Embed Size (px)
Citation preview

NOTE TO USERS
This reproduction is the best copy available.
UMI


Rhodium Catalysed Asymmetric Ring Opening of Oxabicyclic Aikenes and Diastereoselective Ring Opening of Epoxides 115th Hctcroatom Nuclcophiles
Keith Fagnou
- A thesis submitted in conformity with the requirements For the degree of Master of Science Graduate ~epartrnent of ~ h e r n i s t r ~
'~niversity of Toronto
8 Copyright by Keith Fagnou (2000)

National Library I*I of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques 395 Wellington Street 395. nie Wellington Ottawa ON K1A ON4 Ottawa ON KIA ON4 Canada Canada
Your Votre niwBnee
Our fik, Notre refdnmce
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Libtary of Canada to Bibliothèque nationale du Canada de reproduce, ban, distribute or sel1 reproduire, prêter, distribuer ou copies of this thesis in microfom, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/nlm, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fkom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

Objectives
The overall objective was to develop an asymrnetric ring opening reaction of
oxabenzonorbomadienes with heteroatom nucleophiles to generate enantiopure
dihydronaphthalenes.
The first objective was to conduct studies on the rhodium catalysed ring opening of
oxabenzonorbomadiene. To this end, initial investigations focused on establishing the
generality of the reaction towards a variety of alcohols. Regioselectivity issues and an
intramolecular version were studied. Subsequent work dealt with establishing conditions
to effect the ring opening with alcohol nucleophiles in high enantiomeric excess.
The second objective was to extend this methodology to other heteroatom nucleophiles
such as phenols, amines and carboxylates. As a consequence, subsequent transformations
of these hydronaphthalene products could be investigated.
The third objective was to extend this methodology to other substrate classes such as
vinyl epoxides.

Abstract
Oxabenzonorbornadienes were shown to undergo asymmetnc ring opening with a vanety
of alcohol, phenol and activated amine nucleophiles in the presence of catalytic amounts
of [Rh(COD)C1I2 and chiral diphosphine ligands. The product dihydronaphthalenols
were produced in 73->99%ee. Extension of this methodology to O-halophenols required
changing the rhodium source to [Rh(CO)zCl]2. These products were applied to the
synthesis of benzodihydrofiirans.
In order for carboxylates and unactivated amines to induce ring opening, the addition of a
proton source was required. Enantioselectivities of up to 74% and 81% were observed
for the amine and the carboxylate ring opened products respectively. These products
were applied towards the synthesis of 1,4-dihydronaphthalenols.
Vinyl Epoxides were show to undergo diastereoselective ring opening reaction with
alcohols and aromatic amine nucleophiles in the presence of catalytic amounts of
[Rh(C0)2C1]2. The tram- 1,2-alkoxyalcohols and aminoalcobols were produced in >80%
yield and >20: 1 diastereoselectivity.

iii
Acknowledgements
1 would like to thank my supervisor, Mark Lautens, not only for his support, guidance
and instruction throughout the last two yean, but also for giving someone with a
background in highschool teaching a chance to do chemistry. 1 am extremely grateful for
the tremendous oppomuiity which he has offered me.
A number of people in the Lautens group, past and present, deserve special th&: Tom
Rovis, on whose bench this project originated a ~ . d under whose guidance 1 learned not
only the technical aspects of chernistry, but also how to approach a chemical challenge;
Greg Hughes (with whom 1 shared a comrnon appreciation of country music - much to
the chagrin of the other people in the lab), for daily discussions about chemistry and an
endless supply of helpful advice; Mark Taylor with whorn 1 shared an enjoyable
collaboration; and al1 the othen who make daily lab life more stimulating and exciting.
1 would also like to thank my farnily for their support and love. Most importantly, 1 want
to thank my wife, Danielle, who provided me with al1 the love and encouragement (and
even a remarkable level of interest given the emotional s c m suffered by a typical pre-
med experience with organic chemistry) that I could have ever wanted.

Words of uWisdom" After Two Years in the Lab
It is NEVER too early to begin characterizing your compounds.
A week in the lab saves an hour in the library.
Sometirnes the best solution for a problem cannot be found in the library.
With regards to rnethodological studies (like target shooting): Shoot first and cal1 what you hit the target.
With regards to total synthesis studies (like golf): 90% of short putts don? go in.
With regards to mechanistic studies (like much of life): Anyone who isn't confùsed doesn't really understand the situation.

Table Of Contents
1 GENERAL INTRODUCTION -
1.1 AIlvlic Alkvlation. Amination and Etherification -
1.1.1 Palladium Catalvsis -
1.1.1.1 General Concepts
1 . 1 . 1 2 Enantioselective Catalvsis
1.1-1.3 Dvnamic Kinetic Asvmmetric Transformations
1.1 .Z Rhodium Catal~sis
1.1.3 Other Transition Metal Catalvsts
1 1 3 1 Indium Catalvsip
l.1,3 2 Molvbdenum Catalvsis
1.1,3,3 Nickel Catalvsis
L U 4 -3
1 . 1.3 5 Jron Catalvsis
1.1.3.6 Tun~sten Catalysis
1.1 -3 -7 Cobalt Cataiysis
Asvmmetric Rin O~eniny of Orabicvclic Al kenes
1 .X 1
1.2.2 Nucleophilic Rin Opening of Oxabicvclic Alkenes
1 +2.2.1 Concepts and Strategip
L 2 2 2

Goals and Targets
1.3.1 l t
i .3.1.1 Svnthetic Precedent
2 RHODIUM CATALYSED ASYMMETRIC RING OPENlNG OF -
m s
a Alcohol Nucleophile~
2.1.1 Initial investigation^
2.1.1.1 Establishment of Relative Stereochernistrv and Scooe of Alcohol
ucleo~hi les
2,L1.2 Substttution Effects - .
2.1,1.3 JntramolecuIar~anant
2.1.2 Develooment of an Asvmrnetric Variant
2.1.2.1 Effect ofAdded Phosphines
2.122 cons e of AlcohoI Nucleo~hiles
2.1.2.4 Effect of S-mmetric Arvl Substitution on the Substrate
2.1.2.5 Conct usion

2.1.3 Expenmental
2.1 -3.1 General Expenrnental
2.1,3.2 Oxabicylic Startine Materials
2.1.3.3 Ring Ooenine with Alcohols
2.2 Phenol Nucleo~hiles
vii
5 1
5 1
53
60
73
Introduction 73
Investigation of the Conditions 73
Scope of 4-Substinited Phenols as Nudeophiles 74
Develooment of New Catalvst Svstem for O-Halo Phenols 76
&piication of Rine Opened Products Towards the Preparation of Benzofunns
78
Conclusion 79
80
23 Nitropen Nucleophile~
2.3.1 Introduction,
2.3.2 AR0 with Activated Nitrogn Nucleophiles
2 3 A R 0 with Jnactivated Ali~hatic Nucleophilq
2.3.4 Çonclusion
23.5 Expenrnental
19 Carboxylate Nucleo~hi le~
2.4.1 Jntroduction
2.4.2 AR0 with Carbox~late Nucleophiles

viii
2.4.3 &plication of R i n ~ Opened Products in the Preparation of 1 A-disubstituted-
d v 116
2.4.3 Co - nclusion 1 16
4 . 5 Expenmental 117
3 RHODIUM CATALYSED ALCOHOLYSlS AND AMINOLYSIS OF VINYL -
EPOXIDES 125
$J Introduction 125
33 Rhodium Catal~sed Alcoholvsis and Aminolysis of Vinvl Eaoxides 128
3.2.1 Alcohol Nucleophile~ 128
LZZ m ~ m i n e ~ h i l e s 130
3 2.3 Preliminarv Mechanistic Studies 133
Conclusion 134

List of Abbreviations
[al D
Ac
Anal.
Ar
BWAP
Bn
Boc
Calcd
COD
CP
DBU
de
dr
DMAP
DME
DMF
dppb
dppe
depf
ee
specific rotation measured at 589nm
acety 1
analysis
ary 1
2,2'-bis(dipheny1phosphino)- 1.1 '-binaphthyl
benzyl
tert-butylcarbonyl
calculated
1,s-cyclooctadiene
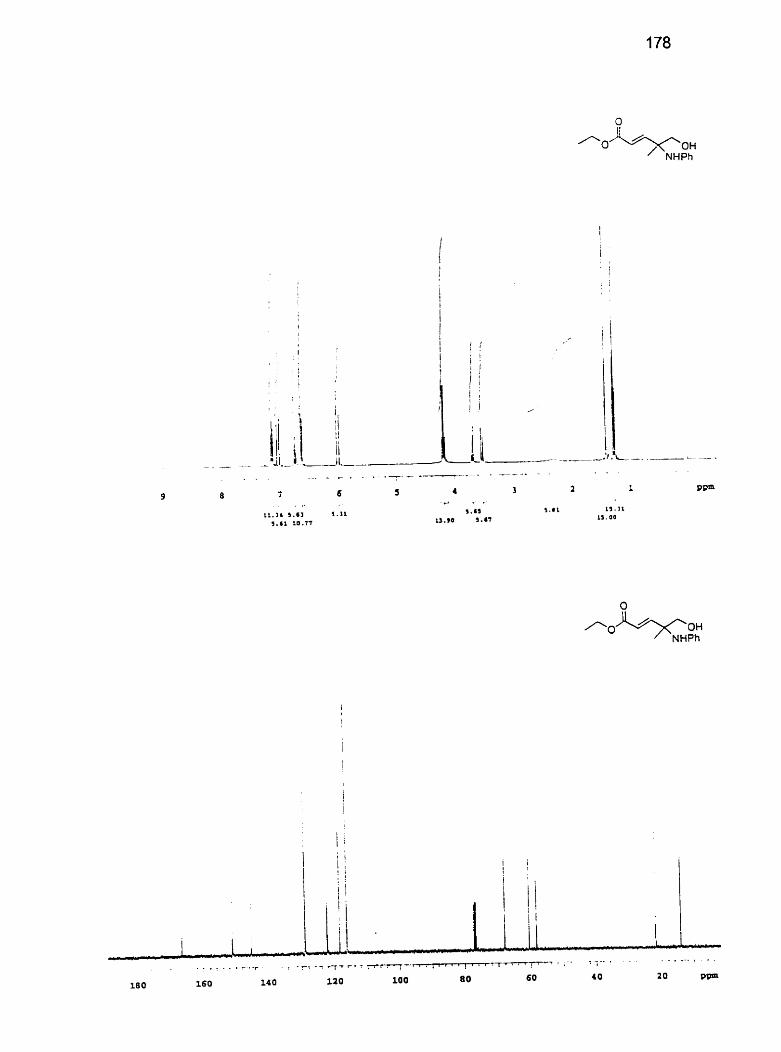
cyciopentadieny 1
( 1,8)diazabicyclo[5.3 .O]undec-7-ene
diastereomeric excess
diastereomeric ratio
4-(dimethy1arnino)pyridine
dimethoxyethane
dimethyl formamide
1,4-bis(dipheny lphosphino)butane
1,4-bis(dipheny1phosphino)ethane
1,4-bis(diphenylphosphino) ferrocene
enantiomeric excess
equivalent

Et
FT
GC
HRMS
imid
iPr
IR
L
M
MCPBA
Me
NMR
NOE
Ph
PMI3
PY
R
rt
TBAF
TBDMS
THF
TLC
ethy 1
Fourier tram form
gas chromatography
high resolution mass spectrum
irnidazole
isopropy 1
infared
ligand
generic metal
meta-chloroperbenzoic acid
methyl
nuclear magnetic resonance
nuclear Overauser effect
phenyl
para-methoxybenzy 1
p yridine
generic akyl group
room temperature
tetrabutylammoniurn fluoride
tert-bu~ldimethylsil y1
terahy dro furan
thin layer chrornatography

List of Tables
Table 2.1: Rhodium Catalvsed Ring Openine with Various Aicohols 37
Table 2 2 : Effect of Bridgehead Subtituents and Temperature on the lntramolecular Rinq
of Oxabicvclic Alkenes 42
Table 2.3: Effect of Phosohines on Reactivity with IRIi(CO1~Cll~ 43
Table 2.4: E ffect of Phos~horous Donor Ligands on [Rh(COD)CIl? 44
Table 2.5: E h c t of Bidentate Ligands on Reactivity with TFWCODICIL 45
Table 2.6: Atmos~here and Solvent Effects 47
Table 2.7: Solvent and Temperature Effect~ 48
Table 2.8: Scope of the Rhodium Catalysed A R 0 with Alcohols 49
Table 2.9: Effects of Number of Eauivalents of Phenol 74
Table 2.10: Scow of AR0 with p-Substituted Phenols 75
Table 2.1 1 : Effect of Phenoi Substitution Pattern 76
Table 2.17. Effect of Chanein~ the Rhodium Source 77
Table 2.13: Rhodium Catalvsed R i a Openine with Nitroeen Nucleophiles 96
Table 2.14: Rhodium Catalvsed AR0 with Nitrogen Nucleophiles 97
Table 2.15: Effect of Aliphatic Amines on React ivi~ 98
Table 2.16: Effect of an Added Proton Source on the Reactivity of Aliphatic Amines 99
Table 2.17: I Ise of a Proton Source to Induce Rino Opening with Aliphatic Amines LOO
Table 2.18: AR0 of Oxabenzonorbomadiene with Nitrogen and Carboxylate
Nucieo~hiies 101
Table 2.19: Effect of an Added Proton Source on Acetate Reactiviw 114

xii
Table 2.20: Scope of the Rhodium Catalysed Carboxvlate Ring Openinp Reaction 1 15
Table 2.2 1 : AR0 with Carboxylate Nucleophiles 115
IV 128
Ta 2 2 n j le 2:
130
Table 3.3: Scope of Rhodium Catalvsed Rine open in^ Of Vinyl Epoxides with Aromatic
Amines 131

1 General Introduction
Transition metal catalysed transformations of allylic hnctionalities have corne to the
forefiont of spthetic organic chemistry. The mildness and selectivity of these reactions
have allowed chemists easy access to structures, ofien in enantioemiched fom, which
were previously impossible or very dificult to obtain. Methods have been developed
which enable a wide range of allylic leaving groups to react with a variety of nudeophile
classes. Different, oAen complementary, selectivities have been obsemed for several
transition metals. Since the studies of this thesis include advances in the area of allylic
amination and etherification on oxabicyclic aikenes and vinyl epoxides, recent advances
in this fieId were reviewed.
1 Ailylic Alkylation, Amination andEtherification
1.1.1 Palladium Catalysis
1.1.1.1 General Concepts
Application of x allyl palladium chemistry to organic synthesis has made signi ficant
advancements and remains an area of intense research.' Catalytic versions based on the
pioneering work of ~ s u j i * and ~ r o s r ' have received the most focus. The basis of the
catalytic cycle, involves formation of r -allyl palladium complexes via oxidative addition
of allylic compounds to Pd(0) and subsequent reaction with various nucleophiles to
regenerate the catalytically active Pd@) species (Scheme 1.1):
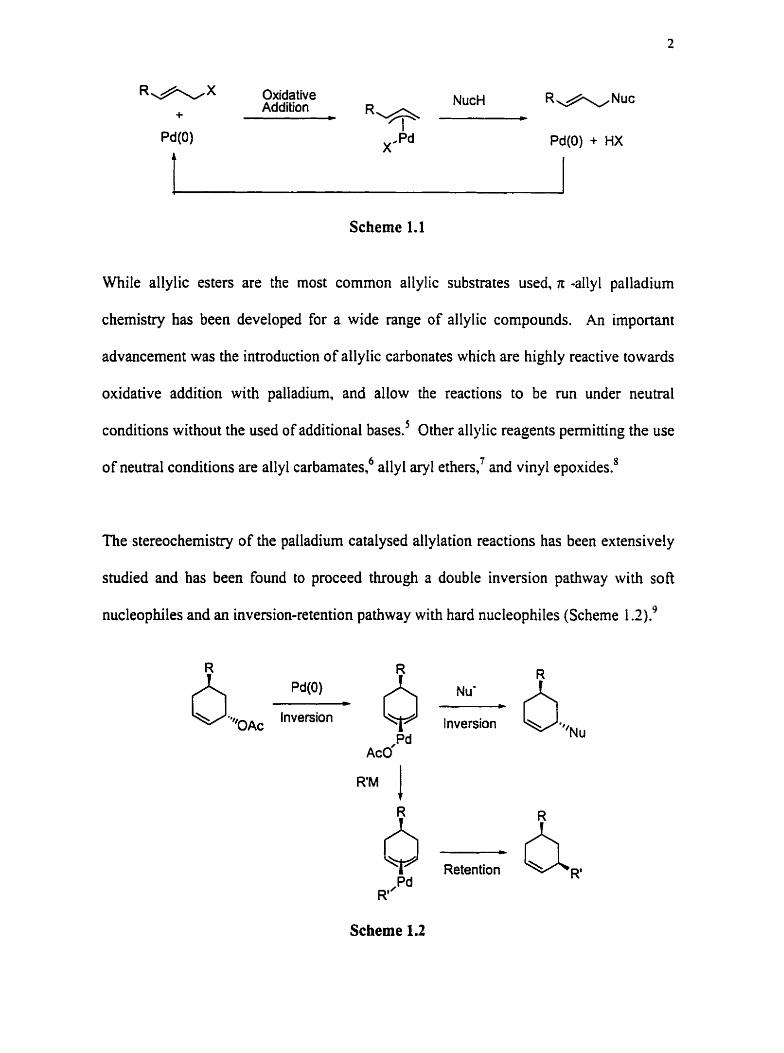
R&X Oxidative Addition NucH R A N u c
+ C m
Scheme 1.1
While allylic esters are the most common allylic substrates used, r allyl palladium
chemistry has been developed for a wide range of allylic compounds. An important
advancement was the introduction of allylic carbonates which are highly reactive towards
oxidative addition with palladium, and allow the reactions to be mn under neutral
conditions without the used of additional bases? Other allylic reagents permitting the use
of neutral conditions are allyl carbarnates: allyl aryl ethers,' and vinyl epoxides.'
The stereochemistry of the palladium catalysed allylation reactions has been extensively
studied and bas been found to proceed through a double inversion pathway with soft
nucleophiles and an inversion-retention pathway with hard nucleophiles (Scheme 1 3 . 9
Scheme f .2

Evidence for the oxidative addition step occming with inversion was obtained by
reactioo of chiral (S)-(0-3-acetoxy- 1 -phenyl- 1-butene 1 with Pd(O)(dppe) followed by
treatment with NaBF4. The resulting complex (IR, 2S, 39-2 is produced with 81%
stereoselectivity (Eq. 1.1). 'O
Proof for the double inversion pathway with sofl nucleophiles was found by reaction of
enantiomencally e ~ c h e d (S)-(E)-1 with Pd(0) and dimethylmalonate. The products, (9-
( 0 - 3 and (S)-(Q-4 (92:8), are produced in 30%ee (Eq. 1 .2).11
Pd(O).d~pe Me \ Ph Me / + NaCH(C02Meh -
OAc 96% Y- CH(C02Me)* + -fPh CH(C02Me)2 (1 -2)
Hard nucleophiles react with allyl substrates with overall inversion of stereochemistry.
Retention of configuration occurs at the final step arising from coordination of the
nucleophile to the Pd center. For exarnple, allylic acetate 1 reacts with phenylzinc
bromide to give 5 (Eq. 1 .3).12

The development of enantioselective transformations continues to be a major focus in
organic chemistry, and transition metal catalysed reactions are among the most powerful
tools in this regard. Asyrnrnetric catalysis involving palladium has been a fi-uitfil area of
research in recent years. Unlike most asymrnetnc transformations which distinguish
between the two faces of a r qstem, allylic akylations do not rely on a single
mechanism as a source of a~~rnme t ry . ' ~ While one possible source of induction can aise
from facial selectivity of the olefin, defining the exact source is complicated by the fact
that one or more of the other steps in the catalytic cycle may be the enantiodiscnminating
step(s). Despite such limitations, a "working model" for regio- and enantioselective
allylic alkylation of unsymmetrical substrates has been proposed.'4
Much of the advances made in this area have emerged fiom the research group of B.M.
Trost and rnethodologies involving the asymmetric allylation of P-ketoesters," ketone
enolates16 and azalactonesL7 have been recently reported.
Asymmetric 0- and C- allcylation of phenols has also been developed.I8 While standard
allylic carboxylates failed due to their propensity to undergo an acyl shift and form
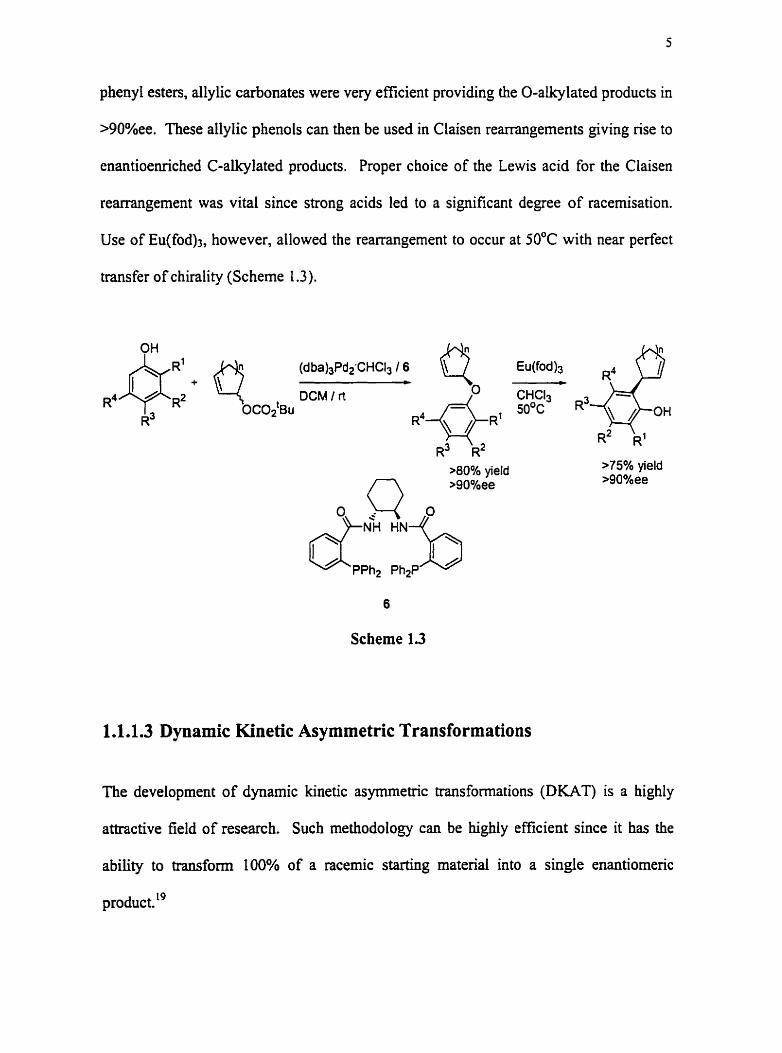
phenyl esters, allylic carbonates were very efficient providing the O-akylated products in
>90%ee. These allylic phenols can then be used in Claisen rearrangements giving rise to
enantioe~ched C-alkylated products. Proper choice of the Lewis acid for the Claisen
rearrangement was vital since strong acids led to a significant degree of racemisation.
Use of E~(fod)~, however, allowed the rearrangernent to occur at 50°C with near perfect
transfer of chirality (Scheme 1.3).
OH (dba)3Pd2-CHC13 / 6
R~ DCM / rt
>80% yield >75% yield >90°hee
6
Scbeme 1.3
1.1.1.3 Dynamic Kinetic Asymmetric Transformations
The development of dynamic kinetic asymmetric transformations ( D U T ) is a highly
attractive field of research. Such methodology can be highly escient since it has the
abiiity to transform 100% of a racemic starhg material into a single enantiomenc
product. ''

h 1998, Trost and CO-worken reported a two-component catalyst system for the
asymmetnc allylic aikylation of alcohol pronucleophiles providing an elegant method for
the preparation of enantioenriched vinyl glycidols via a deracemisation of vinyl
epoxides.'O In order to overcome the known poor reactivity of alcohois, the authors
revealed a remarkable ability of trialkylboranes to promote their nucleophilic addition. In
order for a D U T to be efficient, two key conditions must be met. Firstly, equilibration
of the intermediates must be fast compared to the rate of nucleophilic attack. Secondly,
one of the diastereomeric intermediates (in the presence of chiral ligands) must undergo
reaction faster than the other. Thus, decreasing the arnount of the borane to catalytic
levels, using triethylborane instead of the more reactive trimethylborane, and generating
the reactive diethylalkoxyborane in situ resulted in yields of ~80% and in ee's exceeding
90% (Scheme 1.4).
racemic
R'=H or
R3B (cat.) ROH l DCM l rt
>90%ee for a variety of 1' ROH
equilibration of pi-allyt-Pd interrned. must be fast compared to nucleophilic attack for high ee
Scherne 1.4
Subsequently, inorganic carbonates were found to undergo a similar type of reaction to
produce enantioenriched vinyl glycidols." The combination of these methodologies
should enhance the utility of these products as asymrnetric building blocks.
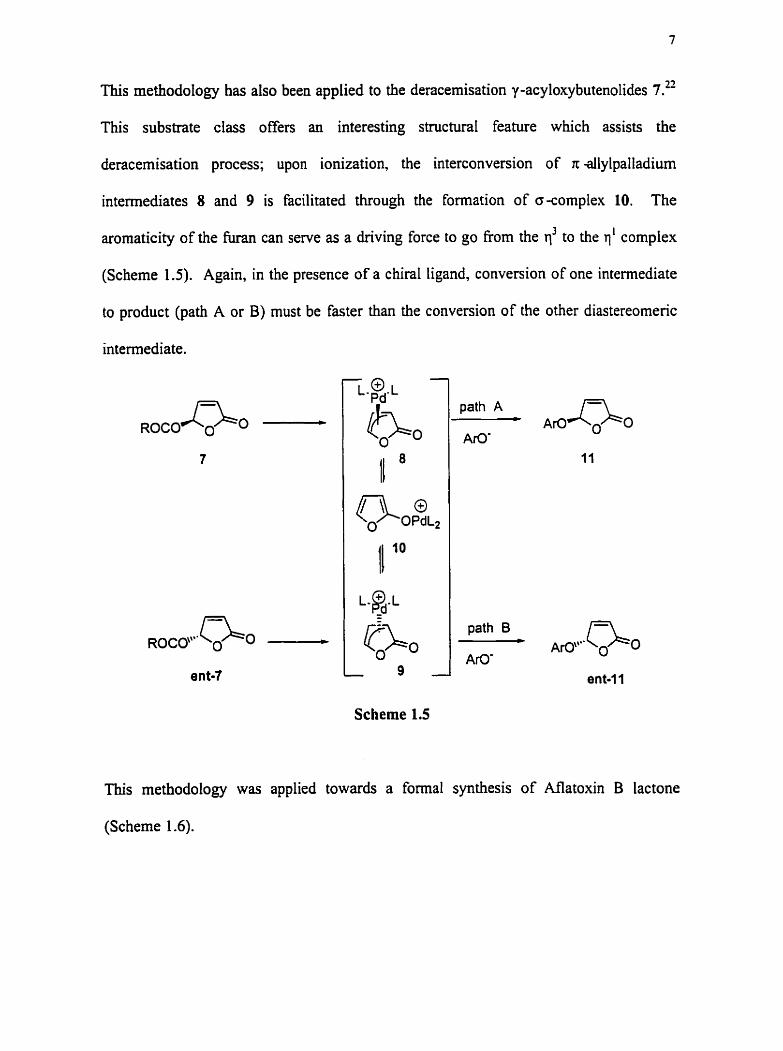
This methodology has also been applied to the deracemisation y -acyloxybutenolides 7.22
This substrate class offers an interesting structural feature which assists the
deracemisation process; upon ionkation, the interconversion of n allylpalladium
intermediates 8 and 9 is facilitated through the formation of o-complex 10. The
arornaticity of the hiran c m serve as a driving force to go from the r13 to the rl' complex
(Scheme 1.5). Again, in the presence of a chiral ligand, conversion of one intermediate
io product (path A or B) m u t be faster than the conversion of the other diastereomenc
intexmediate.
Scheme 1.5
path B - Am-
Thk methodology was applied towards a formal synthesis of Aflatoxin B lactone
(Scheme 1 -6).

"O% / (cibahp2CHCi \
Me0 OH + CsC03 1 DCM I rt MeO O . . ' O H
Afiatoxin 8
Scheme 1.6
Very recently, D U T methodology has been used to deracemise Baylis-Hillrnan adducts
(Scheme 1.7)."
CH302C0 (dbahPd2-CHC13 1 12 oAr R%EwG + ArOH
DCM l rt - R ~ E w G
EWG = C02Et = CN
Scheme 1.7
1.1.2 Rhodium Catalysis
The fint efficient rhodium catalysed allylation of carbonucIeophiles was reported by
Tsuji in 1984. Using 5 mol% RhH(PPh3)4 and 5 mol% PBu3, a vaiiety of allyl carbonates

were found to react with P-keto esters, malonates, cyanoacetates, Fdiketones, and silyl
en01 ethers in good yield. hportantly, high regioselectivity was observed for the
formation of the more highly substituted products (Eq. 1.4). This selectivity is
complementary to that typically observed with palladium.
+ dioxane 1 1 Oo°C
86% one isorner by GC
Durhg the coune of our studies in the area of rhodium catalysed AR0 reactions, P. A.
Evans reported a rhodium catalysed allylic alkylation reaction using an in siru modified
Wilkinson's catalyst which shows high levels of regioselectivity favouring the formation
of the more highly substituted product.'4 Furtherrnore, the reaction was shown to
proceed with overall retention. Treatment of R-13 under standard conditions gives R-14
with essentially complete retention of stereochemistry (Eq. 1.5). Subsequent mechanistic
studies led the authors to attribute this regioselectivity to the presence of an enyl
organorhodium intermediate (vide infra)."
Ri~ (PPh~)~c l (cat.) y(C02Me12
The mechanistic hypothesis is outlined in scheme 1.8. The key steps involve a back side
SN2' displacement of the carbonate leaving group giving nse to an enyl rhodium species

with inversion. A second SN2' displacement of the rhodium metal by the incorning
nucleophile again with inversion gives rise to an overall retention of stereochemistry.
The stereochemical and regiochemical outcome arises from the relative rates of
nucleophilic attack compared to the rates of isomerisation of the enyi rhodium
intermediates. Thus, with kz > kl and k3 > ki, an overall retention of absolute
configuration is observed.
k2 path A 1
~ h ' Ln NU -R - N u ~ R LG k3
path B
Scheme 1.8
The experirnental results that led to this mode1 are as follows. Firstly, it was found that
nucleophilic attack occurs predominantly at the position to which the leaving group was
attached regardless of the relative stenc bulk at either end of the allylic system. For
example, treatment of 15a under rhodium catalysis gave 16a, and 16b in 83% yield and
97:3 regioselectivity in favour of 1 6 a . ~ ~ When 15b was subjected to the sarne reaction
conditions, 16b was formed preferentially in a 97:3 ratio and in 87% overall yield. In
contrast, treatment of both isomeric carbonates 1Sa and 15b under analogous conditions
with a catalytic amount of Pd(PPh3)4 furnished 16a in >19: 1 regioselectivity regardless
of the regiochemistry of the starting material.

0C02Me Rh(PPh&CI (cat.) CH(C02Me)2 CH(C02Me)2
R (Me0)3P i 30°C Me Ahpr Me d i p r
NaCH(C02Me)2 16a 16b
15a: R = ~ e ; R ' = ' P ~ 97 3
Scheme 1.9
Deuteriurn labeling studies shed additional light on the nature of the mechanism. When
17 (on which both allylic positions are stencally equivalent) is reacted with the sodium
salt of dimethylmalonate and a catalytic arnount of phosphite rnodified WiUcinsonYs
catalyst, 18a is produced in N9:1 selectivity. This result indicates that the alkylation
proceeds through a o-complex in which the o-r -O i s o ~ t i o n is slow compared to
nucleophilic attack rather than the more commonly observed r cornplex (Eq. 1.6).
0C02Me Rh(PPh3)3CI (cat.) CH(C02Me)2 O CH(C02Me)*
Me *Me (MeQP i 3bC Me + uMe D D Me (1.6)
NaCH(C02Me)2 17 18a
>lg: l 18b
Evidence for the presence of an enyl intermediate was obtained fiom the reaction outlined
in Eq. 1.7. Since a pure cr-complex produced by an SN2' displacement by the rhodium
metal would have given rise to a large degree of racemisation through rapid C-C bond
rotation after oxidative addition, it was concluded that coordination of the rhodium meta1
to the olefin must be occwrhg.

QC02Me Rh(PPh3)3CI (cat.) @WozMe)z ,,A/
- (MeOhP 1 3o0C m M (1 -7)
97%ee NaCii(C02Me)2
95%ee
Furthermore, oxidative addition via a direct SN2 type process was deemed unlikely since
it was observed that increased alkene substitution in a series of primary allylic carbonates
lead to decreased reactivity (Scheme 1.9).
-OCO*M~ *OCO*M~ Ph LOC02Me
(RT, 4hr ) (heating, 7hr) (NR, 7hr)
Scheme 1.9
Evans has also extended this methodology to the preparation of allyl amines and allyl aryl
ethers. Again, use of the in situ rnodified Wilkinson's catalyst with unsyrnmetrical
acyclic enantiomerically enriched carbonates 19 with the lithium anion of N-tosyl
benzylamine affords the secondary allylamine 20 in high yield and retention of absolute
configuration (eq. 1.8).27 This methodology has been applied in the preparation of
0C02R Rh(PPh3)3CI (cat.) NTsBn
(MeOhP I BnNLqs * P r Prn pi-" (1 -8) 19 THF 1 30°C 20
>99%ee >99 1 >9g0hee

Similarly, use of sodium aryloxides under analogous conditions gave the secondary allyl
aryl ethers in high yields and regoiselectivities? The tolerance of the rhodium catalyst
for the presence of aryl halides makes this method of aryl ether bond formation
Rh(PPh3)3CI P (OW3
OAr
ArONa (1 -9) THF / 0°C to rt
94%ee ~ 8 0 % yield
11 to >99 : 1 regioselectivty
1.1.3 Other Transition Metal Catalysts
1.1.3.1 Iridium Catalysis
Iridium compounds have been shown to be effective catalysts in allylic alkylation
reactions. Takeuchi has show that [Ir(COD)C1I2 / P(OPh), is an efficient systern for the
regioselective allylic alkylation at the more highly substituted allylic carbon (Eq. 1.1 o).~'
[I r(COD)C1I2/ P(OP~I)~ CH(C02Me)2 / A/+
Pr /+-.
OCOIMe NaCH(C02Me)2 Pr CH(CQMe)2 (1.1 0) THF >85% yield
R = "Pr, R' = H 21 R = H, R' = " ~ r 22
96:4 from 21 955 from 22
The nature of the phosphite was shown to have profound effects on the regioselectivity of
this transformation. P(OPh)3 was found to be the most effective Ligand, giving complete
conversion to the desired product in three hours at mom temperature with a 96:4 dr in

favour of the more highly substituted product. As the ligand becomes more electron
donating, the regioselectivty drops as does the yield. The authors explain the
regioselectivity and reactivity by proposing thatn accepting ligands promote carbonium
character at the more substituted allylic terminus of thex -allyliridium intermediate and
thus direct the nucleophilic aaack to this position.
1.1.3.2 Molybdenum Catalysis
Molybdenum(0) will also catalyse allylic alkylation reactions. Complernentary to
palladium catalysis, the introduction of rnalonate anions and some Pketo esters occur at
the more hindered terminus of the allyl unit (Scheme 1. !O)." These reactionç were
s h o k to proceed with net retention of configuration.
OAc M e 0 2 C ~ C o 2 M e CH(C02Me)2
&w R R' 4 Rn R
Mo(O) R'
Scheme 1.10
Kocovsky later offered evidence that an unprecedented double retention pathway was
O C C ~ ~ . ' ~ Oxidative addition with retention has been unambiguously dernonstrated in
the stoichiometric reaction of Mo(CO)~(CH~CN)~ with both a ~ ~ c l i c ' ~ and cyclic3" allylic
substrates.

The complementary regioselectivity observed with Mo catalysts has made the
development of an asyrnmeûic version desirable. The realization of this goal has been
elusive, due in part to the increased number of binding modes available to ligands on the
octahedral molybdenum metal. Trost and Hachiya have recently reported a catalyst
system which shows significant promise.35 By using 15 mol% ligand 27 and 10 mol%
(EtCN)3Mo(CO)3, a mixture of two regioisorners results with the major product being
that arising fiom attack at the more sterically hindered allylic position in a >30: 1 ratio.
Excellent enantioselectivities are observed, typically >95%ee (Scheme 1.1 1).
(EtCN)3M~(C0)3 / 27 b Ar&CR(C02Meh
NaCR(C02Me)* THF CR(C02Me)2
R = H, Me, AIIyi 25 26
typically >30:1
(7 up to 993
typically >95%ee for 25
& ~ i HN$
/ N N /
27
Scheme 1.11
Since both 23 and 24 are suitable substrates for this reaction, it is likely that the
enantiodifferentiation o c c m from selective nucleophilic attack on one of the
diastereomenc n dlylmolybdenum complexes whic h exist in dynamic equilibrium.

This catalyst system has also been applied to the enantioselective alkylation of polyenyl
esters which are known to be problematic with palladium ca ta~~s i s . '~ Here too,
nucleophilic attack occurs selectively at the more highly substituted allylic position
without isornerisation of the other conjugated units of unsaturation. For example, when
28 is treated with 15mol% ligand 27 and lOmol% (EtCN)3Mo(CO)3, two regioisomers 29
and 30 are produced in a 10:l ratio. High enantioselectivities are obtained, typically
>95%ee (Eq. 1.1 1).
- -
28 THF
98%ee for 29
1.1.3.3 Nickel Catalysis
Nickel catalysed substitution reactions of allyIic compounds with soft nucleophiles have
aîtracted less attention than other rnetals. This is likely due to the fact that reaction of
soft nucleophiles under Ni(0) catalysis typically requires harsher conditions and gives
lower yields than do Pd(0) systems. Recently, however, mild conditions for allylic
alkylation, amination and etherification have been reported with very low catalyst
loadings for a narrow range of substrates and nucleophiles (Eq. 1.12):' A drawback of

this methodology is the necessity of using very air sensitive Ni(COD)* as the Ni(0)
source.
p./OA= Ni(dppb)2 (O.Smol%)
t
+ Base -Nu (1.12) NuH rt to 80°C >90% yield
1.1 -3.4 Ruthenium Catalysis
The vast majority ofx dlyl transition metal complexes react as electrophiles. A few
cases exist, however, where these complexes behave as nucleophiles. Ruthenium n -allyl
complexes are one of the rare exarnples where arnbiphilic character is observed. Work
by Watanabe and CO-worken has led to the development of catalytic allylation of both
aldehydes and amines (Scheme 1.1 2).38
- * Et3N I CO
OAc piperidine THF / O°C
v n
Scheme 1.12

In catalytic and stoichiomeûic allylation of aldehydes, the addition of amines is essential
for catalytic activity and high yields. It is believed that the amine acts as a ligand for the
active Ru intermediate as well as serves as a hydrogen source via hydnde extraction
through the intermediacy of a metallaazacyclopropane or an imminiurn ion.
1.1.3.5 Iron Catalysis
B U ~ ~ ~ ( C O ) ~ N O ] has been found to catalyse the alkylation of allylic carbonates with
malonate anions?g The reaction proceeds with good regioselec tivity, wi th the
nucleophilic attack occurring predominantly at the carbon where the leaving group was
attached (Eq. 1.13). Retention of configuration of the stereocentre undergoing
nucleophilic attack was established, and retention of configuration of the double bond
during the course of the reaction was observed. On the basis of the regio- and
stereochemical results, the authors suggest the involvement of a o-iron complex.
1.1 A 6 Tungsten Catalysis
1:99 from 31 93:f from 32
As with molybdenum, the regioselectivity observed with tungsten is also complementq
to that observed with palladium. For this reason, the development of asymmetric reaction
based on these metals is an attractive pursuit. Pfaltz has reported an asymmetric

tungsten-catalysed allylic w l a t i o n of malonates with terminal allylic phosphonates
giving high ee's for a narrow range of substrates (Scheme 1.~3).~*
O Il
(EtOkP,o W (O) catalyst (10molX) CH(C02Me)2 C
X = MeCN
Scheme 1.13
Similar to the Mo(0) systems, W(0) favours nucleophilic attack at the more high
substituted allylic position in moderate to good ratios.
1.1.3.7 Cobalt Catalysis
A cobalt catalysed allylic alkylation reaction has also been reported.'" The
regioselectivity was found to depend on the nature of the 1,3-dicarbonyl nucleophiles.
The highest selectivities were observed for acetylacetone which the authors propose can
be delivered by the cobalt intmnolecularly.
1.2 Asymmetric Ring Opening of Oxabicyclic Aikenes
1.2.1 Use of Oxabicyclic Alkenes in Organic Synthesis
Oxabicyclo [3.2.1] and [2.2.1] alkenes are useful intermediates in synthesis and have
$7
been the focus of a comprehensive review.'- Much of their utility cornes kom the iarge
number of stereochemically defined centres which can be accessed through a variety of
ring opening reactions.
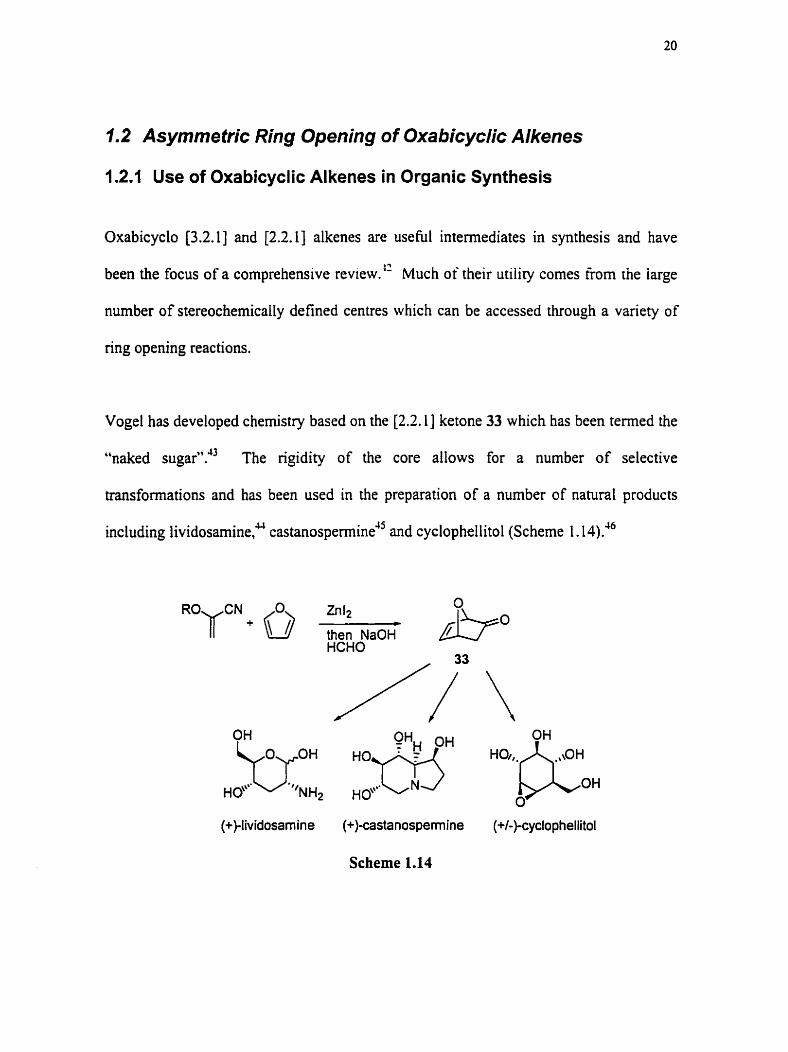
Vogel has developed chemistry based on the [2.2.1] ketone 33 which has been termed the
"naked sugar"." The rigidity of the core allows for a number of selective
transformations and has been used in the preparation of a nurnber of natural products
including lividosarnine? castan~s~ermine"~ and cyclophellitol (Scheme 1.14)?
"OfN + 0 Zn l2 C
then NaOH HCHO
(+)-Iividosam ine (+)-castanospemine (+/-)-cyclo phellitol
Scheme 1.14

The [2.2.1] oxabicyclic akene, 34, readily available fiom a cycloaddition between furan
and vinylene carbonate, has been employed in the preparation of a number of cyclitols.
For example, au-selective dihydroxylation, deprotection of the carbonate,
polyacetylation followed by acidic ring opening gives neo-inositol (Scheme 1-15)."'
1 ) O S O ~ ACOH QH
2) NaOH H20 .,\OH
H2S04 HO '"/OH
OAc OH
Scheme 1.15
Derivatives of D- and L-ribose c m also be obtained fiom 34.* Thus, dihydroxylation,
acetonide formation, cleavage of the carbonate and oxidative cleavage of the resulting
di01 to the acid gives 35. The anhydride is then formed and treated with TMSN3 and an
alcohol which gives ribose derivative 36. The enantiomers of 36 can be separated by
treatment of the anhydride with isopropanol and resolution of the monoacid with brucine.
1) 0 s 0 4
2) Acetone I H* 3) i3a(OHh 'ho 4) KMnOl
34 35
resolve 1
Scheme 1.16

The [3.2.1] oxabicyclo core has also been utilised in synthesis. For example, Baeyer-
Villiger oxidation of 37 was a key step in the preparation of the C-nucleoside
showdomycin.Jg Andagously, a Beckmann rearrangement of 38 leads to a synthesis of a
muscarin analogue (Scheme 1.1 7).50
1 ) O S O ~ CF3C03H 0-0
CuS04 acetone
S howdowmycin
Muscarin analogue
Scheme 1.17
Olefin metathesis is also emerging as a powerful tool in the synthesis of cornplex organic
molecules. While 7-oxanorbomenes are known to undergo ring opening metathesis to
generate a variety of functional polymers, the intermolecular ring opening metathesis of
these compounds had not been investigated until recently. In 1999, Agona and Plumet
reported the regioselective ring opening and cross coupling metathesis reaction of 7-
oxanorbornenes as a route towards trisubstituted tetrahydrofurans.sl Treatment of 39
with Grubbs catalyst in the presrnce of allyl acetate gave the desired ring opened
products as a mixture of the E/Z isomers. Hydrogenation with palladium over carboo
gave the desired teûahydrofurans 40 and 41 in 70 to 80% combined yield and in up to a

4:l dr depending on the stenc bulk of the X, Y functionalities on the oxabicyclic alkene
(Scheme 1.18).
Ha 1 PdIC 1 - -.- CIL
H2 1 PdlC I
Scheme 1.18

1.2.2 Nucleophilic Ring Opening of Oxabicyclic Alkenes
1.2.2.1 Concepts and Strategies
The most comrnonly employed methods for cleavage of the oxabicyclic core are typically
acid or base induced. While these methods reveal the latent stereochemistry existing in
the oxabicyclic skeleton, no new stereocentres are generated. Nucleophile induced ring
openings, however, can create a new stereocentre upon ring opening.
Our group and others have been interested in the developrnent of nucleophilic methods
for cleaving oxabicyclic alkenes, and highly efficient and enantioselective reactions have
been discovered (vide infra). The products of these ring opening reactions are useful
intermediates in organic synthesis. For example, ring opening of oxabenzonorbomadiene
produces 1,2-disubstituted-3,4-dihydronaphthalene compounds. Nuc leophilic ring
opening of [2.2.1] and [3.2.l] oxabicyclic aikenes generates highly substituted
cyclohexenols and cycloheptenols. Oxidatative cleavage of these products produces
stereochemically complex linear arrays which can be elaborated into polyproprionate and
polyacetate anays (Scheme 1.19).

Scheme 1.19
Given the ability of a nucleophilic ring opening reaction to create several differentiated
stereocentres in one step, the development of asymmetric nucleophilic ring opening
reactions would be a desirable goal.
1.2.2.2 Reductive AR0 Methodology
Mile a reductive ring opening reaction will create no new stereocentre during the ring
opening step, the product cyclohexenols and cycloheptenols are usehl synthetic building
blocks. One route to the hydndic ring opening is through a transition metal catalysed
asymmetric hydrometallation (Scheme 1.20).

H-M*
Scheme 1.20
Successfbl realization of this approach was reported in 1995 by Lautens, Chiu, Ma and
~ovis.'' They used a nickel catalysed hydroalurnination reaction to induce ring opening
in excellent ee (typically >95"/oee) (Eq. 1.14). Evidence for a two-step process involving
nickel-catalysed hydrometallation followed by transmetallation and ring opening of the
organoalane was provided when the reactions were run at room temperature.
During studies aimed at improving the efficiency of these reactions with the less reactive
[3.2.1] oxabicyclic aikenes, an intriguing temperature effect was 0bserved.5~ At higher
temperatures, less over-reduction was observed and the ee increased drarnatically. The
authon explain these observations by proposing that at higher temperatures the
hydronickelation step becomes reversible and that the ring opening step occurs kom the
organonickel species rather than fkom the organoalane.

This methodology was used in the total synthesis of Sertraline, a commercial
antidepressent (Scheme 1.2 l)."
Ni(COD)2 SINAP
DIBAL-H THF
- OH 9 steps
38% overall 87% yield yield 98% ee
A NHMe
Scheme 1.21
1.2.2.3 Alkylative AR0 Methodology
Prior to the initiation of this work, methodology involving ~ r ~ a n o l i t h i u r n , ~ ~ and
organocuprateS6 nucleophiles had been developed. ~ e ~ i o s e l e c t i v i t ~ ~ ~ and
enantioselecti~i$~ issues had been investigated and an intramolecular versions9 had been
snidied. A highly enantioselective reaction had not yet been forthcoming, however.
During the course of these studies. research in our group in the area of carbometallation
reactions was undertaken. The mechanism of these reactions parallel that of the reductive
AR0 reaction, so the development of an asyrnrnetric version was again feasible (Scheme
1.22).

R-M*
1
Scheme 1.22
In 2000, Lautens, Renaud and Hiebert reported a palladium catalysed enantioselective
alkylative ring opening reactiun of oxabenzonorbomadiene, [2.2.1] and [3.2.1]
oxabicyclic aikenes giving the ring opened products in good yields and excellent ee's
(Scheme 1.23):' An important aspect of this chemistry was the choice of a nucleophile
that was unreactive in the absence of catalyst. Dialylkzincs were chosen since they were
known to react slowly with even the most activated system 42 to give a mixture of
products. The optimal catalysts were found to be Pd(l1) diphosphine complexes.
Intriguingly, tol-BMAP was found to be the optimal ligand for ethyl addition white
'PrPOX gave the best results when methyl was added (Scherne 1.23).
Pd(CH3CN)&I2 (5rnol%) l L'
R2fn / CH2CI2 I rt - R J@q"J /
42 OH R = Me; L' = 'PrPOX; 90%ee R = Et; L' = toi-BINAP; 96%ee
Scheme 1.23

Ring opening of [2.2.1] and [3.2.1] oxabicyclic alkenes required more forcing conditions.
Thus, by heating the reaction in DCE and using DIPOF as the ligand, the cyclohexenol
and cycloheptenol products were obtained in good yields and ee's >90% (Scheme 1.24).
OPMB
Pd(CH3CN)$& (5~01%) 1 L* mOPMB Me2Zn I. DCE 1 heat
MezZn 1 DCE 1 heat
84%, 95%ee
DIPOF
Scheme 1.24
This allcylative and reductive AR0 methodology has found application in an approach
towards the total synthesis of the calcium ionophore ionomycin which is the focus of
ongoing research in our group.6' Three of the four components can be accessed fkom the
oxabicyclo [3.2.1] octene core 43 by application of these newly developed AR0
methodologies (Scheme 1.25).

lonom ycin
Scheme 1.25
1.3 Goals and Targets
1.3.1 The Oevelopment of an AR0 with Heteroatom Nucleophiles
1.3.1.1 Synthetic Precedent
In 1973, Hogeveen and Middelkoop reported the [RIi(CO)2C1]2 catalysed ring opening
reaction of 44a and 44b with methano1 giving methoxycyclohexandienol products 45a
and 45b r e ~ ~ e c t i v e l ~ ~ ~ (scheme 1.26). Ashworth and Berchtold laîer showed that die
stereochemistry of the incorporated methoxy substituent was cis to the resulting hydroxyl
group by formation of the Diels-Alder adduct 46.63 In chloroform as the solvent, and in

the absence of methanol, the oxabicycle reananges to give the hydroxyfuIvene product
47?
[ W C 0 )2Cll2 O C02Me
* &CO2Me - MeOH
Me0 OH
I i = f i ' = M e 442 R=R '=H 44b R=R'=Me 45a R = R ' = H 45b
46
47
R' Scherne 1.26
While no definitive mechanism was elucidated for the methanol ring opening reaction,
the authors proposed either endo coordination and rearrangement, or rhodium
coordination to the bridgehead oxygen acting as a Lewis acid, thus activating it as a
leaving group. A tentative mechanism was proposed for the rearrangement to the
hydroxyfulvene product (scheme 1.27). The first step involves a rhodium(1) catalysed
rearrangement to the dipolar intermediate 48. The existence of this intemediate is
supported by the observation that when both bndgehead positions are substituted with
methyl groups, the reaction rate increases. They propose that this rate enhancement is
due to a stabilization of the carbocation.

1.3.1.2 Adaptation of the Hogeveen-Middelkoop Conditions to Oxabenzonorbornadiene and Development of an Asymmetric Version
Of the myriad of molecular architectures present in pharmacological agents, certain
structures emerge with a higher frequency than othen. Among these privileged
structures is the hydronaphthalene skeleton which can be found in a wide range of
compounds possessing diverse biological activities. Exarnples include se r t ra~ ine~~ 49, an
antidepressant, Dupont analesic 50, compound 5 1 ~ ~ which shows dopamine agonist
properties, homochelidonine 52, a naturally occumng alkaioid, dihydrexidineb7 53, which
shows antiparkinsonian character, and etoposide" 54, which is used in the treatment of
various cancers. In addition, C N S immunoregulatory agents,70 antibiotics7' and
antitumor agents7* contain variations of this framework. Given the prevalence of the
hydronaphthalene skelelton and the diversity of bioactivity of the compounds in which it
can be found, we sought to develop a new method which would permit access to this core
structure in enantioenriched and highly hinctionaiised form.

Dupont analgesic CI
qg NHM~.HCI Sartraline \
(anti-depressant)
Etoposide (anti -cancer)
54
(anti-parkinson's)
L0 5, dopamine agonist
OMe O M ~
Homochelidonine
Scherne 1.28
in spite of the recent advances in the field of AR0 chemistry (vide supra), no AR0
reactions on oxabicyclic alkenes have been reported involving heteroatom nucleophiles.
Given the prevalence of hydronaphthalene compounds and their diverse biological
activities, we sought to extend the scope of the AR0 methodology such that heteroatomic
nucleophiles would be incorporated in to the molecule during the ring opening step
(Scheme 1 .D).

RXH / M'
Scheme 1.29
Prier to the initiation of these studies, preliminary work perfomed by Rovis reavealed
that no reaction occurred when 42 was subjected to the Hogeveen and Middelkoop
conditions. The formation or a prccipitate which did not dissolve even with heating was
noted and it was reasoned that by using a more highly polarking solvent this problem
could be avoided. Changing the solvent system to a 1: 1 mixture of trifluoroethanol
(TFE):methanol and by increasing the reaction temperature to 60°C gave the desired
product 55 in 70% yield (Scheme 1.30). The relative stereochemistry of the vicinal
dioxygen functionality was assumed to be cis based on the assignment of Ashworth and
Berchtold for 45a, but no studies had yet been performed to prove this cis relationship. It
was at this point that my work began.
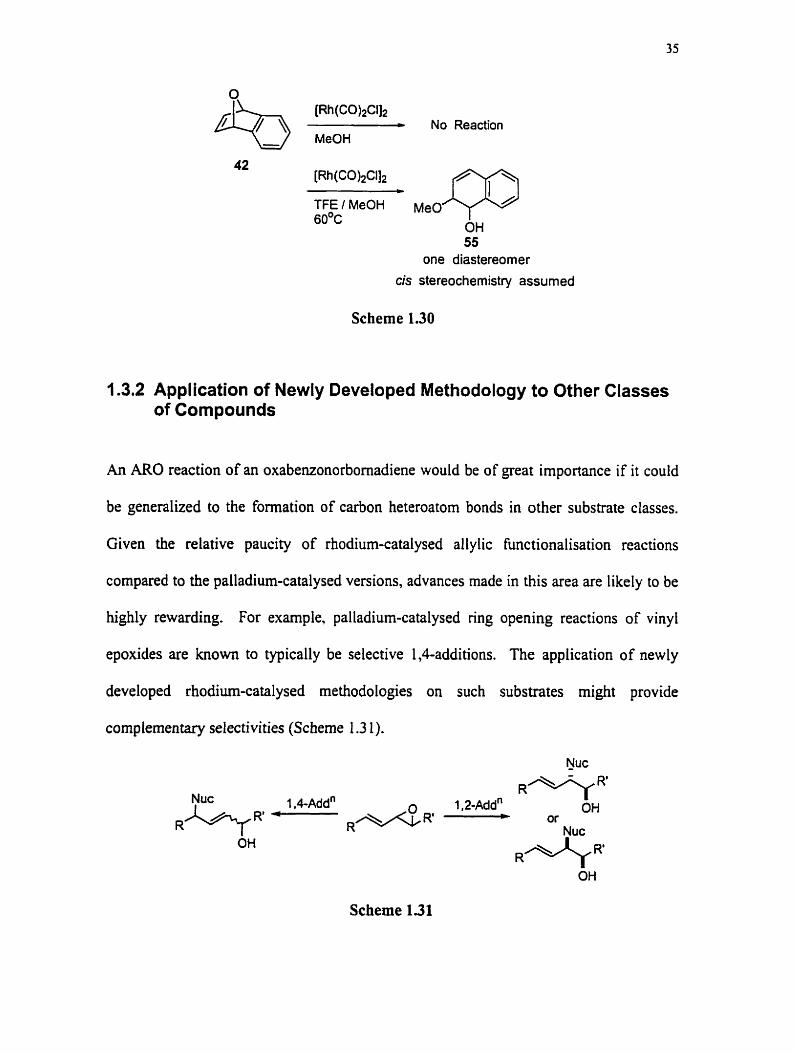
55 one diastereomer
cis stereochemistry assurned
Scheme 1.30
1.3.2 Application of Newly Developed Methodology to Other Classes of Compounds
An AR0 reaction of an oxabenzonorbomadiene would be of great importance if it could
be generalized to the formation of carbon heteroatom bonds in other substrate classes.
Given the relative paucity of rhodium-catalysed allylic fùnctionalisation reactions
compared to the palladium-catalysed versions, advances made in this area are likely to be
highly rewarding. For example, palladium-catalysed ring opening reactions of vinyl
epoxides are known to typically be selective 1,4-additions. The application of newly
developed rhodium-catalysed methodologies on such substrates might provide
cornplementary selectivities (Scheme 1.3 1).
Scheme 1.31

2 Rhodium Catalysed Asymmetric Ring Opening of Oxabicyclic Alkenes with Heteroatom Nucleophiles
2.1 Alcohol Nucleophiles
2.1.1 Initial Investigations
2.1.1.1 Establishment of Relative Stereochernistry and Scope of Alcohol Nucleophiles
Initial studies focused on determining the relative stereochemistry of the vicinal dioxygen
functionality. Application of the conditions established by Rovis for the rnethanolic ring
opening of 42 gave 55 in 70% isoiated yield. To our surprise, however, the
stereochemistry of 55 was proven to be tram by cornparison to authentic sarnples of both
stereoisorners of dimethoxytetrahydronaphthalene 56.7' The authentic cir isomer of 56
was prepared by reaction of 1,2-dihydronaphthalene with 0 s 0 4 followed by
dimethylation with dimethylsulfate. Trans-56 was prepared by epoxidation of 1,2-
dihydronaphthalene followed by ring opening with sodium hydroxide and dimethylation
with DMS.
Scheme 2.1

Given that this stereochemical result is opposite to that obtained on the Hogeveen-
Middelkoop substrate we verified the cis stereochemistry of 45a by X-ray
crystallography.
These reaction conditions were found to be generally applicable for a variety of alcohol
nucleophiles. EtOH, 'P~OH, and 2-TMS ethanol c m al1 be added in acceptable yield
(Table 2.1). In contrast, 2-chloroethanol did not induce any ring opening. We reason
that this lack of reactivity is due to Cl-0 bidentate binding of the 2-chloroethanol on the
rhodium thus poisoning the catalyst.
Table 2.1: Rhodium Catalysed Ring Opening with Various Alcohols
Entry Alcohol Product Yield
1 MeOH 55 70%
2 EtOH 57 61 %
3 'PrOH 58 63%
4 TMSCH2CH20H 59 52%
5 CICH2CH20H - NR
2.1.1.2 Substitution Effects
An investigation of the effects of different substitution patterns on the oxabicyclic aikene
on the outcome of the reaction would provide information on the nature of reactive
interinediates as well as allowing for the preparation of synthetically useful

hydronaphthalene products. If the ring opening step is ionic in nature, cleavage of the
carbon-oxygen bond should occur preferentially at the bridgehead position most able to
stabilize the developing positive charge and thus give rise to a regioselective ring
opening. For example, when one of the bridgehead positions is substituted with a methyl
group, C-O cleavage would hypothetically be favoured at the more highly substituted
carbon (Scheme 2.2).
Favoured Disfavoured Scheme 2.2
Indeed, when 60 is reacted with MeOH in TFE and a catalytic arnount of [R~I(CO)~C~]~,
oniy one regioisomer is produced 61 where C-O bond cleavage and ring opening has
occurred at the more highly substituted bndgehead carbon, as one would predict if the
reaction were arising fiom an ionic pathway (Eq. 2.1)
61 66% (one isomer)
In order to minimize any stenc effects that can corne into play with bridgehead
substitution, more remote substitution patterns were sought. One method to
preferentially stabilize positive charge at the bndgehead carbons is to add electron
donating groups to the aromatic ring. Thus if the substitution is not symmetric, one
bridgehead carbon should preferentially stabilize positive charge more than the other.

For example, in 62, positive charge at one bndgehead carbon is stabilized by resonance,
whereas the other is not (Scheme 2.3).
Resonance Stabilisation
Favoured Scheme 2.3
Disfavoured
When 62 was reacted with MeOH under rhodium catalysis, only one regioisomer was
produced indicating that the remote substitution does indeed effect the ring opening step
(Eq. 2.2). The regiochemistry of 63 was supported by NOE studies, but additional data is
required for this assignment to be conclusive.
OMe
63 68% (one isomer)
These two resuits indicate that, at least for the [Rh(C0)2C1I2 catalyst, the reaction rnay
proceed through an ionic intermediate, and that it is the formation of the more highly
stabilized ionic intermediate that dictates the regiochemical outcome of the reaction.

2.1.1.3 Intramolecular Variant
With conditions established for the intermolecular addition of various alcohols to 42, we
conducted preliminary studies in the development of an intramolecular variant. Such a
reaction could produce t icyc l ic structures with clearly defined stereochemis~. Lessons
learned from our studies on the effect of unsymmeüic substitution patterns, however,
indicated that problems might be encountered due to preferential C-O bond scission at the
bridgehead carbon possessing the tether (Scheme 2.4). This pathway would result in
spiro compounds or, more likely, aromatisation to generate undesired naphthol products.
One potential solution to this problem is to add a second bridgehead substituent so as to
make C-O bond cleavage possible at either position. An altemate strategy would be to
choose a tether which would disfavour the development of positive charge at that position
and thus result in the desired products. Suice this option would likely decrease the
overall reactivity of the system, we opted to conduct initial studies in the effects of
having a second bridgehead substituent.
C-O bond cleavage Would an appropriate ~referred at this position substituent favourise R
cleavage at this position ???
Scheme 2.4

The starting materiais were prepared as follows. Furan (or methylfuran) was treated with
BuLi then with ethylene oxide to produce the corresponding alcohol which was protected
as its silyl ether 66 (or 67) under standard conditions. Cycloaddition with benzyne
generated £tom bromochlorobenzene and BuLi proceeded in acceptable yields and
subsequent deprotection of the TBS eihers gave the oxabicyclic akene alcohols 70 and
71 (Scheme 2.5).
' b j 0 f i yJJoTBS BuLi / BCIC6H4 Ra 2) TBSCl I then 66 or 67
imid / DMAP R=H,64 R = Me, 65
R = H, 66 (61%) R = Me, 67 (66%)
/ TBSO / R = H, 68 (42%)
TBAF/ R = Me, 69 (48%)
0 R /
R=H,?0(97%) f i R = Me, 71 (99%)
Scheme 2.5
Ring opening reactions with 70 and 71 supported our predictions. Indeed, when no
second bridgehead substituent was present, only naphthol products were observed (Table
2.2, Entries 1 and 2). When a methyl group was present small amounts of the desired
tncyclic compound was obtained (Entry 3). The intramolecular reaction was
significantly more rapid than the intermolecular versions, so we investigated whether
lowering the temperature would improve the yield. Unlike the intermolecluar reactions,
heating is not required when the nucleophilic attack is intramolecular. At room
temperature the yield improved to 41%, and cooling to 0°C resulted in a 51% yield

(Entries 4 and 5). The stereochemistry of the tricyciic product was assumed to be tram
based on the results of the intennoIecular reaction.
Table 2.2: Effect of Bridgehead Subtituents and Temperature on the Intramolecular Ring Opening of Oxabicyclic Alkenes
a NMR yield, remainer is naphthol byproduds
It is clear that more work is required as many questions remain unanswered. For
example, ody one substituent was studied. It seems Iikely that the choice of a group
which will be better able to stablise any cationic character present along the reaction
pathway will resdt in increase yields. In addition, only one tether length was used, and
others should be analysed.

2.1.2 Development of an Asymmetric Variant
2.1.2.1 Effect of Added Phosphines
Before an asymmetric variant of this ring opening reaction could be developed, the effect
of added ligands needed to be studied. Since the most cornmonly used ligands for
asymmehic catalysis are phosphine based, we focused our attention on this class of
ligand.
Initial experiments using [ R ~ I ( C O ) ~ C ~ ] ~ as the rhodium source with a variety of
phosphines were not encouraging. In each case, an insoluble red precipitate resulted
upon mixing of the rhodium and the ligand, and no ring opened product was observed
(Table 2.3). This precipitate could not be dissolved by funher heating or proionged
stimng.
Table 2.3: Effect of Phosphines on Reaetivity with [Rh(C0)2Cl12
Entry Ligand Yield
- -
a Insoluble precipiate resulted on mixing of Rh with phosphine

In order to overcome the problem of precipitation of the rhodium, we himed to
[R~I(COD)C~]~ as the rhodium source. Initial experiments focused on phosphorous
Iigands which possess a greater degree of n acid character so as to mimic the carbonyls
of the [Rh(C0)2C1]2 catalyst. For this reason, phosphites were chosen as potential
candidates. Indeed, some reactivity was observed with [Rh(COD)CII2 and phosphites
(Table 2.4, Entries 1-3). We also noted that homogeneous solutions were produced on
mixing of the rhodium and the ligands. To determine if phosphine ligands were
compatible, PPh, was also used and gave sirnilar levels of reactivity (Entry 4). This last
result was significant because it indicated that phosphine ligands did not poison the
catalyst. Since many chiral ligands contain arylphosphine donor atoms, the development
of an asymmetric version appeared feasible.
Table 2.4: Effect of Phosphorous Donor Ligands on [Rh(COD)C1I2
Entry Ligand Yield
a NMR yield. Only product was naphthol. lsolated yield.
We next turned to bidentate ligands to determine the effect of bite angle on the reactivity
of the system. Indeed, not ail ligands showed the same type or level of reactivity. For

example, dppe did not produce any desired product; only the dimerisation of the
oxabicycle was observed (Table 2.5, Entry 1). Dppb, which possesses a largrr bite angle,
showed increased reactivity compared to PPh3 (Entry 2). Increasing the bite angle further
through the use of dppf gave the best results, giving 88% yield at 60°C (Entry 3). The
reactivity dropped when the reaction was run at room temperature.
Table 2.5: Effect of Bidentate Ligands on Reactivity with [Rh(COD)CI]?
Entry Ligand Temp.('C) Yleld
- -- .
a Only dimensation of the oxabicycle was observed. Rernainder is starüng material.
One advantage of dppf being a good ligand for this reaction is that a number of chiral
analogues are known.
2.1.2.2 Establishment of AR0 Conditions
Of the chiral ligands studied, PPF-P'Bu~ 72 gave the best results, producing 55 in 84%
yield and 86%ee at 60°C. We found that the ee could be significantly improved to 97%

when the temperature was increased to reflux. Lautens, Chiu and Rovis made similar
observations of ee vs. temperature in their enantioselective hydroalumination study."
These reactions were typically mn as a 1: 1 mixture of Me0H:TFE under a nitrogen
atrnosphere which gave 55 accompanied by srna11 amounts of naphthol. In neat
trifluoroethanol under nitrogen atmosphere, naphthol is the major product with less than
5% conversion to the trifluoroethanol ring opened product. Given the low nucleophilicity
of TFE, this result is not surprising. Interestingly, however, this is not the case when a
carbon monoxide atmosphere is used in place of nitrogen. Under a CO atmosphere in
neat FE, the reaction proceeds to cornpletion in 30 minutes giving a 70% yield of the
trifluoroethanol ring opened product 73 (Table 2.6). Although we have not investigatd the
effect of CO in detail, we note a colour change of the solution, from yellow to red
indicating that the CO was interacting with the rhodium metal. When the reaction was
performed under asymrnetric conditions using PPF-PtBu2, 73 was obtained in 70% yield
and 98%ee indicating that occupation of an additional coordination site by CO does not
affect the degree of asyrnmetnc induction.

Table 2.6: Atmosphere and Solvent Effects
OPPF (1 eq. to Rh) Solvent 1 TFE 80% (or reflux)
-- -
N2 TFE (neat) 4% after 15 hr
CO TFE (neat) 70% after 30 min
N2 THF (5 eq TFE) 85%
It has been well doc~rnented'~ that rnany diphosphine ligands, when reacted with
[Rh(C0)2C1]2, show a pronounced preference for bridging hvo Rh atoms to yield dimeric
species. This could account for the insoluble precipitates that were observed in our initial
catalyst studies. If the diphosphines are combined with [Rh(COD)ClIt, the cis-complex
would result. The resulting monomeric carbonyl rhodium diphosphine species could
account for the observed increase in reactivity.
M e r our initial investigations using TFE as the solvent, we examined other solvents to
determine their suitability for this reaction. THF was found to work equally well in the
case of ring opening with methanol. While in neat TFE, a CO atmosphere was required
to achieve TFE incorporation, this was found to not be the case when THF is the solvent.
In W, only five equivalents of TFE are required to give 73 in good yield and excellent

ee under either nitrogen and CO atmospheres (Table 2.6). We opted to use the more
operationally simple nitrogen atmosphere for al1 subsequent studies.
An investigation on the effect of Iowenng the catalyst loading revealed that the reaction
proceeds eficiently with only 0.25 mol% rhodium-phosphine complex with excellent
enantioselectivity (Table 2.7).
Table 2.7: Soivent and Temperature Effects
PPF-P'BU~ (1 eq. to Rh) MeOH I Solvent (1 :1)
Entry ~ 0 1 % Cat.' Temp('C) Solvent Yield(o/æe)
3 1% reflux THF 96%(97%ee)
4 0.5% reflux THF 95%(97%ee)
5 0.25% reflux THf 96%(97%ee)
a Mol% Rh l ligand monomer. Plus naphthol byproduct.
2.1.2.3 Scope of Alcohol Nudeophiles
An investigation of the scope of the reaction with regards to the alcohol nudeophile
revealed that a wide varieV of alcohols are compatible. Each of the alcohols added in
good yield and excellent enantioselectivity (Table 2.8). Even the very weakly
nucleophilic hexafluoroisopropano1 (HFIP) added under these reac tion conditions (Entry

9). We were also able to use very low catalyst loadings typically as low as O.l25mol%
[Rh(C0D)C1l2 and 0.25mol% PPF-PcBu2.
Table 2.8: Scope of the Rhodium Catalysed AR0 with Alcohofs
O 0.125mo1°h [Rh(COD)C1l2
I L 0.25 mol% 73 * rn ROH (4-5 eq.)
42 THF / 8 0 ' ~ (or reflux) OH
E m ROH Product Yield(%) ee(%lb -- -
M ~ O H ~
E ~ O H ~
' ~ 1 0 l - l ~
Allyl Alcohol
TMS ~ t h a n o l ~
Benzyl Alcohol
PMB Alcohol
TFE
HFlP
a These reacîions were perfomed under unoptimised conditions using 10 eq. ROH
ee detennined by formation of Moshers ester or by HPLC analysis with a Chiralcel OD colurnn

2.1.2.4 Effect of Symmetric Aryl Substitution on the Substrate
h order to investigate the effects that substituents on the aromatic ring of 42 would have,
difluoro 78, methylene dioxy 79, and dimethyldibromo 80 substrates were prepared and
reacted under the standard conditions. Al1 gave the corresponding ring opened products
81, 82, and 83 in good yields and excellent ee's indicating that this reaction is not
sensitive to electronic effects on the srornatic ring.
2.1.2.5 Conclusion
We have developed a new rhodium catalysed AR0 reaction of oxabenzonorbomadienes.
This reaction produces a new carbon-oxygen bond via a net intermolecular allylic
displacement of the bridgehead oxygen with a wide range of alcohols. This reaction
occun under neutral reaction conditions, and no activation of the alcohol nucleophile is
required. It proceeds with complete regio- and diastereoselectivity, and gives excellent
enantioselectivity (up to 99%ee). The reaction can be carried out using low catalyst
loadings, typically 0.25 mol % of the catalytically active rhodium species. To the best of
Our knowledge, this constitutes the first exarnple of rhodium catalysed enantioselective
carbon-oxygen bond formation.

2.1.3 Experimental
2.1.3.1 General Experimental
The following general experimental details apply to al1 following reactions.
-411 flash wwe flame-dried under a Stream of nitrogen or argon and cooled before use.
Solvents and solutions were transferred with syringes and cannulae using standard inert
atmosphere techniques.
'H NMR spectra were recorded at 200 MHz using a Varian Gemini NMR spectrometer or
at 400 MHz using a Varian XL400 spectrometer with CDC13 as reference standard (6 7.24
ppm) or some other suitable solvent. Spectral features are tabulated in the following
order: chemical shift (6, ppm); number of protons; multiplicity (s-singlet, d-doublet, t-
triplet, q-quartet, m-complex multiplet, br-broad); coupling constants (J, Hz). 13c NMR
spectra were recorded at 400 MHz with CDCl, as reference standard (6 = 77.0 ppm) or
some other suitable solvent.
IR spectra were obtained using a Nicolet DX FT-SR spectrometer as a KBr pellet or neat
film between KBr plates. High resolution mass spectra were obtained From a VG 70-250s
(double focusing) mas spectrometer at 70 eV. Combustion analyses were submitted to
Canadian Microanalytical Service Ltd., BC. Optical rotations were measured on a Perkin-
Elmer Model 243 Polarirneter using the sodium D line with spectro-grade CHC13 in a 1
dm cell. Melting points were taken on a Fisher-Johns melting point apparatus and are
uncorrected.

Gas chrornatography was performed on a Hewlea Packard 5890 gas chromatopph using
an Advanced Separation Technologies G-TA or B-TA chiral columns. HPLC analysis
was perforrned on a Waters 600E with Chiralcel OD or OJ columns. Analytical TLC was
performed using EM Separations precoated silica gel 0.2 mm layer UV 254 fluorescent
sheets. Column chromatography was perfomed as "Flash Chromatography" as reported
by sti1f6 using (200-400 mesh) Merck grade silica gel.
Diethyl ether, THF, benzene and toluene were distilled From sodium
benzophenone ketyl immediately prior to use. CH2C12 was distilled fiom calcium hydride.
DME was distilled fiom sodium benzophenone ketyl and stored. DMF was dried and
stored over activated molecular sieves. Furan was distilled pnor to use. 43-
dibromobenzodioxole was prepared by brornination of benzodioxole in acetic acid in the
presence of NaOAc. Mosher's acid chloride was prepared by refluxing Mosher's acid
(MTPA, obtained fiom Aldrich) in thionyl chloride in the presence of a catalytic amount
of NaCl for 60 h and purified by bulb to bulb distillation. The PPF-PtBu2 ligand was
donated by Novartis. Al1 other reagents were obtained from Aldrich and used as received
unless otherwise stated.

2.1.3.2 Oxabicyclic S tarting Materials
1,4-epoxy-1,4-dihydronaphthalene (42): To furan (100 mL, 1.18 mol) in DME (100
rnL) at 50 O C in a fiame dried 3-neck flask with a reflm condenser and 2 addition fumels
attached was simultaneously added a solution of anthranilic acid (27.5 g, 200 mrnol) in
DME (100 rnL) and a separate solution of isoamylnitrite (40 mL, 298 mrnol) in DME (50
mL). The addition took about 2 h. The reaction was allowed to stir at 50 OC for 30 mm
until no further gas was evolved. The reaction was subjected to distillation at 80 O C until
about half the initial volume rernained. After cooling, the reaction was partitioned
between Et20 and saturated &CO3 and the aqueous layer was extracted 3 times with
EtzO. nie combined organic layers were washed with brine, dned over MgSO4 and
concenûated. Bulb to bulb distillation yielded 42 (18.5 g, 64%) a white solid. The
spectral data correspond well with the literature data?

1-methyl-1,4-epoxy-l,4-dihydronaphthalene (60): To 2-methyl furan (20 mL, 0.19
mol) in DME (20 rnL) at 50 OC in a £'lame dned 3-neck flask with a reflux condenser and
2 addition funnels attached were simultaneously added a solution of anthranilic acid (7 g,
5 1 mmol) in DME (1 5 mL) and a separate solution of isoamylnitrite ( 10 mL, 74 mmol) in
DME (IO mL). The addition took about 2 h. The reaction was allowed to stir at 5 0 ' ~ for
30 mm until no futher gas was evolved. The reaction was subjected to distillation at
80'~ until about half the initial volume remained. After cooling, the reaction was
partitioned between Et20 and 5 M NaOH and the aqueous layer was extracted 3 times
with Et20. The combined organic layers were washed with brine, dried over MgS04 and
concentrated. Buib to bulb distillation yielded 60 (4.32 g, 49%) a colourless oil, The
spectral data correspond well with the literature data.78
6,7-dinuoro-l,4-epoxy-1,4-dihydronaphthalee (78): To 3,4-di fiuoro- 1,2-
dibromobenzene (0.75 g, 2.78 rnrnol) and furan (1 mL, 12.7 rnmol) in EtzO (15 mL) was

added BuLi (1.1 rnL, 2.5 M in hexanes, 2.75 mrnol) dropwise. The reaction was s h e d
for 2 hours at -78 OC and then allowed to warm to rt. AAer 16 h, the reaction mixture was
poured into water. The organic layer was separated and the aqueous layer extracted three
times with Et20. The combined organic layers were washed with brine, dried over
MgSOc concentrated and chromatographed (4: 1 hexanes:EtOAc) on silica gel to yield 78
(350 mg, 70%), a colourless oil. EU? = 0.21 on silica gel (20% Et0Ac:hexanes); bp 40 'C
@ 0.5 mmHg; IR (neat) 30 17 (m). 1624 (s), 1465 (s), 1365 (s), 12% (s), 1 190 (m), 1040
(s), 857 (s) cm-'; 'H NMR (400 MHz, CDC 13) 8 7.06 (2H, dd, pF = 7.7, 7.7 Hz), 7.0 1
(2H. s), 5.67 (ZH, s); 1 3 ~ NMR (100 MHz, CDCI,) G 147.2 (dd, f - F = 247.9, 14.5 Hz),
145.1 (dd, f - F = 4.3, 4.3 HZ), 143.1, 1 1 O.8(rn), 82.1. HRMS calcd for C loH60F2 (M)-:
t 80.1508. Found: 180.1 502.
5,&epoxy-5,8-dihydronap htho [2,3-dl[ lJ 1 dioxole (79). To 3,4-dibroinobenzo- l,3-
dioxolane (1.54 g, 5.50 mrnol) and furan (4 g, 58.8 m o l ) in PhMe (55 mL) was added
BuLi (2.2 mL, 2.5 M in hexanes, 5.5 mmol) dropwise. The reaction was s h e d for 2
hourç at -78 O C and then allowed to warm to rt. Afier 16 h, MeOH (2 mL) was added and
the reaction mixture poured into water, The organic layer was separated and the aqueous
layer extracted three tirnes with Et20. The combined organic layers were washed with
brine, dried over MgS04, concentrated. Recrystallization nom hexanes yielded 79 (560

mg, 54%), as white crystals. Rf = 0.47 on silica gel (30% Et0Ac:hexanes); mp 1 1 1-1 12OC
(hexanes); IR (KBr) 2895, 1455, 1292, 1138, 1038, 10 14, 848 cm-'; 'H NMR (400 MHz,
CDCh) G 7.02 (2W dd, J= 0.9,O.g Hz), 6.82 (2H, s), 5.92 (lH, d, J = 1.5 Hz), 5.87 (LH,
d, 1 = 1.5 Hz), 5.62 (2H, s); '-'c NMR (100 MHz, CDC13) 6 144.3, 143.3, 143.2, 103.9,
101.1, 82.4. HRMS calcd for CiiHs03 (M)': 188.0473. Found: 188.0463.
5,6-dibromo-4,74imethyl-~,4-epoxy-1,4-dihydronaphthalene (80). To tetrabrorno
para-xylene (2.1 g, 5.0 mmol) and furan (4 g, 20.6 mrnol) in PhMe (50 mL) was added
BuLi (2.2 mL, 2.5 M in hexanes, 5.5 rnmol) dropwise over the course of 1 h. The reaction
was stirred for 2 houn at -78 OC and then allowed to warm to rt. After 16 h, MeOH (2
mi,) was added and the reaction mixture was poured into water. The organic layer was
separated and the aqueous layer extracted three times with EtzO. The combined organic
Layen were washed with bnne, dned over MgSO4, and concentrated. Column
chromatography on silica gel yielded 80 (840 mg. 51 %), a white solid. Spectral data
correspond well with the literature data.79

3-~hlorobmethox~-ll-oxa-tric~clo(6.2.1.0~*~undeca-2(,3,5,9tetraene (62): To a
dry round bottomed flask was added Furan (3.8mL,56mrnol), 3,J-dichloroanisole ( 1 g,
5.6mol) and THF. After cooling to -78°C. BuLi (2mL 2.5M in hexanes) was added
and the reaction was allowed to w m to rt. AAer 1 hour, the mixture was quenched with
water, extracted with EtOAc, dried with MgSQ, and concentrated in vaaio. Flash
chromatography (10% Et0Ac:hexanes) gave 185.2mg (1 6%) of 62, white crystals. Rf=
0.34 on silica (1 0% ethyl acetate:hexanes). IR (neat, cm") 16 13, 1474, 1280, 1 180. 1 1 17,
1001, 891, 795, 724; 'H NMR (400MHz, CDCl,) 8 7.2 1-7.16 (2H, rn), 6.97 (1 H, d, .J=
8.8 Hz), 6.73 (lH, d, J= 8.8 Hz), 5.99 (lH, dd, J= 1.8, 1.1 Hz), 5.87 (lH, dd, J= 1.8, 1.1
Hz), 3.87 (3H, s); ' 3 ~ NMR (400MHz, CDC13) 6 152.7, 150.4, 144.3, 143.1, 138.8,
127.6, 118.8, 113.8, 82.0, 81.5. 56.6. HRMS calcd for CliH,O2CI (M3: 210.0216
Found: 2 10.0267.
22.2mmol) and 2mL THF added to a dry round bottomed Bask and cooled to O°C. 1Orn.L
2.5M BuLi added and allowed to react for 2 hours. condensed ethylene oxide (1.10g,
25mmol) added via cannula. Allowed to react for 1 hour then quenched with water.

Standard workup (EtOAc extraction) and flash chromatography (30%
ethy1acetate:hexanes) gave 2-(5-methyl-h-2-yl)-ethanol (2.370, 8 1 % yield) which
was camed on to the next step. Imidazole (2.450, 36mmol) and DMAP (1 22mg, 1 mmol)
were dissolved in 8 mL CH2C12 in a dry round bottomed flask. To this, the crude alcohol
from the previous step (2.370, 18mmoi) was added by cannula followed by dropwise
addition of TBDMSC1 (3.0 lg, 20mmol) in 3rnL CHzClz by cannula. ARer 1 hour, the
reaction was quenched with water, extracted with CH2C12, dned with MgSOd, and
concentrated Ni vacuu. Flash chromatography (5% EtOAchexanes) gave 65 (3.56g,
82%) , a clear oil. RF 0.43 on silica (hexanes). IR (neat, cm-') 2928, 1571, 1472, 1255,
1 104, 836, 776; 'H NMR (400MHz, CDC13) 6 5.9 1 (lH, d, J= 2.9 HZ), 5.84 (1 H, dd, J=
2.9, 1.1 Hz),3.83 (2H, t,.J=7.0Hz), 2.79(2H, t, J= 7.0 Hz), 2.24 (3H, s), 0.88 (9H,s),
0.02 (6H, s); I3c NMR (400MHz, CDCI,) 6 151.4, 150.4, 106.8, 77.4, 77.1, 76.8, 61.9,
32.0, 25.9, 25.8, 18.4, 13.5. HRMS calcd for Ci&02Si (M3: 240.4205, Found:
240.4209.
1-(2-Tert bu tyldimethyIsiloxy-ethy1)-8-methyl l-oxa-tricylco i6.2.1 .02*'1 undeca-
2(7) ,3,5,9-tetraene (69): 65 (1.75g,7.28mmol) and 1 ,2-bromochlorobenzene (1.39g,
7.28mmol) were added to a dry round bottomed flask along with 10 rnL toluene and
cooled to -7g°C. 1.77mL 2.5M BuLi added slowly dropwise. Solution was allowed to
warm slowly to room temperature over 1 h o u (a precipitate formed at -5°C). The

reaction was quenched with water, extracted with EtOAc, dried with MgS04, and
concentrated in vacuo. Flash chromatography (2% Et0Ac:hexanes) gave 1.1 1 (48%) of
69. RF 0.43 on silica (20% ethyl acetate:hexanes). iR (neat, cm-') 3071, 2929, 1471,
1255, 1097, 837, 776, 692; 'H b&fR (400MHzT C6D6) 8 7.15-7.09 (2H, rn), 6.96-6.94
(2H, m), 6.83 (lH, d, J= 5.5 Hz), 6.71 (lH, d, J= 5.2 Hz), 3.97 (2H, ddd, J= 7.3 6.2 1.1
Hz), 2.62-2.55 (1H, m), 2.47-2.40 (IH, m), 1.87 (3H, s), 0.89 (9H, s), 0.06 (6H, s); I3c
NMR (400MH2, C6Do) 6 152.9, 152.3, 146.3, 146.1, 124.8. 124.7, 119.0, 118.4, 90.2,
88.6. HRMS calcd for CisHu02Si (m: 3 16.1858, Found: 3 16.1858.
(928.4mg7 2.93mmol) was added to a dry round bottomed flask. To this 3.23m.L 1M
TBAF in THF was added by syringe and was allowed to react for 8 hours at room
temperature. The reaction was extracted with EtOAc, dried with MgS04, and
concentrated in vacuo. Flash chromatography (50% Et0Ac:hexanes) gave 71 (585.3rng
99%). RF O. 15 on silica (30% ethyl acetatezhexanes). IR (neat, cm-') 3426, 293 1, 1643,
1451, 1305, 1037, 857,763; 'H NMR (400MHz. CDCli) S 7.14 (2H, m), 6.98-6.95 (2H,
m), 6.80 (lH, d, .i= 5.5 Hz), 6.77 (lH, d, J= 5.5 Hz), 3.95 (2H, dd, J= 5.5, 5.5 Hz). 2.62-
2.46 (2H, m), 1.88 (3H, s); HRMS calcd for Ci&Iis02 (m: 202.0994, Found:

2.1.3.3 Ring Opening with Alcohols
OMe
~e 0'"' @- ,' OH CI
63
8-Chloro-2,5-dimethoxy-1,2-dihydro-naphthlen--ol (63): To a flarne dned round
bottom flask was added 62 (30mg, O. ISrnmoI), [ R ~ I ( C O ) ~ C ~ ] ~ (4mg, 0.0 lmmol), MeOH
( 1 rnL) and TFE ( ImL). The mixture was heated at 60°C for 3 hours. Concentration and
flash chrornatography (10% ethy1acetate:hexanes) gave 63 (23rng, 68% yield), white
crystals RF 0.28 on silica (30% ethyl acetate:hexanes). IR (neat, cm-') 3410,2937, 1636,
1470, 1259, 1082, 951, 799; 'H NMR (400MHz, acetone-d) 6 7.24 (IH, d, J= 8.8 Hz),
6.98 (lH, d, J= 10.0 Hz), 6.95 (IH, d, J=9.0 Hz), 6.17 (lH, ddd, J= 10.0, 5.4, 1.3 Hz),
5.00 (lH, ddd, J= 5.9, 1.7, 1.2 Hz), 4.20 (IH, d, J= 5.8 Hz), 3.84 (3H, s), 3.30 (3H, s),
2.84 (1 H, d, J= 16.6 Hz; I3c NMR (400MHz, acetone-d) 6 154.5, 133.2, 129.5, 123.9,
123.9, 112.4, 75.6, 65.8, 56.6, 56.0. HRMS calcd for CirHi303CI (M3: 242.0534,
Found: 2 10.0267.
61
2-Metho~y4methyl-l,2-dihydro-naphthalen-1-0 (61) : To a round bottom ffask was
added 60 (30rng, 0.19mmol) and m(C0)2C1]2 (4mg, 0.Oimmol) followed by 1rnL

MeOH and 1m.L TFE. The mixture was heated at 60°C for 2 hours. Concentration and
flash chromatography ( 10% ethy1acetate:hexanes) gave 6 1 ( 1 3.9mg, 42% yield), red
crystals. RF 0.20 on silica (10% ethy1acetate:hexanes). IR (neat, cm-') 3281, 2933,
1806, 1451, 1379, 1284, 1132,804,746; 'H NMR (400MHz, C a s ) 6 7.89 (IH, ddd, J=
7.3, 1.5, 1.5 Hz), 7.14 (IH, dd, J= 15.0, 1.5 Hz), 7.08 (IH, dddd, J= 15.0, 7.3, 1.5, 0.8
Hz), 7.02 (lH, dd, J= 7.3, 1.5 Hz), 5.65 (lH, dd, J= 1.5, 1.4 Hz), 4.91 (1H. d, J= 9.0 Hz),
3.90 (LH, dq, J= 11.0, 2.2 Hz), 3.09 (3H, s), 2.61 (lH, d, J= 2.9 Hz), 1.79 (3H, dd, J= 2.2,
1.5 Hz); "C NMR (~OOMHZ, C a 6 ) 8 137.5, 134.2, 133.1, 128.2, 128.1, 125.4, 124.9,
123.5, 83.1, 73.3, 56.4, 19.1. HRMS calcd for C12H& (m: 190.2442, Found:
190.2440.
55
(1S,2S)-2-Methoxy-1,2-dihydro-naphthalen-l-o (55): To a flame dried round bottom
flask, w(COD)Cl]2 (8.6 mg, 0.017 rnrnol), (R)-(S)-PPF-P'BU~ (19 mg, 0.035 mmol) and
42 (500 mg, 3.47 rnrnol) were added followed by addition of THF (1 mL) and rnethanol
(1 mL). The mixture was heated for 15 hours and the solvents were removed in vacuo.
The resulting solid was purified by flash chromatography (20% ethyl acetate in hexanes)
to give 55 a white crystalline solid (586 mg, 96%). The ee was detemined to be 97%
using HPLC analysis on a CHIRALCEL OD column, h= 486 nm. Retention times in 4%
isopropanol in hexanes were 10.1 min (major) and 1 1.1 min. RF 0.29 on silica gel (10%
ethyl acetate:hexanes); mp 86-87' (EtD); [afSD= -208' (c= 10.1, CHCls); RF 0.39 on

silica (20% ethyl acetate:hexanes). IR (KBr, cm*') 3277 (br), 2971 (m), 1466(m),
1285(m), 1 114(s), 1048(m), 979(m), 775(s); 'H NMR (400bfHz, CDC13) 6 7.60-7.62
(lH, m), 7.30-7.21 (2H, m), 7.13-7.1 1 (IH, m), 6.50 (IH, dd, J= 9.9,1.8 Hz), 6.04 (lH,
dd, J=9.9, 2.2 Hz),4.85 (1H. dd,J=9.9, 6.2 Hz), 3.50 (3H,s),2.89 (IH, d,J= 12.8 Hz);
I3c NMR (400MHz, acetone-d) 6 138.5, 133.2, 129.1, 128.4, 128.3, 128.2, 126.8, 126.3,
83.1,73.O, 57.1. HRMS calcd for Ci i H i 2 0 2 (M3: 176.0837. Found: 176.0835.
(lS,ZS)-f-(Etholry)-l,2-dihydro-naphthalen-O (57): : To a flarne dried round bottom
flask, [Rh(COD)C1I2 (8.6 mg, 0.0 17 mrnol), (S)-(K)-PPF-P'BU~ (1 9 mg, 0.035 mmol) and
42 (500 mg, 3.47 mmol) were added followed by addition of ethanol (1.5 mL) and THF
(1.5 mL). The mixture was heated to reflux for five hours and the solvent was removed
in vacuo. The resulting solid was purified by flash chromatography (20% ethyl acetate in
hexanes) to give 57 as a white crystalline solid (553 mg, 84%). The ee was determined to
be 97% using HPLC analysis on a CHIRALCEL OD column, )c = 254 nm. Retention
times in 1 .5% isopropanol in hexanes were 13.6 min and 14.2 min (major). RF 0.26 on
silica gel (20% ethyl acetate:hexanes); mp 33" (EtzO); [a]250= 185.9" (c= 9.6, CHCL);
iR (KBr, cm-') 3601 (br), 3040 (m), 2977 (s), 1454 (s), 1396 (m), 1185 (s), 1104 (s); 'H
NMR (400MHz, CDCb) 8 7.59-7.57 (lH, m), 7.27-7.20 (2H, m), 7.07-7.05 (lH, m), 6.43
(lH, dd, J= 9.9, 2.2 Hz), 6.01 (LH, dd, J= 9.9, 2.2 Hz), 4.90 (lH, d, J= 10.6 Hz), 4.18
(IH, ddd, J= 10.6,2.2,2.2 Hz), 3.79 (lH, AB, dq, J= 9.4, 6.9 Hz), ), 3.58 (lH, AB, dq, J

= 9.4, 6.9 Hz), 2.65 (lH, s), 1.27 (3H, t, J= 6.9 Hz),; 13c NMR (400MHz, CDC13) 6
135.9, 131.9, 128.0, 127.8, 127.8, 126.1, 124.9, 80.7, 72.5, 64.6, 15.5. HRMS calcd for
C IZH1402 0: lgO.0994. Found: 190.0993.
(1S,2S)-2-(Isopropoxy)-1,2-dihydro-aaphthaen-l-o1 (58): : To a flame dned round
bonom flask, [Rh(COD)C1I2 (8.6 mg, 0.0 L 7 mmol), (S)-(R)-PPF-PtBu2 ( 19 mg, 0.0 17
mmol) and 42 (500 mg, 3.47 mmol) were added followed by addition of THF (2 mL) and
isopropanol(2 mL). The mixture was heated to 80°C for two hours and the solvent was
removed in vacuo. The resulting oil was purified by flash chromatography (10% ethyl
acetate in hexanes) to give 58 as a colourless oil(666 mg, 94%). The ee was determined
to be 92% using HPLC analysis on a CHIRALCEL OD column, A = 486 nrn. Retention
times in 1.5% isopropanol in hexanes were 9.7 min (major) and 10.7 min. RF 0.42 on
silica gel (10% ethyl acetatehexanes); +154.0° (c= 12.6, CHC13); IR ( U r , cm'')
7.61-7.58 (1H, m), 7.27-7.19 (2H, m), 7.06-7.04 (lH, m), 6.40 (IH, dd, J= 9.9, 2.0 Hz),
5.95 (1H, dd, J= 9.9, 2.2 Hz), 4.87 (1 H, d, J= 10.8 Hz), 4.24 (LH, ddd, J= 10.8, 2.2, 2.2
Hz), 3.85 (1H, h, J = 6.2 Hz), 2.98 (1H, s), 1.25 (6H, dd, J= 8.8, 6.2 Hz); ')c
71.1, 23.5, 22.4. KRMS calcd for Ci3His02 (m: 204.1 150. Found: 204.1 150.

(1S,2S)-3-(1-propenyloxy)-1,2-dihydro-naphthalen-1-ol(74): : To a flame dned round
bottom flask, [RLi(COD)C1l2 (9.1 mg, 0.0 18 rnrnol), (S)-(R)-PPF-PIBuz ( 15 mg, 0.028
mrnol) and 42 (1.06 g, 7.35 mmol) were added followed by addition of THF (1.5 mL) and
allyl alcohol (2 mL, 29.4 mmol). The mixture was heated to 80°C for two h o u and the
THF was removed in vacuo. The resulting oil was purified by flash chromatography
(10% ethyl acetate in hexanes) to give 74 as a colourless oil (898mg, 60%) which
solidified on sitting. The ee was determined to be >99% using HPLC analysis on a
CHIRALCEL OD column, h = 486 nrn. Retention times in 1.5% isopropanol in hexanes
were 15.2 min and 16.3 min (major). RF 0.17 on silica gel ( 1 0% ethyl acetate:hexanes);
mp 25-26' (EtzO); [alBo= +M. 1' (c= 1 1.5, CHC13); IR (KBr, cm") 3435(br), 3037(m),
2857(s), 1454(s), 1 l65(s), 1083(s); 'H NMR (400MHz, CDC13) S 7.6 1-7.58 ( 1 H, m),
7.27-7.20 (2H, m), 7.08-7.05 (1H, m), 6.44 (IH, dd, J= 9.9,2.0 Hz), 6.00 (1H, dd, J= 9.9,
2.4 Hz), 6.00-5.92 (fH,m), 5.32 (1 H, ddd, J= 17.2, 3.3, 1.6 Hz), 5.21 (lH, ddd, J= 10.4,
2.9, 1.3 Hz), 4.94 (lH, d , J = 10.2 Hz), 4.27 (lH, ddd, J = 10.3, 2.2, 2.2 Hz), ), 4.23 (LH,
dddd, J = 12.8,5.5, 1.5, 1.5 Hz), 4.12 (IH, dddd, J = 12.8, 5.9, 1.5, 1.5 Hz), 3.09 (lH, s);
I3c NhlR (400MHz, CDC13) 6135.8, 134.5, 131.8, 128.1, 127.7, 127.6, 127.4, 126.1,
125.0, 1 17.5, 80.1, 76.7, 72.4, 70.2. HRMS calcd for CIa14O2 (M+):202.0994. Found:

dned round bottorn flask, [Rh(COD)C1I2 (8.6 mg, 0.017 mmol), (S)-(R)-PPF-P'BU? (19
mg, 0.035 mmol) and 42 (500 mg, 3.47 mmol) were added followed by addition of TW
(3 mL) and trimethylsilylethanol (4 mL). The mixture was heated to reflux for two hours
and the THF was removed in vacuo. The resulting oil was purified by flash
chromatography (10% ethyl acetate in hexanes) to give 59 as a colourless oi! (482 mg,
53%). The ee was determined to be 95% using HPLC analysis on a CHIRALCEL OD
column, h = 486n.m. Retention times in 0.5% isopropanol in hexanes were 17.9 min and
18.5 min (major). RF 0.25 on çilica gel (10% ethyl acetate:hexanes); [alZSD= + 1 19.2"
(c= 13.0, CHCI,); IR (KBr, cm-" 3447(br), 3037(m), 2972(s), 1454(m), 1381(m),
1 1 18(s), 1078(s); 'H NMR (400MHz, CDC13) 6 7.59-7.57 (1 H, m), 7.28-7.2 1 (2H, m),
7.08-7.06 (lH, m), 6.43 (1H. dd. J= 9.9,2.0 Hz), 6.03 (IH, dd, J= 9.9,2.2 Hz), 4.89 (1 H,
d, J= 10.6 Hz), 4-18 (lH, ddd, J= 10.6, 2.2, 2.2 Hz), 3.85-3.78 (2H, m), 3.63-3.56 (2H,
m), 2.79 (lH, s), 1.05-0.97 (2H, m), 0.36 (9H, m); "C NMR (400MHz, CDC13) 6. 135.9,
132.0, 127.9, 127.9, 127.8, 127.6, 126.1, 124.9,80.4,72.6,66.5, 18.6, -1.4. HRMS calcd
for CisHuOzSi (m: 262.1389. Found: 262-1388.

a BnO -
(1S,2S)-2-Benzyloxy-1,2-dihydro-aaphthalenl-o1(75): To a fiame dried round bottom
flask, m(COD)C1]2 (4.3 mg, 0.009 mmol), (S),(R)-PPF-P'BU~ (9.4 mg, 0.017 mmol),
and 42 (1.00 g, 6.94 mmol) were added followed by addition of THF (1.8 mL) and
benzylalcohol(3.6 rnL, 34.7 mmol) and heating to 80°C for 24 hours. The THF was then
removed in vacuo and the resulting oil was purified by flash chromatography (10% ethyl
acetate in hexanes) to give 75 as a crystalline solid (1.22 g, 70%). The ee was determined
to be >98% using HPLC analysis on a CHIRALCEL OD column, h = 486 nm. Retention
tirnes in 1.5% isopropanol in hexanes were 29.0 min and 32.5 min (major). RF 0.34 on
silica gel (20% ethyl acetate:hexanes); mp 52-54' (EtzO); [ c c ] ' ~ ~ = +167.3' (c= 10.0,
CHC13); IR (KBr, cm-') 3305 (br), 3020 (w), 2876 (w), 1496 (m), 1352 (m), 128 1 (m),
1 169 (m), 1050 (s), 777 (s); 'H NMR (400MHz, CDC13) G 7.58-7.56 (1 H, m), 7.41-7.22
(7H, m), 7.22-7.07 (lH, m), 6.46 (lH, dd, J= 9.9,2.1 Hz), 6.05 (IH, dd, J= 9.9, 2.1 Hz),
4.98 (lH, d, J= 10.4 Hz), 4.78 (lH, d, J= 11.7 Hz), 4.63 (lH, d, P 11.7 Hz), 4.33 (IH,
ddd, J= 10.4, 2.2, 2.2 Hz), 2.61 (lH, s); "C NMR (400MHz, CDC13) 6 138.0, 135.9,
131.9, 128.5, 128.3, 128.1, 127.9, 127.9, 127.8, 127.4, 126.2, 125.1, 80.4, 72.6, 71.3.
HRMS calcd for CI7HL602 (M'): 252.1 150. Found: 252.1 148.

PMBO -
(lS,2S)-2-(4-rnethoxybenzyloxy-1,2-dihydro-nap hthe -1 -01 (76): To a flame dried
round bottom flask, [Rh(COD)C1I2 (2.2 mg, 0.005 mmol), (s),(R)-PPF-Pt~u2 (4.7 mg,
0.008 mmol), and 42 (693 mg, 4.8 1 mmol) were added foollowed by addition of THF (1.5
rnL) and anisyl alcohol (3.0 mL, 24.1 mmol) and heating to 80°C for 24 houn. . The
THF was then removed in vacwo and the resulting oil was punfied by flash
chromatography (20% ethyl acetate in hexanes) to give 76 as a crystalline solid ( 1.18 g,
87%). The ee was detemined to be 97% using HPLC analysis on a CHIRALCEL OD
column, h = 486 m. Retention times in 1.5% isopropanol in hexanes were 37.1 min and
42.1 min (major). R ç 0.53 on silica gel (30% ethyl acetate:hexanes); mp 63-64" (EtIo);
[a]"~= + 138.5" (c= 1 O S , CHCI,); IR (KBr, cm-') 3435(br), 3035(m), 2836(s), 16 1 2(s),
15 l3(s), 1454(m), 1249(s), 1082(s); 'H NMR (400MHz, CDC13) 6 7.59-7.57 ( 1 H, rn),
7.32 (2H, ddd, J= 8.7, 2.8, 1.9 Hz), 7.28-7.22 (lH, m), ), 6.90 (2H, ddd, J= 8.7, 2.8, 1.9
Hz), 6.46 (lH, dd, J= 9.9, 2.1 Hz), 6.04 (lH, dd, J= 9.9, 2.4 Hz), 4.96 (IH, d, J= 10.1
Hz), 4.64 (lH, dd, P 57.1, 11.4 Hz), 4.32 (lH, ddd, J= 10.2, 2.2, 2.2 Hz), 3.80 (IH, s),
2.96 (lH, s); 13c NMR (4OOMHz, CDC13) G 159.2, 135.9, 131.9, 129.9, 129.5, 128.1,
127.8, 127.6, 127.5, 126.1, 125.0, 113.8, 80.0, 72.5, 70.9, 55.1. HRMS calcd for
Ci7Hi& (M3: 252.1 150. Fouad: 252.1148.

(lS,2S)-2-(2,2,2-Trifluoro-ethoxy)-lf -dihydro-naphthalen-1-01 (73): : To a flame
dried round bonom flask, [Rh(COD)Cl]2 (2.1 mg, 0.004 mmol), (S)-(R)-PPF-P'BU? (4.7
mg, 0.0087 mmol) and 42 (500 mg, 3.47 rnrnol) were added followed by addition of
ûifluoroethanol(l.3 mL, 1 7.4 mrnol) and THF (4 mL). The mixture was heated to reflux
for three hours and the solvent was removed in vaaio. The resulting solid was purified
by flash chromatography (10% ethyl acetate in hexanes) to give 73 as a white crystailine
solid (594 mg, 70%). The ee was determined to be 98% using HPLC analysis on a
CHIRALCEL OD column, )L = 254 nm. Retention times in 4% isopropanol in hexanes
were 1 1.3 min (major) and 13.3 min. RF 0.4 1 on silica gel (20% ethyl acetate: hexanes);
mp 79-80' (EtzO); [a12'~= 145.4' (c= 12.6, CHCI,); iR (KBr, cm") 3354 (br), 3036 (w),
2939 (w), 1455 (w), 1275 (s), 1 169 (s), 1050(m), 977 (m); 'H NMR (400MHz, CDCl]) 8
7.57-7.55 (lH, m), 7.30-7.23 (2H, m), 7.10-7.08 (IH, m), 6.48 (IH, dd, P 9.9, 2.0 Hz),
5.94 (lH, dd, J= 9.9, 2.4 Hz), 4.96 (1 H, d, J= 2.2 Hz), 4.38 (lH, ddd, P 9.9, 2.4, 2.2
Hz), 4.03 (2H, q, y-F= 8.6 Hz), 2.55 (IH, s); NMR (400MHz, CDC13) G 135.5,
131.7, 129.2, 128.3, 128.1, 126.6, 125.9, 125.2, 122.4,83.0, 72.8, 67.0 (q, PF= 34.4 Hz).
HRMS calcd for C i2Hi ,02Fi (M3: 244.07 1 1. Found: 244.0720.

77
(1S,2S)-2-(2,2,2-Trüluoro-l-t~fluoromethyl-ethoxy)-l,2-dihydro-naphthalen-l-ol
(77): To a flame dried round boaom flask, [Rh(COD)C1l2 (1.7 mg, 0.003 mmol),(S)-(R)-
PPF-P'BU~ (3.8 mg, 0.007 mmol) and 42 (500 mg, 3.47mmol) were added followed by
addition of THF (2.0 mL) and hexafluoroisopropanol (1.8 mL. 17.4 m o l ) . The mixture
was heated to reflux for two hours and the solvent was removed in vacuo. The resulting
solid was ptuified by flash chromatography (10% ethyl acetate in hexanes) to give 77 as a
white solid (974 mg, 90%). The ee was detemined to be 93% using HPLC analysis on a
CHIRALCEL OD column. h = 486 m. Retention tirnes in 1.5% isopropanol in hexanes
were 11.3 min and 17.6 min (major); RF 0.28 on silica gel (10% ethyl acetate:hexanes);
mp 88.5-90' (EtzO); [alZsD= + 10 1 .go (c= 10.9, CHCI,); IR (KBr, cm-') 3 19 1 (br), 2937
(m), 1379 (s), 1280 (s), 1247 (s), 1194 (s), 1100 (s), 954 (s), 753 (m); 'H NMR
(400MHz, CDC13) 8 7.55-7.53 (1 H, m), 7.3 1-7.26 (2H, m), 7.1 1-7.09 ( I H, m), 6.49 ( 1 H,
dd, J= 9.9,2.1 Hz), 5.92 (lH, dd, J= 9.9,2.4 Hz), 5.07 (lH, dd, J= 9.7, 5.0 Hz), 4.63 (IH,
ddd, J= 9.9, 1.5, 1.5 Hz), 4.58 (LH, h , f e F = 6.1 Hz), 2.50 (IH, d, J= 4.2 Hz); "C NMR
(400MHz, CDC13) G 135.2, 131.5, 129.7, 128.5, 128.3, 126.7, 125.2, 122.9, 120.1, 85.4,
75.4 (h, pF= 32.2 Hz), 73.5. HRMS calcd for Ci3Hio02F6 (w: 312.0585 Found:
3 12.0574.

round bottom flask' W(COD)Cl]? (2.5 mg. 0.005 mmol), (S)-(R)-PPF-P'Bu~ (5.4 mg,
0.0 10rnmol) and 6,7-difluoro- 1,4-epoxy- l,4-dihydronaphthalene (72 mg, 0.40 rnmol)
were added followed by addition of THF ( 1 .O mL) and methanol (1 .O mL). The mixture
was heated to reflux for 1 hour. The solvents were then removed in vacuo. The resulting
solid was purified by flash chromatography (20% ethyl acetate in hexanes) to give 81 as a
white crystalline solid (74.9 mg, 88%). The ee was determined to be 96.4% using HPLC
analysis on a CHIRALCEL OD column, h = 486 nm. Retention times in 4% isopropanol
in hexanes were 8.9 min and 10.1 min (major). RF 0.27 on silica gel (30% ethyl
3269 (br), 2937 (w), 1597 (m), 1503 (s), 1306 (s), 1 103 (s), 893 (s); 'H NMR (400MHz,
CDCb) G 7.40 (IH, ddd, f-F= 10.8, 7.8 Hz, pH=0.6 HZ), 6.85 (lH, dd, 10.9, 7.8
Hz), 6.31 (IH, dd, J= 10.0, 2.0 Hz), 6.05 (1H, dd, J= 10.0, 2.0 Hz), 4.79 (lH, d, J= 11.0
Hz), 4.05 (1H, ddd, J= 11.0, 2.0, 2.0 Hz), 3.49 (3H, s), 2.94 (1H, d, J= 2.2 Hz); "C
NMR (400MHz, CDCli) G 15 1 .O (d, y*F= 12.5 HZ), 148.5 (dd, PF= 12.5,2.9 Hz), 133.2
(dd, f-F= 5.2, 3.6 Hz), 128.9 (dd, pF= 6.6, 4.4 Hz), 128.0 (d, 4/.- 2.2 Hz), 126.5 (dd,
fdF= 2.2, 1.5 HZ), 115.1 (d, pF= 18.3 Hz), 114.8 (d, fSF= 19.8 Hz), 82.3, 72.0, 57.0.
HRMS calcd for Ci IH1002F2 (m: 2 12.0649. Found: 2 12.0658.

(lS,2S)-6-Methoxy-5,6-dihydro-naphtho [ 2 , 3 [l,3 ldioxol-5-ol(82): To a fiame dried
round bottom flask, [R.h(COD)C1I2 (1.7 mg, 0.0035 mmol), (s)-(R)-pPF-PtBu2 (3.8 mg,
0.0069 mmol) and 5,8-epoxy-5,8-dihydronaphtho[2,3-d][l,3]dioxole (100 mg, 0.694
rnmol) were added followed by addition of THF (1 .O mL) and methanol (1 .O mL) and
heating to reflux for 30 minutes. The solvents were then removed Ni vacuo. The
resulting solid was purified by flash chromatography (30% ethyl acetate in hexanes) to
give 82 as a white crystalline solid (127.5 mg, 90%). The ee was determined to be 95%
using HPLC analysis on a CHIRALCEL OD column, h = 486 nrn. Retention times in 4%
isopropanol in hexanes were 19.2 min (major) and 22.6 min. RF 0.24 on silica (30%
ethyl acetate:hexanes); mp 1 17- 1 19' (EtzO); [alZso= +298.7' (c= 1 1.1, CHCI,); IR (KBr,
cm') 3248 (br), 2926 (s), 1600 (rn), 1483 (s), 1260 (s), 11 13 (s), 941 (s), 876 (s); 'H
NMR (400MH~, CDC13) 6 7.10 ( lH, s), 6.59 (lH, s), 6.34 (lH, d, J= 9.9 HZ), 5.92-5.99
(3H, m), 4.77 (lH, d, J= 9.9 Hz), 4.04 (lH, d, J= 10.1 Hz), 3.49 (3H, s), 2.2 (1Hs);
NMR (400MHz, CDC13) 6 147.2, 146.9, 130.4, 128.1, 126.0, 124.8, 107.1, 106.7, 101.0,
82.0, 72.3, 56.7. HRMS calcd for C i t H t r 0 4 (M'): 220.0736. Found: 220.0684.

(1S,2S)-6,7-Dibromo-2-methoxy-5,8-dimethyl-1,2-dihydro-aaphthalen-l-ol (83j: To
a flame dried round bonom flask, [Rh(COD)C1I2 (1.5 mg, 0.0029 mmol), (R)-(S)-PPF-
PCBu2 (3.2 mg, 0.0059 mmol) and 5,6-dibromo-4,7-dimethyl-14-epoxy-l,4-
dihydronaphthalene (195 mg, 0.59 mmol) were added followed by addition of
trifluoroethanol (1.0 mL) and methanol (1.0 d). The mixture was heated to reflux for
20 houn. The solvents were then removed in vacuo. The resulting solid was purified by
flash chromatography (50% ethyl acetate in hexanes) to give 83 as a white crystalline
solid (171.6. mg, 79%). The ee was determined to be 97% using HPLC analysis on a
CHIRALCEL OD column, h = 486 m. Retention times in 4% isopropanol in hexanes
were 16.8 min (major) and 19.3 min. R p 0.39 on silica gel (50% ethyl acetate:hexanes);
mp 1 14-1 16' (EtzO); [alZD= -1%'. 1" (c= 10.0, CHCls); IR (KBr, cm-') 3349 (s), 290 1
CDCb) 6 6.96-6.93 (lH, m), 6.23-6.19 (lH, m), 4.89 (lH, s), 3.96-3.90 (lH, m), 3.38-
3.35 (3H, m), 2.61-2.57 (3H, m), 2.54 (3H, s), 1.82-1.54 (lH, m); I3c NMR (400MHr,
20.6. HRMS calcd for C13H1602Br2 (m: 36 1.9518. Found: 36 1.9335.

2.2 Phenol Nucleophiles
2.2.1 Introduction
Given the success obtained with alcohols, we sought to expand the scope of this
rhodium catalysed AR0 to include substituted phenols. While phenol nucleophiles have
been successfdly been uscd in a few transition metal satalysecl ~ ~ s t e r n s , ~ ~ rhodium had
not previously been used as a catalyst with this class of nucleophile and we had no
previous results indicating that phenols were sufficiently nucleophilic to participate in
oxabicyclic ring opening reactions. The extension to include phenol nucleophiles would
allow facile access to a broad range of enantioe~ched dihydronaphalene products which
could be fùrther transforrned into substituted benzohrans which are themselves
pharmaceutically interesting ~orn~ounds.~ ' Some of the experiments in this section were
perfomed by Mark Taylor as a collaboration on this project.
22.2 Investigation of the Conditions
Our initial experiments using the previously reported [Rh(C0D)C1l2 / PPF-PLB u2
catalyst system with 10 equivalents of phenol gave the desired product 84 in near
quantitative yield and outstanding enantioselectivity (>99%ee). Subsequent experiments
revealed that the mount of phenol could be lowered to five equivalents with no
deleterious effects (Table 2.9). Below five equivalents, however, the reactions did not go
to cornpletion even after prolonged reaction times, although the enantioselectivity was
not advenely affected.

Table 2.9: Effects of Number of Equivalents of Phenol
THF I 80%
Phenol Equiv. Reaction Time yielda eeb
2.5 5 d 35%= >99%
1.5 5 d 1 o0/oC >99%
a lsolated yield. Ee determined by CSP HPLC. Remainder is unreacted starting material.
Because no improvement in reactivity was observed above five equivalents of phenol,
five equivalents was adopted as the standard conditions.
2.2.3 Scope of 4Substituted Phenols as Nucleophiles
Using these conditions, various para-substituted phenols were shown to add in high
yields and excellent enantioselectivity (Table 2.10). The reaction proceeded well even
when aryl bromides and iodides were used indicating that the rhodium insertion into the
aryl halide bond is slow compared to ring opening. This selectivity permits the
preparation of various halo aryl ethen with which further coupling reactions could be

perfonned. An X-ray crystal structure of 87 proved the regiochemistry, the relative
stereochemistry, and the absolute configuration of the ring opened products.
Table 2.10: Scope of AR0 withp-Substituted PhenoIs
THF I 80°C
Phenol (X) Product Yield(%)' ee(o/lb
F 85 92% 97%
Cl 86 89% 92%
Br 87 94% 98%
I 88 92% 98%
COCH3 89 91 Oh >9g0h
CF3 90 87% 95%
CH3 91 60% 91%
-C N 92 88% 97%
OMe 93 85% 95%
a lsolated yields. Ee datemined by CSP HPLC or formation Mosher's Ester.
We obsewed significant differences in the relative rates of reaction, with the more
acidic phenols adding faster. Similar observations were made by Sinou of the reactivity
of phenols in allylic ethenfication under palladium ~ a t a l ~ s i s . 8 ~ In order to quanti& these
observations, we conducted a competition experirnent using equholar amounts 4-
hydroxyacetophenone and Phydroxyanisole. At 68% conversion we observed a 16: 1

ratio of 89:93 (Scheme 2.6) confming that the presence of an electron withdrawing
group on the aromatic ring accelerates the rate of addition.
0.5 mol% [Rh(COD)C1I2
1 mol% PPF-P'BU* -xy--& 0" p 4-MeOCC6H40H (Seq.) OH 4-MeOC6H40H (5eq.) 89: X = COMe 64% THF 1 80°C 93: X = OMe 4%
68% Conversion
Scheme 2.6
2.2.4 Development of New Catalyst System for O-Halo Phenols
We next examined the effect of other substitution patterns on the reactivity of the
phenol. In the case of a bromo substituent, 3- and 4-bromophenol added in high yields
and excellent enantioselectivity (Table 2.1 I j, but 2-bromophenol did not give satisfactory
Table 2.11: Effect of Phenol Substitution Pattern
THF 1 80°C
Phenol Product Yield(%)= ee(%lb
a lsolated yield. Ee determined by CSP HPLC. After 24hr reaction time. Remainder is unreacted starting material.

results adding in only 17% yield after prolonged reaction tirnes. Despite the low
conversion, the enantioselectivity was still very hi&, with 95 being produced in 97%ee.
Changing the rhodium source to [R~I(CO)~CI]~, increased the yield of the reaction with
2-bromophenol to 92% and had no detrimental effects on the enantioselectivity (Table
2.12). It is noteworthy that [Rh(C0)2C1]2 is not compatible with DPPF under these
conditions due to the formation of an insoluble precipitate on mixing the phosphine and
the rhodium together."
Table 2.12: Effeceet of Changing the Rhodium Source
THF i 80°C
Rh Source Ligand Reaction Tirne ~ield'(~hee~)
[Rh(cm )al2 OPPF 24 hr 40%
[Rh(C0)2Clb OPPF 24hr No ~eaction*
[ W W z C l h PPF-P'BU~ 24 hr 92% (97%ee)
a lsolated yield. ' Ee detenind by CS? HPLC. ' Remainder is unreacted starting material. An insoluble precipitate multed upon mixing DPPF with the Rh source which did not disotve.
The enantioselectivity with [Rh(COD)Cll2 I PPF-PtBu2 with 2-bromophenol is similar
to that observed for 3- and 4bromophenol which suggests that the catalytically active
complex is the same in each case. We reasoned that the poor yield with [Rh(COD)C1I2

might be due to the rhodium being sequestered from the catalytic cycle by reversible
bidentate binding of the 2-halophenol through the oxygen and the bromine atoms. Such a
binding pattern has been invoked by ~ o ~ o r i " for the rutheniun catalysed asymmetric
hydrogenation of O-bromoacetophenone and by ~ i l l s ' ~ for the rhodium catalysed
asymmetric hydrosilation of O-chloroacetophenone and O-bromoacetophenone (Scheme
2.7).
RU (X)2(B INA?)
ux diphosphine ligand Ph2SiH2 1 O'C
X = H No Reaction X = Br 97%{92%ee) m, p-Br also gave no rxn
Scheme 2.7
[Rh(C0)2C1]2, upon mixing with certain classes of diphosphines, is known to produce
complexes in which one of the carbonyls remains bound? As a result, the [Rh(C0)2C1]2 /
PPF-ptBu2 catalyst system might have one less vacant coordination site compared to the
[Rh(COD)C1I2 system. This difference could serve to disfavour bidentate phenol binding
that occurs and thus increase the amount of the catalytically active species.
2.2.5 Application of Ring Opened Products Towards the Preparation of Benzofurans

The ring opened products 95 should be usefül for M e r transformations such as
coupling reactions and cyclisations. To demonstrate this concept, 95 was treated with
B u & H and AIBN under high dilution conditions to provide the cyclised bernofuran
product 96 in 86% yield as one diastereomer (stereochemistry not confirmed) (Eq. 2.3).
The benzohiran skeleton can be found in several biologically interesting pharrnaceutical
agents and such radical cyclisation reactions provide a facile and attractive route to these
skeletons in enantioenriched form.
9 O" (2.3) PhH
Br OH 55Oc H OH
96 86% (one diastereorner)
2.2.6 Conclusion
In conclusion, we have demonstrated that phenols are a usefûl class of nucleophiles for
the rhodium catalysed AR0 of oxabenzonorbomadiene. While 3- and 4-substituted
phenols can be added using the previousiy reported [Rh(COD)C1I2 1 PPF-PtBu2 catalyst
system, the extension to 2-halophenols required a change in the rhodium source to
w(C0)2C1]2. We propose that the presence of the additional CO ligand on the rhodium
prevents the poisoning of the catalyst caused by reversible bidentate binding of the
halophenol. The hydronaphthalene and the tetracyclic benzofuran products both belong
to biologically important classes of compounds.

2.2.7 Experimental
84
(1S,2S)-2-Phenoxy-l,2-dihydro-naphthalen1-01 (84): To a flame dned round bottom
flask, [Rh(COD)C1]2 (1.7 mg, 0.0035 mmol), (S)-(R)- PPF-PLBu2 (3.8 mg, 0.0069 mmol,
and 42 (100 mg, 0.694 rnrnol) were added. THF (2 mL) and phenol(327 mg, 3.47 mmol)
were then added followed by heating to 80°C for 1.5 hours. The reaction mixture was
then poured in to ether and washed three times with 5% aqueous NaOH. The aqueous
layen were combined and back extracted three times with ether. The organic layers were
combined, washed with bnne, dried over Na2SO4, and concentrated in vacuo. The
resulting solid was purified by flash chromatography (20% ethyl aceiate in hexanes) to
give 84 as a white crystalline solid (130.7 mg, 83%). The ee was determined to be 99.2%
using HPLC analysis on a CH[RALCEL OD column, h= 486 m. Retention times in 4%
isopropanol in hexanes were 15.2 min (major) and 17.8 min. Rf = 0.26 on silica gel (10%
ethyl acetate:hexanes); mp 10% 1 1 O°C (EtzO); [alZo= +204.7' (c= 10.1, CHC13); IR
(KBr, cm*') 3337 or), 3029 (w), 2866 (w), 1600 (m), 1496 (s), 1249 (s), 1062 (s); 'H
NMR (400 MHz, CDCl3) 6 7.65-7.63 (lH, m), 7.33-7.25 (4H, m), 7.13-7.1 1 (lH, m),
7.01-6.95 (3H, m), 6.51 (lH, dd, J= 9.9, 1.6 Hz), 6.02 (lH, dd, J= 9.9,2.2 Hz), 5.19 (lH,
d, J= 10.4 Hz), 5.11 (1H, ddd,J= 10.4,2.0, 2.0 Hz), 2.66 (lH, s); ' 3 ~ NMR (400 MHz,

CDCb) 6 157.4, 135.5, 131.9, 129.7, 129.0, 128.2, 128.0, 126.4, 126.1, 125.2, 121.5,
115.9,79.1, 72.4. HRMS calcd for C&14O2 (hll): 238.0994. Found: 238.0984.
(1S,2S)-2-(4-fluorophenoxy)-1,2,-dihydro-naphthalen-l-ol (85): To a flame dried
round-bottomed flask, [Rh(C0D)C1l2 ( 1.7 mg, 0.003 5 rnmol), (S)-(R)- PPF-PtBu2 (3.8
mg, 0.0069 mmol) and 42 (100 mg, 0.694 mmol) were added followed by addition of
THF (2.5 mL) and Cfluorophenol(389 mg, 3.47 mmol). The mixture was heated at 80°C
for 5 hours, then poured into diethyl ether and extracted 3 times with IO% aqueous
sodium hydroxide solution. The aqueous extracts were combined and back-extracted
three times with diethyl ether. The combined ether extracts were washed with brine and
dried with anhydrous sodium sulfate. The solvents were removed N, vacuo, yielding a
solid which was purified by flash chromatography on silica gel (10% ethyl acetate in
hexanes) giving a white crystalline solid 85 (163 mg, 92%). The ee was determined to be
97% by HPLC anaiysis on a CHIRALCEL OD column, h= 486 m). Retention times in
1.5% isopropanol in hexanes were 28.1 min (major) and 29.5 min. Rr = 0.39 on silica
(20% ethyl acetate in hexanes); mp 127-12g°C (Et20); = +216' (c = 9.5, CHCI,).
IR (KB~, cm') 3309 @), 3071 (w), 2864 (w), 1504 (SI, 1284 (ml, 1052 (s), 781 (s), 692
(m); 'H NMR (400 MHz, mCl3): 6 7.63-7.6 1 (IH, m), 7.3 1-7.26 (2H, m), 7.12-7.10

(IH, m), 7.00-6.95 (2H, m), 6.92-6.88 (2H, m), 6.51 (IH, dd, J= 2.1, 9.9 Hz), 5.98 (lH,
dd, J= 2.2, 9.9 Hz), 5.15 (lH, dd, J= 3.6, 10.0 Hz), 5.01 (lH, ddd, J= 2.1,2.1, 10.1 Hz),
2.54 (lH, d, J= 3.8 HZ); "C NMR (400 MHz, CDC13): 6 157.6 (d, FF= 239 HZ), 156.4,
153.4, 135.4, 13 1.8, 129.1, 128.2, 126.5, 125.7, 125.2, 117.5 (d, pF= 8 HZ), 116.1 (d, f- - 23.5 Hz);. KRMS calcd for (M?) (C,,H,,02F): 256.08 10. Found: 256.09 1 1.
86
(1S,2S)-2-(4-chlorophenoxy)-1,2,-dihydro-phthaIen-o (86): To a flame dried
round-bottomed flask, [Rh(COD)C1I2 (1.7 mg, 0.0035 mmol), (S)-(R)- PPF-P'Bu? (3.8
mg, 0.0069 mmol) and 42 (100 mg, 0.694 mmol) were added followed by addition of
THF (2.5 mL) and 4-chlorophenol (446 mg, 3.47 rnrnol). The mixture was heated at
80°C for 6 hours, then poured into diethyl ether and extracted 3 tirnes with 10% aqueous
sodium hydroxide solution. The aqueous extracts were combined and back-extracted
three times with diethyl ether. The combined ether extracts were washed with brine and
dried wiîh anhydrous sodium sulfate. The solvents were removed in vacuo, yielding a
solid which was purîfied by flash chrornatography on silica gel (5% ethyl acetate in
hexanes) giving a white crystalline solid 86 (169 mg, 89%). The ee was determined to be
92% by formation of Mosher's ester. Rr = 0.47 on silica (20% ethyl acetate in hexanes);
rnp 125-1255°C (&O); [a12'~ = +150° (c = 10.6, CHCb). IR ( D r , cm-') 3302 @r),
3064 (w), 2874 (w), 1590 (m), 1489 (s), 1362 (w), 1230 (s), 1052 (m), 890 (w), 846 (rn),

778 (s), 663 (m); 'H NMR (400 MHz, CDC13): 6 7.65-7.64 (lH, m), 7.33-7.26 (4H, m),
7.16-7.13 (lH, m), 6.91(1H, ddd, J= 2.0,2.0,8.9 Hz), 6.55 (lH, dd, J= 1.8,g.g Hz), 5.99
(lH, dd, J= 2.2,9.9 Hz), 5.19 (IH, dd, J= 3.8, 10.0 Hz), 5.07 (lH, ddd, J= 2.0, 2.0, 10.1
Hz), 2.56 (lH, d, J= 4.0 Hz); "C NMR (400 MHz, CDCI,): 6 155.8, 135.2, 131.7, 129.5,
129.3, 128.2, 128.1, 126.5, 126.2, 125.3, 125.2, 116.9, 79.2, 72.1. HRMS calcd for (M-
~ ~ 0 ) ' (C',Hl ,OCl): 254.0498. Found: 254.0499.
87
(1~2R)-2-(4-Bromo-phenoxy)-1,2-dihydro-naphthalen-lsl (87): To a flarne dned
round bottom flask, [Rh(COD)C1]2 (2.1 mg, 0.0043 mmol), (R)-(S)- PPF-PLBu2 (4.6 mg,
0.0085 rnmol, and 42 (122 mg, 0.85 mmol) were added. ). THF (2 mL) and p-
bromophenol(734 mg, 4.245 rnrnol) were then added followed by heating to 80°C for 1.5
hours. The reaction mixture was then poured in to ether and washed three times with 5%
aqueous NaOH. The aqueous layers were combined and back extracted three times with
ether. The organic layers were combined, washed with brine, dried over Na2S04, and
concentrated Ni vacuo. The resulting solid was purified by flash chromatography (20%
ethyl acetate in hexanes) to give 87 a white crystalline solid (239.7 mg, 90%). The ee
was determined by debrominating 87 (40 mg, 0.1 1 mmol) by reaction with t-BuLi (0.32
mL, 1.7M) in diethyl ether (2 mL) at -78OC followed by quenching with isopropanol.
Extraction with ether fiom water, washing with brine, drymg over anhydrous sodium

sulfate and removal of the solvents in vacuo gave a white crystalline solid 84 (24 mg,
92%). The ee was determined to be 96.8% by HPLC analysis on a CHIRALCEL OD
column, h= 48611x11. Retention times in 4% isopropanol in hexanes were 15.2 min and
17.5 min (major). RF 0.26 on silica gel (10% ethyl acetate:hexanes); mp 145-146'
(Et2O); [a12'o= - 135.7" (c= 10.2, CHC13); IR (KBr, cm") 3290 (br), 3060 (rn), 2870 (w),
1583 (m), 1484 (s), 1227 (s), 1052 (m), 980 (s), 776 (s); 'H NMR (400MHz, CDCl,) 6
7.70-7.65 (IH, m), 7.44-7.42 (2H, m), 7.35-7.32 (2H, m), 7.18-7.16 (1H, m), 6.88-6.86
(2H, m), 6.56 (lH, dd, J= 10.0, 2.0 Hz), 6.00 (IH, dd, J= 9.7, 2.2 Hz), 5.20 (lH, dd, J=
9.7, 3.6 Hz), 5.09 (lH, ddd, J= 10.0, 2.0, 2.0 Hz), 2.70 (lH, d, J= 3.9 Hz); I3c NMR
(400MHz. CDC13) 6 156.5, 135.3, 132.5, 131.7, 129.3, 128.3, 128.1, 126.5, 125.3, 117.6,
113.7, 79.4, 72.2. HRMS calcd for Ci& [OBr (M-H20)' 297.9994. Found: 297.9995.
(1SJS)-2~4-iodophenoxy)-1,2,-dihydro-naphthalen-l-o1 (88): To a flame dried
round-boîîomed flask, m(COD)C1]r (1.7 mg, 0.0035 mmol), (S)-(R)- PPF-P'BU~ (3.8
mg, 0.0069 mmol) and 42 (100 mg, 0.694 mmol) were added followed by addition of
THF (2.5 mL) and 4-iodophenol(763 mg, 3.47 mmol). The mixture was heated at 80°C
for 12 hours, then poured into diethyl ether and extracted 3 times with 10% aqueous
sodium hydroxide solution. The aqueous extracts were combined and back-extracted
three times with diethyl ether. The combined ether extracts were washed with brine and

dried with anhydrous sodium sulfate. The solvents were removed in vacuo, yielding a
solid which was purified by flash chromatography on silica gel (10% ethyl acetate in
hexanes) as a white crystalline solid 88 (193 mg, 73%). The ee was determined by
deiodinating 88 (40 mg, 0.1 1 mmol) by reaction with t-BuLi (0.32 rnL, 1.7M) in diethyl
ether (2 mL) at -78OC followed by quenching with isopropanol. Extraction with ether
from water, washing with brine, drying over anhydrous sodium sulfate and removal of
the solvents in vacuo gave a white crystalline solid 84 (24 mg, 92%). The ee was
detemined to be 98% by HPLC analysis on a CHIRALCEL OD column, X= 256 m.
Retention tirnes in 4% isopropanol in hexanes were 15.2 min (major) and 17.9 min; Rf =
0.44 on siiica (20% ethyl acetate in hexanes); mp 160- 1 6Z0C (Et20); = + 107' (c =
9.7, CHCl3). IR (KBr, cm-') 3264 (br), 3050 (w), 2926 (w), 2843 (w), 1581 (m), 1485
(s), 1388 (w), 1279 (m), 1246 (s), 1046 (m), 824 (m), 780 (m), 571 (w); 'H NMR (400
MHz, CDCls): 6 7.63-7.6 1 (1 H, rn), 7.58-7.55 (2H, m), 7.30-7.27 (2H1 m), 7.13-7.1 1 (1 Hl
m), 6.73 (2H, ddd, J= 2.2, 2.2, 9.0 Hz), 6.52 (IH, dd, J= 1.8, 9.8 Hz), 5.96 (lH,dd, J=
2.2, 9.8 Hz), 5.16 (lH, d, J= 10.0 Hz), 5.05 (IH, ddd, J= 2.0, 2.0, 10.0 Hz), 2.54 (lH, s);
13c NMR (400 MHz, CDC13): 6 157.3, 138.5, 135.3, 131.7, 129.4, 128.3, 128.1, 126.6,
125.3, 125.3, 1 18.1, 83.6, 79.2, 72.2. H R M S calcd for (M-HlO)' (C,,H, ,OI): 345.9855.
Found: 345.9849,

COMe
( I S , 2 S ) - 2 - ( 4 - a c y l p h e n o x y ) - 1 , 2 , - d i h y d r o ~ I (89): To a flame dned round-
bottomed flask, [Rh(COD)C1I2 (1.7 mg, 0.0035 mmol), (S)-(R)- PPF-P'BU~ (3.8 mg,
0.0069 mmol) and 42 (100 mg, 0.694 rnrnol) were added followed by addition of THF
(2.5 mL) and 4-hydroxyacetophenone (472 mg, 3.47 mmol). The mixture was heated at
80' C for 2.5 hom, then poured into diethyl ether and extracted 3 times with 10%
aqueous sodium hydroxide solution. The aqueous extracts were combined and back-
extracted three times with diethyl ether. The combined ether extracts were washed with
bine and dried with anhydrous sodium sulfate. The solvents were removed in vaaio,
yielding a solid which was punfied by flash chromatography on silica gel (30% ethyl
acetate in hexanes) giving a white crystailine solid 89 (177 mg, 9 1%). The ee was
determined to be > 99% by formation of Mosher's ester; Ri = 0.28 on silica (30% ethyl
acetate in hexanes); mp 124126°C (EtzO); [ u ] ~ ' ~ = +153" (c = 9.8, CHCIj). IR ( U r ,
(m); 'H NMR (400 MHz, CDC13): 6 7.94 (2H, d, J=8.8 Hz), 7.66-7.64 (IH, m), 7.34-7.27
(2H, m), 7.16-7.14 (1H, m), 6.98 (2H, d, J= 8.8 Hz), 6.57 (LH, d, J= 9.9 Hz), 5.99 (lH,
d, J= 9.9 Hz), 5.21 (2H, s), 2.85 (1H, s), 2.56 (3H, s); "C NMR (400 MHz, CDC13): 6
196.8, 161.4, 135.3, 131.7, 130.7, 130.6, 129.6, 128.3, 128.1, 126.6, 125.4, 125.0, 115.2,
79.0,72.0,26.3. HRMS cakd for (M-HtO)' (Cl8HI4o2): 262.0994. Found: 262.0989.

90
(lS,ZS)- 2-(~~a)-tri8uoro-4-methylphenoxy)-l,2,-dihydro-naphthalen-l-ol (90):
To a tlarne dried round-bottomed flask, [IUi(COD)C1I2 (1.7 mg, 0.0035 mrnol), (S)-(R)-
PPF-PtBu2 (3.8 mg, 0.0069 mrnol) and 42 (100 mg, 0.694 rnrnol) were added followed by
addition of THF (2.5 mL) and (a,a.a)-trifluoro-p-cresol (563 mg, 3.47 mmol). The
mixture was heated at 80' C for 8 houn, then poured into diethyl ether and extracted 3
t h e s with 10% aqueous sodium hydroxide solution. The aqueous extracts were
combined and back-extracted three times with diethyl ether. The combined ether extracts
were washed with brine and dried with anhydrous sodium sulfate. The solvents were
removed in vacuo, yielding a solid which was punfied by flash chrornatography on silica
gel (10% ethyl acetate in hexanes) to give a white crystalline solid 90 (184 mg, 87%).
The ee was determined to be 95% by HPLC analysis on a CHIRALCEL OD column, h=
486 nm. Retention times in 4% isopropanol in hexanes were 14.8 min and 17.3 min
(major). Rr = 0.46 on silica (20% ethyl acetate in hexanes); mp 1 18-1 1 9'C (Et20); [afSD
= +178' (c = 9.6, CHC13). IR (KBr, cm-') 3360 (br), 306 1 (w), 2874 (w), 16 17 (m), 15 18
(m), 1326 (s), 1103 (s), 1051 (m), 839 (m), 782 (m), 745 (w); 'H NMR (490 MHz,
CDCb): 6 7.63-7.54 (lH, m), 7.55 (2H, d, J= 8.6 Hz), 7.33-7.24 (2H, m), 7.14-7.12 (IH,
m), 7.01 (2H, d, J= 8.6 Hz), 6.55 (IH, dd, J= 1.6,g.g Hz), 5.97 (lH, dd, J= 2.0,9.9 Hz),

5.2 1-5-13 (2H, m), 2.47 (1 H, d, J= 3.6 Hz); "C NMR (400 MHz, CDCI,): 6 159.9, 135.2,
131.7, 129.6, 128.4, 128.2, 127.1 (q, f-F= 3.6 Hz), 126.6, 125.4, 124.9, 123.4 (d, fF=
33.0 Hz), 122.9 (d, f-F= 27 1.6 Hz), 1 15.6, 79.1, 72.1; H R M S calcd for (MT
(C,,H,,0,F3): 306.0868. Found: 306.0852.
91
(1S,2S)-2w(4-methylphenoxy)-l,2,-dihydro-nap h thalen-1-01 (91): To a flame dried
round-bottomed flask, [Rh(COD)C1I2 ( 1.7 mg, 0.0035 mmol), (S)-(R)- PPF-PtBu2 (3.8
mg, 0.0069 mmol) and 42 (50 mg, 0.347 mmol) were added followed by addition of THF
(2.5 mL) and p-cresol (188 mg, 1.74 mmol). The mixture was heated at 80°C for 24
houn, then poured into diethyl ether and extracted 3 times with 10% aqueous sodium
hydroxide solution. The aqueous extracts were combined and back-extracted three times
with diethyl ether. The combined ether extracts were washed with brine and dned with
anhydrous sodium sulfate. The solvents were removed in vacuo, yielding a solid which
was purified by flash chromatography on silica gel (5% ethyl acetate in hexanes) giving a
white crystalline solid 91 (57 mg, 65%). The ee was determined to be 91% by HPLC
analysis on a CHIRALCEL OD column, b 2 5 6 nm. Retention times in 1% isopropanol
in hexanes were 33.8 min (major) and 37.1 min. Rr = 0.49 on silica (20% ethyl acetate in
hexanes); mp 80-8 1°C (&O); [a]% = +145' (c = 12.1, CHCG). IR (KBr, cm-') 3303
@r), 3050 (w), 2210 (m), 1598 (s), 1503 (s), 1238 (s), 1025 (m), 859 (m), 778 (m); 'H

NMR (400 MHz, CDCI,): 6 7.67-7.65 (lH, m), 7.33-7.28 (2H, m), 7.14-7.1 1 (3H, m),
6.88 (2H, d, J= 8.4 Hz), 6.51 (lH, dd, J= 1.8, 9.9 Hz), 6.04 (LH, dd, J= 2.0, 9.9 Hz),
5.20 (lH, dd, J= 1.6, 10.2 Hz), 5.09 (lH, ddd, J= 1.8, 1.8, 10.2 Hz), 2.87 (lH, d, J= 2.7
Hz), 2.33 (3H, s). 13c NMR (400 MHz, CDC13): 6 155.0, 135.4, 13 1.8, 130.7, 130.1,
128.8, 128.1, 127.9, 126.4, 126.2, 125.1, 115.6, 79.0, 72.3, 20.5. HRMS calcd for (v (C,,H,,O,): 252.1 150. Found: 252.1 140.
92
(lS,2S)-2-(4-cyanophen0xy)-1,2,-dihydro-naphthaen-l-o1 (92): To a ffame dn'ed
round-bottomed flask, m(COD)C1]2 (1.7 mg, 0.0035 mrnol), (S)-(R)- PPF-P'Bu2 (3.8
mg, 0.0069 rnmol) and 42 (100 mg, 0.694 m o l ) were added followed by addition of
THF (2.5 mL) and Ccyanophenol (413 mg, 3.47 mmol). The mixture was heated at 80°C
for 5 hours, then poured into diethyl ether and extmcted 3 times with 10% aqueous
sodium hydroxide solution. The aqueous extracts were combined and back-extracted
three times with diethyl ether. The combined ether extracts were washed with brine and
dried with anhydrous sodium sulfate. The solvents were removed Ni vacuo, yielding a
solid which was purified by flash chromatography on silica gel (30% ethyl acetate in
hexanes) giving a white crystalline solid 92 (160 mg, 88%). The ee was detennined to be
97% by HPLC analysis on a CHLRALCEL OD column, A= 256 nm. Retention times in
3% isopropanol in hexanes were 35.3 min and 37.7 min (major). Rf = 0.40 on silica (30%

ethyl acetate in hexanes); rnp 140-141°C (Et20); [a]"D = +182.3* (c = 11.2, CHCIi) IR
(KBr, cm-') 3303 (b) 3050 (w) 2210 (m) 1598 (s) 1503 (s) 1238 (s) 1025 (m) 859 (m)
778 (m); 'H NMR (400 MHz, CDCb): 6 7.62-7.57 (3H, m), 7.33-7.27 (3H, m), 7.14-7.12
(1H, m), 6.56 (IH, dd, J= 1.4, 9.7 Hz), 5.93 (1H, dd, J= 1.4,9.7 Hz), 5.20-5.13 (2H, m),
2.25 (1H, s). 13c NMR (400 MHz, CDC13): 6 160.8, 135.0, 134.2, 131.5, 130.0, 128.5,
128.3, 126.7, 125.4, 124.4, 119.0, 116.2, 104.6, 79.2, 72.0. HRMS calcd for (M-H20)'
(C ,,H, ,ON): 245.084 1. Found: 245 .O845.
OMe /
93
(1S,2S)-2-(4-methoxyphenoxy)-1,2,-dihydro-naphthaen--o (93): To a flame dried
round-bottomed flask, [Rh(COD)C1I2 (1.7 mg, 0.0035 mrnol), (S)-(R)- PPF-ptBu2 (3.8
mg, 0.0069 mrnol) and 42 (100 mg, 0.694 mmol) were added followed by addition of
THF (2.5 mL) and Cmethoxyphenol (43 1 mg, 3.47 mmol). The mixture was heated at
80°C for 6 hours, then poured into diethyl ether and extracted 3 times with 10% aqueous
sodium hydroxide solution. The aqueous extracts were combined and back-extracted
three tirnes with diethyl ether. The combined ether extracts were washed with bnne and
dned with anhydrous sodium sulfate. The solvents were removed in vaczio, yielding a
solid which was purified by flash chromatography on silica gel (10% ethyl acetate in
hexanes) as a white crystalline solid 42 (159 mg, 85%). The ee was detemiined to be
95% by HPLC analysis on a CHlRALCEL OD column, h= 256 m. Retention times in

4% isopropanol in hexanes were 22.1 min (major) and 25.9 min. Rr = 0.33 on silica (20%
ethyl acetate in hexanes); mp 91-92OC (Et20); [a]"* = +12g0 (c = 9.9, CHCL); IR (KBr,
cm*') 3349 (br), 3050 (w), 2822 (w), 1508 (s), 1233 (s), 1046 (m), 825 (m), 751 (rn), 695
(w); 'H NMR (400 MHz, CDC13): S 7.66-7.64 (IH, m), 7.30-7.27 (2H, m), 7.12-7.10
(IH, m), 6.91 (2H, ddd, J= 2.3, 2.3, 9.1 Hz), 6.84 (2H, ddd, J= 2.4, 2.4, 9.2 Hz), 6.49
(1H. dd, J=2.0, 9.9 Hz), 6.02 (lH, dd, J= 2.4. 9.9 Hz), 5.17 (lH, dd, J= 3.3, 10.1 Hz),
5.02 (lH, ddd, J= 2.0, 2.0, 10.3 Hz), 3.77 (3H, s), 3.12 (lH, d, J= 3.4 Hz). I3c NMR
(400 MHz, CDC13): 6 154.3, 151.2,135.5, 131.9, 128.7, 128.1, 127.9, 126.4, 126.3, 125.2,
1 17.2, 1 14.8, 80.0, 72.4, 55.7. H R M S calcd for ( M 3 (C,,H,,O,): 250.0994. Found:
94
(1S,2S)-2-(3-bromophenoxy)-l,2,-dihydro-naphthIenl-o1 (94): To a flame dried
round-bottomed fiask, [Rh(COD)C1I2 (1.7 mg, 0.0035 mrnol), (S)-(R)- PPF-PLBu2 (3.8
mg, 0.0069 mmol) and 42 (100 mg, 0.694 mmol) were added followed by addition of
THF (2.5 mL) and 2-bromophenol (0.40 mL, 3.47 rnmol). The mixture was heated at
80°C for 24 hours, then poured into diethyl ether and extracteci 3 times with 10% aqueous
sodium hydroxide solution. The aqueous exûacts were combined and back-extracted
three times with diethyl ether. The combined ether extracts were washed with brine and
dried with anhydrous sodium sulfate. The solvents were removed in vacuo, yielding a

solid which was purified by flash chromatography on silica gel (5% ethyl acetate in
hexanes) as a white crystalline solid 94 (200 mg, 92%). The ee was determined to be
96% by HPLC analysis on a CHIRALCEL OD colurnn, h= 486 nm. Retention times in
1.5% isopropanol in hexanes were 22.8 min and 32.1 min (major). Rr = 0.44 on silica
(20% ethyl acetate in hexanes); mp 120- 122'C (EtzO); [a]25D = +254' (c = 9.2, CHC13).
IR (KBr, cd') 3341 (br), 3071 (w), 2884 (w), 158 1 (m), 1472 (s), 1358 (m), 1237 (s),
1028 (s), 987 (s), 780 (s), 689 (m), 569 (m); [H NMR (400 MHz, CDC13): 6 7.57-7.62
(lH, m), 7.22-7.30 (2H, m), 7.14-7.08 (4H, m), 6.82-6.88 (lH, m), 6.49 (1H, dd, J= 1.4,
9.9 Hz), 5.94 (lH, dd, J= 2.1, 9.9 Hz), 5.13 (lH, dd, J= 2.9, 9.9 Hz), 5.03 (IH, ddd, J=
1.9, 1.9, 9.9 Hz), 2.76 (lH, d, J= 3.6 Hz). NMR (400 MHz, CDC13): 6 158.1, 135.3,
131.7, 130.7, 129.6, 128.3, 128.1, 126.5, 125.3, 125.3, 124.5, 122.9, 119.2, 114.5, 79.3,
72.1. HRMS calculated for ( M - ~ ~ 0 ) ' (C,,H, ,OBI-): 297.9993. Found: 297.9976.
95
(1S,2S)-2-(2-bromophenoxy)-1,2,dihydro-aphtalen-o1 (95): To a flame dried
round-bottomed flask, [Rh(C0)2C1I2 (1.5 mg, 0.0035 m o l ) , (S)-(R)- PPF-PLBu2 (3.8 mg,
0.0069 rnmol) and 42 (100 mg, 0.694 rnmol) were added followed by addition of THF
(2.5 rnL) and 2-bromophenol(0.40 rnL, 3.47 mmol). The mixture was heated at 80°C for
24 hours, then poured into diethyl ether and extracted 3 times with 10% aqueous sodium
hydroxide solution. The aqueous extracts were combined and back-extracted three times

with diethyl ether. The combined ether extracts were washed with brine and dried with
anbydrous sodium sulfate. The solvents were removed NI vacuo, yielding a solid which
was purified by flash chromatography on silica gel (5% ethyl acetate in hexanes) as a
white crystalline solid 95 (206 mg, 94%). The ee was determined to be 97% by HPLC
analysis on a CHIRALCEL OD column, h= 486 nrn. Retention times in 1.5%
isopropanol in hexanes were 22.8 min and 32.1 min (major). Ri = 0.44 on silica (20%
ethyl acetate in hexanes); mp 120-122°C (EtzO); = +254' (c = 9.2, CHCI,). IR
(s), 987 (s), 780 (s), 689 (m), 569 (m); 'H NMR (400 MHz, CDCI,): 6 7.67 (lH, d J= 6.8
Hz), 7.58 (IH, dd, J= 1.5, 7.9 Hz), 7.33-7.23 (3H, m), 7.14-7.12 (lH, m), 6.95(1H, dd,
J= 1.1, 8.2 Hz), 6.92-6.87 (lH, m), 6.52 (lH, dd, J= 2.0, 9.9 Hz), 6.06 (IH, dd, J= 1.8,
9.9 Hz), 5.32 (IH, d, J= 11.0 Hz), 5.10 (IH, ddd, J= 2.0, 2.0, 11.0 Hz), 2.85 (IH, d, J=
3.2 Hz). I3c NMR(400 MHz, CDC13): 6 154.3, 135.4, 133.6, 131.8, 129.1, 128.6, 128.3,
128.0, 126.4, 126.0, 124.9, 122.9, 115.6, 113.5, 82.2. 72.5. HRMS calculated for (M-
HzO)' (C,,H, ,OBr): 297.9993. Found: 297.9976.
5a,6,11,1 la-Tetrahydro-benzo[b] nap htho[2 ,3-d] furan (96): To a flame dried
round bottm fiask fitted with a condenser was added 50mg 95, 65mg tributyltin hydnde
and 26mL benzene. The solution was heated to 55OC then lOmg AIBN was added Mer

5 hours, the reation mixture was cooled, and 50 ml o f a 5% NaOH solution was added.
Extraction with ether, concentration and chromatography (20%EtOAc in Hexanes) gave a
white crystaline solid 96 (31mg, 86%). Rf = 0.33 on silica (20% ethyl acetate in
hexanes); mp 1 74- l7g0C (CDCL); [a]'% = +8 1 (c = 9.2, CHCI,). IR ( D r , cm'') 334 1
(br), 3071 (w), 2884 (w), 1581 (rn), 1472 (s), 1358 (m), 1237 (s), 1028 (s), 987 (s), 780
(s), 689 (m), 569 (m); 'H NMR (400 MHz, CDC13): 6 7.60 (1H. d, J= 7.3 Hz), 7.12-7.35
(SH, rn), 6.90 (lH, m), 6.81 (lH, d, J= 8.0 Hz), 4.87 (1H. d, J= 7.3 Hz), 4.80 (lH, t, J=
10.0 Hz), 3.83 (IH, dt, J= 7.0,9.7 Hz), 3.30 (IH, dd, AB, J= 6.8, 14.5 Hz), 2.74 (lH, dd,
AB, J= 9.9, 14.3 Hz), 2.70 (lH, s). ')c NMR (400 MHz, CDC13): 6 159.2, 136.9, 135.8,
129.8, 128.6, 127.4, 127.0, 126.9, 124.3, 124.2, 120.9, 109.7, 88.1. 71.2. 40.4, 33.4.
HRMS calculated for (M)' (C ,,H,,02): 238.0994. Found: 238.099 1.

2m3 Nitrogen Nucleophiles
2.3.1 Introduction
Pnor to the initiation of our studies in the area of asymmetnc C-N bond formation
through the AR0 of oxabicyclic alkenes, no reports of rhodium catalysed asymmetric C-
N bond formation had been reported. During the course of our studies, P.A. Evans
reported an enantiospecific allylic amination reaction which could produce allyl amines
in >90%ee starting fIom enant ioe~ched allyicarbonates (vide supra). Given the dearth
of chemistry dealing with rhodium catalysed amination reactions, we felt that t h s work
had the potential to be highly rewarding. Furthemore, the aminohydronaphthalene
skeleton is found in several natural and pharmaceutical compounds. An efficient,
enantioselective method for their preparation would be useful.
2.3.2 AR0 with Activated Nitrogen Nucleophiles
It has been previously shown that sulfonarnide salts react with allyl carbonates under
rhodium catalysis to give retention of absolute configuration (vide supra). While these
sulfonamide salts were not compatible with Our conditions (Table 2.13, Entry I), the use
of five equivalents of benzene sulfonarnide gave the desired product in good yield (Entry
2). ui addition to benzene sulfonamide, we found that a wide variety of aromatic amines
are compatible as is phthalirnide. Phthalimide is particularly usefûl since it can be used
to access the free amine. The tram stereochemistry was proven for the N-methylaniline
adduct by X-ray crystalography.

We have conducted prelirninary studies on the AR0 of 42 with nitrogen nucleophiles and
obtained encouraging results. Ring opening with toluene sulfonamide and PPF-P'Bu?
occurs in 86% yield and 95% ee (Table 3). With other nucleophiles, BPPFA 102 gives
better results than PPF-P'BU~ 72 (entries 2 to 4) so we anticipate that further modification
of the ligand will lead to high ee's.
Table 2.13: Rhodium Catalysed Ring Opening with Nitrogen Nucleophiles
[Rh(C00)2C1]2 (2.5mol%) DPPF (1 eq. to Rh)
Z
RORN\" THF 80'~ (or reflux)
Entry Amine Nucleophile Product Yield
3 PhNHMe 98 95%

BPPFA
Tabie 2.14: Rhodium Catiiiysed AR0 with Xitrogro Xucleophiles
[Rh(C00)2Cl]2 (2.5mo1°h) Ligand (1 eq. to Rh)
Nudeophile (5 eq) - N u c'" THF 1 80'~ (or reflux)
Entry Nucleophile PPF-P'BU~ BPPFA
1 PhS02NH2 86% (95%ee) - 2 Phthalimide 83% (45%ee) 64% (74%ee)
3 lndole 87% (73%ee) 81 % (79%ee)
4 PhNHMe 96% (74% ee) 94% (74%ee)
Thus, benzene sulfonamide can be added in 95%ee and aromatic amines can be used with
ee's up to 79%. We are currently performing screening experiments to detennine the
optimal ligand for this transformation.
2.3.3 AR0 with Unactivated Aliphatic Nucleophiles
While aromatic amines and sulfonamides efficiently induce ring opening of
oxabenzonorbomadiene 42, aiiphatic amines fail under similar conditions. This poor
reactivity was found to be me for a wide range of aliphatic amines. For example, while

N-methylaniline is a good nucleophile giving 98 in 94% yield, pyrrolidine does not
induce ring opening (Table 2.15). A cornpetition experiment sheds light on the nature of
the poor reactivity of aliphatic amines. When equimolar amounts of N-methylaniline and
pyrrolidine are added, neither nucleophile reacts indicating that the more basic aliphatic
amine poisons the catalyst. Since amines are known to be good ligands for rhodium,87 it
seems likely that the catalyst is being "poisoned" by irreversible binding of the amine to
the metal center. This mode of binding is either inefficient or is revenible with the less
basic aromatic amines.
Table 2.15: Effect of Aliphatic Amines on Reactivity
[R~I(COD)~CI]~ (2.5mol%) - DPPF (1 eq. to Rh) R.
THF / 80% (or reflux) 42
a OH
Entry Nucleophile Product Yteld
1 PhNHMe (5eq) 98 94%
2 Pyrrolidine (5eq) NR
PhNHMe (5eq) and . Pyrmlidine (5eq) NR
After significant experimentation, we found that the addition of a protodhaiide source to
the reaction mixture produced beneficial effects. For example, use of equimolar amounts
of pyrrolidine and tnethylarnine hydrochlonde, showed dramatic irnprovernents, giving
103 in greater than 85% yield (Table 2.16). Analogously, the use of equimolar amounts
of pyrroliche hydrochloride and triethylamine (Entry 3) or pyrrolidine hydrochloride and
pyrrolidine (Entry 4) showed similar reactivity, again giving the desired product in 83

and 89% yield respectively. In contmst, pyrrolidine hydrochloride alone gave no reaction
indicating that not al1 the amine can be tied up as the hydrochloride salt. Other rnethods
to effect this same transformation are to add HCI or TsOH to an excess of the amine
nucleophile. The reaction with TsOH is significantly slower than those where chloride
ions are present indicating that both the halide and the protons may be involved in the
reversal of catalyst poisoning. PPTS is not a compatible proton source, likely due to
catalyst poisoning arising fom pyridine binding to the rhodium metal.
Table 2.16: Effect of an Added Proton Source on the Reactivity of Aliphatic Amines
DPPF (1 eq. to Rh) Nudeophile Proton Source
42 THF 1 80°C (or reff ux)
Entry Nucleophile Yield
2 Pyrrolidine (5eq) l Et3N.HCI(5eq) 85%
4 Pyrrolidine (5eq) 1 Pyrrolidine.HCI (Seq) 89%
5 Pyrrolidine (1 Oeq) 1 HCI (Seq)
6 Pyrrolidine (1 Oeq) / TsOH (5eq) 81 %
Pyrrolidine (1 Oeq) l NR PPTS (5eq)
This effect of an added protonhalide source was found to be generally applicable for the
addition of a wide varïety of secondary aliphatic mines giving (Table 2.17). The tram
relative stereochemistry was proven for 105 by X-ray diffraction. Reactions with

benylamine and p-methoxybenzylamine did not proceed to completion even after
prolonged reaction times (Entries 4 and 5). We surmise that N,O-bidentate binding of the
ring opened product to rhodium can occur when a prirnary amine in used thus
sequestenng the rhodium fiom the catalytic cycle.
Table 2.17: Use of a Proton Source to Induce Ring Opening with Aiiphatic Amines
DPPF (1 e q to Rh) Amine (5 eq) R .,,,.w & N . H ~ (5-eq)
42 THF I 8 0 ' ~ (or reflux) a ÔH
Entry Nucleophile Product Yield
3 Piperidine 1 06 83%
"~emainder is unreactad starting malenal.
The asymmeûic version of this reaction was also explored and the prelirninary results are
encouraging. In our previously reported studies of alcohol and phenol nucleophiles, we
found that the use of PPF-P'BU~ 72 gave the best results. With aliphatic amines and
carboxylate nucleophiles, BPPFA 102 gave much better results (Table 2.18). Through
further ligand tuning, higher ee's should be possible.

Table 2.18: AR0 of O~abenzonorbornadiene with Nitrogen and Carboxylate Nucleop hiles
[Rh(COD)2Cf12 (2.5rnol%) Ligand (1 eq. to Rh)
e
N u c"' THF 8 0 ' ~ (or reflux) OH
Bn2NH - 83%(72%ee)
2 Piperidine 68%(8%ee) 82%(74%ee)
Conditions: 5 equiv. amine nucleophile with 5 equiv. ESN.HCI
2.3.4 Conclusion
In conclusion, we have shown that sulfonamides and aromatic amines are compatible
nucleophiles for the AR0 of oxabenzonorbomadiene. We have also discovered
conditions which take advantage of a previously observed pKa eflect and permit the use
of aliphatic amines which are otherwise unreactive nucleophiles.
2.3.5 Experimental
97
(1S,2S)-N-(l-Hydroxy-1,2-dihydro-naphthaIen--y)beene sulfonamide (97): To a
flame dried round bottorn flask, [Rh(COD)C1I2 (4.3 mg, 0.0087 rnmol), (S)-(R)-PPF-

P'BU~ (9.4 mg, 0.0173 mmol), benzenesulfonamide (545 mg, 3.47 mmol) and 42 (100
mg, 0.69 rnrnol) were added. THF (2 rnL) was then added, followed by heating to 80°C
for 12 hours. The reaction mixture was then poured into water and extracted three times
with ethyl acetate. The organic layee were combined, washed with brine dried over
Na2S04, and concentrated in vacuo. The resulting solid was purified by flash
chromatography (30% ethyl acetate in hexanes) to give 97 a white crystalline solid (223
mg, 96%). The ee was detemined to be 95% by Mosher's ester formation and HPLC
analysis on a CHIRALCEL OD colurnn, )c= 486nm. Retention times in 10% isopropanol
in hexanes were 26.6 min (major) and 39.4 min. RF 0.22 on silica gel (30% ethyl
acetate:hexanes); mp 128- 1 30° (dec); [alBD= 70 O (c= 8.3, CHU3); R (KBr, cm-') 3462
(br), 3200 (m), 2957 (w), 1447 (m), 1329 (m), 1329 (m), 1164 (s), 1093 (m). 'H NMR
(400MHz, CDC13) 6 7.9 1-7.90 (2H, m), 7.62-7.58 ( 1 H, m), 7.54-7.50 (2H, rn), 7.47-7.45
(iH, m), 7.27-7.23 (2H, m), 6.40 (lH, dd, J= 9.7, 1.7 Hz), 5.55 ( lH, dd, J= 9.7, 3.1 Hz),
5.26 (IH, s), 4.77 (lH, d, J= 8.8 Hz), 4.13-4.07 (iH, m), 2.91 (IH, s); "C NMR
(400MHz, CDCl,) G 140.2, 134.9, 132.9, 131.3, 129.5, 129.2, 128.4, 128.4, 127.1, 126.4,
126.0, 72.0,56.3. HRMS calcd for Cia i5NOsS (m: 30 1 .O773. Found: 301 .O769.
98
(1~2R)-2-(Methyl-phenyl-amino)-1,2aihydronphaIen--ol (98): To a Bame
dned round bottorn flask, [Rh(COD)C1]2 (3.5 mg, 0.007 mmol), (R)-(S)-BPPFA (7.7 mg,
0.014 mmol), N-methylanihe (372 mg, 3.47 mmol), 42 (105 mg, 0.728 mmol) and THF

(3 mL) were added followed by heating to reflux for 3 hours. The solvent was then
removed in vacuo and the resulting oil purified by flash chromatography (5% ethyl
acetate in hexanes) to give 98 a white crystalline solid (176.3 mg, 96%). nie ee was
determined to be 74% using HPLC analysis on a CHIRALCEL OD column, h= 254x1111.
Retention times in 10% isopropanol in hexanes were 1 1.1 min (major) and 13.3 min. RF
0.41 on silica gel (20% ethyl acetatehexanes); mp 55-56' (EtzO); 50.4' (c= 1 1.8,
CHC13); IR (KBr, cm") 3594 (br), 3037 (m), 2884 (m), 1596 (s), 1503 (s), 1463 (m),
1 186 (m), 935 (m). 'H NMR (400MHz, CDC13) 6 7.57-7.55 (lH, m), 7.3 1-7.26 (4H, m),
7.15-7.13 (lH, m), 6.99-6.97 (2H, m), 6.84-6.81 (1H, m), 6.61 (IH, dd, J= 9.8, 2.6 Hz),
5.94 ( lH, dd,J= 9.7.2.9 Hz), 5.1 1 (lH, d, J= 9.8 Hz), 4.76 (lH, ddd, J= 9.7,2.6, 2.6 Hz),
2.85 (3H, s), 2.50 (lH, s); "C NMR (400MHz, CDC13) 6 150.1, 136.4, 131.9, 129.6,
129.2, 128.0, 127.8, 127.7, 126.4, 125.5, 118.0, 114.5, 70.0, 63.3, 33.3. E-iRMS calcd for
Ci7Hi7N0 (M?): 251.1310. Found: 251.1307.
99
(1~2R)-2-(3,4-Dihydro-ZH-quinoLin-I-y1)-I,2-dihydro-naphtha1en-l-o1 (99): To a
flame dned round bonom flask, m(COD)C1]2 (4.3 mg, 0.0087 mmol), (R)-(S)-BPPFA
(9.6 mg, 0.0 173 mmol), tetrahydroisoquinoline (23 1 mg, 1.735 mmol), 42 (60 mg, 0.4 16
mmol) and THF (2.5 mL) were added followed by heating to reflux for 3 hours. The
solvent was then removed in vacuo and the resulting oil purified by flash chromatography
(5% ethyI acetate in hexanes) to give 99 a colourless oi1 (1 14.1 mg, 98%). The ee was

determined to be 65% using HPLC analysis on a CHIRALCEL OD column, A= 254nrn.
Retention tjrnes in 10% isopropanol in hexanes were 10.3 min (major) and 1 1.2 min. RF
0.30 on silica gel (10% ethyl acetatchexanes); -30.0' (c= 13.8, CHC13); [R (KBr,
cmi) 3588 (br), 3037 (w), 2932 (w), 1601 (s), 1495 (rn), 1190 (rn). 'H NMR (400MHz,
CDC13) S 7.54-7.52 (IH, m), 7.31-7.29 (2H, m), 7.17-7.14 (1H. m), 7.10-7.09 (lH, m),
7.06-7.04 (lH, m), 6.94-6.93 (IH, m), 6.68-6-67 (lH, m), 6.65 ( lH, dd, J= 9.4, 2.2 Hz),
5.96 (1H, dd, J= 9.9,3.3 Hz), 5.13 (IH, d, J= 8.8 Hz), 4.78 (lH, ddd,J= 8.8, 2.5,2.5 Hz),
3.31-3.26 (lH, m), 3.14-3.08 (lH, m), 2.81-2.80 (2H, in), 2.30 (IH. s), 1.95-1.89 (2H,
m); I3c NMR (400MHz, CDCIi) 6 145.1, 136.5, 131.9, 129.7, 129.5, 128.0, 128.0,
128.0, 127.9, 127.0, 126.5, 125.9, 124.0, 116.8, 112.2, 69.5, 60.9, 44.1, 28.1, 22.5.
HRMS calcd for C isH i9N0 (M3: 277.1467. Found: 277.1463.
100
(lR,2R)-2-IndoI-l-yl-1,2-dihydro-naphthalen-01 (100): To a £lame dried round
bottom Bask, [IUi(COD)C1I2 (4.3 mg, 0.009 mmol), (R)-(S)-BPPFA (9.6 mg, 0.017
mmol), indole (407 mg, 3.47 mmol) and 42 (100 mg, 0.69 mmol) were added. THF (4
mL) was then added, followed by heating to 80°C for 3 days. The reaction mixture was
then concenîrated in vocuo. The resulting oil was puified by flash chromatography (30%
ethyl acetate in hexanes) to give 100 a colourless oil (147 mg, 81%). The ee was
determined to be 79% using HPLC analysis on a CHIRALCEL OD column, h= 254~1 .
Retention times in 10% isopropanol in hexanes were 28.5 min (major) and 30.1 min. RF

0.26 on silica gel (30% ethyl acetate:hexanes); [alZso= -46.7' (c= 1 1.3, CHU3); IR (KBr,
c d ) 3485 (br), 3059 (rn), 1592 (rn), 1455 (s), 1414 (s), 1245 (m), 1091 (rn), 908 (m);
1 H NMR (400MHz, CDCL) S 8.13 (lH, s), 7.79 (IH, d, J= 7.8 Hz), 7.42 (IH, d, J= 7.3
Hz), 7.34-7.19 (6H, m), 6.85 (IH, d, J= 2.2 Hz), 6.69 ( lH, dd, J= 9.5, 2.0 Hz), 6.20 (lH,
dd, J= 9.5, 3.8 Hz), 5.06 (lH, d, J= 7.9 Hz), 4.12-4.08 (lH, m), 2.35 ( lH, s); ' 3 ~ NhlR
(400MHz, CDC4) 6 136.5, 135.9, 132.5, 130.1, 128.0, 127.7, 126.9. 126.5, 126.4, 126.2,
122.6, 122.0, 119.3, 119.2, L 13.9, 11 1.4, 72.7, 41.0. HRMS calcd for CisHlsNO (M3:
261.1 154. Found: 261.1 141.
101
( l R , 2 R ) - 2 - ( l - h y d r o x y - 1 , 2 - d i h y d r o - n a p h t ~ 1 , 3 - d i o n e (101): To a
flame dried round bottom flask, [Rh(COD)C1I2 (5.4mg, 0.0 1 1 rnrnol), (R)-(S)-BPPFA
(12.2 mg, 0.022 mmol), phthalimide (5 10 mg, 3.47 mmol) and 42 (100 mg, 0.69 m o i )
were added. THF (4 mL) was then added, followed by heating to 80°C for 3 days. The
reaction mixture was then poured in to water and exûacted three times with ethyl acetate.
The organic layers were combined, washed with bnne dned over Na2S04, and
concentrated Ni vacuo. The resuiting soiid was purified by flash chromatography (30%
ethyl acetate in hexanes) to give 101 as a white crystalline solid (103.5 mg, 52%). The ee
was deterrnined to be 74% using HPLC anaiysis on a CHIRALCEL OD column, A=
486nm. Retention times in 10% isopropanol in hexanes were 2 1.1 min (major)and 29.1

min. RF 0.36 on silica gel (30% ethyl acetate:hexanes); mp 175476" (dec); [alBo= -
6. 1' (c= 12.9, CHCb); IR (KBr, cm-') 3536 (br), 3067 (w), 292 1 (w), 1772 (m), 1693 (s),
1388 (s), 1084 (m), 955 (m), 719 (s); 'H NMR (400MHz, CDC13) 8 7.78-7.75 (2H, m),
7.68-7.64 (2H, m), 7.57-7.55 (IH, m), 7.26-7.22 (2H, m), 7.09-7.07 (lH, m), 6.51 (lH,
dd, J= 9.7, 2.7 Hz), 5.84 (IH, ddd, J= 9.7, 2.7, 2.2 Hz), 5.48 (lH, d, J= 12.8 Hz), 5.12
(1H, ddd, J= 12.8, 2.5, 2.4 Hz), 2.82 (IH, s); "C NMR (400MHz, CDC13) 6 168.6,
137.3, 134.2, 132.6, 132.1, 128.7, 128.2, 128.1, 126.9, 126.5, 124.4, 123.5, 70.9, 55.3.
HRMS calcd for ClsHl ,NO2 (M'-H20): 273.2939. Found: 273 .OZU.
(1R*,ZR*)-2-Pyrrol id in- l -y l -1 ,2-d ihydro~1-01 (103): To a flame dned
round bottom flask were added [Rh(COD)C1I2 (4.3mg, 0.009 mmol), DPPF (9.6 mg,
0.0 17 mmol), pyrrolidine (146 mg, 3.47 mmol), triethylamine hydrochloride (478 mg,
3.47 mmol) and 42 (125 mg, 0.865 mmol) followed by addition of THF (3 rnL) and
heating to reflux for 8 hours. The solvent was then removed in vacuo and the resulting
mixture purified by flash chromatography (10% methanol in acetone) to give 103 a white
crystalline solid (1 19 mg, 80%). RF 0.14 on silica gel (10% methanol in acetone); mp
97-98' (Et20); IR (KBr, cm") 3496 @r), 3035 (rn), 2967 (s), 1454 (rn), 1193 (s), 11 17
(m), 1048 (s). 'H NMR (400MHz, CDC13) 8 7.56 (1 H, d, J= 7.1 Hz), 7.29-7.2 1 (2H, m),
7.08-7.06 (lH, m), 6.57 (IH, dd, J= 9.9,2.4 Hz), 6.05 (lH, dd, J= 9.9,2.4 Hz), 4.83 (lH,
d , J= 11.3 Hz), 3.66 (lH, ddd, J= 11.3, 2.4, 2.4 Hz), 3.57 (IH, s), 2.81-2.79 (2H, m),
2.73-2.71 (2H, m), 1.84-1.80 (4H, m); NMR (400MHz, CDC13) G 136.9, 131.8,

129.6, 127.7, 127.3, 126.1, 125.4, 124.7, 69.8, 63.3, 48.7, 23.8. HRMS calcd for
Ci4Hi7N0 0: 215.13 10. Found: 215.1314.
(1R*,2R*)-2-Diethylamino-1,2-dihydro-naphthaIen-l-o1 (104): To a flame dried
round bottom flask were added [Rh(COD)ClIr (4.3 mg, 0.009 mmol), DPPF (9.6 mg,
0.0 1 7 m o l ) , diethylamine hydroc blonde ( 1 9 1 mg, 1.74 mmol), triethylamine (242 PL,
1.74 mmol) and 104 (50 mg, 0.347 rnmol) followed by addition of THF (3 mL) and
heating to reflux for 12 hours. The solvent was then removed in vacuo and the resulting
mixture purified by flash chromatography (70% ethyl acetate in hexanes) to give 104 a
white crystalline solid (62 mg, 82%). RF 0.15 on silica gel (70% ethyl acetate:hexanes);
IR (KBr, cm-') 3482 @r), 3036 (w), 2937 (s), 2853 (m), 1453 (s), 1193 (s), 1 109 (s), 1046
(s) 'H NMR (400MHz, CDC1,) 6 7.57 (lH, d, J= 7.3Hz), 7.27-7.18 (2H, m), 7.04-7.03
(lH, m), 6.47 (lH, dd, J= 9.9, 2.8Hz), 6.04 (lH, dd, J= 9.9, 2.2Hz), 5.80 (IH, d, J=
12+7Hz), 3.73 (lH, s), 3.58 (lH, ddd,J= 12.7, 2.4, 2.4Hz), 2.80 (2H, dq, J= 7.3, 5.9Hz),
2.57 (2H, dq, J= 7.3, 5.9Hz), 1.09 (6H, t, J= 7.2Hz); ')c NMR (400MHz, CDC13) G
137.1, 131.8, 129.2, 127.7, 127.1, 125.8, 124.2, 68.9, 64.2, 44.7, 14.4. HRMS calcd for
Ci&Ii9NO 0: 217.1467; Found: 217.1460.

10s
( l R * , 2 R * ) - 2 - D i b e a z y l a m i n o - l , 2 - d i h y d r o p l s l (105): To a flame dned
round bottom flask were added [Rh(COD)C1I2 (4.3 mg, 0.009 mmol), DPPF (9.6 mg,
0.01 7 mrnol), dibenzylamine (303 mg, 1.74 rnmol), triethylamine hydrochloride (240mg,
1.74 mrnol) and 42 (50 mg, 0.347 mmol) followed by addition of THF (3 mL) and
heating to reflux for 12 houn. The solvent was then removed in vacuo and the resulting
mixture purified by flash chromatography (40-70% ethyl acetate in hexanes) to give 105
a colourless oil which crystaliçed on sitting (92 mg, 84%). R p 0.30 on silica gel (70%
ethyl acetate:hexanes); IR (KBr, cm") 3482 (br), 3036 (w), 2937 (s), 2853 (m), 1453 (s),
1 193 (s), 1 109 (s), 1 O46 (s) 'H NMR (400MHz, CDC13) G 7.50 ( 1 H, d, J= 7.1 Hz), 7.28-
7.34 (SH, m), 7.15-7.25 (4H, m), 7.00-7.04 (IH, m), 6.53 (IH, dd, J= 2.3, 9.8Hz), 6.12
(lH, dd, J= 2.3,9.9Hz), 5.01 (IH, d? J= 11.7Hz). 3.95 (2H, d, AB, J= 13.6Hz), 3.65 (lH,
d, J= 1 1.7Hz), 3.57 (2H, d, AB, J= l3.6Hz), 3.07 (1 H, s); j3c MUR (400MHz, CDCI,) 6
139.0, 136.7, 131.7, 129.6, 128.9, 128.5. 127.8, 127.3, 127.2, 126.0, 125.5, 124.8, 68.8,
62.3, 54.7. HRMS calcd for C2.&N0 (m: 34 1.1780; Found: 34 1.1786.

106
(IR*,2R*)-2-Pipendin-l -yl-l,2-dih ydro-napht halen-1-01 (106): To a flame dried
round bottom flask, [Rh(COD)C1I2 (4.3 mg, 0.0087 mrnol), DPPF (9.6 mg, 0.0173
m o l ) , pipendine hydrochloride (422 mg, 3.47 mrnol), triethylamine (350 PL, 2.51
mmol) and 42 (100 mg, 0.69 mrnol) were added followed by THF (3 mL) and heating to
80°C for 12 hours. The reaction mixture was then concentrated in vacuo and punfied by
flash chromatography (50% ethyl acetate, 48% hexanes, 2% methanol) to give 106 a
white crystalline solid (130 mg, 82%). RF 0.24 on silica gel (50% ethyl acetate, 48%
hexanes, 2% methanol); mp 62-64' (Et20); IR (KBr, cm") 3482 (br), 3036 (w), 2937 (s),
2853 (m), 1453 (s), 1193 (s), 1109 (s), 1046 (s). 'H NMR (400MHz, CDC13) G 7.57 ( l H ,
d, J= 7.1 Hz), 7.27-7.18 (ZH, m), 7.05 (IH, dd, J= 6.9,O.g Hz), 6.49 (1H, dd, J= 9.9, 2.6
Hz), 6.12 (IH, dd, J= 9.9,2.4 Hz), 4.87 (lH, d, J= 12.2 Hz), 3.58 (IH, s), 3.37 (1H, ddd,
J= 12.2,2.4,2.4 Hz), 2.79-2.73 (2H, m), 2.48 (2H, m), 1.67-1.57 (4H, m), 1.56-1.46 (2H,
m); I3c NMR (400MHz, CDCL) S 137.4, 131.8, 128.8, 127.1, 125.9, 125.2, 124.4, 68.2,
67.6,50.4,26.5,24.6. HRMS caIcd for Ci5HisN0 (M*-H): 228.1388. Found: 228.13 18.

(1 R*,2R*)-2-(3.4-Dih ydro-4H-isoquinoh-2-yi) -2-dihydro-na phthalen sl (1 07):
To a flarne dned round bottom flask were added [Rh(COD)C1]2 (4.3 mg, 0.009 rnmol),
DPPF (9.6 mg, 0.0 17 rnmol), tetrahydroisoquinoline (23 1 mg, 1.74 rnmol), triethylamine
hydrochloride (240mg, 1.74 m o l ) and 42 (50 mg, 0.347 rnmol) followed by addition of
THF (3 rnL) and heating to reflux for 12 hours. The solvent was then removed in vacuo
and the resulting mixture purified by flash chromatography (40-70% ethyl acetate in
hexanes) to give 107 a colourless oil (78 mg, 81%). RF 0.25 on silica gel (70% ethyl
(s), 1109 (s), 1046 (s) 'H NMR (400MHz, CDCI,) G 7.61-7.60 (lH, m), 7.30-7.23 (2H,
m), 7.18-7.13 (3H, m), 7.1 1-7.09 (lH, m), 7.06-7.04 (IH, m), 6.58 (lH, dd, J= 9.9,
2.6Hz), 6.15 (IH, dd, J= 9.9, 2.4Hz), 5.02 (1H, d, J= 12.1Hz), 4.02 (IH, d, AB, J=
14.8Hz), 3.80 (lH, d, AB, J= 14.8Hz), 3.66 (lH, ddd, J= 12.1, 2.5, 2.5Hz), 3.12-3.18
(IH, m), 2.90-3.00 (2H, m), 2.77-2.84 ( lH, m); '.'c NMR (400MHz, CDCI,) 6 .
124.6, 124.5, 68.0, 67.6, 52.0, 46.9, 29.9; HRMS calcd for Ci9Hi9N0 (M+): 277.1467,
Found: 277.1462.

(1R*,ZR*)-2-Benzylamino-1,2-dihydro-naphthaen-l-o (108): To a flame dried round
bottom flask, [RIi(COD)C1I2 (4.3 mg, 0.009 mmol), DPPF (9.6 mg, 0.017 mmol),
benzylamine hydrochlonde (279 mg, 1.74 mmol), triethylamine (242 pL, 1.74 mmol) and
42 (50 mg, 0.347 m o l ) followed by addition of THF (3 mL) and heating to reflux for 3
days. The solvent was then removed in vacuo and the resulting mixture punfied by flash
chromatography (50% ethyl acetate in hexanes) to give 108 a white crystalline solid (26.9
mg, 3 1%). RF 0.44 on silica gel (50% ethyl acetate, 48% hexanes, 2% methanol); rnp
1 15- 1 17' (dec) (&O); IR (KBr, cm-') 3528 (br), 3030 (w), 2849 (w), 1455 (s), 1 190
(m), 11 12 (m), 1048 (m). 'H NMR (400MHz, CDC13) G 7.47-7.45 (lH, m), 7.29-7.24
(4H, m), 7.24-7.17 (3H,m), 7.02-7.01 (lH, m), 6.41 (lH, dd,J=9.7, 2.0 Hz), 6.00 (IH,
dd, J= 9.7,2.5 Hz), 4.64 (IH, d, J= 9.0 Hz), 3.94 (IH, AB,& 13.0 Hz), 3.75 (IH, AB, J=
13.0 Hz), 3.42 (lH, ddd,J= 11.0,2.4,2.4 Hz), 2.44 (1H, s); 13c NMR (400MKz, CDC13)
59.7,50.7. H R M S calcd for Ci7HlrN0 (hf): 25 1.13 10. Found: 25 1.13 16.

109
(1R*,2R*)-2-(4-Methoxy-benzylamino)-1,2-dihydro-naphthaienl-o1 (109): To a
flarne dried round bonom flask, [Rh(COD)C1I2 (4.3mg, 0.009 mmol), DPPF (9.6 mg,
0.0 17 mmol), p-methoxybenzylarnine (23 8 mg, L -74 mrnol), triethylamine hydrochloride
(239 mg, 1.74 rnmol) and 42 (50 mg, 0.728 mmol) followed by addition of THF (3 mL)
and heating to reflux for 3 days. The solvent was then rernoved NI vacuo and the
resulting mixture purified by flash chromatography (50% ethyl acetate in hexanes) to
give 109 a white crystalline solid (43 mg, 44%). RF 0.27 on silica gel (50% ethyl
acetate, 48% hexanes, 2% methanol); mp 96-98' (dec) (Et20); IR (KBr, cm-') 3528 (br),
3033 (w), 2835 (m), 1612 (m), 1512 (s), 1455 (m}, 1248 (s), 1040 (m). 'H NMR
(400h@b, CDC13) G 7.52-7.50 (lH, m), 7.26-7.22 (4H, m), 7.08-7.06 (1 H, m), 6.85 (2H,
d,J=9.0 Hz), 6.47(1H,dd,J=9.7,2.0 Hz),6.05 (lH,dd,J=9.9,2.6 Hz),4.68 (1H,d,J=
11.0 Hz), 3.95 (lH, d, J= 12.9 Hz), 3.79 (3H, s), 3.75 (lH, d, J= 2.9 Hz), 3.46 (lH, ddd,
P 1 1.0, 2.4, 2.4 Hz), 3.0-2.0 (2H, s (br)); 13c NMR (400MHz, CDCl,) 6 158.7, 136.7,
132.1, 131.9, 129.4, 128.9, 127.9, 127.7, 127.5, 126.0, 124.9, 113.9, 72.1, 59.6, 55.2,
50.1. HRMS calcd forClsHi9N02 0: 281.1416. Found: 281.1403.

2.4 Carboxylate Nucleophiles
2.4.1 Introduction
The use of carboxylic acid nucleophiles is desirable since the reaction would create an
allylic carboxylatc with to td contro! of stereochemistry. Such functionality has found
use in organic chemistry in a variety of SN2' displacements and rearrangement reactions.
The carboxylate ring opened products could thus grant access to a variety of other
naphthalene substitution pattern not directly available from ring opening methodologies.
2.4.2 AR0 with Carboxylate Nucleophiles
Initial experiments focused on determining conditions under which carboxylate
nucleophiles would induce ring opening. Indeed, neither acetic acid nor potassium
acetate induced any ring opening (Table 2.19). Gratiaingly, addition of equimolar
amounts of acetic acid and triethylamine or equimolar amounts of sodium acetate and
triethylamine hydrochloride gave 110 in good yield.
This methodology is generally applicable for a wide range of carboxylic acids or
carboxylate salts. (Table 2.20).

Table 2.19: Effect of an Added Proton Source on Acetate Reactivity
DPPF (1 eq. to ~ h ) Nudeophile ACO"' Proton Source A
nu THF 1 80'~ (or reflux)
--
Entry Nucleophile Y ield
1 AcOH (5eq) NR
2 AcONa (5eq) NR
3 AcOH (5eq) 1 Et3N (5eq)
83%
4 AcONa (5eq) 1 89% Et3N.HCI (5eq)
The relative stereochemisüy for 110 was proven by deprotection of the acetate moiety,
dimethylation with dimethylsulfate and cornparison to trans-
dimethoxydihydronaphthalene prepared via an alternative method.
Preliminary experiments dealing with the AR0 of 42 with carboxylate nucleophiles have
been performed using the BPPFA 102 and Cl-Femphos 111 ligands, which gave the
same level of induction in the two cases examined (Table 2.21). We are currently
conducting studies to determine the optimal ligand for these transformations.
C2-Ferrfp hos 111

Table 2.20: Scope of the Rhodium Catalysed Carboxylate Ring Opening Reaction
[Rh(COD)Clh (2.5mol%) DPPF (1 eq. to Rh)
Conditions A or B THF 80°C (or reflux)
Entry Nucleophile Conditions Product Yield
2 JOX A 112 66%
O
Condltlons A; 5 equiv. wrboxyiic acid with 5 equiv. Et3N. Conditions 8: 5 equiv. cahxylate sait wiîh 5 equiv. Et3N.HCI
Table 2.21: AR0 with Carboxylate Nucleophiles
[Rh(COD)Clj2 (2.5mol%) Ligand (1 eq. to Rh)
0
N uc"" THF 1 8 0 ' ~ (or reflux)
OH
Entry Nucleophile p f + p t ~ u Z BPPFA C2-Ferriphos
1 AceticAcid 71%(32°hee) 74°h(810hee) 72°/o(81%ee)
2 Benzoic Acid - 70%(76%ee) 73%175%ee)
Candithns C: S equiv. carboxylic acid with 5 equiv. Et3N.

2.4.3 Application of Ring Opened Products in the Preparation of 1,4- disu bstituted-dihydronaphthalenols
These products are useful compounds en route to other synthetically useful intermediates.
By taking advantage of the allylic carboxylate functionality present in the carboxylate
ring opened products, palladium coupling reactions can be performed. For example, after
protection of 115 as the TBS ether 117, a palladium catalysed Claisen rearrangement and
subsequent decarboxylation produces the 1,4-dihydronaphthalene product 1 18 in good
yield (Scheme 2.8).
Scheme 2.8: Preparation of 1,4=DihydronaphaIenes
115 R = H
117R = TBDMS 2 90%
L OTBDMS A OTBDMS
2.4.4 Conclusion
In conclusion, the rhodium catalysed AR0 reaction of oxabicyclic alkenes has been
extended to the less reactive carboxylate nucleophiles. Under the previously established
conditions, carboxylates faiied to induce ring opening. We found, however, that the
addition of a proton source had beneficial effects, allowing the ring opening to occur with
a wide variety of carboxylate nucleophiles. Such transformations are synthetically useful
not only since they grant access to the hydronaphthalene core, but they also create an

allylic carboxylate functionality which cm be used to access hydronaphthalenes not
readily available frorn ring opening reactions. This has been realized through the
application of a palladium catalysed Claisen rearrangement giving 1,4-
dihydronaphthalenes products.
2.4.5 Experimental
(1 R*,ZR*)-Acetic acid 1-hydroxy-1,2-dihydro-nap htha1en-2-yl-ester (1 10): To a
flame dried round bottom flask, [Rh(COD)C1I2 (4.3 mg, 0.008 mm01 DPPF (9.6 mg,
0.017 mmol), 42 (50 mg, 1.39 mmol), and sodium acetate (142 mg, 1.74 rnrnol) were
added followed by addition of THF (2 mL) and triethylarnine hydrochloride (239 mg,
1.74 mmol). The mixture was heated at reflux for 3 hours and the solvents were removed
in vacuo. The resulting mixture was purified by flash chromatography (30% ethyl acetate
in hexanes) to give 110 as a crystalline solid (41 mg), 63%). R ç 0.26 on silica gel (20%
ethyl acetate:hexanes); mp 67-68' (Et2O); IR (KBr, cm-') 'H NMR (400MHz, CDCla) 8
7.54-7.53 (IH, m), 7.29-7.24 (2H, m), 7.10-7.08 (1H, m), 6.50 (lH, dd, J= 3.9, 1.3 Hz),
5.85 (IH, dd, J= 9.9, 3.1 Hz), 5.59 (lH, ddd, J= 9.0, 2.8, 1.9 Hz), 4.92 (lH, d, J= 9.0
Hz), 2.64 (lH, s), 2.12 (3H, s); 13c NMR (400MHz, CDCh) 6 171.3, 135.2, 131.5,

129.5, 128.3, 126.7, 126.0, 125.4, 75.3, 71.7, 21.2. HRMS calcd for C12H1203 (m: 204.0786. Found: 204.079 1.
(1 R*,ZR*)-Propionic acid 1-bydroxy-l,2-dihydro-naphthalen-2-yl-ester (1 12): To a
flame dried round bottom flask, [Rh(C0D)C1l2 (4.3 mg, 0.0087 mmol), DPPF (9.6 mg,
0.017 rnrnol) and 42 (50 mg, 0.347 mmol) were added followed by addition of THF (2.5
mi,), triethylarnine (242 pL, 1.735 rnmol) and propionic acid (1 30 pL, 1.735 rnmol). The
mixture was heated at reflux for 3 hours and the solvents were removed in vacuo. The
resulting mixture was purified by flash chromatography (20% ethyi acetate in hexanes) to
give 113 a white crystalline solid (50 mg, 66%). RF 0.24 on silica gel (% 20 ethyl
acetate:hexanes); mp 55-56" (EtzO); IR (KBr, cm-') 349 1 (br), 3048 (w), 2984 (w), 1739
(s), 1454 (m), 1363 (w), 1182 (s), 1083 (m). 'H NMR (400MHz, CDC13) 8 7.55-7.52
(W m), 7.29-7.24 (2H, m), 7.11-7.08 (lH, m), 6.50 (1 H, dd, P 10.0, 2.0 Hz), 5.85 (IH,
dd, J= 12.8, 2.8 Hz), 5.61 (lH, ddd, J= 9.2, 2.8, 2.0 Hz), 4.93 (lH, d, J= 9.2 Hz), 2.40
(2H, qd, J= 7.6, 1.2 Hz), 1.16 (3H, t, J= 7.6 Hz); "C NMR (400MHz, CDC13) 6 174.8,
135.3, 131.5, 129.4, 128.3, 128.3, 126.7, 125.9, 125.5, 75.2,71.9, 27.7,g.O. HRMS calcd

113
(lR*,2R*)-2-Methyl acrylic acid 1-hydroxy-1,2-dihydro-naphthalen-2-yl-ester (1 13):
To a flarne ciried round bottom flask, [Rh(COD)C1I2 (4.3 mg, 0.0087 mmol), DPPF (9.6
mg, 0.017 mmol) and 42 (50 mg, 0.347 mmol) were added followed by addition of THF
(2.5 mL), triethylamine (242 PL, 1.735 mmol) and methacrylic acid (147 PL, 1.735
mmol). The mixture was heated at reflux for 3 houn and the solvents were removed in
vacuo. The resulting mixture was punfied by flash chromatography (30% ethyl acetate in
hexanes) to give 113 a white crystalline solid (50 mg, 63%). RF 0.32 on silica gel
(20% ethyl acetate:hexanes); rnp 80-82' (Et20); IR (KBr, cm-') 3450 (br), 3030 (w), 2928
(w), 1722 (s), 1637 (m), 1454 (m), 1289 (m), 1 163 (s); 'H NMR (400MHz. CDCl,) 6
7.56-7.55 (lH, m), 7.29-7.24 (2H, m), 7.10-7.09 (lH, m), 6.51 (IH, dd, J= 9.9, 1.9 Hz),
6.15 (1H, s), 5.87 (lH, dd, J= 9.9, 3.0 Hz), 5.67 (IH, ddd, J= 9.3, 2.1,2.1 Hz), 5.61 (lH,
s), 5.01 (lH, dd, J= 9.0, 5.7 Hz), 2.74 (lH, d, J= 6.1 Hz), 1.96 (3H, s); 13c NMR
(400MHz, CDCL) G 167.6, 135.9, 135.3, 131.5, 129.4, 128.3, 128.2, 126.6, 126.4, 125.8,
125.5,75.9,7 1.9, 18.3. . HRMS calcd CL4H1102 (MT-H20): 2 12.0837. Found: 2 12.083 1.
(lR*,2R*)-Formic acid 1-hydroxy-l,2-dihydro-naphthalen-2-yI-es (114): To a
flame dned round bottom flask, [RIi(COD)C1I2 (4.3 mg, 0.0087 mmol), DPPF (9.6 mg,

0.0 17 mmol), 42 (100 mg, 0.694 rnmol), and ammonium formate (219 mg, 3.47 mmol),
were added followed by addition of THF (5 mL). The mixture was heated at reflux for 3
hours and the solvents were rernoved M vacuo. The resulting mixture was purified by
flash chromatography (30% ethyl acetate in hexanes) to give 114 a white crystalline solid
(84 mg, 64%). RF 0.25 on silica gel (30% ethyl acetate:hexanes); mp 133-1 35' (EtzO);
IR (KBr, cm-') 3 146 (br), 2935 (w), 1720 (s), 1482 (w), 1186 (s), 1049 (m), 968 (m); 'H
NMR (400MHz, CDC13) 8 8.17 (1 H, d, J= 0.8 Hz), 7.52-7.50 ( lH, m), 7.29-7.27 (2H, m),
7.13-7.1 1 (lH, m), 6.54 (lH, dd, J= 9.6, 1.6 Hz), 5.88 (lH, dd, J= 9.6,2.8 Hz), 5.71-5.68
(IH, m), 4.96 (IH, d, J= 8.8 Hz), 2.8 (lH, s); I3c NMR (400MHz, CDC13) 6 160.9,
134.8, 131.4, 130.0, 128.5, 126.9, 126.1, 124.6, 74.8, 71.4. HRMS calcd for Ci iHlo03
(M'): 190.0630. Found: 190.0625.
(lR*JR*)-Malonic acid ethyl ester (1-hydrory-1,2-dihydro-naphthalen-t-yl) ester
(1 15): To a flarne dried round bottom flask, [Rh(COD)C1]2 (8.6 mg, 0.0 17 mrnol DPPF
(19.2 mg, 0.035 mmol), 42 (200 mg, 1.39 mmol), ethyl malonate potassium salt (590 mg,
3.47 mmol), and triethylamine hydrochloride (478 mg, 3.47 mrnol) were added followed
by addition of THF (8 mL). The mixture was heated at reflux for 3 hours and the
solvents were removed in vacuo. The resulting mixture was purified by flash
chrornatography (30% ethyl acetate in hexanes) to give 115 a colourless oil (300 mg),
79%). RF 0.29 on silica gel (30% ethyl acetatezhexanes); IR ( D r , cm-') 3470 (br),

2983 (w), 173 1 (s), 1453 (w), 1370 (m), 1 150 (s), 103 1 (m); 'H NMR (400MHz, CDC13)
6 7.56-7.54 (lH, m), 7.27-7.21 (ZH, m), 7.08-7.06 (lH, m), 6.48 (1H, dd, J= 9.9, 2.1 Hz),
5.83 (lH, dd,J= 9.7, 2.8 Hz), 5.70 (1H, ddd, J= 9.7, 2.5, 2.2 Hz), 4.97 (lH, d, J= 9.5 Hz),
4.18 (2H, q, J= 7.2 Hz), 3.43 (2H, dd, J= 23.6, 15.9 Hz), 3.21 (lH, s), 1.25 (3H, t, J= 7.1
Hz); ' 3 ~ NMR (400MHz, CDC13) 6 167.1, 166.5, 135.0, 131.5, 129.6, 128.3, 128.1,
126.6, 125.6, 125.1, 77.0, 71.6, 61.9, 41.6, 14.0. HRMS calcd for Ci5Hi404 (M--H20):
258.0892. Found: 258.0899.
116
(lR,ZR)-Benzoic acid 1-bydroxy-1,2-dihydro-naphthalen-2-yl-este (1 16): To a
flame dried round bottorn flask, [Rh(COD)C1I2 (4.3 mg, 0.0087 mmol), (R)-(S)-BPPFA
(9.6 mg, 0.0 17 mrnol) and 42 (100 mg, 0.694 mrnol) were added followed by addition of
THF (4 mL), rriethylamine (483 PL, 3.47 mmol) and benzoic acid (424 mg, 3.47 mrnol).
The mixture was heated at reflux for 6 hours and the solvents were removed in vacuo.
The resulting mixture was purifiecf by flash chromatography (20% ethyl acetate in
hexanes) to give 116 a white crystalline solid (129 mg, 70%). The ee was determined to
be 76% using HPLC analysis on a CHIRALCEL OD column, 10% isopropanol in
hexanes, h=254 nm. Retention times were 10.0 min (major) and 12.9 min. Rr 0.3 on
silica gel (10% ethyl acetate:hexanes); mp 107- log0 (Et20); -298.4' (c= 1 1.3,
CHC13); IR (KBr, cm-') 3619 @r), 3071 (w), 2977 (w), 1724 (s), 1451 (rn), 1324 (m),
1265 (s), 1 1 10 (s). 'H NMR (400MHz, CDCb) G 8.10 (2H, d, J= 7.6 Hz), 7.64-7.59 (2H,

m), 7.48-7.45 (2H, m), 7.34-7.32 (2H, m), 7.13-7.1 1 (lH, rn), 6.55 (IH, d, J= 10.0 Hz),
5.97 (lH, dd, J= 9.8,2.9 Hz), 5.86 (lH, ddd, J= 9.8,2.0,2.0 Hz), 5.1 1 (lH, d. J= 9.0 Hz),
2.84 (IH, s); ')c NMR (~OOMHZ, CDC13) S 166.9, 135.3, 133.3, 131.6, 129.9, 129.8,
129.7, 128.4, 128.4, 128.4, 126.8, 126.1, 125.5, 76.1, 71.9. HRMS calcd for Cl7Hl4o3
(m: 266.0943. Found: 266.0938.
117
(1Rf,2R*)-Malonic acid (1-tert- butyldimethylsiloxy-l,2-dihydro-naphthalen-2-yl)
ester ethyl ester (117): To a dried round bottorn flask, 115 (270 mg, 0.98 m o l ) ,
imidazole (134 mg, 1.96 mrnol), dimethylarninopyridine (6 mg, 0.05 mmol) were
dissolved in dichloromethane (4 mL). Tert-butyldimethylsilyl chloride (222 mg, 1.47
mmol) was then added portionwise and allowed to react for 24 hours. The reaction was
then quenched with water, extracted with dichloromethane, dried over Na2S04 and
concentrated in vocuo. Flash chromatography (10% ethyl acetate in hexanes) gave a
colourless oil 117 (343 mg, 90%). RF 0.48 on silica gel (10% ethyl acetate:hexanes. IR
( D r , cm-1) 2983 (w), 1731 (s), 1453 (w), 1370 (m), 1150 (s), 1031 (m); 'H NMR
(400MHz, CDCl3) 6 7.41-7.39 (IH, m), 7.24-7.22 (2H, m), 7.07-7.05 (lH, m), 6.47 (1H,
dd, J= 9.9, 1.8 Hz), 5.83 (lH, dd, J= 9.7, 2.7 Hz), 5.60 (lH, ddd, J= 9.3, 2.9, 2.0 Hz),
5.00 (lH, dd, J= 9.3, 0.5 Hz), 4.22-4.15 (2H, m), 3.40 (2H, dd, J= 19.6, 16.0 Hz), 1.57
(LH, s), 1.25 (3H, t, J= 7.1 Hz), 0.92 (9H, s), 0.13 (3H, s), 0.09 (3H, s); 13c NMR

(400MHz, CDC13) G 166.3, 166.2, 136.2, 132.1, 129.4, 128.0, 127.9, 126.5, 125.9, 125.7,
76.4, 71.6, 61.6, 41.7, 25.8, 18.1, 14.0, 3.3, -4.5. HRMS calcd for Ci7H2105Si (w- C4H9): 333.1 158. Found: 333.1 149.
(1S*,2S*)-(4-Tert-butyldimetbylsiloxy- 1,4-dihydro-nap hthaien-2-yl) acetic acid
ethyl ester (17): To a dried round bonom flask, 117 (100 mg, 0.256 rnmol) was dissolved
in THF (4 mL). Potassium hydnde (1 1.3 mg, 0.28 rnmol) was then added portionwise
and allowed to react for five minutes at room temperature. Triphenylphosphine (34.1 mg,
0.13 mmol) was then added followed by Pd(PPh3)4 (14.8 mg, 0.0 13 mmol). The reaction
was then heated to reflux for two hours. The solvent was then removed in vacuo and the
resulting oil purified by flash chromatography (5% ethyl acetate in hexanes) giving 118 a
colourless oil(54 mg, 61%). RF 0.27 on silica gel (5% ethyl acetate:hexanes); IR (KBr,
cm-') 3036(w), 2956(s), 173 5(s), l472(rn), 1 Z i ( s ) , 1 O77(s); ' H NMR (400MHz, CDC13)
8 7.54-7.52 (IH, m), 7.30-7.23 (3H, ni), 6.09 (IH, ddd, J= 2.4, 4.6, 10.2 Hz), 6.02 (IH,
ddd, J= 10.2,2.0,0.5 Hz), 5.22-5.21 (1H, m), 4.15 (2H, q, J= 7.2 Hz), 3.92-3.87 (lH, m),
2.62 (lH, dd, J= 15.7, 5.7 Hz), 2.39 (lH, dd, J= 15.2, 9.0 Hz), 1.25 (3H, t, J= 7.2 Hz),
0.98 (9H, s), 0.21 (3H, s), 0.15 (3H, s); "C NMR (400MHz, CDCI,) 6 171.7, 138.3,

136.1, 131.8, 128.2, 127.2, 127.0, 126.9, 126.6, 65.3, 60.5,42.7, 36.5, 25.9, 18.2, 14.2, -
4.2, -4.5. HRMS calcd C17H210SSi (M"+-CJH~): 289.1260. Found: 289.1257

3 Rhodium Catalysed Alcoholysis and Aminolysis of Vinyl Epoxides
Vinyl epoxides are commonly used starting matenals in organic ~ ~ n t h e s i s . ~ ~ Among
their most important applications is as eiectrophiles in transition metal catalysed
reactions. The most fiequently used cataiysts are palladium based which permit the use of
a wide variety of n u c l e ~ ~ h i l e s . ~ ~ A cornrnon trend in al1 these reactions is a preference
for the nucleophilic addition to occur in a lT4-manner syn to the leaving group via a net
SN2' addition with retention of configuration ansing from a double inversion pathway
(vide supra).
Transition metal catalysed reactions giving 1,2-addition products are far fewer in number.
To obtain 12-addition with palladium, the nucieophile is typically delivered in an
intmmolecular fashion via a tether to give the cis product. By trapping the alkoxide
produced upon epoxides opening with an appropriate eiectrophile and subsequent
cyclisation leads to five membered ring formation and a net syn-1,2-addition. For
example, the use of isocyanates generates oxazolidinones,w carbon dioxide gives cyclic
carbonates:' and aldehydes react to produce cyclic acetalsg2 (Scheme 3.1).

Scheme 3.1
Tin alkoxides have also been found to undergo 1,2-addition with vinylepoxides and
palladium catalysis. This is due to the coordination of the tin species to the intermediate
alkoxide generated upon ring opening. This temporary tin tether then delivers the
alkoxide nucleophile to generate the syn- 1,2-product (Scheme 3.2).93
Scheme 3.2
In 1998, Trost and CO-worken reported a two-component catalyst system for the
asymmetric allylic alkylation of alcohol pronucleophiles providing an elegant method for
the preparation of enantioe~ched vinyl glycidols via a deracemisation of vinyl

epoxides? The use of catalytic amounts of allcylboranes serve to activate the alcohol
nucleophiles by forming a tether to the 7-c palladium alkoxide and delivering the alcohol
intramolecularly. Under optimal conditions, the desired products are produced in >80%
yield and with ee's exceeding 90% (vide supra).
While trans- 1,2-addition products c m sometimes be obtained through stoichiometric or
catalytic use of Brmsted and Lewis acids, these methods suffer fiom poor functional
group compatibility. In addition, such methods generally fail with amine nucleophiles
due to their high ba~icity.~' Given the synthetic utility of the 1,Zaddition products,
particularly 1,2-alkoxyalcohols and 1,2-aminoalcohols, the development of a mild and
selective method for their preparation from vinyl epoxides would be a desirable goal
(Scheme 3.3).
Scheme 3.3

3.2 Rhodium Catalysed Alcoholysis and Aminolysis of Vinyl Epoxides
3.2.1 Alcohol Nucleophiles
Our initial experiments focused on finding an active catalyst system with alcohol
nucleophiles (Table 3.1). As a consequence of Our ongoing studies on the asymmeûic
ring opening (ARO) reaction of oxabicyclic alkenesg6 we selected rhodium as the metal
to promote the reaction with epoxides. While neither the catalyst developed for our
oxabicyclic alkene studies (entry l), nor the modified Wilkinson's catalyst used by P.A.
Evans in his allylic aminatiodalkylation studiesg7 (entry 4) showed any reactivity with
1 19, [Rh(C0)2C1]2 with trimethy lphosphite ligands showed slightly improved results.
Table 3.1: Effect of Rhodium Catalysis on Reactivity
Rh Catalyst System
MeOH (10eq.) THF I rt
Rhodium Catalyst Yield
[Rh(COD)C1)2 12DPPF NR
[Rh(COD)C1]2 1 4P(OMe)3 NR
[Rh(C0)2CIJ2 14P(OMe)3 NR
Wilkinson's Cat. / 4P(OMe)3 NR
a lsolated yield. Stereochemistry pmven by hydrogenation of the olefin and compatison of spedra to titerature

[Rh(C0)2C1]2 in the absence of any added ligand (entry 6) produced the best results,
giving the 1,Zaddition product 120 in 94% isolated yield with >20:1 diastereo- and
regiselectivity.
The regiochernistry of the ring opened product was determined by oxidation of 121 and
isomerisation of the olefin into conjugation to give 122 .'"he relative stereochemistry
was determined by hydrogenation of the olefin of 120 to give 123 which was proven to
be the tmns isomer by cornparison to literature spectral data (Scheme 3.4):'
1 ) Dess-Martin Oxidation - - m"
Scheme 3.4
An investigation into the reactivity of other alcohol nucleophiles with this substrate
revealed that both primary and secondary alcohols are effective while phenol gives
poorer yields and benzyl alcohol is ineffective, giving very little of the desired product.
The reaction is general for a wide variety of vinyl epoxides (Table 3.2). It was found
however that when the ring opened products contained a primary alcohol, a significant

130
Table 3.2: Scope of Rhodium Catalysed Ring Opening Of Vinyl Epoxides wÎth
Alco hols
1 Vinvl Eooxide 1
'O L O
TBSO
Product 1 Cond.
TBSO +l A
Yield (R) IProduct
Conditions A: 1-2 mol% m(C0)2C1]2, 10 eq. ROH in THF.
Conditions B: 1-2 mol% [Rh(CO)2C1]2, neat ROH.
amount of dimeric and oligomeric byproducts were produced. By running the reactions
in neat alcohol, however, the isolated yields were increased to greater than 90%. These
reactions are very rnild, occming under neutral conditions and at room temperature. In
al1 cases the diastereo- and regioselectivity was greater than 20: 1.
3.2.2 Arornatic Amine Nucleophiles

In order to study the scope of the reaction a variety of amines were examined. We were
gratified to find that although aliphatic amines are not reactive, aromatic amines are
Table 3.3: Scope of Rhodium Catalysed Ring Opening Of Vinyl Epoxides with Aromatic Amines
TBSO -+??
Product
NMePh
Ph &OH
Eto-oH NHR
n&J TBSO N M ~ P ~
91% lndole 133 81 %
Conditions: 1-2 mol% [Ilh(C0)2CI]2, 3-5 eq. Arornatic amine, THF
highly reactive nucleophiles, providing the arnino alcohols arising f?om attack at the
allylic position with selectivities exceeding 20: 1. The reaction occun with a wide variety
of aromatic amines, varying in both stenc bulk and in basicity (e-g. both p-anisidine and
p-nitroaniline are compatible nucleophiles) (Table 3.3).

Ln the case of linear vinyl epoxides, the regiochernistry was determined for both alcohol
and aromatic amine nucleophiles by oxidation of the primary alcohol resulting from the
ring opening step. In both cases, generation of the aldehyde proved that nucleophilic
attack had occurred at the more sterically hindered allylic carbon atom (Eq. 3.1).
O Dess-Martin Oxida tion
Nuc = OMe 129 = NHC6H4Br 136
Nuc = OMe 142 = NHC6H4ûr 143
The relative stereochemistry was established by formation of the oxazolidinone 144
and NOE analysis (Scheme 3.5).
O
139 1 44
Scheme 3.5
Aliphatic amines fail to react under these reaction conditions, likely due to their binding
strongly to the rhodium metal resulting in catalyst poisoning. Since aromatic amines are
less basic than aliphatic amines, this mode of binding is significantly reduced. Evidence
for this hypothesis is obtained by perfoming the reaction with five equivalents of both N-
rnethylaniline and benzylamine (Eq. 3.2). Ln the presence of the more basic amine, the

aromatic amine addition pathway is shut down, indicating that the rhodium is being
sequestered frorn the catalytic cycle.
4Ao [Rh(C0)2Cl12 -
Ph - no reaction
BnNH2 (Seq) (3.2) PhNHMe (5eq) THF 1 rt
3.2.3 Preliminary Mechanistic Studies
There are two possible roles that rhodium might be playing; it can act as a mild Lewis
acid, or it can insert into the carbon-oxygen bond to produce a x dlyl or enyl rhodium
intermediate. Preliminary mechanistic studies have shown that the presence of an olefin
is required for the reaction to occur since cyclohexene oxide did not react under these
conditions. Furthemore, styrene oxide did not react even after prolonged reaction times
indicating that the rhodium is likely not acting as a Lewis acid.
When vinyl epoxides possessing a teminal olefin are used, a mixture of 1,4- and 1,2-
addition products is produced, analogous to the results obtained by Evans in the allylic
amination studies of allyl carbonates. In contnist to previous reports on rhodium
catalysed allylic alkylation, amination and etherification which occur with retention of
absolute confliguration, @Rh(C0)2C1]2 promotes the ring opening of vinyl epoxides with
inversion of stereochemistry allylic position undergoing nucleopIülic attack. In our
shidies of AR0 reactions of oxabicyclic alkenes, inversion is also observed, but the

reaction proceeds via a net SN2' displacement contrary to the net SN2 reaction which
occurs with vinyl epoxides. Curent studies are focussed on elucidating the source of
these intriguing differences.
3.3 Conciusion
In conclusion, we have demonstrated that [Rh(C0)2C1]2 is an effective catalyst for the
ring opening of vinyl epoxides with alcohols and aromatic amines under neutral
conditions at r o m temperature. The reaction occun with excellent diastereo- and
regioselectivity (>20:1) giving the ».ans-1,2-addition products for a wide range of
substrates. The regio- and stereoselectivity is complementary to that typically observed
with palladium catalysis, and the stereoselectivity is opposite that of previously reported
rhodium catalysed allylic alkylation reactions The simplicity and mildness of this
rnethodology make it an attractive option whenever substrates possess acid sensitive
groups.

3.4 Experimental
General Procedure (A): For reaction of cyclic substrates with alcohol nucleophiles. A
round bottom flask was charged with cyclohexadiene monoxide (SOmg, 0.52mmol) and
m(CO)1C1]2 (2mg, 0.005mmol). THF (0.5rnL) and MeOH (0.5mL) were then added
producing a light yellow solution which was stirred at room temperature for one hour.
AAer one hour the solvents were removed in vacuo and the resulting oil chromatographed
(30% ethyl acetate:hexanes) to give 1 a colourless oiI(62mg, 93%).
General Procedure (B): For reaction of cyclic substrates with phenol nucleophiles. A
round bottom flask was charged with cyclohexadiene monoxide (SOmg, 0.52mmol) and
phenol (244mg, 2.6rnmol) and THF (ImL). [Rh(C0)2C1]2 (2mg, 0.005mmol) was then
added producing a light yellow solution which was stirred at room temperature for two
hours. M e r the reaction was completed, the reaction was poured into ether and washed
three times with 5% aqueous NaOH. The aqueous layers were combined and back
extracted with ether. The organic layen were combined. washed with brine, dried over
MgS04, and concentrated in vacuo. The resulting oil was then chromatographed (20%
ethyl acetate:hexanes) to give 4 a white crystalline solid (83mg, 84%).
General Procedure (C): For reaction of cyclic or acyclic substrates with aniline
nucleophiles. A round bottom flask was charged with cyclohexadiene monoxide (50mg,
0.52mmol), N-methylaniline (278mg, 2.6mmol) and [R~I(CO)~CL]~ (2mg, 0.005mmol).
THF ( I d ) was then added producing a light yellow solution which was stined at roorn
temperature for one hou. Mer one hour the THF was removed in vacuo and the

resulting oil chrornatographed (10% ethyl acetate:hexanes) to give 5 a colourless oil
(96mg, 9 1 %).
General Procedure 0): For reaction of acyclic substrates with alcohol nucleophiles. A
round bottom fiask was charged with 2-styt-yl-oxirane (50rng 0.34mmol) and
[Rh(C0)2Cl]2 (2mg, 0.005mmol). MeOH (1m.L) was then added producing a light
yellow solution which was stirred at room temperature for one hour. AAer one hour the
solvents were removed in vamo and the resulting oil chromatographed (30% ethyl
acetate:hexanes) to give 7 a colourless oil(62mg 93%).
120
2-Methoxy-cycloher-3-en01 (120): Following general procedure (A), cyclohexadiene
monoxide (SOmg, 0.52mmol) and w(C0)2C1]2 (2mg, 0.005mmol) were reacted in THF
(0.5mL) and MeOH (0.5mL) for one hour. Concentration and chromatography (30%
ethyl acetatehexanes) gave 120 a colourless oiI (62rng, 93%). Rf = 0.13 on silica gel
(30% ethyl acetate:hexanes); IR (neat, cm-') 342 I, 3027, 2926, 1652, 139 1, 1086. 'H
NMR (400MHz, CDC13) G 5.74-5.8 l(1 H, m), 5.68-5.73 ( 1 H, m), 3.68-3.78 (2H, m), 3.45
(3H, s), 2.58 (lH, s (br)), 2.16-2.22 (2H, m), 1.92-2.00 (1H, m), 1.60-1.72 (LH, m); I3c
NMR (400MHz, CDC13) S 129.6, 124.6, 82.1, 70.5, 56.2, 28.0, 24.4; H R M S calcd for
C7Hl2O2 (hlt): 128.0837 Found: 128.0835.

124
2-Ethory-cyclohex-3-en01 (124): Following general procedure (A), cyclohexadiene
monoxide (50mg, 0.52rnmol) and w(C0)2C1]2 (2mg, 0.005rnmol) were reacted in THF
(OSmL) and EtOH (0.5m.L) for one hou. Concentration and chromatography (30% ethyl
acetate:hexanes) gave 124 a colourless oil (68mg, 92%). RF 0.17 on silica gel (30%
ethyl acetate:hexanes); iR (neat, cm*') 3421, 3027, 2972, 165 1, 1440, 1090. 'H NMR
(400MHz, CDC13) S 5.65-5.79 (2H, m), 3.68-3.80 (3H, m), 3.50-3.60 (lH, m), 2.40 (1 H,
s (br)), 2.12-2.22 (1H, m), 1.92-2.02 (IH, m), 1.58-1.72 (IH, m), 1.24 (3H, r, J= 7.0 Hz);
13c NMR (400MHz, CDC13) 8 129.3, 125.5, 80.7. 70.8, 64.2, 27.9, 24.4, 15.7; HRMS
calcd for C8Hl4O2 (M3: 142.0994 Found: 142.099%
2-Isopropory-cyclo hex-3-enol(12 1): Following general procedure (A), cyclohexadiene
monoxide (50mg, 0.52mrnol) and [Rh(C0)2C1]2 (2mg, 0.005mmol) were reacted in THF
(0.5mL) and 'P~OH (0.5mL) for 90 minutes. Concentration and chromatography (20%
ethyl acetate:hexanes) gave 121 a coIourless oil (76mg, 94%). RF 0.19 on silica gel
(20% ethyl acetate:hexanes); IR (neat, cm-') 3408, 3028, 2929, 1648, 1394, 1088. 'H
NMR (400MHz, CDCL) G 5.69-5.75 (1 H, m), 5.6 1 (1 H, ddd, J= 10.1,4.0,2.0 Hz), 3.78-
3.85 (2H, m), 3.64-3.71 (lH, m), 2.39 (IH, s), 2.12-2.18 (2H, m), 1.94-2.02 (lH, m),

1.60-1.70 (IH, m), 1.2 1 (6H, dd, J= 6.2, 2.2 Hz); I3c NMR (400=, CDC13) 6 1298.9,
126.6, 78.4, 71.1, 70.3, 27.8, 24.4, 23.4, 22.4; HRMS calcd for CgH& (m: 156.1 150
Found: 156.1 149.
2 - ~ h e n o x ~ - c ~ e l o heu-3-eaol (1 25): Following general procedure (B), c yclohexadiene
monoxide (SOmg, 0.52mmol), phenol (244mg, 2.6mrnol) and [Rh(C0)2Cl]2 (2mg,
0.005mmol) were reacted in THF ( I d ) for two hours. Extraction and chromatography
(30% ethyl acetate:hexanes) gave 125 a colourless oil (83mg, 84%). RF 0.26 on silica
gel (20% ethyl acetate:hexanes); IR (CH2CI2, cm-') 3410, 3030, 2997, 1648, 1390, 1090.
'H NMR (400MHz, CDC13) 6 7.26-7.3 1 (2H, m), 6.93-6.99 (3H, m), 4.70 (IH, ddd, J=
6.8, 2.1, 2.1 Hz), 3.95-4.03 (IH, m), 2.41 (lH, s), 2.20-2.28 (2H, m), 2.02-2.12 (lH, m),
1.70-1.84 (lH, m); 13c NMR (400MHz, CDC13) 6 157.7, 130.5, 129.6, 124.2, 121.2,
115.8, 78.8, 70.6, 27.8, 24.4; HRMS calcd for Ci2Hlr02 (M3: 190.0994 Found:
2-(MethyCphenyI-amino)-cyclohex-34nol (132): Following general procedure (C),
cyclohexadiene monoxide (50mg, 0.52mrnol), N-methylaniline (278mg, 2.6mmol) and

[Rh(C0)2C1I2 (2mg, 0.005mmol) were reacted in THF (0.5mL) for 1 hou. Concentration
and chrornatography ( 10% ethyl acetate:hexanes) gave 132 a colourless oil (96mg, 9 1 %).
RF 0.22 on silica gel (20% ethyl acetate:hexanes); IR (neat, cm") 3408, 3014, 2929,
1644, 1595, 1500, 1356, 1205, 1067,898. 'H NMR (400MHz, CDCI,) G 7.20-7.26 (2H,
m), 6.90 (2H, d,J= 8.1 Hz), 6.75 (IH, t,J= 7.1 Hz), 5.75-5.85 (1H. m), 5.45 (lH, dd,J=
9.9, 1.5 Hz), 4.26-4.34 (IH, m), 3.83-3.93 (lH, m), 2.76 (3H, s), 2.41 (lH, s), 2.16-2.23
(IH, m), 2.03-2.10 (lH, m), 1.70-1.8 1 (lH, m); "C NMR (400MHz, CDC13) 8 150.8,
130.2, 129.1, 126.5, 117.2, 114.2, 68.6, 64.2, 32.3, 29.2, 24.7; HRMS calcd for
C13Hi7N0 (M'): 203.1310 Found: 203.1308.
133
2-Indol-1-yl-cyclohex-3-en01 (133): Following general procedure (C), cyclohexadiene
monoxide (50mg, 0.52rnmol), indole (278rng, 2.6mmol) and [Rh(CO)2CI]2 (2mg,
0.005rnmol) were reacted in THF (0.5mL) for 2 hours. Concentration and
chromatography (10% ethyl acetate:hexanes) gave 133 a colourless oil (96mg, 91%).
R ~ 0 . 2 0 on silica gel (20% ethyl acetate:hexanes); IR (neat, cm-') 'H NMR (400MHz,
CDC13) 6 8.13 (lH, s), 7-67 (IH, d, P 7.9 HZ), 7-33 (lHT d, J= 8.1 HZ), 7.23-7.16 (lH,
m), 7.12-7.06 (lH, m), 7.01 (lH, d, J= 2.4 Hz), 5.86-5.79 (lH, m), 5.74-5.68 (1H. m),
4.01-3.94 (1% m), 3.59-3.53 (lH, m), 2.33-2.26 (2H, m), 2.08-1.99 (2H, m), 1.80-1.67
(lH, rn); 13c NMR (400MHz, CDCL) G 136.7, 128.6, 126.9, 126.6, 122.5, 122.2, 119.6,

119.4, 116.8, 11 1.3, 71.9, 43.0, 29.1, 24.4; HRMS calcd for Ci&NO (m: 213.1 154
Found: 213.1 155.
OMe
Ph AOH
2-Methoxy-4-phenyl-but-3-en-l-ol (126): Following general procedure (D), 2-styryl-
oxirane (SOmg, 0.34mrnol) and [Rh(CO)2C1]2 (Zmg, 0.005rnmol) were reacted in MeOH
(0.5mL) for 30 minutes. Concentration and chromatography (40% ethyl acetate:hexanes)
gave 126 a colourless oil (65mg, 92%). R ~ 0 . 1 9 on silica gel (30% ethyl
acetate:hexanes); IR (neat, cm-') 3429, 302 1 , 2936, 1493, 1447, 1 106, 1067, 9 13. 'H
NMR (400MHz. CDC13) 6 7.38-7.42 (2H, m). 7.31-7.36 (2H, m), 7.24-7.29 (lH, m),
6.65 (lH, d, P 16.1 Hz), 6.05 (lH, dd, J= 16.1, 8.8 Hz), 3.86-3.92 (1H, m), 3.61-3.68
(2H, m), 3.40 (3H, s), 2.20 (1H, s); I3c NMR (400MHz, CDC13) S 136.1, 134.3, 128.6,
128.0, 126.5, 125.8, 83.0, 65.5, 56.6; HRMS calcd for CI1Hi4O2 (m: 178.0994 Found:
2-Ethoxy-4-phenyl-but-3sn-li>l (127): Following general procedure (D), 2-styryl-
oxirane (SOmg, 0.34mrnol) and j3h(C0)2C1]2 (2mg, 0.005mrnol) were reacted in EtOH
(0.5mL) for 30 minutes. Concentration and chromatography (20% ethyl acetate:hexanes)

gave 127 a colourless oil (67mg, 89%). R~0.27 on silica gel (30% ethyl
acetate:hexanes); IR (neat, cm*') 3422, 2971, 1493, 1447. 1085, 969. 'H NMR
(400MHz, CDC13) 6 7.37-7.41 (2H, m), 7.30-7.35 (2H, m), 7.23-7.28 (1H, m), 6.63 (lH,
d, J= 15.9 Hz), 6.07 (IH, ddd, J= 15.9, 7.7, 0.9 Hz), 3.97-4.03 (lH, m), 3.57-3.72 (3H,
m), 3.41-3.49 (fH, m), 2.29 (IH, dd, J= 6.8, 6.8 Hz), 1.24 (3H, dt, J= 6.8, 1.1 Hz); 13c
NMR (400MHz, CDC13) 6 136.2, 133.6, 128.6, 127.9, 126.6, 126.5, 81.2, 65.5, 64.2,
15.3; HRMS calcd for Ci2Hia02 ( ~ 3 : 192.1 150 Found: 192.1 153.
2-Isopropolry-4-phenyi-but-3-en-1-01(128): Following general procedure (D), 2-styryl-
oxirane (SOrng, 0.34mrnol) and [Rh(C0)2C1]2 (2mg. 0.005mmol) were reacted in 'P~OH
(0.5rnL) for 45 minutes. Concentration and chromatography (20% ethyl acetate:hexanes)
gave 128 a colourless oil (72mg, 90%). R ~ 0 . 2 on silica gel (20% ethyl
acetate:hexanes); IR (neat, cm-') 3422, 2975, 1490, 1447, 1080. 'H NMR (400MHz,
CDCI,) 6 7.37-7.41 (2H, m), 7.30-7.34 (2H, m), 7.23-7.27 (lH, m), 6.62 (lH, d, J= 16.1
Hz), 6.08 (IH, dd, J= 16.1,7.5 Hz), 4.08-4.14 (IH, m), 3.76 (lH, h, J= 6.0 Hz), 3.54-3.65
(2H, m), 2.22 (IH, dd, J= 8.0, 4.4 Hz), 1.19 (6H, dd, J= 6.0, 3.1 Hz); "C NMR
2 1.6; HRMS calcd for Ci3Hls02 0: 206.1307 Found: 206.1307.

NPhMe
Ph AOH 134
2-(MethyCphenyCamino)4phenyl-b~t~3-en-I-ol (134): Following general procedure
(C), 2-styryl-oxirane (50mg, 0.34mrnol), N-methylaniline (182mg, 1.7mrnol) and
p(C0)2CL]2 (Zmg, 0.005nmiol) wer3 reacted in TW (0.5mL) for I hou . Concentmtion
and chrornatography (10% increasing to 30% ethyl acetate:hexanes) gave 134 a
colourless oil (SOmg, 93%). RF on silica gel (% ethyl acetate:hexanes); IR (neat, cm'
') 3394, 302 1, 2887, 1595, 1496, 1384, 1032, 747. 'H NMR (400MH2, CDC13) 8 7.17-
7.33 (7H, m), 6.93 (2H, d, J= 0.9 Hz), 6.80 (IH, ddd, J= 0.9, 1.9, 8.3 Hz), 6.16 (IH, dd,
J= 1.3, 16.2 Hz), 6.12 (1H, dd, J= 5.9, 16.2 Hz), 4.504.60 (1H. m), 3.76-3.89 (2H. rn),
2.82 (3H, s), 2.25 (IH, s); ' 3 ~ NMR (400MHz. CDC13) 6 150.8, 136.2, 132.7. L29.1.
128.5, 127.7, 126.3, 124.5, 118.4, 115.0, 63.2, 62.1, 31.9. HRMS calcd for Ci7H19N0
(m: 253.1467, Found: 253.1466.
142
4 ,&Dimethy l -2 - ( rne thyCphenyL-adno) -nona~ l (142): Following general
procedure (C), 2-(2,6-dimethyl-hepta-1,s-dienyl)-oxirane (50mg, 0.30rnrnol), N-
methylaniline (182mg, 1.7rnmol) and [Rh(C0)2CLI2 (2mg, 0.005rnrnol) were reacted in
THF (0.5mL) for I hour. Concentration and chromatography (10% increasing to 30%
ethyl acetate:hexanes) gave 142 a colourIess oil (71mg, 87%). RrO. 15 on silica gel
(15% ethyl acetate:hexanes); IR (neat, cm-') M O 1,29 15, 1658, 1598, 1500, 1447, 1 103,

1025, 747. 'H NMR (400MHz, CDC13) 6 7.22 (2H, d, J= 8.0 HZ), 6.92 (2H, d, J= 8.1
Hz), 6.79 (IH, d, J= 7.1 Hz), 5.08 (lH, d, J= 8.1 Hz), 5.02-5.00 (lH, m), 4.58-4.50 (LH,
m), 3.67 (lH, t,J= 11.8 Hz), 3.60-3.52 (lH, m), 1.67 (3H, s), 1.57 (3H, s), 1.48 (3H. s);
"C NMR (400MHz, CDC13) 6 151.4, 142.0, 131.7, 129.0, 123.8, 119.1, 118.4, 115.3,
62.5, 60.1, 39.7, 31.5,26.3, 25.7, 17.7. 17.1; HRMS calcd for CIBH17N0 (M3: 273.2093
Found: 273 .îO9O.
129
5-Hydroxy-4-methoxy-4-methyCpent-2-enoic acid ethyl ester (129): Following
general procedure (D), 3-(2-methyl-oxiranyl)-acrylic acid ethyl ester (SOmg, 0.32mmol)
and m(C0)2C1]2 (2mg, 0.005mrnol) were reacted in MeOH (0.5m.L) for 5 hrs.
Concentration and chromatography (50% ethyl acetatehexanes) gave 129 a colourless oil
(57mg, 94%). Rr=0.34 on silica gel (50% ethyl acetate:hexanes); IR (neat, cm") 3464,
2936, 1715, 1655, 1461, 1366, 1292, 1 176, 1064. 'H NMR (400MHz, CDC13) S 6.86
(lH, d, J= 16.1Hz), 5.99 (IH, d, J= 16.1Hz), 4.21 (ZH, q, J= 7.1Hz), 3.51 (2H, d, J=
6.2Hz), 3.24 (3H, s), 2.52 (lH, t, J= 6.3Hz), 1.32 (3H, s), 1.31 (3H, t, J= 7.1Hz); "C
NMR (400MHz, CDC13) G 166.0, 148.7, 122.6, 77.6, 68.3, 60.5, 50.7, 18.2, 14.1. HRMS
calcd for C9HiaOs (MC): 188.1049 FOUI^: 188.1042 .

4-Methoxy-4-rnethyi-5-0x0-pent-2-enoic acid ethyl ester (143): To a round bottom
flask was dissolved 129 (30mg, 0.16mmol) and Dess-Martin periodinane (64mg,
O. lbmrnol) in 1m.L dichloromethane. The reaction was stirred for 1 hr at room
temperature. The mixture was the concentrated and chromatographed ( 10% ethy 1
acetate:hexanes) gave 143 a colouriess oil (28mg, 94%). Rf =0.27 on silica gel (10%
ethyl acetate:hexanes); IR (neat, cm") 2985, 1729, 145 1, 1370, 1303, 1 180. 'H NMR
(400MHz, CDC13) 6 9.48 (lH, s), 6.78 (1H, d, J= 16.0Hz), 6.15 (1H, d, J= 16.OHz), 4.22
(ZH, q,J= 7.OHz), 3.36 (3H, s), 1.43 (3H, s), 1.30 (3H. t. J= 7.OHz); "C NMR (400MHz,
CDC13) 6 165.5, 143.7, 124.3, 83.1, 60.7, 52.2, 18.4, 14.1, 12.7. HRMS calcd for
C9Hi4O4 (If'): 186.0892 FOUII~: 186.0895.
~(4-Bromo-phenylamino)-5-hydroxy~ethyI-pent-2-enoic acid et hyl ester (1 36):
Following general procedure (C), 3-(2-methyl-oxirany1)-acrylic acid ethyl ester (50mg,
0.32mmol), 4-bromoaniline (1 72mg, I .6mmol) and [Rh(C0)2C1]2 (2mg, 0.005mmol)
were reacted in THF (0.5mL) for 8 hours. Concentration and chromatography (10%

increasing to 30% ethyl acetate:hexanes) gave 136 a colourless oil (97mg, 93%).
RpO. 1 1 on silica gel (30% ethyl acetate:hexanes); R (neat, cm") 340 1, 297 1, 1704,
1655, 1496, 1363, 1303, 1180, 1036,909. 'H NMR (400MHz, CDC13) G 7.20 (2H, d, J=
Hz), 2.47 (IH, s), 1.40 (3H, s), 1.28 (3H, t, J= 7.1Hz); NMR (400MHz, CDC13) G
166.3, 150.4, 144.3, 131.6, 122.5, 117.5, 110.1,68.2, 60.7, 58.3,21.4, 14.1. HRMS calcd
for Ci&IisBrN03 (m: 327.0470 Found: 327.0472.
- 0 W O H NHPh
5-Hydroxy-4-methyl4phenylamino-pent-t-en acid ethyl ester (135): Following
general procedure (C), 3-(2-methyl-oxirany1)-acrylic acid ethyl ester (50mg, 0.34mmol),
N-methylaniline ( 182mg, 1.7mmol) and [Rh(CO)F1l2 (2mg, 0.005rnmol) were reacted in
THF (0.5m.L) for 9 hours. Concentration and chromatography (10% increasing to 30%
ethyl acetate:hexanes) gave 135 a colourless oil (80mg, 93%). R ~ 0 . 4 0 on silica gel
(50% ethyl acetate:hexanes); IR (neat, c d ) 3300(br), 2980, 3007, 17 15, 1655, 1450; 'H
NMR (400MHZ CD(%) 6 7- 10-7.15 (2H, m), 7.02 ( 1 H, d, J= 16.OHz), 6.72-6.76 (1 H,
m), 6.63 (lH, d, J= 8.6Hz), 6.00 (lH, d, J= 16.OHz), 4.18 (2H, q, J= 7.1Hz), 3.69 (lH, d,
AB, J= 11.0 Hz), 3.53 (lH, d, AB, J= 11.0 Hz), 2.34 (lH, s), 1.41 (3H, s), 1.28 (3H, t, J=
7.lHz); "C NMR ( ~ O O M H Z , CDC13) 8 166.4, 151.0, 145.2, 128.9, 122.1, 118.5, 116.2,

68.1, 60.6, 58.3, 2.8, 14.1. HRMS calcd for C l a l 9 N o 3 (hlt): 249.1365 Found:
249.13 62.
O
136
5-Hydro~uy4methyl4(naphthalen-l-ylamino)pent2-enoic acid ethyl ester (136):
Following general procedure (C), 3-(2-methyl-oxirany1)-acrylic acid ethyl ester (50mg,
0.32mmol), 1-aminonaphthalene (228mg, l.6mmol) and N(CO)ICI]~ (2mg,
0.005mmol) were reacted in THF (0.5mL) for 10 hom. Concentration and
chromatography (10% increasing to 30% ethyl acetate:hexanes) gave 136 a colourless oil
(80mg, 87%). R~0.20 on silica gel (30% ethyl acetate:hexanes); IR (neat, cm")
3400(br), 30 10, 2990, 17 15, 1660, 1460; 'H NMR (400MHz, CDCli) G 7.87-7.9 1 (IH,
m), 7.76-7.79 (lH, m), 7.41-7.46 (2H, m), 7.23-7.26 (2H, m), 7.08 (IH, d, J= 16.OHz),
6.59-6.63 (IH, m), 6.02 (lH, d, J= 16.OHz), 5.04 (lH, s), 4.16 (2H, q, J= 7.1Hz), 3.79
(IH, d, AB, J= 10.8 Hz), 3.63 (lH, d, AB, J= 10.8 Hz), 2.40 (lH, s), 1.51 (3H, s), 1.25
(3H, t, J= 7.lH~); "C NMR (~OOMHZ, CDC13) 6 166.4, 150.8, 139.9, 134.4, 128.7,
125.9, 125.6, 124.9, 122.4, 122.4, 120.0, 118.2, 109.2, 68.8, 60.6, 58.3, 21.2, 14.1.
HRMS calcd for C isH2iN03 0: 299.1 52 1 Found: 299.1526.
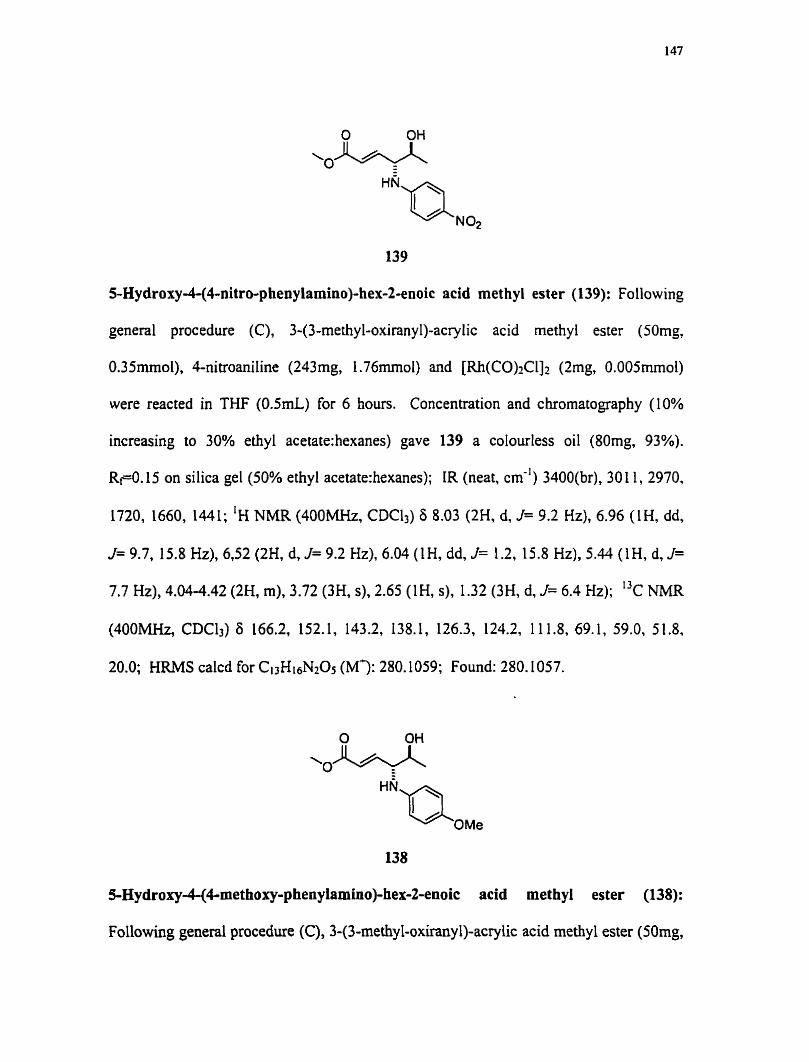
139
5-Hydroxy-4-(4-nitro-phenylamino)-hex-2-enoic acid methyl ester (139): Following
general procedure (C), 3-(3-methyl-oxirany1)-acrylic acid methyi ester (50mg,
0.35mrnol), Cnitroaniline (243mg, 1.76rnmol) and [Rh(C0)2C1]2 (2mg 0.005mrnoI)
were reacted in THF (0.5mL) for 6 hours. Concentration and chromatography (10%
increasing to 30% ethyl acetate:hexanes) gave 139 a colourless oil (80mg, 93%).
RFO. 15 on silica gel (50% ethyl acetate:hexanes); IR (neat, cm-') 3400(br), 301 1 , 2970,
1720. 1660, 144 1; 'H NMR (400MHz, CDC13) 8 8.03 (ZH, d, J= 9.2 Hz), 6.96 (IH, dd,
J= 9.7, 15.8 Hz), 6.52 (2H. d, J= 9.2 Hz). 6.04 (lH, dd, J= 1.2, 15.8 Hz), 5.44 (lH, d, J=
7.7 Hz), 4.04-4.42 (2H, m), 3.72 (3H, s), 2.65 (1H. s), 1.32 (3H, d, J= 6.4 Hz); ')c NMR
(400MHz, CDC13) 6 166.2, 152.1, 143.2, 138.1, 126.3, 124.2, 1 1 1.8, 69.1, 59.0, 51.8,
20.0; H R M S calcd for Ci3Hi6NzOs (m: 280.1059; Found: 28O.IOSï.
138
5-Hydrory4(4-methory-phenylamino)-hex-2-enoic acid methyl ester (138):
Following general procedure (C), 3-(3-methyl-oxirany1)-acrylic acid methyl ester (50rng,

0.35mmol), p-anisidine (216mg, 1.76mmol) and [Rh(C0)2C1]2 (2mg, 0.00Smrnol) were
reacted in THF (OSmL) for 1 hour. Concentration and chromatography (10% increasing
to 30% ethyl acetate:hexanes) gave 138 a colourless oil (80mg, 93%). RFO. 10 on silica
gel (40% ethyl acetate:hexanes); IR (neat, cm*') 3300(br), 2940, 1720, 1660, 1470; 'H
NMR (400MHz, CDC13) 8 6.93 (lH, dd, J= 6.4, 15.8 Hz), 6.77 (2H, d, J= 9.0 Hz), 6.57
(2H, d, J= 9.0 Hz), 6.02 (lH, dd, J= 1.0, 15.8 Hz), 1.05-4.12 (lH, m), 3.90-3.95 (lH, m),
3.73 (3H, s), 3.71 (3H, s), 1.80-2.20 (LH, br s), 1.24 (3H, d, J= 6.6 Hz); ')c NMR
5 1.6, 19.7; HRMS calcd for Ci4HisNOr (M3: 265.13 14; Found: 265.13 10.
3-[S-Methyl-3-(4-nitro-phenyi)-2-0x0-oxamMb4yl] acrylic acid methyl ester
(145): To a round bonom Bask was added 139 (30mg, O. lmmol), 200 triethylamine
and 1mL THE Carbonyldiimidazole (20mg, 0.125rnrnol) was then added and the
reaction stined at room temperature for 2 hours. The solution was then concentrated and
chromatographed (50% ethyl acetate:hexanes) to give 144 (28mg, 90%). R~0.15 on
silica gel (40% ethyl acetate:hexanes); mp 155'; IR (CCb, cm-') 30 10,2940, 1770, 1660,
1450; 'H NMR (300MHz, CDCb) 8 8.21 (2H, d, J= 9.2Hz), 7.68 (2H, d, J= 9.2Hz), 6.86
(IH, dd, J= 6.6, 15.8Hz), 6.05 (1H, dd, J= 0.8, 15.8Hz), 5.08-4.93 (2H, m), 3.75 (3H, s),
1.44 (3H, d, J= 6.4Hz); 13c NMR (400MHZ, CDCI,) 8 164.8, 153.8, 143.5, 142.8, 139.4,

126.6, 124.9, 1 18.7, 73.5, 60.4, 52.1, 15.8. HRMS calcd for CI*H14NZ04 (m: 306.0852;
Found: 306.0850.
TBSO T 145
te~-Butyl-dimethyl-[3-(3-methyl-oxiranyl)-allyioy]-silane (145): To a flame dried
round bottom fiask was added TBSCl(595mg, 3.95rnmol), imidazole (4 10mg, 6.Omrnol),
DMAP (1 Omg), and 300mg 3 -(3-methyl-oxiranyl)-prop-2-en- 1-ollW (3 OOmg, 2.63 mmol).
DCM (3mL) was then added and the reaction was stirred at room temperature for 30
minutes. The solution was then concentrated and chromatographed (2.5% ethyl
acetate:hexanes) to give 145 a colourless oil (475mg, 80%). R ~ 0 . 5 on silica gel (5%
ethyl acetate:hexanes); IR (neat, cm-') 3010, 2985, 1565, 1275; 'H NMR (400MHz,
CDC13) 8 5.96 (lH, ddd, J= 4.6,4.6, 15.5Hz), 5.44 (lH, dddd, J= 1.8, 1.8, 7.9, 15.5Hz),
4.17 (2H, dd, J= 1.8, 4.6Hz), 3.06 (LH, dd, J= 2.1, 7.9Hz), 2.90 (lH, ddd, J= 2.1, 5.2,
10.4Hz), 1.32 (3H, d, J= 5.2Hz), 0.89 (9H, s), 0.06 (6H, s); '.'c NMR (400MHz, CDC13)
6 134.4, 127.2, 62.9, 59.1, 56.4, 25.9, 18.4, 17.5, -5.3. HRMS calcd for C12H2J02Si
0: 228.1546 Found: 228.15444.
5-(tert-Butyl-dimethyl-silanoxy)-2-metho- (131): Following general
procedure @), 145 (SOmg, 0.22mmo 1) and [Rh(CO)2C1]2 (2rng, 0.005mmol) were reacted

in MeOH (0.5mL) for 15 minutes. Concentration and chromatography (30% ethyl
acetate:hexanes) gave 131 a colourless oil (5 h g , 90%). Rr0.15 on silica gel (30%
ethyl acetate:hexanes); IR (neat, cm-') 3400@r), 3010, 2990, 1565, 1275; 'H NMR
(400MHz, CDC13) 6 5.8 1 (1 H, ddd, J= 4.6,4.6, 15.6Hz), 5.53-5.64 (1 H, m), 4.22 (2H, d,
J= 3.2Hz), 3.78-3.87 (LH, m), 3.51 (1H, dd, J= 3.8, 8.OHz), 3.30 (3K, s), 2.19 (IH, s),
1.1 1 (3H, d, J= 6.6Hz), 0.90 (9H, s), 0.07 (6H, s); '-'c NMR (400MHz, CDCI,) S 135.5,
125.7, 85.7, 69.4, 63.0, 56.4, 25.9, 18.4, 17.8, -5.2. HRMS calcd for Ci3H2s03Si (M3:
260.1808 Found: 260.180 1.
TBSO &
Following general procedure (C), 145 (SOmg, 0.34rnrnol), N-methylaniline (182mg,
1 .immol) and [Rh(C0)2C1]2 (2mg, 0.005rnrnol) were reacted in THF (0.5m.L) for 1 hou.
Concentration and chromatography (1 0% increasing to 30% ethyl acetate:hexanes) gave
140 a colourless oil (gomg, 93%). R~0 .40 on silica gei (20% ethyl acetate:hexanes); iR
(neat, cm-') 3400(br), 3014,2930, 1644, 1590, 1210; 'H NMR (400MHz, CDCI,) 8 7.22
(2H, dd, J= 7.4, 8.8Hz), 6.79 (2H, d, J= 7.9Hz), 6.72 (IH, t, J= 7.2Hz), 5.90 (IH, dddd,
J= 1.4, 1.4, 6.1, 15.6Hz), 5.78 (lH, ddd, J= 4.1, 4.1, 15.6Hz), 4.19 (2H, d, J= 4.1Hz),
3.95-4.16 (2H, m), 2.80 (3H, s), 1.77 (IH, s), 1.21 (3H, d, J= 5.7Hz), 0.89 (9H, s), 0.05
(6H, s); I3c NMR (400MHz, CDC13) 6 150.3, L34.2, 129.1, 124.9, 117.1, 113.5, 68.5,

66.5, 63.2, 33.2, 25.9, 20.4, 18.4, -5.2. HRMS calcd for CI9HaNO2Si (M?): 335.2281
Found: 335.2278.

' For a review on allylic alkylation, see: Trost, B.M.; Van Vranken, D.L. Chem. Rev.
1996,96,395 and pertinent references therein; For a review on ailylic amination, see:
Iohannsen, M.; Jorgensen, KA.; Chem. Rev. 1998,98, 1689 and pertinent references
therein; For a general reference on the chemistry of palladium, see: Palladium reagents
and Caraiysis, Tsuji, J., Witey and Sons Ltd., Toronto, i 995.
' Tsuji, J.; Takahashi, H.; Morikawa, M. Tetradehron Lett. 1965,4387; Tsuji, J. Acc.
Chem. Res. 1969,2, 144.
Trost, B.M.; Fullerton, T.J. J. Am. Chem. Soc. 1973,95, 292.
J Hata, G.; Takahashi, K.; Miyake, A. Chem. Commun. 1392 (1970); Hata, G.; Takahashi,
K.; Miyake, A. Chem. Commun Bull. Chem. Soc. Jpn. 1972,45, 230; Atkins, K.E.;
Waker, W.E.; Manyik, R.M. Tetrahedron Lett. 1970,382 1.
Tsuji, J.; Shimizu, 1.; Minami, 1.; Ohashi, Y. Tetrahedron Lett. 1982,23,4809; Tsuji,
I.; Shimizu, 1.; Minarni, 1.; Ohashi, Y .; Sugiura, T.; Takahashi, K. J. Org. Chem., 1985,
50, 1523; For reviews, see: Tsuji, J.; Shimizu, 1.; Minarni, 1. Acc. Chem. Res. 1987,20,
140; Tsuji, I. Tetrahedron, 1986,42,436 1.
Tsuji, J.; Shimizu, L; Minami, L; Ohashi, Y. Tetrahedron Lett. 1982,23,4809; Tsuji, J.;
Shimizu, 1.; Minami, 1.; Ohashi, Y.; Sugiur;; T.; Takahashi, K. J. Org. Chem., 1985,50,
1523.
7 Hata, G.; Takahashi, K.; Miyake, A. Chem. Commun. 1392 (1970); Hata, G.; Takahashi,
K.; Miyake, A. Bull. Chem. Soc. Jpn. 1972,45, 230; Tsuji, J.; Kobayashi, Y.; Kataoka,
H.; Takahashi, T. Tetrahedron Lett., 1980,21, 1475.
Tsuji, J.; Kataoka? H.; Kobayashi, Y. Tetrahedron Lett., 1981,22,2575; Trost, B.M.;
Molander, GA. J. Am. Chem. Soc. 1981,103,5969.

Trost, B.M.; Weber, L. J.Am. Chem.Soc. 1975,97, 161 1; Trost, B.M.; Weber, L.;
Strege, P.E.; Fullerton, T.J.; Dietsche, T. J. Am. Chem. Soc. 1978, 100, 34 16; Trost,
B.M.; Verhoeven, T.R. Am. Chern. Soc. 1980,100,3435; Trost, B.M.; Verhoeven, T.R.
J. Org. Chem. 1976,41,3215; Akermark, B.; Backvall, J.E.; Lowenborg, A.; Zetterberg,
K. J. Organomet. Chem. 1981,2I 7 , C4 1 ; Matsushita, H.; Negishi, E. Chem. Commun.
1982, 160; Temple, AS.; Riediker, M.; Schwartz, J. J. Am. Chem. Soc. 1982, 104, 13 10.
10 Hayashi, T.; Hagihara, T.; Konishi. M.; Kumada, M. J. Am. Chem. Soc. 1983, I M,
7767; 'O Hayashi, T.; Konishi, M.; Kumada, M. Chem. Commun. 1984, 107.
" Hayashi, T.; Yamamoto, A.; Hagihara, T. J. Org. Chem. 1986,5l, 723.
'' Ibid. l 3 Trost, B.M.; VanVranken, D.L. Chem. Rev. 1996,96,395; Trost, B.M. Acc. Chem.
Res. 1996,29,355.
'' Trost, B.M.; Toste, F.D. J. Am. Chem. Soc. 1999, 121,4545.
" Trost, B.M.; Radinov, R.; Grezner, E.M. J. Am. Chem. Soc. 1997, 1 19, 7879.
l6 Trost, B.M.; Schroeder, G.M. J. Am. Chem. Soc. 1999,121,6759.
l7 Trost, B.M.; Ariza, X. J. Am. Chem. Soc. 1999, 121, 10727.
'' Trost, B.M.; Toste, F.D. J. Am. Chem.Soc. 1998,129,8 15.
l9 For a review on dynamic kinetic transformations, see: Ward, R.S. Tetrahedron:
Asyrnmetry 1995,6,1475.
Trost, B.M.; McEachem, E.J.; Toste, F.D. J. Am. Chem. Soc. 1998,120, 12702.
2' Trost, B.M.; McEachem, E.J. J. AM. Chem.Soc., 1999,12 1,8649.
* Trost, B.M.; Toste, F.D.J. Am. Chem. Soc. 1999,121,3543.
Trost, B.M.; Tsui, H.-C.; Toste, F.D. J. Am. Chem. Soc. 2000,122,3534.

- - . - -
" Evans, P.A.; Nelson, J.D. Tetrahedron Lett. 1998,39, 1725.
Evans, P.A.; Nelson, J.D. J. Am. Chem. Soc. 1998, I20,5581.
26 Ibid.
" Evans, P.A.; Robinson, LE.; Nelson, J.D. J. Am. Chem. Soc., 1999, (21.676 1.
Evans, P.A.; Robinson, J.E. Org. Lett., 1999,1, 1929.
29 Evans, P.A.; Leahy, D.K. J. Am. Chem. Soc. 2000, ASAP article.
'O Talceuchi, R.; Kashio, M. Angew. Chem. Int. Ed. Engl. 1997,36,263.
3 1 Trost, B.M.; Lautens, M. J. Am. Chem. Soc. 1987,109, 1469; Trost, B.M.; Merlic. C.A.
J. Am. Chem. Soc. 1990,110,9590.
32 Dvorak, D.; Stary, L; Kocovsky, P. J. Am. Chem. Soc. 1996, 118.897.
33 Faller, J. W.; Linebarrier, D. Organorne~ailics 1988, 7, 1670.
34 Rubio, A.; Liebeskind, L.S.; Bombmn, A. J. Am. Chem. Soc. 1993, I I5,89 1.
'' Trost, B.M.; Hachiya, L J. Am. Chern. Soc. 1998,170, 1 104.
36 Trost, B.M.; Hildebrand, S.; Dogra, K. J. Am. Chem. Soc. 1999,121, 104 16.
" Bricout, H.; Carpentier, J.-F.; Mortreux, A. J. Chem. Soc.. Chem. Commun. 1995,
1863.
38 Kondo, T.; Ono, H.; Satake, N.; Mitsudo, T-a.; Watanabe, Y. Organometallics, 1995,
14, 1945.
39 Xq Y.; Zhou, B. J. Org. Chem. 1987,52,974.
40 Lloyd-Jones, G.C.; Pfaltz, A. Angew. Chem. Int. Ed. EngZ. 1995,34,462.
'' Bhatia, B.; Reddy, M.M.; Iqbal, J. Tetrahedron Lett. 1993,34,630 1.
42 Chiu, P.; Lautens, M. Top. Cuw. C'hem. 1997, 1.
" Vogel, P.; Fattori, D.; Gasparini, F.; Drian, C.L. Synlett-1990, 173.

44 Guchteneere, E.D.; Fattori, D.; Vogel, P. Tetrahedron 1992,48, 10603.
" Reymond, J.-L.; Pinkerton, A.A.; Vogel, P. J. Org. Chem. 1991,56,
Moritz, V.; Vogel, P. Tetrahedron Lat. 1992,33,5243.
47 Kowarski, C.R.; Sarel, S. J. Org. Chem. 1973,38, 1 17.
48 Schmidt, R.R.; Liebemecht, A. Angew. Chem. Inr. Ed. Engl. 1978,!7,769.
" Noyon, R.; Sato, T.; Hayakawa, Y. J. Am. Chem. Soc. 1978,100,256 1 .
50 Cowling, A.P.; Mann, J.; Usmani, A.A. J. Chem. Soc. Perkin Tram. 2 1981, 21 16.
5' Arjona, O.; Leon, M.; Plumet, I. J. Org. Chem. 1999,64,272.
52 Lautens, M.; Chiu, P.; Ma, S.; Rovis, T. J. Am. Chem. Soc. 1995, 11 7, 532.
Lautens, M.; Rovis, T. J. Am. Chem.. Soc. 1997, 119, 11090.
" Lautens, M.; Rovis, T. J. Org. Chern. 1997, 62, 5246.
55 Arjona, O.; Pradilla, R.F.d.1.; Garcia, E.; Martin-Domenech, A.; Plumet, J. Tetrohedron
Lett. 1989,30,6437; Lautens, M.; Smith, A.C.; Abd-El-Aziz, A.S.; Huboux, A.H.
Tetrahedron Lett. 1990,3 1,3253.
56 Lautens, M.; Felice, CD.; Hubow, A. Tetrahedron Lett. 1989,30, 68 17.
57 Lautens, M.; Chiu, P.; Tetrahedron Lett. 1993,34, 773.
58 Lautens, M.; Gajda, C.; Chiu, P.J. Chem. Soc. Chem. Commun. 1993, 1193.
59 Lautens, M.; Kumanovic, S. J. Am. Chem. Soc. 1995,l 2 7, 1 954.
60 Lautens, M.; Renaud, J.-L.; Hiebert, S. J. Am. Chem. Soc. 2000,122, 1804.
61 Coluci, J. Total Synthesis of Ionomycin, University of Toronto, 1997.
'* Hogeveen, H.; Middeikoop, T.B. Tetrahedron Lett. 1973,367 1.
" Ashworth, R. W.; Berchtold, G.A. Tetrahedron Lett. 1977,339.
Bmggink, A.; Hogeveen, H. Tetrahedron Lett. 1972,496 1.

65 Johnson, B. M.; Chang, P.-T. L. Anaiytical Profiles of Dnrg Substances and Excipients
66 Jones, J.H.; Anderson, P.S.; Baldwin, J.J.; Clineschmidt, B.V.; McClure, D.E.;
Lundell, G.F.; Randall, W.C.; Martin, G.E.; Williams, M.; Hirshfield, J.M.; Smith, G.;
Lurnma, P.K. J. Med. Chem. 1984,27,1607.
'' Snyder, S. E. JMed Chem. 1995,38,2395.
68 Karnal, A.; Gayatri, L. Tetrahedron Lett. 1996,3 7,3359.
* Welch, W.M.; K R S ~ ~ , A-R; Sargcs, R; Keo, B.K J. Med. Chem. 1984, 77. 1508; Wyrick. S.D.; Booth. R.G.; Mytrs. AM.; Owens, C.E.; Kula. n.S.; Batdessarini, M.; McPhail. A.T.; Mailmrin, RB. J. Med. Chem. 1993, 36. 15-42; lohansson. A M . ; Arvidssan, LE.; H;ickseIl, U.; Nitsson. I.L.G.; Svcnsson. K.; Carlsson, A. J. Med. Chem. 1987.30.602; Snydcr. S.E.; Avilcs-Gmy. FA.; Ch;iknboni, R; Nichols D.E.; Watts. VJ.; Mailm. RB. J. Med. Chem. 1995. 38. 2395; Permne. R.; Benrdi. F.: Colûbufo. NA.; Lcopoldo, M.; Tonorcila, V,; Fiorcntini. F.; Olgiani, V.; Ghiglicrï, A.; Giovoni. S. J. Med. Chem. 1995, 38. 932; New. S.J.; Yevich, IF.; Eison, M.S.; Taylor, D.P.; Eison. AS.; Riblet. L.A.: VanderMaelen, C.P.; Temple Ir. D.L. J. .\fed. Chem. 1986. 29, 14c6: HaIlim. K.O.: Honda. T. Termhedmn 1995.51.1221 1.
Saiio, A.; Kayoma, Y.; Watanabe. T.; Fukushima, H.; H a T. J. Md. Chem. 1980,IJ. 1364. " Sobti, A; Kim, K.; Sulikowski. I.A. J. Org. Chem. 1996.61.5: Davis. FA.; Clnrk C.; Kumar, A.; Chcm. 8 . C J. Org. Chem.
I R * 59.1184. Kancko. T.; Wong, H.; Terruhdron Lerr. 1987 28.5 17: Kamal, A.; Gayatn, N.L. Terrahedmn Leu. 1996.37.3359.
73 The cis isomer of 15 was prepared by reaction of 1,2-dihydronaphthalene with 0 s 0 4
followed by dimethylation with dimethylsuifate. Trans-15 was prepared by epoxidation
of 1,2-dihydronaphthalene followed by ring opening with hydroxide and dimethylation
with DMS.
74 Lautens, M.; Chiu, P; Ma, S; Rovis, T J. Am. Chern. Soc. 1995,Ii 7,532.
" Comprehensive Organometallic Chemistry Vol. 5, Wilkinson, G. Permagon Press,
1982. pages 296-3 1 1.
76 Still, W.C.; Kahn, M.; Mitra, A. J. Org. Chem. 1989,54,5667.
Stiles, M.; Miller, RG. J. Am. Chem. Soc. 1960,82,3802.
'' Feiser, L.F.; Haddadin, M. Can. J. Chem. 1965,43, 1599.
79 Hart, H.; Lai, C.-Y.; Nwokogu, G.C.; Sharnouilian, S. Tetrahedron 1987,43,5203.
For palladium catalyzed allylic etherification reactions, see: Muzart, J.; Genet, J.P.;
Denis, A. J. Organomet. Chem. 1987,326, C23. For enantioselective ring opening of

epoxides with Cr(sa1en) complexes, see: Ready, J.M.; Jacobsen, EN. J. Am. Chem. Soc.
1999,12 1,6086. For the enantioselective ring opening of epoxides with Ga
heterobimetallic complexes, see: Iida, T.; Yamamoto, N.; Matsunaga, S.; Woo, H.-G.,
Shibasaki, M. Angew. Chem. Int. Ed. 1998, 37,2223. For desymmetrization of Baylis-
Hillman adducts with palladium, see: Trost, B.M.; Tsui, H.C.; Toste, F.D. J. Am. Chem.
Soc. 2000, 122,3534.
" Benrofurans, Mustafa, A. Wiley Intencience, Toronto, 1974.
" GOW, C.; Lhoste, P.; Sinou, D. Synlett, 1992, 725.
83 In our initial studies with alcohol nucleophiles, we found it necessary to change from
[R~I(CO)~C~]~ to [R~I(COD)C~]~ as the rhodium source since insoluble precipitates
resulted on mixing [Rh(C0)2C1]2 with phosphine ligands, which is the case with DPPF in
this study. We surmise that this precipitate is due to the formation of dimeric and
oligomeric rhodiurdphosphine complexes. FomiitousIy, this is not the case with the
PPF-PtBu2 ligand.
" Kitamura, M.; Ohkuma, T.; houe, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, S.;
Ohta, T.; Takaya, K.; Noyon, R. J. Am. Chem. Soc. 1988,110,629.
85 Brenchley, G.; Fedouloff, M.; Merifield, E.: Wills, M. Tetrahedron: Asymmetv 1996,
2809.
" Comprehensive Organometallic Chemistry, Vol. 5, Wilkinson, G. (Ed.), Permagon
Pens, Toronto, 1982, pages 307-3 1 1.
87 Comprehensive Organomeîallic Chemistry Vol. 5, Wilkinson, G. Permagon Press,
1982.

Comprehensive Organic Synthesis, Vol. 6, Trost, B.M.; Flemming, 1. (Eds.), Permagon
Press, New York, 199 1.
89 For a review on allylic alkylation, see: Trost, B.M.; Van Vranken, D.L. Chem. Rev.
1996, 96,395 and pertinent references therein; For a review on allylic amination, see:
Johannsen, M.; Jorgensen, K.A.; Chem. Rev. 1998,98, 1689 and pertinent references
therein; For a general reference on the chemistry of palladium, see: Pnlladium reagents
and Catalysis, Tsuji, J., Wiley and Sons Ltd., Toronto, 1995.
90 Trost, B.M.; Sudhakar, A.R. J. Am. Chem. Soc. 1987, 109,3792.
91 Trost, B.M.; Angle, S.R. J. Am. Chem. Soc. 1985,107,6 123.
9' Suzuki, S.; Fujita, Y.; Kobayashi, Y.; Sato, F. Tetrahedron Leti. 1986,27, 69.
93 Trost, B.M.; Tenaglia, A. Tetrahedron Lett., 1988,29,293 1; Keinan, E.; Sahai, M.;
Roth, 2.; Nudelman, A.; Heaig, S. J. Org. Chem. 1985,50,3558.
94 Trost, B.M.; McEachem, E.J.; Toste, F.D. J. Am. Chem. Soc. 1998, 120, 12702.
95 For use of diethylaluminum amides, see: Oveman, L.E.; Flippin, L.A. Tetrahedron
Lett. 1981, 195. For use of amino silanes and stannanes, see: Papini, A.; Ricci, A.;
Taddei, M. J. Chem. Soc. Perkin T m . 1 1984,2261; Fiorenza, M.; Ricci, A.; Taddei,
M.; Tassi, D. Synthesis, 1983,640. For the use of lithium, magnesiurn and zinc salts,
see: Chini, M.; Crotti, P.; Macchia, F. Tetrahedron Lett. 1990,3 1,466 1. For the use of
amino lead reagents, see: Yamada, I.; Yumoto, M.; Yamamoto, Y. Tetrahedron Lett.
1989,30,4255. For the use of cobalt catalysed aminolysis, see: Iqbal, S.; Pandey, A.
Tetrahedron Lett. 1990,3l, 575. For the use of lanthanide catalysts, see: Fu, X-L.; Wu,
S-H. S ' e t i c Comrnunicutions, 1997,27, 167%

96 Lautens, M.; Fagnou, K.; Rovis, T. Submitted for publication.; Lautens, M.; Fagnou,
K.; Taylor, M. Accepted to appear in Org. Lett.
'' Evans, P.A.; Nelson, J.D. J. Am. Chem. Soc. 1998,120,5581; Evans, P.A.; Robinson,
LE.; Nelson, J.D. J. Am. Chem. Soc. 1999,121,676 1
'* Ponaras, A.A.; Meah, M.Y. Tetrahedron Lett. l986,2 7,4953.
" Shibata, L; Yoshida, T.; Kawakami, T.; Baba, A.; Matsuda, H. J. Org. Chem. 1992,57,
4049.
'O0 Ley, S.V.; Bruckhart, S.; Cox, L.R.; Meek, G. JCS Perkin 1, 1997,3327.

Appendiv 1:
Selected Spectra of Representative Compounds











I 1 i - L ILL- - - -- J
- .- -- I I * e t * u I l .1



TBSO