Embed Size (px)
Citation preview

NormalizationIntro to R
Carol BultThe Jackson Laboratory
Functional Genomics (BMB550)Spring 2012
February 7, 2012

Biological questionDifferentially expressed genesSample class prediction etc.
Testing
Biological verification and interpretation
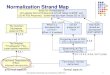
Microarray experiment
Estimation
Experimental design
Image analysis
Normalization
Clustering Discrimination

Microarray Data Analysis
Microarray experiment Image
Analysis
Gene level expression values
UnsupervisedAnalysis – clustering
Networks & Data Integration
Supervised Analysis
Normalized Data
Decomposition techniques

Why normalize?
Microarray data have significant systematic variation both within arrays and between arrays that is not true biological variation Accurate comparison of genes’ relative expression within and across conditions requires normalization of effects Sources of variation:
Spatial location on the array Dye biases which vary with spot intensity Plate origin Printing/spotting quality Experimenter

Sources of Systematic Bias
• Individual Factors– Print (20% - 30%)– Experimenter (20% -
30%)– Organism (3% - 10%)– Date (5%)– Software (2%)– Number of tips (3%)
• Interactions– Print - Experimenter (40%)– Print - Date (40%)– Experimenter - Date (40%)
(slide from Catherine Ball)
(based on ~4,600 experiments in Stanford Microarray Database analyzed by ANOVA)

KO #8
Probes: ~6,000 cDNAs, including 200 related to lipid metabolism. Arranged in a 4x4 array of 19x21 sub-arrays.
Clearly visible plate effects

Spatial Biases

Spatial plots: background from two slides

Log transform
Typical Affymetrix probe intensity distribution
After log-transform

#Create a boxplot of the normalized databoxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue")
#To save the boxplot as a jpeg filejpeg("normal_boxplot.jpg")boxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue")dev.off()

Normalization• There are many different ways to normalize
data– Global median, LOWESS, LOESS, RMA etc– By print tip, spatial, etc
• BUT: don’t expect it to fix bad data!– Won’t make up for lack of replicates– Won’t make up for horrible slides
Assignment: Read about normalization (Quackenbush and RMA article)http://functionalgenomics.wordpress.com/2010/01/01/lecture-9-normalization-carol-bult/

RMA Processing
• Background correction• Corrects for background noise and processing effects• Adjusts for cross hybridization• Adjusts for estimated expression values to fall on proper scale
• Normalization• Reduces unwanted variation across chips. • Ensures the quantiles of each chip are equal. Give same distribution to
each chip.
• Probe summarization– Once probe-level PM values have been background-corrected and normalized,
they need to be summarized into expression measures, so that the result is a single expression measure per probe-set, per chip.

• Using R• RMA processing
Log into the cloud.(don’t forget to have X windows server running)At the prompt, type “R” to invoke R...To quit R, type q() or quit() at the prompt.Save workspace.

You can invoke on-line help with the help.start() command.
Note: R is a case sensitive language! What happens if you type Help.Start() at the command prompt?

At the prompt (>) type the command , library(), to see which packages are available in your R installation.
A library is a location where R goes to find packages.
What is listed for you may differ from what is shown here.

•The caret “>” is the command prompt•‘library()’ is the command•the parantheses are for defining specific operations associated with the function.
You might find this R reference card helpful….Use it as a stub to create your own reference card!
http://www.psych.upenn.edu/~baron/refcard.pdf

Use this command to get a list of functions associated with the stats package.

Running this command will load all of the data sets that come with your R installation.

In this series of commands, we load the BOD (Biological Oxygen Demand) data set and then print out the data to the screen.

Data Frames in R
• The data sets in R are objects called data frames– You can think of a data frame as a table where the
columns are variables and rows are observations

Types of Objects in R
• Vector– One-dimensional array of arbitrary length. All members of a vector must be of the
same type (numeric, alpha, etc.)• Matrix
– Two –dimensional array with an arbitrary number of rows and columns. All elements in a matrix must be of the same type.
• Array– Similar to a matrix but with an arbitrary dimension
• Data frame– Organized similar to a matrix except that each column can include its own type of
data. Not all columns in a data frame need to contain the same type of data.• Function
– A type of R object that performs a specific operation. R contains many built-in functions.
• List– An arbitrary collection of R objects

Create the BOD Data Frame from Scratch1. Create a vector object for time using the c() function.
2. Create a vector object for demand.
3. Use the data.frame() function to create the data frame object

Use the read.table() function to read in data from a text file and use it as a data frame in R.
Other input functions for R include:read.csv()read.delim()
Reading in Data from Files
If you had data in an Excel spreadsheet, how could you import it into R?
<- can be used as an assignment operator, but = should also work.

How would you find out more about the c() function?

Writing Output to Files
Want to save the data in your data frame as a text file on your computer?
Use the write.table() function to output the MyBOD data frame to a text file.
This file will be saved to the directory that R is working from.

Writing Output to Files
Use the write.csv() function to output the MyBOD data frame to a comma separated file (which can be opened easily in Excel).
This file will be saved to the directory that R is working from.

Editing Data
For smaller data files, you can edit the file using the edit() function. This launches an R Editor window.
Always write the edited file to a new object name!
In this case we will edit the newdata object and store the results as an object called Mynewdata. To store the edited object as a file on your computer, use the write.table() function.

Exploring What is In a Data Frame
The names() and str() commands let you get an overview of what is in a data frame.
The names() function allows you to access the column names and edit them. In this code snippet, the first [1] and second [2] column names are changed from lower case to sentence case.

Accessing Data in a Data Frame
Use the name of the data frame (MyBOD2), a dollar sign ($) and the name of the variable (Time or Demand) to see a list of all of the observation values.
To access a specific value, you simply indicate the position in the vector…for example, MyBOD2$Demand [2] will access the second value for that variable which is 10.3.
If you “attach” the data frame using the attach() command you can access the variables and observations without the cumbersome need to specify the name of the data frame or the $.
Using what you know…how could you change the value of Demand[2] from 10.3 to 10.5? (be careful that you don’t make such changes to the original data frames!)

Adding columns to a Data Frame
You can add and delete columns from a data frame.
Here we add a column for the sex of whatever it is we are measuring oxygen demand for.
Oops!!! We have a data entry error. The value for sex should all be female (F). How would you fix this?

Deleting columns from a Data Frame
You can delete columns from a data frame.
Here we deleted the column for sex that we just created from the MyBOD2 data frame.

Note: When you are done using a data frame it is a good practice to “detach” it.

Displaying Data in RR comes with an incredible array of built in data analysis tools for exploring, analyzing, and visualizing data.
Here is a plot of the Time and Demand variables for the MyBOD2 data frame using the plot() command.
Note that because we “attached” this data frame we can just use the names of the variables to access the observation data.
Use help(plot) to look up the details of this command. Figure out how to change the command to add a title to the plot.

Displaying Data in R
Here is a box plot of the Demand variables for the MyBOD2 data frame using the boxplot() command.

Analyzing Data
The summary() command provides summary statistics for a data frame.

Analyzing DataHere are a series of commands to generate some basic statistics for the Demand variable in the MyBOD2 data frame.
The data frame has been attached so that the variable names can be used directly.
Remember that the case of the variable names were changed relative to the original BOD data set (Time vs time; Demand vs demand)!

Examples of stats functions in R
• mean()• median()• table() – there is no function to find the mode of a data set
but the table() function will show how many times a value is observed.
• max()• min()• There is no built in function for midrange so you have to
construct a formula to calculate this based on the values from the max() and min() functions.

Measuring data spread
Remember that the case of the variable names were changed relative to the original BOD data set (Time vs time; Demand vs demand)!
Here are a series of commands to generate some basic statistics related to the spread of measurements for the Demand variable in the MyBOD2 data frame.
The data frame has been attached so that the variable names can be used directly.

More examples of stats functions in R
• var()• sd()• There is no built in function for calculating the standard error
of the mean (sem) so you have to create a formula to calculate this.
• There is no built in function for calculating the range so you have to construct a formula to calculate this based on the values from the max() and min() functions.

What is meant by mode?
What do the variance, standard deviation and standard error of the mean tell us about a data set?

Your Turn
Create a data frame for age and frequency using the data on this slide.
Calculate the cumulative frequency and add it as a column to the data frame.
Save the data frame as a comma separated text file and then open it in Excel.
Plot age versus cumulative frequency.
What are mean and median age?
What is the variance, standard deviation, and standard error mean for frequency?


Affymetrix Data Files
• CEL file = results of the intensity calculations on the pixel values of the DAT file (the DAT file is the image file)
– http://www.stat.lsa.umich.edu/~kshedden/Courses/Stat545/Notes/AffxFileFormats/cel.html
• CDF = chip description file– Describes the layout for an Affymetrix gene chip array
I have put some CEL files and the related CDF file in my home directory for you to work with.

1. Log into the Maine Innovation Cloud 2. Copy the CEL files from my home directory to your directory2. Also copy a file called MoGene-1_0-st-v1.cdf to your directory3. Create a subdirectory called TestData4. Move the CEL files and cdf file into TestData5. Change directory to TestData6. Start R at the command line7. After R starts, type the commands on the next slide at the prompts in R

library(affy)library(makecdfenv)
CELData=ReadAffy()
Array.CDF=make.cdf.env("MoGene-1_0-st-v1.cdf")CELData@cdfName="Array.CDF"
rma.CELData=rma(CELData)rma.expr=exprs(rma.CELData)
rma.expr.df=data.frame(ProbeID=rownames(rma.expr), rma.expr)write.table(rma.expr.df, "rma.expr.dat", sep="\t", row=F, quote=F)
Performing RMA and saving the normalized data file

#CELData boxplot - plot CEL data intensities (non-normalized)jpeg("boxplot.jpeg")boxplot(CELData, names=CELData$sample, col="blue")dev.off() #CELData histogram - density histogram of CEL data (non-normalized)jpeg("histogram.jpeg")hist(CELData)dev.off() #print out a key to sample order (for plots and such)pData(CELData) #save R workspace to file (OPTIONAL)save.image("CEL files.Rdata")
Create a plot of the non-normalized intensities

Read data back in….create plot of normalized intensities
Open the rma.expr.dat text file using Excel. Save as a new file (tab delimited text format) such as “mydata.dat”
#Create an R object and read your tab delimited text file into itmydata <- read.table(“mydata.txt”, header=T, sep=”\t”) summary(mydata) #Exclude probe informationsummary(mydata[-1]) #Create a boxplot of the normalized databoxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue") #To save the boxplot as a jpeg filejpeg("normal_nobg_boxplot.jpg")boxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue")dev.off()

#Create a boxplot of the normalized databoxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue")
#To save the boxplot as a jpeg filejpeg("normal_boxplot.jpg")boxplot(mydata[-1], main = "Normalized Intensities", xlab="Array", ylab="Intensities", col="blue")dev.off()