Primary tumor samples were collected for in situ hybridizationanalysis immediately after finishing five doses of i.v. injection of virus.
In situ hybridization. Slides were prehybridized for 30 minutes at37jC. Hybridization was carried out overnight at 42jC with 1 Ag/mL
biotinylated viral fiber oligonucleotide probe complimentary to thefiber coding region (5V-GGAACTGGCCTTAGTTTTGACAGCACAGGTG-CCATTACAG-3V). An alkaline phosphatase–conjugated anti-biotinantibody (Boehringer Mannheim, Mannheim, Germany) was addedto bind hybridized probe. The slides were then incubated withnitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate for 30minutes and counterstained once with nuclear fast red.
Statistical analysis. The cell culture data from at least threeindependent experiments were expressed as means F SD and examined
Fig. 2. Replication of M1in normal cells and tumor cells.A, M1, wt-Adv5, and replication-defectiveAdv-TK were tested for replication-dependent lysis in nonproliferatingMVEC (Q-MVEC), proliferating MVEC (P-MVEC), human normal hepatocytes (h NHeps), MDA-MB-231breast cancer cells (p53 mutant and Rb wild type), A2780 ovariancarcinoma cells (wild-type p53 and mutant Rb), HeLa cervical carcinoma cells (p53 inactivated by HPVE6 and mutant Rb), and A549 lung cancer cells (wild-type p53 andmutant Rb). Cells in six-well plates were infected with virus mutants at a MOI from 0.01to 500 and cultured for the indicated days before cytopathic effect assay.B, viralreplications ofM1, wt-Adv5, and Adv-TKwere determinedby viral replication assays in cancer (A549 andMDA-MB-231) andnormal cells (Q-MVEC andproliferatingMVEC).Columns, logarithms of viral titers [plaque-forming units (pfu)/mL)]. C, M1was tested for quantitative cytotoxicity on HeLa, Q-MEVC, or proliferating MVEC cells using3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide assay.The cells in 96-well tissue culture plateswere infectedwithM1at aMOI from 0.1to1,000 and cultured for96 hours before cytopathic effect assay. Columns, mean growth inhibition rate of three independent experiments.D, M1-, wt-Adv5-, orAdv-TK-infected A549 and Q-MVECwere tested for levels of viral fiber mRNA by real-time quantitative PCR. Columns, mean logarithms of the ratio toGAPDH of three independent experiments. Control,amplification done in cells without any treatment.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(23) December1, 2005 8434
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
by one-way ANOVA followed by the Student-Newman-Keuls test. Forin vivo data, the cumulative probability of survival was determined bythe Kaplan-Meier method, and the significance of differences wasdetermined with the log-rank test. Complete tumor regression (CR)rates and metastasis rates were compared with Fisher’s test. All Ps weretwo sided. SPSS v11.5 software was used for all statistical.
Results
Construction and confirmation of M1. Two artificial featureswere combined into one wt-Adv5 genome to generate M1mutant adenovirus (Fig. 1A). First, a 27-bp sequence, from wt-Adv5 bases 920 to 946, was deleted to generate Adv5/dE1A.This deletion did not produce stop codon, and the Adv5/dE1Acould express a mutant E1A protein lacking the CR2 domainnecessary for pRb binding. Second, E3 transcription unit ofAdv5/dE1A genome, corresponding to wt-Adv5 bases 28530to 29360 known to encode E3 6.7K and gp19K protein, wasexcised and substituted by a fragment of reverse plk1 cDNA(bases 960-161) with a ClaI restriction site introduced at eachend to generate M1. The genomic structure of M1 was verifiedby ClaI restriction and appearance of a 0.8-kb-long band
verified the presence of the inserted plk1 cDNA and deletionin the E1A region (Fig. 1B). In M1, antisense plk1 cDNA wasfused to the 5V portion of the E3 ADP gene and transcribe asingle chimeric transcript, which was detected in A549 lungcancer cells 24 hours after infected with M1 at a MOI of 5(Fig. 1C).
M1 selectively replicated in and lysed tumor cells. Cytopathiceffect assays were done to determine whether M1 selectivelylysed tumor cells in a replication-dependent fashion (Fig. 2A).All adenoviruses tested exhibited complete cytopathic effect in293 cells. Although Adv-TK had no detectable cytopathic effecton tumor cells wt-Adv5 and M1 yielded complete lysis at lowMOI irrespective of Rb and p53 status. The cytopathic effectpotency of M1 was 1 to 100 times lower than that of wt-Adv5,depending on the cancer cell line tested. In nonproliferatingprimary normal cells that would be exposed to high levels ofadenovirus after i.v. administration (i.e., Q-MVEC and humannormal hepatocytes), wt-Adv5 caused complete cytopathiceffect 12 days after infection at a MOI of 0.1. In contrast, M1exhibited no detectable cytopathic effect on Q-MVEC or humannormal hepatocytes even at 5,000-fold higher MOI. Inproliferating MVEC, a 5,000-fold higher MOI exhibited only
Fig. 3. Effects of M1on plk1expression and function in tumor and normal cells.A, viral E3 14.7K cDNAwas amplified by real-time PCR to assess the relative activitiesof E3 promoters of M1versus wt-Adv5 orAdv-TK in cancer (A549 and HepG2) or normal cells (Q-MVEC). Columns, mean logarithms of the ratio to GAPDH for threeindependent experiments.B, M1was testedbyWesternblot for potency in depleting endogenous levels of plk1protein inprimary normal Q-MVEC andHeLa cervical carcinomacells. Chk1, checkpoint kinase1. C, M1- orAdv5/dE1A-infected HeLa cells were checked for the function of centrosomematuration and movement by confocal laserscanning microscopy analysis. DNA (red arrow), centrosomes (yellow arrow).
Targeting Plk1with an Oncolytic Adenovirus
www.aacrjournals.org Clin Cancer Res 2005;11(23) December1, 20058435
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
15% cytopathic effect. In viral production assays, M1 yielded atleast 1,000 times increase of viral titers in A549 and MDA-MB-231, which was 10 to 20 times lower but comparable withthat of wt-Adv5. In Q-MVEC, however, M1 did not produceincreased viral titers, which was similar to that of ADV-TK. Inproliferating MVEC, M1 replication yielded only five timesincrease of titers (Fig. 2B). The results of cytopathic effect andreplication assays were further confirmed by quantitative 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromideassay as depicted in Fig. 2C, which showed M1 preferentiallykilled HeLa cells in every viral titers tested. To further confirmrestriction of replication of M1 to cancer cells, real-time PCRwas done to determine transcript levels of viral fiber, a latestructural viral gene whose expression is dependent on activeviral replication. Although fiber transcripts were nearly unde-tectable in Adv-TK-infected A549 cells 24 hours after infection,given their lack of viral replication, in M1-infected A549 cells,they were detected at levels similar to those obtained with wt-Adv5 infection. In Q-MVEC, fiber transcripts from M1 weremuch lower than with wt-Adv5, indicating lack of replication ofM1 in Q-MVEC (Fig. 2D).M1 selectively depleted plk1 protein in cancer cell. We first
determined the activity of E3 promoters of M1 in tumor andnormal cells by quantifying transcripts of the viral 14.7K(a gene located in the E3 region) using real-time PCR. Theabundance of 14.7K mRNA from M1 was similar to that fromwt-Adv5 in A549 and HepG2, indicating that E3 promoter was
highly active in cancer cells. In Q-MVEC, however, the 14.7KmRNA was undetectable, indicating lack of E3 promoteractivity (Fig. 3A). We next assessed the effects of M1 on plk1protein. Infection of cancer cells (HeLa, A2780, or MCF-7) withM1 at a MOI of 1 resulted in a substantial loss of plk1 protein24 hours after infection and lasted up to the 96-hour timepoint, whereas infection of Q-MVEC with M1 at 100 timeshigher MOI did not reduce levels of plk1 protein. M1, on theother hand, did not affect levels of other endogenous proteins,checkpoint kinase 1 and h-actin, indicating that the decreaseof plk1 protein was plk1 specific and not a nonspecific effect ofcellular toxicity (Fig. 3B). To determine whether depletionof plk1 protein gave rise to abnormalities in centrosomematuration and movement, HeLa cells 24 hours after infectedwith either Adv5/dE1A or M1 were subjected to confocal laserscanning microscopy analysis. Adv5/dE1A-infected HeLa cellsexhibited mitotic progression with normally separated centro-somes as labeled by staining for g-tubulin. In contrast, M1-treated HeLa cells either failed to form centrosomes orexhibited abnormally oriented centrosome pairs (Fig. 3C).M1 showed antitumoral efficacy superior to Adv5/dE1A. To
evaluate the therapeutic benefits of antisense plk1 cDNA, apanel of cancer cells (HeLa, A2780 and MCF-7) was infectedwith various virus mutants at MOI of 1 and cultured for 24hours to deplete plk1 protein (M1) or leave plk1 proteinunaffected (Adv5/dE1A). The cells were then treated by DNAdamage agents as described in Materials and Methods. M1
Fig. 4. Evaluation of the benefits ofinclusion of antisense plk1cDNA inM1.A, relative killing effects of M1versus Adv5/dE1A onnormal cells [Q-MVEC andproliferating MVEC (P-MVEC)] or tumorcells (HeLa, A2780, or MCF-7) weredetermined. A panel of cancer cells (HeLa,A2780, andMCF-7) was infected withvarious virus mutants at MOI of1andcultured for 24 hours to deplete plk1protein(M1) or leave plk1proteinunaffected (Adv5/dE1A). MVEC (both nonproliferating andproliferating) was included to assess thetherapeutic window. Cells were then treatedby DNA damage agents (A2780 treatedwith cisplatin, all other type of cells treatedwith irradiation) and cultured for another 46hours with HeLa, 24 hours with MCF-7, 28hours with A2780, or 24 hours with MEVCbefore subjected to apoptosis analysis.Columns, means for three independentexperiments. **, P < 0.01, M1plus DDPcompared with Adv5/dE1A plus IR/DDP.IR, irradiated at a dose of 6 Gy. DDP, treatedwith10 Amol/L cisplatin. IR/DDP, treatedwith IRor DDP. B, i.t. viral replicationwasassayed by in situ hybridization staining forviral fiber 40 days after i.t. injection of M1orAdv-TK. Cells that contained replicativevirions were stained dark blue (arrows) inM1-treated tumor tissue. C, M1, ADV-TK,orAdv5/dE1Awas tested for ability to inhibitin vivo plk1protein expression. Forty daysafter i.t. injection, 30 Ag of total protein fromtumor tissueswere isolated and subjected toWestern blot analysis.D, Kaplan-Meiersurvival curves following i.t. injection ofM1,Adv5/dE1A, Adv-TK, or PBS (negativecontrol) in BALB/c athymic s.c. humantumor-bearingmice. Each group included12animals. E, tumor CRin each group treatedbyvarious virus mutants was evaluated at thetime of study termination. Columns, %micewith CR.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(23) December1, 2005 8436
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
showed sensitizing effects significantly superior to Adv5/dE1Ain all cancer cells tested. Antisense plk1 cDNA was thusresponsible for the sensitizing. On the other hand, M1 did notsignificantly sensitized Q-MVEC or proliferating MVEC toirradiation-induced apoptosis and featured a good therapeuticwindow (Fig. 4A). In BALB/c athymic tumor-bearing mice,active viral replication in s.c. tumors and consequent reductionof plk1 protein were evident when assayed 40 days after i.t.
injection of M1 (Fig. 4B and C). HeLa tumor-bearing micetreated with M1 exhibited survival superior to Adv-TK (P <0.0001) or Adv5/dE1A (P = 0.125; Fig. 4D). Tumor CR wassignificantly common following treatment with M1 (10 of 12)than Adv-TK (0 of 12; P < 0.0001) or Adv5/dE1A (4 of 12; P =0.036; Fig. 4E).
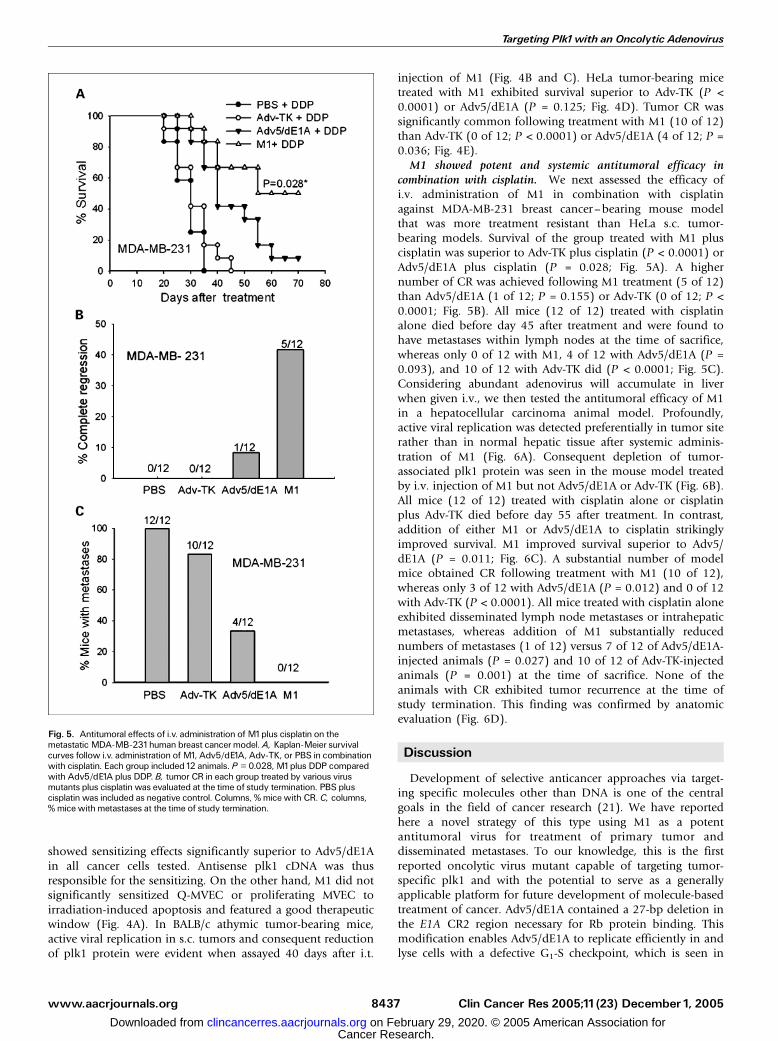
M1 showed potent and systemic antitumoral efficacy incombination with cisplatin. We next assessed the efficacy ofi.v. administration of M1 in combination with cisplatinagainst MDA-MB-231 breast cancer–bearing mouse modelthat was more treatment resistant than HeLa s.c. tumor-bearing models. Survival of the group treated with M1 pluscisplatin was superior to Adv-TK plus cisplatin (P < 0.0001) orAdv5/dE1A plus cisplatin (P = 0.028; Fig. 5A). A highernumber of CR was achieved following M1 treatment (5 of 12)than Adv5/dE1A (1 of 12; P = 0.155) or Adv-TK (0 of 12; P <0.0001; Fig. 5B). All mice (12 of 12) treated with cisplatinalone died before day 45 after treatment and were found tohave metastases within lymph nodes at the time of sacrifice,whereas only 0 of 12 with M1, 4 of 12 with Adv5/dE1A (P =0.093), and 10 of 12 with Adv-TK did (P < 0.0001; Fig. 5C).Considering abundant adenovirus will accumulate in liverwhen given i.v., we then tested the antitumoral efficacy of M1in a hepatocellular carcinoma animal model. Profoundly,active viral replication was detected preferentially in tumor siterather than in normal hepatic tissue after systemic adminis-tration of M1 (Fig. 6A). Consequent depletion of tumor-associated plk1 protein was seen in the mouse model treatedby i.v. injection of M1 but not Adv5/dE1A or Adv-TK (Fig. 6B).All mice (12 of 12) treated with cisplatin alone or cisplatinplus Adv-TK died before day 55 after treatment. In contrast,addition of either M1 or Adv5/dE1A to cisplatin strikinglyimproved survival. M1 improved survival superior to Adv5/dE1A (P = 0.011; Fig. 6C). A substantial number of modelmice obtained CR following treatment with M1 (10 of 12),whereas only 3 of 12 with Adv5/dE1A (P = 0.012) and 0 of 12with Adv-TK (P < 0.0001). All mice treated with cisplatin aloneexhibited disseminated lymph node metastases or intrahepaticmetastases, whereas addition of M1 substantially reducednumbers of metastases (1 of 12) versus 7 of 12 of Adv5/dE1A-injected animals (P = 0.027) and 10 of 12 of Adv-TK-injectedanimals (P = 0.001) at the time of sacrifice. None of theanimals with CR exhibited tumor recurrence at the time ofstudy termination. This finding was confirmed by anatomicevaluation (Fig. 6D).
Discussion
Development of selective anticancer approaches via target-ing specific molecules other than DNA is one of the centralgoals in the field of cancer research (21). We have reportedhere a novel strategy of this type using M1 as a potentantitumoral virus for treatment of primary tumor anddisseminated metastases. To our knowledge, this is the firstreported oncolytic virus mutant capable of targeting tumor-specific plk1 and with the potential to serve as a generallyapplicable platform for future development of molecule-basedtreatment of cancer. Adv5/dE1A contained a 27-bp deletion inthe E1A CR2 region necessary for Rb protein binding. Thismodification enables Adv5/dE1A to replicate efficiently in andlyse cells with a defective G1-S checkpoint, which is seen in
Fig. 5. Antitumoral effects of i.v. administration of M1plus cisplatin on themetastatic MDA-MB-231human breast cancer model. A, Kaplan-Meier survivalcurves follow i.v. administration of M1, Adv5/dE1A, Adv-TK, or PBS in combinationwith cisplatin. Each group included12 animals. P = 0.028, M1plus DDP comparedwith Adv5/dE1A plus DDP. B, tumor CR in each group treated by various virusmutants plus cisplatin was evaluated at the time of study termination. PBS pluscisplatin was included as negative control. Columns, % mice with CR. C, columns,% mice with metastases at the time of study termination.
Targeting Plk1with an Oncolytic Adenovirus
www.aacrjournals.org Clin Cancer Res 2005;11(23) December1, 20058437
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from
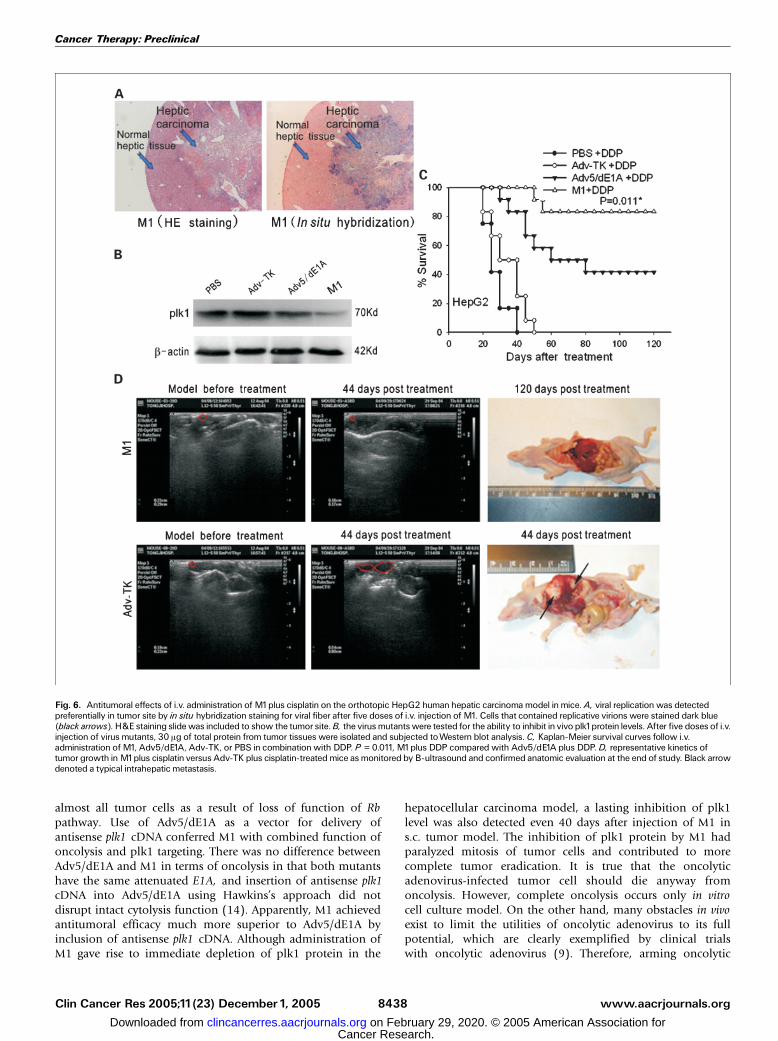
almost all tumor cells as a result of loss of function of Rbpathway. Use of Adv5/dE1A as a vector for delivery ofantisense plk1 cDNA conferred M1 with combined function ofoncolysis and plk1 targeting. There was no difference betweenAdv5/dE1A and M1 in terms of oncolysis in that both mutantshave the same attenuated E1A, and insertion of antisense plk1cDNA into Adv5/dE1A using Hawkins’s approach did notdisrupt intact cytolysis function (14). Apparently, M1 achievedantitumoral efficacy much more superior to Adv5/dE1A byinclusion of antisense plk1 cDNA. Although administration ofM1 gave rise to immediate depletion of plk1 protein in the
hepatocellular carcinoma model, a lasting inhibition of plk1level was also detected even 40 days after injection of M1 ins.c. tumor model. The inhibition of plk1 protein by M1 hadparalyzed mitosis of tumor cells and contributed to morecomplete tumor eradication. It is true that the oncolyticadenovirus-infected tumor cell should die anyway fromoncolysis. However, complete oncolysis occurs only in vitrocell culture model. On the other hand, many obstacles in vivoexist to limit the utilities of oncolytic adenovirus to its fullpotential, which are clearly exemplified by clinical trialswith oncolytic adenovirus (9). Therefore, arming oncolytic
Fig. 6. Antitumoral effects of i.v. administration of M1plus cisplatin on the orthotopic HepG2 humanhepatic carcinoma model inmice. A, viral replicationwas detectedpreferentially in tumor site by in situ hybridization staining for viral fiber after five doses of i.v. injection of M1. Cells that contained replicative virions were stained dark blue(black arrows). H&E staining slidewas included to show the tumor site.B, the virus mutantswere tested for the ability to inhibit in vivo plk1protein levels. After five doses of i.v.injection of virus mutants, 30 Ag of total protein from tumor tissues were isolated and subjected toWestern blot analysis.C, Kaplan-Meier survival curves follow i.v.administration ofM1, Adv5/dE1A, Adv-TK, or PBS in combinationwith DDP. P = 0.011, M1plus DDP compared with Adv5/dE1A plus DDP.D, representative kinetics oftumor growth inM1plus cisplatin versus Adv-TK plus cisplatin-treatedmice as monitored by B-ultrasound and confirmed anatomic evaluation at the end of study. Black arrowdenoted a typical intrahepatic metastasis.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(23) December1, 2005 8438
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from

adenovirus with plk1-targeting function would provide bestchance for tumor eradication. It should be pointed out thatviral replication of M1 would robustly amplify the copies ofantisense plk1 in tumor cells and allow spread of viralinfection to nearby tumor cells, which would further enhancethe potency of plk1 targeting. In present design, we had madeM1 with a 3-bp more deletion than D24 for safety consi-deration because a recent study suggested that D24 exhibitedefficient replication in normal keratinocytes in an in vitroorganotypic keratinocytes culture (22). Our findings hadshown that this extra deletion did not attenuate the potencyof tumor-selective replication, and Adv5/dE1A exhibitedsimilar antitumoral potency to D24 as previously reported intumor model mice (10, 11).Although current methods of chemotherapy, radiotherapy,
and surgical resection effectively eradicate local primarytumors, they are less effective in controlling metastatic disease.Thus, one of our most exciting findings is that cisplatin-refractory tumor xenograft models responded to i.v. adminis-tration of M1 plus cisplatin with reduced incidence ofmetastases and substantially improved survival. The resultsobserved with the orthotopic HepG2 human hepatocellularcarcinoma model are even more encouraging. It is well knownthat hepatocellular carcinoma is one of the most commonmalignant tumors, and hepatic metastases from other malig-nancies are frequent. Very few patients can be cured due todifficulty in controlling disseminated metastases (21–26). M1may be useful in combination with current therapies incontrolling not only local primary tumors but also disseminat-ed metastases. Whether the present findings can be translatedinto clinical benefits should be determined in clinical trials.Because most adenovirus will accumulate in liver when giveni.v., the findings presented here are encouraging enough towarrant a clinical evaluation (27).
A number of questions arise regarding the ultimate clinicalefficacy and safety of M1. First, because no appropriate animalmodel is now available to mimic the response of humans tooncolytic adenovirus, well-designed clinical trials will beneeded to answer the clinical safety of M1. However, theoverall safety of use of oncolytic adenovirus has been shownin a number of clinical trials, and the valuable lessons leanedin such trials will aid in development of M1 into a clinicallyuseful anticancer agent (28–34). Second, concern exists thatreplication and targeting of plk1 might also occur inhematopoietic cells with physiologically inactivated G1-Scheckpoint. This is unlikely to occur because hematopoieticcells are resistant to adenovirus infection (35). Third, thedifferences in potency of M1 against the orthotopic MDA-MB-231 cancer model and the orthotopic HepG2 carcinomamodel might reflect variability in distribution of virus after i.v.injection. For effective tumor eradication, precise targetingof M1 to tumor sites is required. Further modification ofM1 is therefore required to overcome the native tropism ofadenovirus and retarget it to tumor sites to achieve full thera-peutic efficacy. Lastly, silencing of multigenes is possible bymodifying the present virus mutant by inserting more thanone antisense cDNA into the E3 6.7K/gp19K site. Whether thisstrategy will further improve antitumor efficacy is aninteresting question. Our present findings lay the conceptualadvance for the development of new generation of potentoncolytic adenoviruses that are generally applicable to a widerange of molecule-based therapeutics.
Acknowledgments
We thank Dr. Jing Chen for single-photon emission computed tomographyexamination, Dr. Qiling Ao for reviewing histology data, and Dr. Jerry Y. Niederkornfor inspiration and discussions.
References1. DaiW, Cogswell JP. Polo-like kinases and the micro-tubule organization center: targets for cancer thera-pies. Prog Cell Cycle Res 2003;5:327^34.
2.WeichertW, Denkert C, SchmidtM, et al. Polo-like ki-nase isoform expression is a prognostic factor in ovar-ian carcinoma. BrJCancer 2004;90:815^21.
3. Gumireddy K, Reddy MV, Cosenza SC, et al.ON01910, a non-ATP-competitive small molecule in-hibitor of Plk1, is a potent anticancer agent. CancerCell 2005;7:275^86.
4. Reagan-Shaw S, Ahmad N. Silencing of polo-likekinase (Plk) 1via siRNA causes induction of apoptosisand impairment of mitosis machinery in human pros-tate cancer cells: implications for the treatment ofprostate cancer. FASEB J 2005;19:611^3.
5.Winkles JA, Alberts GF. Differential regulation ofpolo-like kinase1, 2, 3, and 4 gene expression inmam-malian cells and tissues. Oncogene 2005;24:260^6.
6. Van Vugt MA, Medema RH. Getting in and out ofmitosis with Polo-like kinase-1. Oncogene 2005;24:2844^59.
7. Kirn D. Replication-selective oncolytic adenoviruses:virotherapy aimed at genetic targets in cancer. Onco-gene 2000;19:6660^9.
8. Chu RL, Post DE, Khuri FR,Van Meir EG. Use of rep-licatingoncolytic adenoviruses in combination therapyfor cancer. Clin Cancer Res 2004;10:5299^312.
9. Khuri FR,NemunaitisJ, GanlyJ, et al. A controlled trialof intratumoral ONYX-015, a selectively-replicatingadenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neckcancer. NatMed 2000;6:879^85.
10. Fueyo J, Gomez-Manzano C, Alemany R, et al. Amutant oncolytic adenovirus targeting theRbpathwayproduces anti-glioma effect in vivo. Oncogene 2000;19:2^12.
11.Heise C,HermistonC, Johnson L, et al. AnadenovirusE1Amutant that demonstrates potent and selective sys-temic anti-tumoral efficacy. NatMed 2000;6:1134^9.
12. Stubdal H, Perin N, Lemmon M, et al. A prodrugstrategy using ONYX-015-based replicating adenovi-ruses to deliver rabbit carboxylesterase to tumor cellsfor conversion of CPT-11to SN-38. Cancer Res 2003;63:6900^8.
13.NandaD,Vogels R, HavengaM, AvezaatCJ, Bout A,Smitt PS.Treatment of malignant gliomas with a repli-cating adenoviral vector expressing herpes simplexvirus-thymidine kinase. Cancer Res 2001;61:8743^50.
14. Hawkins LK, Johnson L, Bauzon M, et al. Genedelivery from the E3 region of replicating human ade-novirus: evaluation of the 6.7K/gp19K region. GeneTher 2001;8:1123^31.
15. Gary K, Boyer J. Isolation, growth, and purificationof defective adenovirus deletion mutants. In: WoldWSM, editor. Adenovirus methods and protocols.Totowa (NJ): Humana Press; 1999. p. 25^32.
16. HermistonTW,Tollefson AE,WoldWSM. Construc-tion of mutations in the adenovirus early region 3(E3) transcription units. In:WoldWSM, editor. Adeno-virus methods and protocols. Totowa (NJ): HumanaPress; 1999. p.11^24.
17.MaD,Gerard RD, Li XY, et al. Inhibitionofmetastasisof intraocular melanomas by adenovirus-mediatedgene transfer of plasminogen activator inhibitor type1
(PAI-1) in an athymic mouse model. Blood 1997;90:2738^46.
18. Zhou J, Gurates B,Yang S, et al. Malignant breastepithelial cells stimulate aromatase expression via pro-moter II in human adipose fibroblasts: an epithelial-stromal interaction in breast tumors mediated byCCAAT/enhancer binding protein h. Cancer Res2001;61:2328^34.
19. Xu G, Arregui C, Lilien J, et al. PTP1B modulatesthe association of h-catenin with N-cadherinthrough binding to an adjacent and partially over-lapping target site. J Biol Chem 2002; 277:49989^97.
20. GaoYS, Chen XP, Li KY, et al. Nude mice model ofhuman hepatocellular carcinoma via orthotopic im-plantation of histologically intact tissue.World J Gas-troenterol 2004;10:3107^11.
21.Von Eschenbach AC. Avision for the national cancerprogram in theUnited States. Nat Rev Cancer 2004;4:820^8.
22. Balague¤ C, Noya F, Alemany R, et al. Human papil-lomavirus E6 E7-mediated adenovirus cell killing:selectivity of mutant adenovirus replication in organo-typic cultures of human keratinocytes. JVirol 2001;75:7602^11.
23.Baron M, LewisV. Images in clinical medicine. Livermetastases from lung cancer. N Engl J Med 2001;345:180.
24. Lorenz M, Hochmuth K, Muller HH. Hepatic arterialinfusion of chemotherapy for metastatic colorectalcancer. NEnglJMed 2000;342:1525^6.
25. Kemeny N, Huang Y, Cohen AM, et al. Hepatic
Targeting Plk1with an Oncolytic Adenovirus
www.aacrjournals.org Clin Cancer Res 2005;11(23) December1, 20058439
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from

arterial infusion of chemotherapy after resection ofhepatic metastases from colorectal cancer. N EnglJ Med 1999;341:2039^48.
26. Mazzaferro V, Regalia E, Doci R, et al. Liver trans-plantation for the treatment of smallhepatocellular car-cinomas in patients with cirrhosis. NEnglJMed1996;334:693^700.
27. Groot-WassinkT, Aboagye EO, Glaser M, et al. Ade-novirus biodistribution and noninvasive imaging ofgene expression in vivo by positron emission tomog-raphy using human sodium/iodide symporter as re-porter gene. HumGeneTher 2002;13:1723^35.
28. Fabra A, Parada C, Vinyals A, et al. Intravascularinjections of a conditional replicative adenovirus(adl118) prevent metastatic disease in human breastcarcinoma xenografts. GeneTher 2001;8:1627^34.
29. Habib NA, Sarraf CE, Mitry RR, et al. E1B-deleted
adenovirus (dl1520) gene therapy for patients withprimary and secondary liver tumors. Hum GeneTher2001;12:219^26.
30.Vasey PA, Shulman LN, Campos S, et al. Phase I trialof intraperitoneal injection of the E1B-55-kd-gene-deleted adenovirus ONYX-015 (dl1520) givenondays1through 5 every 3 weeks in patients with recurrent/refractory epithelial ovarian cancer. JClin Oncol 2002;20:1562^9.
31. Freytag SO, Stricker H, Pegg J, et al. Phase Istudy of replication-competent adenovirus-mediateddouble-suicide gene therapy with conventional-dosethree-dimensional conformal radiation therapy forthe treatment of newly diagnosed, intermediate- tohigh-risk prostate cancer. Cancer Res 2003;63:7497^506.
32. Nemunaitis J, Khuri F, Ganly I, et al. Phase II trial of
intratumoral administration of ONYX-015, a replica-tion-selective adenovirus, in patients with refractoryhead and neck cancer. JClin Oncol 2001;19:289^98.
33. Nemunaitis J, Ganly I, Khuri F, et al. Selective repli-cation and oncolysis in p53 mutant tumors with ON-YX-015, an E1B-55kD gene-deleted adenovirus, inpatients with advanced head and neck cancer : aphase II trial. Cancer Res 2000;60:6359^66.
34.ReidT,Galanis E, AbbruzzeseJ, et al. Hepatic arterialinfusionof a replication-selective oncolytic adenovirus(dl1520): phase II viral, immunologic, and clinical end-points. Cancer Res 2002;62:6070^9.
35.ColinM,Renaut L,Mailly L, et al. Factors involved inthe sensitivity of different hematopoietic cell lines toinfection by subgroup C adenovirus: implication forgene therapy of human lymphocytic malignancies.Virology 2004;320:23^39.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(23) December1, 2005 8440
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from

Correction
Correction: Novel Oncolytic AdenovirusSelectively Targets Tumor-AssociatedPolo-Like Kinase 1 and Tumor Cell ViabilityJianfeng Zhou, Qinglei Gao, Gang Chen, Xiaoyuan Huang,YunpingLu,KanyanLi,DaxingXie, LiangZhuang, JingniuDeng,and Ding Ma
In the original version of this article (1), the HeLa and MDA-MB-231 well plates forwt-Adv5 and replication-defective Adv-TKwere unintentionally duplicated in Fig. 2A.This error has been corrected in the latest online PDF version of the article. Theauthors regret this error.
Reference1. Zhou J, Gao Q, Chen G, Huang X, Lu Y, Li K, et al. Novel oncolytic adenovirus selectively targets
tumor-associated polo-like kinase 1 and tumor cell viability. Clin Cancer Res 2005;11:8431–40.
Published online December 15, 2019.Clin Cancer Res 2019;25:7611doi: 10.1158/1078-0432.CCR-19-3370�2019 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org 7611

2005;11:8431-8440. Clin Cancer Res Jianfeng Zhou, Qinglei Gao, Gang Chen, et al. ViabilityTumor-Associated Polo-Like Kinase 1 and Tumor Cell Novel Oncolytic Adenovirus Selectively Targets
Updated version
http://clincancerres.aacrjournals.org/content/11/23/8431
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/11/23/8431.full#ref-list-1
This article cites 33 articles, 12 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/11/23/8431.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/11/23/8431To request permission to re-use all or part of this article, use this link
Cancer Research. on February 29, 2020. © 2005 American Association forclincancerres.aacrjournals.org Downloaded from




![Oncolytic virus immunotherapy: future prospects for oncologyInducing Ligand Tumor Downregulation Induction of NK cell apoptosis by TRAIL-R2 binding [31, 32, 38, 39, 43] FAS CD95 Tumor](https://img.pdfslide.us/doc/110x75/611df3952340b5255074a0a6/oncolytic-virus-immunotherapy-future-prospects-for-oncology-inducing-ligand-tumor.jpg)
























