Embed Size (px)
Citation preview

Nitric Oxide–Dependent Bone Marrow ProgenitorMobilization by Carbon Monoxide Enhances Endothelial
Repair After Vascular InjuryBarbara Wegiel, PhD; David J. Gallo, BS; Kathleen G. Raman, MD; Jenny M. Karlsson, MS;
Brett Ozanich, BS; Beek Y. Chin, PhD; Edith Tzeng, MD; Shakil Ahmad, PhD; Asif Ahmed, PhD;Catherine J. Baty, PhD; Leo E. Otterbein, PhD
Background—Carbon monoxide (CO) has emerged as a vascular homeostatic molecule that prevents balloon angioplasty–induced stenosis via antiproliferative effects on vascular smooth muscle cells. The effects of CO on reendothelializationhave not been evaluated.
Methods and Results—Exposure to CO has diametrically opposite effects on endothelial cell (EC) and vascular smooth musclecell proliferation in rodent models of carotid injury. In contrast to its effect of blocking vascular smooth muscle cell growth,CO administered as a gas or as a CO-releasing molecule enhances proliferation and motility of ECs in vitro by �50% versusair controls, and in vivo, it accelerates reendothelialization of the denuded artery by day 4 after injury versus day 6 inair-treated animals. CO enhanced EC proliferation via rapid activation of RhoA (Ras homolog gene family, member A),followed by downstream phosphorylation of Akt, endothelial nitric oxide (NO) synthase phosphorylation, and a 60% increasein NO generation by ECs. CO drives cell cycle progression through phosphorylation of retinoblastoma, which is dependentin part on endothelial NO synthase–generated NO. Similarly, endothelial repair in vivo requires NO-dependent mobilizationof bone marrow–derived EC progenitors, and CO yielded a 4-fold increase in the number of mobilized green fluorescentprotein–Tie2–positive endothelial progenitor cells versus controls, with a corresponding accelerated deposition of differen-tiated green fluorescent protein–Tie2–positive ECs at the site of injury. CO was ineffective in augmenting EC repair and theensuing development of intimal hyperplasia in eNOS�/� mice.
Conclusions—Collectively, the present data demonstrate that CO accelerates EC proliferation and vessel repair in a mannerdependent on NO generation and enhanced recruitment of bone marrow–derived endothelial progenitor cells. (Circulation.2010;121:537-548.)
Key Words: angioplasty, balloon � endothelium � nitric oxide synthase � signal transduction � stenosis
Proliferation of vascular smooth muscle cells (VSMCs)and their acquisition of a proinflammatory phenotype are
central events in the pathogenesis of vascular lesions, includ-ing vein occlusion, postangioplasty restenosis, and transplantarteriosclerosis.1–3 Restenosis rates at 1 year approach 30%without stents versus 5% in patients who receive stents. Morethan 95% of percutaneous coronary interventions involvestenting.4–6 Although stents hold great promise, there contin-ues to be a need for advances in current therapies. Drug-eluting stents are now used as novel drug-delivery devices,including rapamycin/sirolimus-coated devices that furtherreduce stenosis by interfering with VSMC proliferation.7,8
Drug-eluting stents, however, are complicated by in-stentthrombosis that results from delayed endothelialization ifantiplatelet therapies are stopped. Appropriate reendotheli-
Clinical Perspective on p 548alization of a stent or denuded vessel becomes crucial foreffective vascular homeostasis. In the present study, wehypothesized that carbon monoxide (CO) administered as aninhaled gas or via a CO-releasing molecule would providevascular protection by facilitating endothelial cell (EC) repairand preventing intimal hyperplasia. We demonstrated previouslythat a 1-hour exposure to CO at low, nontoxic concentrationsbefore injury, with no further treatment, prevented the develop-ment of intimal hyperplasia caused by balloon angioplasty viadirect effects on VSMC proliferation.9 Growth arrest in thesecells occurred via a pathway that sequentially involved cGMP,p38 mitogen-activated protein kinase, and p21. Interference atany point in this cascade resulted in abrogation of the effects of
Received June 19, 2009; accepted November 24, 2009.From the Transplantation Institute (B.W., B.Y.C., L.E.O.), Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School,
Boston, Mass; Department of Surgery (K.R., B.O., E.T.) and Department of Cell Biology and Physiology (J.K., C.B.), University of Pittsburgh Schoolof Medicine, Pittsburgh, Pa; Alfama Inc (D.J.G.), Beverly, Mass; and Department of Reproductive and Vascular Biology (S.A., A.A.), Institute forBiomedical Research, University of Birmingham, Birmingham, United Kingdom.
Drs Wegiel and Raman contributed equally to this article.The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/121/4/887695/DC1.Correspondence to Leo E. Otterbein, PhD, Associate Professor, Beth Israel Deaconess Medical Center, Harvard Medical School, 3 Blackfan Circle
EC/CLS-603, Boston, MA 02215. E-mail [email protected]© 2010 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.109.887695
537
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

CO. CO, like nitric oxide (NO), is pleiotropic in its effects,modulating cellular behavior and physiology in diverse waysdepending on the cell type, circumstance, and model beingevaluated.10,11 The end result, however, is that CO functions toreestablish vascular stability.
We hypothesized that the endothelium and more specifi-cally the ECs that are physically injured through denudationtrauma during the balloon procedure are a target by which COwould exert beneficial effects. Heme oxygenase-1 (HO-1),the cytoprotective enzyme responsible for the generation ofendogenous CO, was shown to regulate EC proliferation invitro12; however, the role and the mechanism of action ofHO-1 and CO in EC repair after trauma have not beenevaluated. HO-1–deficient mice show an exaggerated re-sponse to vessel injury. We reasoned that CO, in addition tohaving an antiapoptotic role in ECs,13 would enhance prolif-eration and migration of ECs and promote vessel repair byfacilitating mobilization of endothelial progenitor cells(EPCs). The knowledge that NO promotes the survival andproliferation of ECs and mobilization of EPCs14–16 promptedus to investigate the hypothesis that endothelial NO synthase(eNOS) may play a role in the effects of CO on ECs.
In the present study, we describe the effects of both CO anda CO-releasing molecule (CORM) on the augmentation of ECproliferation and migration and EPC mobilization to theinjured site in well-established rodent models of vasculartrauma.3 We show that CO accelerates reendothelialization ofthe injured vessel, and we identify eNOS and NO as essential forthe CO effect. In ECs, activation of Akt, eNOS, and retinoblas-toma (Rb) protein dominate to accelerate cell cycle progressionand EC migration. The diametrically opposite effects of CO onproliferation of ECs and VSMCs are clear evidence of thepleiotropic homeostatic effects of CO, in this case preventing thedevelopment of the intimal lesion by acting through disparatemodes of action on these 2 vascular cell types.
MethodsCell Culture and Pharmacological ReagentsRat primary aortic ECs (RAECs) were purchased from VEC Technol-ogies (Rensselaer, NY) and maintained in MCDB-131 Complete me-dium with antibiotics and growth factors (VEC Technologies) on 0.2%gelatin-coated plates. Cells were used between passages 3 and 10. Bonemarrow–derived EPCs from Tie2–green fluorescent protein (GFP)mice (Tie2 receptor is expressed in endothelial lineage cells17) werecultured in Endothelial Basal Medium (EBM-2) supplemented withEGM-2 BulletKit (Clonetics-Lonza, Rockland, ME) as described pre-viously.18 Cells were exposed to 250 ppm CO, 5% CO2, 21% O2,balance N2 as described previously.19 CORM (ALF421/CORMA-1,Alfama Inc, Cambridge, Mass) was used at a concentration of 1 to20 �mol/L unless otherwise stated. We used an inactive CORM(iCORM) as a negative control where indicated. VSMCs were isolatedfrom mouse aortas and grown in Dulbecco’s Modified Eagle Medium(DMEM; Invitrogen, Carlsbad, Calif) containing 15% FBS. Pharmaco-logical agents used included the selective inhibitor of NO synthase,NG-nitro-L-arginine methyl ester (100 �mol/L; Sigma-Aldrich, St Louis,Mo), and the phosphatidylinositol-3-kinase (PI3K) inhibitor LY294002(10 mmol/L; Sigma-Aldrich), which was used at 10 �mol/L. RAECs werepretreated for 1 hour with inhibitors, followed by CO or air treatment.
Adenoviral TransductionAdenovirus containing a dominant-negative mutant RhoA (Rashomolog gene family, member A) and constitutively active RhoAwere a kind gift from Dr James Bamburg, as described previously.20
Y5 and GFP adenoviruses were used as controls. All of the viruseswere used at 10 multiplicities of infection for infection of RAECs.Adenoviruses were prepared in serum-free medium and added to thecells for 6 hours. Cells were then washed with PBS, and freshserum–containing medium was added to the cells, which were thenincubated for 48 hours before use.
Plasmids and TransfectionsDominant-negative mutant Akt and control constructs were a kindgift from Dr Alex Toker (Beth Israel Deaconess Medical Center,Boston, Mass). For transfection experiments, RAECs were trans-fected with SuperFect transfection reagent (Qiagen, Valencia, Calif)according to the vendor’s protocol. Transfection efficiency wasevaluated by GFP and was determined to be on average 40% to 50%.
Proliferation AssaysThe effect of CO on proliferation of RAECs was determined with anonradioactive bromodeoxyuridine-based cell-proliferation assay (perthe manufacturer’s guidelines; Roche, Basel, Switzerland). Thymidineincorporation was measured in growth-arrested VSMCs and ECs stimulatedto proliferate with 10% FBS in the presence of 5 �Ci/mL of 3H-thymidine(NEN, Boston, Mass) for 24 hours with or without CO as indicated.
Cell Motility and MigrationCell motility and migration are described in the Methods section ofthe online-only Data Supplement.
Cell Cycle StudiesRAECs were starved for 20 hours in serum-free medium. Serum-containing medium was then applied together with pharmacologicalagents with or without CO for 6 to 24 hours. After they wereharvested, cells were fixed and permeabilized with 70% ice-coldethanol for 30 minutes, washed twice with 1� PBS, resuspended,and incubated in fluorescence-activated cell sorting (FACS) buffercontaining 5 mg/mL propidium iodide (Sigma), 100 mmol/L sodiumcitrate, pH 7.3 and 0.05 mg of RNase A (Sigma) for 30 minutes at4°C. Cell fluorescence was measured by FACS.
RhoA ActivationActivity of the small GTPase RhoA was determined by use of anEZ-Detect Rho Activation Kit (Pierce, Rockford, Ill) according tothe manufacturer’s protocol.
Immunoblotting and PhalloidinDetails concerning immunoblotting and the use of phalloidin areprovided in the online-only Data Supplement.
NO Generation
FluorescenceRAECs were loaded with the NO-selective fluorophore 4-amino-5methylamino-2�,7�-difluorescein diacetate (DAF-FM; MolecularProbes, Carlsbad, Calif) 20 minutes before exposure to air or CO(250 ppm) for 5 to 60 minutes. Cells were fixed, and fluorescencewas assessed by flow cytometry with excitation/emission of 495/515nm at various time points.
ChemiluminescenceHuman umbilical vein ECs were plated onto 24-well plates andtreated for 1 hour in the presence and absence of CO gas (250 ppm),and NO was measured in cell supernatants with a Sievers chemilu-minescence NO analyzer as described previously.21
Carotid Artery Injury Model in Rats and MiceMale Sprague Dawley rats (weight 250 to 300 g) were purchasedfrom Harlan Laboratories (Indianapolis, Ind), and mice were pur-chased from Jackson Laboratories (Bar Harbor, Me). Balloon angio-plasty and wire injury were performed as described previously.11
Injury of the vessel wall and subsequent pathological analysis wereaccomplished in a manner that was blinded to the treatment group
538 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
and was performed by trained scientists. Male C57BL/6 (JacksonLaboratories, Bar Harbor, ME), eNos�/� mice (7 to 8 weeks old),and Tie2-GFP mice were purchased from Jackson Laboratories.Mice were treated with CO (250 ppm for 1 hour), ALF421 (10 mg/kgIP 1 hour before and 1 hour after), or iCORM. Water or air was usedas a control. All animals were housed in accordance with theguidelines of the American Association for Laboratory AnimalScience. The carotid injury and bone marrow transplantation proto-cols were approved by the Institutional Animal Care and UseCommittee of Beth Israel Deaconess Medical Center.
Immunostaining and Cell PopulationHistomorphometric AnalysisVessels were harvested 1, 3, and 5 days after angioplasty. Rat leukocyteand EC populations were detected with anti-rat macrophage (CD68,ED1), intercellular adhesion molecule-1 (CD54; 1A29), myeloperoxi-dase, and CD31 antibodies. Mouse vessels were stained with hematox-ylin and eosin, Sca1�, and CD31 as described previously.22 Eight to 10images were captured from each injured carotid and analyzed as detailedabove. Intima/media ratio was measured as described previously.9RAECs were seeded on microscopic slides and treated with or withoutCO for 24 hours. Cell staining with P-histone H3 antibodies was appliedas described previously.22
Bone Marrow Transplantation and Generation ofTie2-GFP ChimerasWild-type C57BL/6J (Jackson Laboratories) mice were lethallyirradiated (12 Gy). On the same day, mice were injected with 5�106
bone marrow cells from Tie2-GFP mice (as above). Reconstitutionof bone marrow was determined by flow cytometry 1 month aftertransplantation. Tie2-GFP chimeras were used for the experiments asdescribed above. The average percent reconstitution was determinedto be between 90% and 95%, with 0.9% to 12% Tie2-GFP–positivecells in the marrow 4 to 6 weeks after transplantation.
Colony Outgrowth AssayColony outgrowth assay was performed as described previously.23
C57B6 mice were exposed to CO (250 ppm) or air for 1 hour, and
blood samples were collected. Mononuclear cells were freshlyisolated with Ficoll gradient (20 minutes, 500g) and seeded on the0.2% gelatin-coated plates in EGM endothelial growth mediumsupplemented with an EGM Bullet Kit (Clonetics Lonza, San Diego,Calif). Colonies were counted and photographed 10 days afterisolation of mononuclear cells.
Flow CytometryTo assess the percentage of GFP-positive ECs that were mobilized tothe circulation after CO/air treatment, blood samples were harvestedfrom animals 12 hours after wire injury. Red blood cells were lysedat room temperature for 5 minutes with erythrocyte lysis buffer,followed by washing with PBS. Cells were fixed with 2% parafor-maldehyde and blocked with 1% BSA in PBS followed by stainingwith antibody against GFP (Invitrogen, Carlsbad, Calif) for 1 hour atroom temperature. Secondary antibody conjugated with fluoresceinwas applied for 1 hour at room temperature. Cells were analyzed ona FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ).The chimeric mice were housed in the Beth Israel DeaconessMedical Center facility according to Institutional Animal Care andUse Committee–approved protocols. The average percent reconsti-tution was determined to be 0.9% to 1.2% GFP-Tie2–positive cellsin the marrow 4 to 6 weeks after transplantation.
Statistical AnalysesIn all of the in vitro experiments, the significance of difference wasdetermined with 1- or 2-way ANOVA (with post hoc Tukey test) asdescribed in the figure legends (SPSS Inc, Chicago, Ill). A nonpara-metric Wilcoxon test was used where indicated. Statistical analyseswere applied to all of the independent experiments, with significanceaccepted at P�0.05.
ResultsCO Augments EC ProliferationTo test the effects of CO on EC proliferation, primary RAECswere exposed to CO (250 ppm), and [3H]thymidine or bromode-
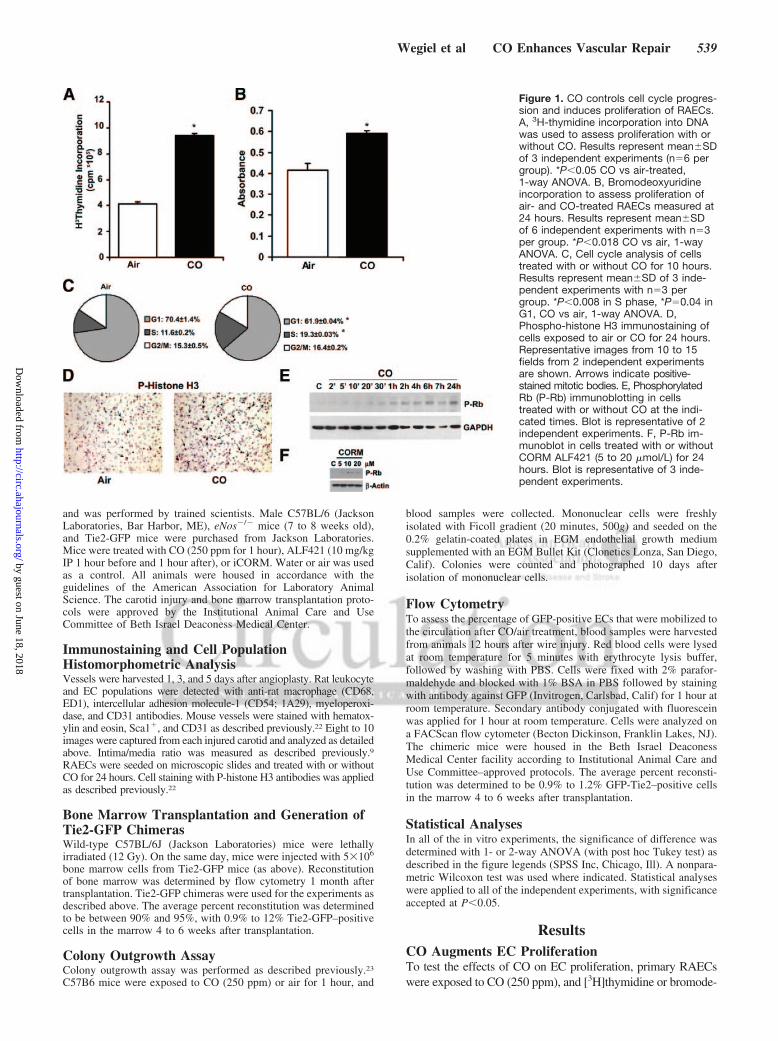
Figure 1. CO controls cell cycle progres-sion and induces proliferation of RAECs.A, 3H-thymidine incorporation into DNAwas used to assess proliferation with orwithout CO. Results represent mean�SDof 3 independent experiments (n�6 pergroup). *P�0.05 CO vs air-treated,1-way ANOVA. B, Bromodeoxyuridineincorporation to assess proliferation ofair- and CO-treated RAECs measured at24 hours. Results represent mean�SDof 6 independent experiments with n�3per group. *P�0.018 CO vs air, 1-wayANOVA. C, Cell cycle analysis of cellstreated with or without CO for 10 hours.Results represent mean�SD of 3 inde-pendent experiments with n�3 pergroup. *P�0.008 in S phase, *P�0.04 inG1, CO vs air, 1-way ANOVA. D,Phospho-histone H3 immunostaining ofcells exposed to air or CO for 24 hours.Representative images from 10 to 15fields from 2 independent experimentsare shown. Arrows indicate positive-stained mitotic bodies. E, PhosphorylatedRb (P-Rb) immunoblotting in cellstreated with or without CO at the indi-cated times. Blot is representative of 2independent experiments. F, P-Rb im-munoblot in cells treated with or withoutCORM ALF421 (5 to 20 �mol/L) for 24hours. Blot is representative of 3 inde-pendent experiments.
Wegiel et al CO Enhances Vascular Repair 539
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

oxyuridine incorporation was evaluated 24 hours later. CO gassignificantly increased EC proliferation over that of air-treatedcells as assessed by DNA synthesis (Figures 1A and 1B).Similarly, a CORM induced significant proliferation of RAECsin a concentration-dependent manner (online-only Data Supple-ment Figure IA). FACS analyses by propidium iodide stainingshowed that cells exposed to CO had a greater accumulation inthe S phase than air-treated cells (Figure 1C). Importantly, theeffect of CO or a CORM was unlikely to be due to hypoxia,because exposure of RAECs to hypoxia (1% O2) inhibitedproliferation of RAECs (online-only Data Supplement FigureIB). To corroborate the CO effects on the cell cycle, weevaluated cells for phosphorylated histone H3 and Rb expressionby immunostaining and immunoblotting, respectively (Figures1D and 1E; online-only Data Supplement Figure IIA) andobserved increased phosphorylation of both proteins. Treatmentwith a CORM also induced significant phosphorylation of Rb at24 hours versus vehicle control (Figure 1F). These results in ECscontrast directly with the effects of CO on proliferation ofVSMCs, which when exposed to identical conditions showedsignificant growth arrest (8.5�0.1�106 cpm/well in air-treatedcells versus 3.85�0.096 cpm/well in CO-treated cells,P�0.01), as reported previously9 but presented here again forcomparison.9,24,25
CO Increases EC MotilityUsing live cell time-lapse microscopy, we next evaluated theeffects of CO exposure on EC motility. Using the well-described“scratch” model, we disrupted an endothelial monolayer tosimulate EC denudation, to mimic that which occurs duringangioplasty trauma, and exposed the cells to media saturatedwith air or 250 ppm CO. In those cells exposed to CO, weobserved a 2-fold greater motility rate than in air controls (Figure2A). These data demonstrate that CO not only increased prolif-eration of ECs (Figure 1) but also increased their motility.Immunostaining for F-actin showed that ECs exposed to CO hada greater propensity for cytoskeletal organization that correlatedwith motility measurements (Figures 2B and 2C [air] versusFigures 2D and 2E [CO]). CO-treated cells showed a highlyorganized actin distribution, with stress fibers forming denseperipheral and polarized bands at the leading edge, which wasotherwise irregular and punctate in air-treated cells.
CO Activates RhoA and Akt Kinase in ECs,Which Regulates CO-Induced ProliferationWe next attempted to elucidate the signaling mechanisms thatwere influenced by CO to control EC proliferation. Owing to thehigh diffusivity of CO, we hypothesized that CO would elicit arapid effect on the cell initiated at the cell membrane. Weassessed expression of the small GTPase RhoA, which isimportant in cell growth and cytoskeletal organization and whichis an initiator of downstream signaling events. Exposure of ECsto CO resulted in a strong and rapid time-dependent activation ofRhoA (Figure 3A). Downstream of RhoA are the mitogen-acti-vated protein kinases, as well as Akt,26 both of which have beenshown to be modulated by CO.27,28 We first evaluated p38activation, which was decreased modestly (data not shown), indirect contrast to the effects of CO observed in VSMCs.24 Akthas been shown by others to be activated by CO, albeit in an
inconsistent fashion depending on the cellular model. One reportdemonstrated in human ECs that CO inhibits Akt activation,29
whereas another report showed that CO increases Akt in hearttissue from rats undergoing ischemia/reperfusion injury.28 Wetherefore wanted to assess the effects in our model. In RAECsexposed to CO, we observed a time-dependent induction of Aktphosphorylation (Figure 3B; online-only Data Supplement Fig-ure IIB). CO induces proliferation of ECs through activation ofRhoA and Akt (Figure 3C). The effect of CO on Akt activationwas abrogated in the presence of dominant-negative mutantRhoA (data not shown). Using a selective and well-characterizedpharmacological inhibitor of PI3K (LY290024), we evaluatedthe role of PI3K-dependent Akt on CO-induced proliferation ofECs. Blockade of PI3K led to a partial inhibition of the effectsof CO on phosphorylation of Rb (Figure 3D) and EC prolifer-ation (Figure 3E). To strengthen our observation that Akt waslinked to enhanced EC proliferation by CO, we transientlytransfected RAECs with a dominant-negative mutant Akt ex-pression vector (Figure 3F). When both pharmacological andgenetic methodologies were used to block Akt, the effects of CO onaugmentation of proliferation were lost in cells without functional
Figure 2. CO enhances motility and modulates cytoskeletalchanges in RAECs. A, Bar graph of the average speeds of RAECstreated with or without CO. Data are representative of 3 indepen-dent experiments. Results represent the mean distance traveled�SD of 120 cells in each treatment group counted in 3 indepen-dent experiments. Five separate fields were monitored in thecourse of 24 to 48 hours, with images taken of each field every 5minutes. Values from 5 fields of CO- and air-treated cells wereused to calculate mean�SD. *P�0.02, 1-way ANOVA. Note thatCO more than doubled the speed of cell migration after insult.B–E, Phalloidin staining of cells treated with air (A and B) or CO (Dand E) for 24 hours. Arrowheads indicates accumulation of F-actinat the leading edge of cells, indicative of directed growth. Imagesare representative of 2 independent experiments from 6 to 10images per slide in each treatment group.
540 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from
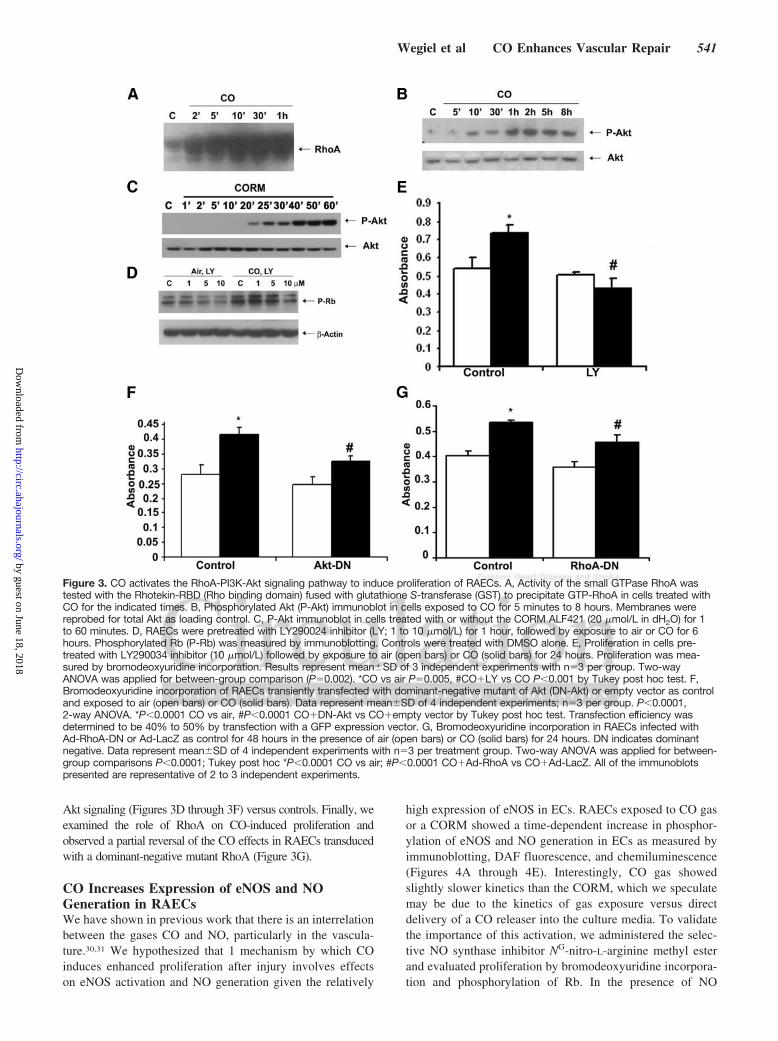
Akt signaling (Figures 3D through 3F) versus controls. Finally, weexamined the role of RhoA on CO-induced proliferation andobserved a partial reversal of the CO effects in RAECs transducedwith a dominant-negative mutant RhoA (Figure 3G).
CO Increases Expression of eNOS and NOGeneration in RAECsWe have shown in previous work that there is an interrelationbetween the gases CO and NO, particularly in the vascula-ture.30,31 We hypothesized that 1 mechanism by which COinduces enhanced proliferation after injury involves effectson eNOS activation and NO generation given the relatively
high expression of eNOS in ECs. RAECs exposed to CO gasor a CORM showed a time-dependent increase in phosphor-ylation of eNOS and NO generation in ECs as measured byimmunoblotting, DAF fluorescence, and chemiluminescence(Figures 4A through 4E). Interestingly, CO gas showedslightly slower kinetics than the CORM, which we speculatemay be due to the kinetics of gas exposure versus directdelivery of a CO releaser into the culture media. To validatethe importance of this activation, we administered the selec-tive NO synthase inhibitor NG-nitro-L-arginine methyl esterand evaluated proliferation by bromodeoxyuridine incorpora-tion and phosphorylation of Rb. In the presence of NO
Figure 3. CO activates the RhoA-PI3K-Akt signaling pathway to induce proliferation of RAECs. A, Activity of the small GTPase RhoA wastested with the Rhotekin-RBD (Rho binding domain) fused with glutathione S-transferase (GST) to precipitate GTP-RhoA in cells treated withCO for the indicated times. B, Phosphorylated Akt (P-Akt) immunoblot in cells exposed to CO for 5 minutes to 8 hours. Membranes werereprobed for total Akt as loading control. C, P-Akt immunoblot in cells treated with or without the CORM ALF421 (20 �mol/L in dH2O) for 1to 60 minutes. D, RAECs were pretreated with LY290024 inhibitor (LY; 1 to 10 �mol/L) for 1 hour, followed by exposure to air or CO for 6hours. Phosphorylated Rb (P-Rb) was measured by immunoblotting. Controls were treated with DMSO alone. E, Proliferation in cells pre-treated with LY290034 inhibitor (10 �mol/L) followed by exposure to air (open bars) or CO (solid bars) for 24 hours. Proliferation was mea-sured by bromodeoxyuridine incorporation. Results represent mean�SD of 3 independent experiments with n�3 per group. Two-wayANOVA was applied for between-group comparison (P�0.002). *CO vs air P�0.005, #CO�LY vs CO P�0.001 by Tukey post hoc test. F,Bromodeoxyuridine incorporation of RAECs transiently transfected with dominant-negative mutant of Akt (DN-Akt) or empty vector as controland exposed to air (open bars) or CO (solid bars). Data represent mean�SD of 4 independent experiments; n�3 per group. P�0.0001,2-way ANOVA. *P�0.0001 CO vs air, #P�0.0001 CO�DN-Akt vs CO�empty vector by Tukey post hoc test. Transfection efficiency wasdetermined to be 40% to 50% by transfection with a GFP expression vector. G, Bromodeoxyuridine incorporation in RAECs infected withAd-RhoA-DN or Ad-LacZ as control for 48 hours in the presence of air (open bars) or CO (solid bars) for 24 hours. DN indicates dominantnegative. Data represent mean�SD of 4 independent experiments with n�3 per treatment group. Two-way ANOVA was applied for between-group comparisons P�0.0001; Tukey post hoc *P�0.0001 CO vs air; #P�0.0001 CO�Ad-RhoA vs CO�Ad-LacZ. All of the immunoblotspresented are representative of 2 to 3 independent experiments.
Wegiel et al CO Enhances Vascular Repair 541
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

blockade, CO was unable to impart proproliferative effects,returning growth patterns to those of air controls (Figure 4F)and preventing increases in phosphorylated Akt and phos-phorylated Rb (data not shown). These data support theinterrelation of these gases in vitro in the regulation ofproliferation of RAECs and suggest that CO not only in-creases eNOS phosphorylation but also influences its activityto generate NO and importantly to drive activation of Akt andRb. The relation between NO, Akt, and Rb in ECs has beendescribed in the literature in other models but to date has notbeen evaluated with CO in ECs.32,33 We describe here thatCO clearly triggers activation of this pathway. NO has beenshown to impart prosurvival effects in ECs.14,34 We concludethat by imparting prosurvival benefits to the EC, NO iscritical in allowing CO to act via a RhoA3Akt3Rb cascadeto augment proliferation.
CO Augments Reendothelialization After BalloonAngioplasty in Rats and Wire Trauma in MiceCO can limit vascular occlusion, an effect driven primarily byreduced intimal thickening over the course of weeks. To date,
the effects of CO treatment on early events that occur afterinjury within the first 3 to 5 days have not been evaluated. Weexposed rats to either air or CO for 1 hour before angioplasty,as described previously, and evaluated the effects on reendo-thelialization after balloon trauma. Importantly, the animalswere not exposed to CO again. We harvested vessels at 1, 3,and 5 days after injury and stained sections for CD31 andintercellular adhesion molecule, markers specific for ECs thatare readily observed in uninjured vessels (Figure 5A). Inanimals exposed to air, the EC monolayer was absent at 1, 3,and 5 days (Figure 5B) after angioplasty but was fullyrestored by 7 days. In contrast, animals exposed to 1 hour ofCO showed a complete restoration of the EC monolayer by 5days (Figure 5C; 5 of 6 animals in the CO group versus 0 of6 animals in the air group, P�0.03). In the same vessels, weevaluated the inflammatory response and observed increasedmacrophages (online-only Data Supplement Figure IIIA) andneutrophils (data not shown) infiltrating the lesion at day 3 to5 after angioplasty, both of which were inhibited by CO. Invitro, both CO and CORM treatment effectively inhibited
Figure 4. NO mediates the proliferative enhancing effects of CO in RAECs. A, Phosphorylated eNOS (P-eNOS) immunoblot ofCO-exposed RAECs. Membranes were reprobed for total eNOS as loading control. RAECs were treated with 250 ppm CO at the indi-cated time points. B, P-eNOS immunoblotting in RAECs treated with or without CORM ALF421 (20 �mol/L) at the indicated timepoints. C and D, NO production in RAECs with or without CO for 5 minutes, measured by FACS with DAF-FM. Representative analyses(C) and quantitation (D) are shown. Data represent mean�SD from 4 independent experiments with n�3 per group. *P�0.05 CO vs air,Wilcoxon nonparametric test. E, Total NO production was measured in the media of human umbilical vein ECs stimulated with CO for 1hour. Data represent mean�SD from 3 independent experiments with n�3 to 6 per group. *P�0.0001 CO vs air, 1 way-ANOVA. F,Bromodeoxyuridine incorporation in RAECs pretreated for 1 hour with 100 �mol/L NG-nitro-L-arginine methyl ester (L-NAME) or dH2Oas vehicle control followed by treatment with air or CO as above. Data represent mean�SD from 3 independent experiments with n�3per group. P�0.002, 2-way ANOVA. *P�0.001 CO vs air, #P�0.001 CO� L-NAME vs CO by Tukey post hoc test. All of the immuno-blots are representative of 3 independent experiments.
542 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

transmembrane migration of U937 monocytes (online-onlyData Supplement Figures IIIB and IIIC). We also evaluatedthe effects of CO on EC restoration in the murine model ofwire trauma, a well-accepted surrogate for angioplasty. Aswas observed in rats, CO enhanced repair of the endotheliumin mice, restoring the EC monolayer by day 4 (Figure 6A),whereas air controls showed little to no EC presence untildays 5 and 6 after injury (data not shown). We also demon-strated that administration of the CORM ALF421 before andjust after wire injury accelerated reendothelialization in thecarotid artery, similar to CO gas (Figure 6B). The carboxy-hemoglobin levels achieved with CORM were 11�2% versus15�3% with 250 ppm for 1 hour. A separate cohort of micewere also treated with iCORM, which failed to enhance ECrepair after injury (Figure 6B).
CO Requires NO to Enhance ReendothelializationOur in vitro studies showing that CO increased NO generation inpart through phosphorylation of eNOS prompted us to evaluatewhether CO would enhance repair in the absence of eNOS invivo. CO was unable to enhance reendothelialization in eNos�/�
mice at 4 days, as opposed to results in CO-exposed wild-typemice (Figure 6C). To begin to assess a link between reendothe-lialization and development of intimal hyperplasia, we alsoevaluated the ability of CO to block the development of intimalhyperplasia in eNos�/� versus wild-type mice in the presence ofCO, having demonstrated previously that CO can inhibit intimalhyperplasia in response to wire trauma.9 We validated that again
here and additionally demonstrated that CO was unable toprevent intimal hyperplasia in the absence of NO (Figures 6Dand 6E). We observed similar effects in rats in which NO wasblocked by the administration of NG-nitro-L-arginine methyl esterwith or without CO (data not shown). Although this could beinterpreted as NO also being absent in VSMCs in these animals, weshowed previously that neither eNOS nor inducible NO synthasewas important in the ability of CO to inhibit growth of VSMCs.9
CO Increases Progenitor Cell Recruitment Intothe Circulation and the Injured VesselReendothelialization of injured vessels is thought to be driven inpart by the influx of undifferentiated progenitor cells. Wetherefore tested the hypothesis that CO targeted and enhancedthe influx of progenitor cells into the denuded site after injury viaexpansion of the bone marrow pool of ECs and mobilization ofEPCs. Immunostaining of vessels from wire-injured miceshowed that CO-exposed animals had a significantly highernumber of sca1-positive cells at the site of injury than didair-treated controls (online-only Data Supplement Figure IV).Interestingly, the sca1-positive cells did not express CD34 at thistime point, and we speculate that this phenotypic change occursrapidly, particularly with CO and likely in the adventitia. Wespeculate that the enhanced recruitment is responsible in part forthe more rapid infiltration and repair. Assessment of outgrowthcolonies showed that CO enhanced the number of colonies by 6-to 8-fold versus control mice (P�0.001). We next exposedprogenitor cells to CO in culture and observed that CO induced
Figure 5. CO monoxide induces reendothelialization after balloon injury in rats. A–C, Immunostaining for CD31 and intercellular adhe-sion molecule (ICAM) in noninjured rat carotids at 3 and 5 days after balloon injury in animals pretreated with air (B) or CO (C). CO wasadministered for 1 hour at 250 ppm. Images are representative of 6 to 8 images from each vessel isolated from 4 to 6 rats per treat-ment group. Note that CO-treated rats have CD31 and ICAM-positive staining at day 5, which was not present in air-treated animals.
Wegiel et al CO Enhances Vascular Repair 543
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

differentiation of progenitors into EPCs/ECs. Bone marrowprogenitor cells purified from Tie2-GFP mice were exposed toCO for 3 days in culture. In these cells, Tie2, which is a specificEC promoter, is linked to GFP; therefore, GFP can be used as asurrogate marker for mature ECs (Figure 7A). Air-treated cellsshowed limited expression of GFP by day 3 in contrast toCO-treated progenitors, which showed a 2- to 3-fold increase inGFP expression versus air-treated cells (Figures 7B through 7D).As a control for specificity of EC proliferation, we also differ-entiated progenitors into macrophages by treating them withmacrophage colony-stimulating factor and observed no effects
of CO on GFP after differentiation (Figure 7B). These data thussupport a direct effect of CO in inducing progenitor cells todifferentiate into ECs. To recapitulate in vivo our observations invitro, we generated Tie2-GFP chimeric mice and analyzed EPCmobilization into the blood after wire injury at 12 hours (Figures8A and 8B). We selected this time point because we observed astrong recruitment of sca1-positive cells to the injured vesselafter CO exposure at 12 hours (online-only Data SupplementFigure IV). Vessel injury alone induced recruitment of 5%GFP-positive EPCs to the circulation (Figures 8A and 8B);however, pretreatment with CO before vessel injury induced a
Figure 6. CO enhances reendothelializa-tion after wire injury in mice that isdependent on eNOS. A and B, Immuno-staining for CD31 3 and 4 days afterwire injury of carotid arteries in miceexposed to air or CO as described.Images were taken at magnification of20� (upper figure; scale bar�50 �m)and 63� (inset below). B, Immunostain-ing for CD31 in injured mice treated withCORM ALF421 or iCORM 10 mg/kg IPas described in Methods. Images repre-sent control-naïve vessel, 4 days injureduntreated, 4 days injured plus iCORM,and 4 days injured plus CORM ALF421.C, eNos�/� mice were treated with air orCO 1 hour before injury. Injured vesselswere harvested at day 4. All of theimages are representative of 8 to 10fields at 40� magnification and are rep-resentative of 4 to 6 mice per group. D,Hematoxylin-and-eosin staining of con-trol and injured vessels 2 weeks afterinjury from wild-type and eNos�/� micepretreated with air or CO. Images arerepresentative of 4 to 6 fields from 3 to5 mice per group. Magnification �20,scale bar�50 �m. Black line indicatesneointima. E, Quantitation of intimalexpansion expressed as intima-media(I/M) ratio from n�4 to 6 mice per group.P�0.02, 2-way ANOVA. *P�0.025 forair�eNos�/� and CO�eNos�/� groups,P�0.364 for CO�eNos�/� vsair�RAECs by Tukey post hoc test.
544 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

further 2- to 5-fold enhancement of GFP-positive EPC mobili-zation into the blood (Figures 8A and 8B). Enhanced mobiliza-tion of bone marrow–derived progenitors after CO exposure for1 hour was further confirmed by the performance of a colonyoutgrowth assay. Blood mononuclear cells were collected frommice exposed to air or CO and then cultured in EC media for 10days after isolation. CO significantly increased colony numbersover air-treated cells, and in some of the wells, we observedtubule formation in response to CO (online-only Data Supple-ment Figure V). Finally, we assessed whether GFP-positiveEPCs in the Tie2-GFP chimeric mice contributed to reendothe-lialization of the vessel after CO treatment. We detected GFP-positive ECs in the injured vessels after CO treatment at day 4(Figures 8C and 8D). We detected no GFP-positive cells inair-treated mice because EC repair was not present at day 4(Figures 8C and 8D). Collectively, these data suggest thatCO-enhanced EC repair occurs in part via enhanced mobiliza-tion and differentiation of EPCs.
DiscussionThe denudation of the endothelium caused by physical balloontrauma, combined with a rapid increase in leukocyte infiltration,leads to increased smooth muscle cell proliferation and forma-tion of the neointima. The loss of the endothelium is perhaps theinitiating element associated with subsequent vaso-occlusion.The direct effects of CO to induce growth arrest of VSMCs andprevent intimal expansion in vivo are clear, driven primarily by
a cGMP–p38-p21 signaling pathway that is NO independent.9,31
We hypothesized that CO treatment must initiate additionalmechanisms involved in repair of the vessel that contribute toinhibition of intimal expansion after trauma. Namely, CO wouldmodulate the acute inflammatory response (ie, leukocyte infil-tration), as well as target the endothelium to facilitate theregeneration/replacement of this barrier lamina. We used boththe clinically relevant balloon-injury model in rats and thewire-injury trauma model in mice, which permitted mechanisticexperiments to test the effects of CO on the early events aftervessel trauma.
The effects of CO in restoring homeostasis continue toimplicate a critical role for NO. NO is a potent prosurvivalfactor in ECs, unlike VSMCs.35,36 We thus hypothesized thatNOSIII/eNOS would be a likely target for CO in ECs toincrease NO through direct binding to NO synthase or viaspecific signal transduction and would contribute to survivaleven if NO did not necessarily promote a proliferativeresponse. Increased survival would then allow other signalingpathways, such as phosphorylation of Rb, to promote growth.The present data suggest that 1 mechanism by which COpromotes cell growth is through phosphorylation of eNOSand activation of Akt and Rb. The ability of CO to increaseeNOS phosphorylation, however, may occur indirectlythough an upstream potassium channel–mediated event thatleads to activation of PI3K and Akt. Blockade of potassiumchannels resulted in a loss of the effects of CO on EC
Figure 7. CO augments EPC expansion in vitro. A,Immunostaining for CD31 (red) and GFP (green)–positive staining of bone marrow–derived EPCsisolated from Tie2-GFP mice treated with or with-out CO for 72 hours in the presence of EC growthfactors. Images are representative of 4 to 6 fieldsfrom 2 independent experiments of n�6 pergroup. Note that CO enhanced EC differentiation.B, Expansion of EPCs by CO is specific. Bonemarrow from Tie2-GFP mice was differentiated intoeither ECs (left and center panels) or macrophages(right panel). CO enhanced GFP expression andtherefore Tie2 expression in EC-differentiated butnot macrophage-differentiated populations. Repre-sentative Tie2-GFP–positive EPCs and Tie2-GFP–negative macrophage colonies are shown. Imagesare representative of 4 to 6 fields from 2 indepen-dent experiments of n�3 per group. C and D,Quantitation of colonies (C) and single GFP-positive cells (D). Results represent mean�SD of 3independent experiments with n�4 to 5 per group.One-way ANOVA: *P�0.0001 CO vs air (C) and*P�0.001 (D). Note that CO enhanced both thenumber of colonies and single-cell populations.Scale bar�50 �m.
Wegiel et al CO Enhances Vascular Repair 545
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

proliferation (unpublished observation), and CO is known tofunction in part through activation of this channel.37
Cyclins and cyclin-dependent kinases induce hyperphos-phorylation of Rb, liberating E2F and other transcriptionfactors such as YY1 that play a pivotal role in the coordinatedtransactivation of cell cycle regulatory genes.24 We speculatethat CO modulates growth and genome transcription at thelevel of histone and chromatin modification. CO-inducedmitosis is blocked by the inhibitor of histone deacetylase,trichostatin (unpublished observation). The present resultsclearly demonstrate that exposure to CO in ECs leads to quickactivation of the small G protein RhoA and acceleratedentrance into the S phase, with increased phosphorylation ofRb that results in enhanced growth; however, we cannotexclude the possibility that CO activates other moleculesinvolved in cytoskeletal organization and signaling. COpromotes migration of the ECs in the scratch assay. The exactmechanism by which CO modulates the cytoskeleton remainsto be fully elucidated but likely involves the RhoA signalingmachinery described herein, which is known to be involved inF-actin cytoskeleton reorganization and stress-fiber forma-tion. The combination of the present observations in vitro andin vivo, including bone marrow progenitor cell recruitment tothe site of injury, supports the concept that CO administeredas a gas or a CORM fosters earlier reendothelialization and
involves recruitment, differentiation, and motility of ECs inan effort to augment repair of the injured vessel, whichultimately contributes to less intimal hyperplasia. The CO-mediated benefit is sustained for more than 21 days despitethe 1-time exposure of the animals to CO, which indicatesthat the process of vascular remodeling is determined in largemeasure early after acute injury. The kinetics of the eventsthat lead to augmentation of repair are multifactorial andclearly reflect decreased inflammation, earlier EC deposition,and ultimately decreased hyperproliferation of VSMCs. In amodel of pulmonary hypertension in rodents, we demon-strated that intermittent exposure to CO, initiated after theestablishment of disease, results in reverse remodeling (ie, areturn to original architecture and function).31 In these ani-mals, CO induced ECs to generate NO, which ultimately ledto restoration of normal artery and vessel size. In thisinstance, CO-induced NO arose from the ECs present in thevessels. In the data presented here, in which ECs were notpresent at the time of CO exposure, the origin of the ECs islikely circulating or recruited endothelial progenitors, giventhe present GFP data, or a significant contribution from theECs immediately adjacent to the denuded lesion that prolif-erated and mobilized into the injured area, perhaps driven byan augmented chemokine gradient elicited by NO, such asstromal cell–derived factor (SDF). SDF has been demon-
Figure 8. CO induces GFP-positive EPCperipheral mobilization and recruitmentto injured vessels in mice. A, Represen-tative flow cytometric analysis of GFP-positive cells in peripheral blood fromTie2-GFP chimeric mice 12 hours afterwire trauma. Isotype control (IgG), wild-type (Wt)–injured, air/Tie2-GFP–injured,and CO/Tie2-GFP–injured samples areshown. B, Quantitation of GFP-positivecells in peripheral blood. Data representmean�SD (n�3 mice per group).*P�0.05 CO vs air, nonparametric Wil-coxon test. C and D, Immunofluorescentstaining for GFP-Tie2–positive ECs invessels 4 days after wire injury per-formed in Tie2-GFP chimeric mice. Rep-resentative images of GFP fluorescencein wild-type (non-Tie2-GFP), Tie2-GFPuninjured, and GFP-Tie2 chimeric micetreated with CO (250 ppm or 1 hourbefore injury) or air. Representativeimages of 8 to 10 sections from n�2 to4 vessel regions per mouse per group;n�4 to 6 mice per treatment group.Magnification �20 (C) and �63 (D).Scale bar�50 �m. Arrows indicate GFP-positive cells. Note that elastin fibers inthe media autofluoresce at thesewavelengths.
546 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

strated to be involved in the ability of HO-1 to regulateangiogenesis.38 HO-1–deficient mice were unable to formcapillary sprouts, which was reversed by administration of aCORM. The mechanism by which HO-1/CO regulates angio-genesis is different from what we describe here and involvesVASP (vasodilator-stimulated phosphoprotein) and PGE2(prostaglandin E2) versus the Akt-eNOS-Rb pathway.12,38
Perhaps this speaks to the vast differences between the processesof neovascularization and EC repair. In this same vein, eNOS/NOSIII is essential for EPC mobilization.39 Neovascularizationdoes not occur in mice lacking eNOS, which strongly supportsthe observations described here that CO requires NO to enhancereendothelialization of denuded vessels.
From a therapeutic standpoint, these results, combinedwith our data showing that CO prevents neointima formationafter trauma, combine 3 actions: (1) Antiinflammatory ef-fects, (2) direct effects on VSMCs to block proliferation, and(3) proproliferative actions on ECs that lead to rapid reendo-thelialization of the denuded EC lesion. As such, exposure toCO results in greater efficacy in preventing neointima forma-tion and stenosis after balloon angioplasty than other ap-proaches aimed at the sole blockade of VSMC prolifera-tion.40–42 One of the principal challenges with stent therapy isthe inability of the stent to become endothelialized becausethe coatings also limit EC proliferation. The data presentedhere offer a potential therapeutic adjuvant involving treat-ment with CO gas or local delivery of a CORM to substituteor complement stent placement, perhaps even impregnating 1of the emerging CORM molecules onto a stent. In principal,either mode of CO delivery would yield the same beneficialresult. If the pulmonary data showing that exposure to CO canreverse intimal expansion without intervention hold true, theneed for angioplasty and stents may be reduced, which wouldbe particularly useful in the peripheral circulation, in whichdrug-eluting stents have not proven as efficacious, withrestenosis rates approaching 30% at 1 year.
In conclusion, we demonstrate a novel function of CO inpromoting reendothelialization that likely is the result ofenhanced proliferation, recruitment, and migration of neigh-boring ECs. Additional experiments are under way to addressboth the detailed mechanism and whether chronic delivery ofCO would be needed to prevent long-term restenosis andreduce the need for reintervention. Of course, with longer-term CO exposure, it will be necessary to carefully accrueadditional safety data. Accelerated reendothelialization addsto the clinical vascular protective benefit achieved by shortexposures to CO before angioplasty. The sooner reendothe-lialization has occurred in the vessel, the sooner the entry ofcirculating monocytes and T cells will be blocked, therebylimiting inflammation and subsequent vaso-occlusion.43 Fi-nally, these data identify, delineate, and add to the growingdatabase of the interrelation between the 2 gas molecules NOand CO, which act in tandem to reestablish homeostasis, invascular proliferative diseases. With careful clinical testing,CO may prove to be a novel therapeutic agent in the treatmentof numerous vascular disease syndromes.
AcknowledgmentsWe thank the Julie Henry Fund at the Transplant Center of the BethIsrael Deaconess Medical Center for their support. We thank Dr
Shiva Gautam from the Beth Israel Deaconess Medical CenterBiostatistics Program and Harvard University Clinical and Transla-tional Science Center for his assistance with biostatistics. We alsothank Carlos Romão and the chemistry unit at Alfama Inc, Portugalfor their input on the CORMs.
Sources of FundingThis work was largely supported by National Institutes of Healthgrants HL-071797 and HL-076167 to Dr Otterbein and in part bygrants from the Medical Research Council (G0600270 andG0601295) to Dr Ahmed.
DisclosuresDr Wegiel is the recipient of an American Heart Association ScientistDevelopment Grant (10SDG2640091). Mr Gallo is employed by andowns stock options in Alfama Inc. Dr Chin owns stock options inAlfama Inc. Dr Otterbein is a basic research consultant for Ikaria. DrsWegiel, Chin, and Otterbein are employed by Beth Israel DeaconessMedical Center, which owns patents related to carbon monoxide. Theremaining authors report no conflicts.
References1. Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the patho-
genesis of human disease. Nat Immunol. 2004;5:975–979.2. Ishikawa K, Sugawara D, Wang X, Suzuki K, Itabe H, Maruyama Y,
Lusis AJ. Heme oxygenase-1 inhibits atherosclerotic lesion formation inLDL-receptor knockout mice. Circ Res. 2001;88:506–512.
3. Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397.4. Nobuyoshi M, Kimura T, Nosaka H, Mioka S, Ueno K, Yokoi H,
Hamasaki N, Horiuchi H, Ohishi H. Restenosis after successful percuta-neous transluminal coronary angioplasty: serial angiographic follow-upof 229 patients. J Am Coll Cardiol. 1988;12:616–623.
5. Serruys PW, Luijten HE, Beatt KJ, Geuskens R, de Feyter PJ, van denBrand M, Reiber JH, ten Katen HJ, van Es GA, Hugenholtz PG. Incidenceof restenosis after successful coronary angioplasty: a time-related phe-nomenon: a quantitative angiographic study in 342 consecutive patients at1, 2, 3, and 4 months. Circulation. 1988;77:361–371.
6. Elezi S, Kastrati A, Neumann FJ, Hadamitzky M, Dirschinger J, SchomigA. Vessel size and long-term outcome after coronary stent placement.Circulation. 1998;98:1875–1880.
7. Degertekin M, Serryus PW, Foley DP, Tanabe K, Regar E, Vos J, SmitsPC, van der Giessen WJ, van den Braund M, de Feyer P, Popma JJ.Persistent inhibition of neointimal hyperplasia after sirolimus-elutingstent implantation. Circulation. 2002;106:1610–1613.
8. Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M,Colombo A, Schuler G, Barragan P, Guagliumi G, Molnar F, Falotico R;RAVEL Study Group. A randomized comparison of a sirolimus-elutingstent with a standard stent for coronary revascularization. N Engl J Med.2002;346:1773–1780.
9. Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A,Stachulak C, Bodyak N, Smith RN, Csizmadia E, Tyagi S, Akamatsu Y,Flavell RJ, Billiar TR, Tzeng E, Bach FH, Choi AM, Soares MP. Carbonmonoxide suppresses arteriosclerotic lesions associated with chronic graftrejection and with balloon injury. Nat Med. 2003;9:183–190.
10. Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine.Bioessays. 2004;26:270–280.
11. Scott JR, Chin BY, Bilban MH, Otterbein LE. Restoring HOmeostasis: isheme oxygenase-1 ready for the clinic? Trends Pharmacol Sci. 2007;28:200–205.
12. Li Volti G, Sacerdoti D, Sangras B, Vanella A, Mezentsev A, ScapagniniG, Falck JR, Abraham NG. Carbon monoxide signaling in promotingangiogenesis in human microvessel endothelial cells. Antioxid RedoxSignal. 2005;7:704–710.
13. Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP.Heme oxygenase-1-derived carbon monoxide requires the activation oftranscription factor NF-kappa B to protect endothelial cells from tumornecrosis factor-alpha-mediated apoptosis. J Biol Chem. 2002;277:17950–17961.
14. Lopez-Farre A, Sanchez de Miguel L, Caramelo C, Gomez-Macías J,Garcia R, Mosquera JR, de Frutos T, Millas I, Rivas F, Echezarreta G,Casado S. Role of nitric oxide in autocrine control of growth and apo-ptosis of endothelial cells. Am J Physiol. 1997;272(pt 2):H760–H768.
Wegiel et al CO Enhances Vascular Repair 547
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

15. Ono Y, Ono H, Matsuoka H, Fujimori T, Frohlich ED. Apoptosis,coronary arterial remodeling, and myocardial infarction after nitric oxideinhibition in SHR. Hypertension. 1999;34:609–616.
16. Cooke JP. NO and angiogenesis. Atheroscler Suppl. 2003;4:53–60.17. Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W,
Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific geneexpression in both embryonic and adult transgenic mice. Proc Natl AcadSci U S A. 1997;94:3058–3063.
18. Suriano R, Chaudhuri D, Johnson RS, Lambers E, Ashok BT, Kishore R,Tiwari RK. 17Beta-estradiol mobilizes bone marrow-derived endothelialprogenitor cells to tumors. Cancer Res. 2008;68:6038–6042.
19. Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, GalloD, Cszimadia E, Bach FH, Lee PJ, Otterbein LE. Hypoxia-induciblefactor 1alpha stabilization by carbon monoxide results in cytoprotectivepreconditioning. Proc Natl Acad Sci U S A. 2007;104:5109–5114.
20. Wiggan O, Shaw AE, Bamburg JR. Essential requirement for Rho familyGTPase signaling in Pax3 induced mesenchymal-epithelial transition.Cell Signal. 2006;18:1501–1514.
21. Ahmed A, Dunk C, Kniss D, Wilkes M. Role of VEGF receptor-1 (Flt-1)in mediating calcium-dependent nitric oxide release and limiting DNAsynthesis in human trophoblast cells. Lab Invest. 1997;76:779–791.
22. Fondevila C, Shen XD, Tsuchiyashi S, Yamashita K, Csizmadia E, LassmanC, Busuttil RW, Kupiec-Weglinski JW, Bach FH. Biliverdin therapy protectsrat livers from ischemia and reperfusion injury. Hepatology. 2004;40:1333–1341.
23. Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, Girardi L, YurtR, Himel H, Rafii S. Vascular trauma induces rapid but transient mobi-lization of VEGFR2(�)AC133(�) endothelial precursor cells. Circ Res.2001;88:167–174.
24. Song R, Mahidhara RS, Liu F, Ning W, Otterbein LE, Choi AM. Carbonmonoxide inhibits human airway smooth muscle cell proliferation viamitogen-activated protein kinase pathway. Am J Respir Cell Mol Biol.2002;27:603–610.
25. Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H,Schafer AI, Durante W. Heme oxygenase-1-derived carbon monoxide isan autocrine inhibitor of vascular smooth muscle cell growth. Blood.2002;99:4443–4448.
26. Basile JR, Gavard J, Gutkind JS. Plexin-B1 utilizes RhoA and Rho kinaseto promote the integrin-dependent activation of Akt and ERK and endo-thelial cell motility. J Biol Chem. 2007;282:34888–34895.
27. Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide differentiallymodulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidyl-inositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway duringanoxia-reoxygenation injury. J Biol Chem. 2005;280:8714–8721.
28. Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H,Yoshida K, Nagai R. Carbon monoxide protects against cardiac ische-mia–reperfusion injury in vivo via MAPK and Akt–eNOS pathways.Arterioscler Thromb Vasc Biol. 2004;24:1848–1853.
29. Batzlsperger CA, Achatz S, Spreng J, Riegger GA, Griese DP. Evidencefor a possible inhibitory interaction between the HO-1/CO- and Akt/NO-
pathways in human endothelial cells. Cardiovasc Drugs Ther. 2007;21:347–355.
30. Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, BachFH, Otterbein LE. Carbon monoxide protects against liver failure throughnitric oxide-induced heme oxygenase 1. J Exp Med. 2003;198:1707–1716.
31. Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J,Shimoda L, Ifedigbo E, Kanno S, Otterbein LE. Carbon monoxide reversesestablished pulmonary hypertension. J Exp Med. 2006;203:2109–2119.
32. Ziche M, Morbidelli L, Masini E, Granger H, Geppetti P, Ledda F. Nitricoxide promotes DNA synthesis and cyclic GMP formation in endothelialcells from postcapillary venules. Biochem Biophys Res Commun. 1993;192:1198–1203.
33. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM.Activation of nitric oxide synthase in endothelial cells by Akt-dependentphosphorylation. Nature. 1999;399:601–605.
34. Dimmeler S, Zeiher AM. Nitric oxide: an endothelial cell survival factor.Cell Death Differ. 1999;6:964–968.
35. Tanner FC, Meier P, Greutert H, Champion C, Nabel EG, Luscher TF.Nitric oxide modulates expression of cell cycle regulatory proteins: acytostatic strategy for inhibition of human vascular smooth muscle cellproliferation. Circulation. 2000;101:1982–1989.
36. Ceneviva GD, Tzeng E, Hoyt DG, Yee E, Gallagher A, Engelhardt JF,Kim YM, Billiar TR, Watkins SA, Pitt BR. Nitric oxide inhibitslipopolysaccharide-induced apoptosis in pulmonary artery endothelialcells. Am J Physiol. 1998;275:717–728.
37. Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, PeersC, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097.
38. Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S,Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J,Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor 1promotes angiogenesis via a heme oxygenase 1-dependent mechanism.J Exp Med. 2007;204:605–618.
39. Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-IhlingK, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxidesynthase for mobilization of stem and progenitor cells [published correctionappears in Nat Med. 2004;10:999]. Nat Med. 2003;9:1370–1376.
40. Morishita R, Gibbons GH, Horiuchi M, Ellison KE, Nakajima M, ZhangL, Kaneda Y, Ogihara T, Dzau VJ. A gene therapy strategy using atranscription factor decoy of the E2F binding site inhibits smooth muscleproliferation in vivo. Proc Natl Acad Sci U S A. 1995;92:5855–5859.
41. Morishita R, Gibbons GH, Ellison KE, Nakajima M, Zhang L, Kaneda Y,Ogihara T, Dzau VJ. Single intraluminal delivery of antisense cdc2 kinaseand proliferating-cell nuclear antigen oligonucleotides results in chronic inhi-bition of neointimal hyperplasia. Proc Natl Acad Sci U S A. 1993;90:8474–8478.
42. Luo Y, Marx SO, Kiyokawa H, Koff A, Massague J, Marks A.Rapamycin resistance tied to defective regulation of p27kip1. Mol CellBiol. 1996;16:6744–6751.
43. Gomes D, Louedec L, Plissonnier D, Dauge MC, Henin D, Osborne-Pellegrin M, Michel JB. Endoluminal smooth muscle cell seeding limitsintimal hyperplasia. J Vasc Surg. 2001;34:707–715.
CLINICAL PERSPECTIVECarbon monoxide (CO) is recognized as a potent therapeutic molecule at low, nontoxic doses. CO is in phase II clinicaltrials to improve kidney function after transplant of a kidney allograft. Inhaled CO is known to block intimal hyperplasiaafter balloon angioplasty. The data presented here offer continued insight into a potential therapeutic adjuvant involvingtreatment with CO gas or local delivery with a CO-releasing molecule to substitute or complement stent placement andprevent intimal hyperplasia. We demonstrate that CO prevents intimal expansion in mice in part by early and enhancedreendothelialization of the injured vessel. CO enhances proliferation, recruitment, and migration of neighboring endothelialcells and potent recruitment of bone marrow–derived progenitor cells to the site of injury. Accelerated reendothelializationadds to the clinical vascular cytoprotective benefits achieved with short exposures to CO. This observation describing theeffects of CO on endothelial cell proliferation is in direct contrast to that with vascular smooth muscle cells, theproliferation of which is blocked by CO. Mechanistically, CO acts via distinct nonoverlapping signal transductionpathways in endothelial cells versus vascular smooth muscle cells. Nitric oxide regulates endothelial cell function, whereasguanylate cyclase regulates vascular smooth muscle cell function. The present studies demonstrate that CO is avasoprotective gas that acts via multiple mechanisms to best reestablish vessel homeostasis. Collectively, our data supportthe use of CO as a therapeutic modality in the treatment of vascular proliferative disorders.
548 Circulation February 2, 2010
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

Y. Chin, Edith Tzeng, Shakil Ahmad, Asif Ahmed, Catherine J. Baty and Leo E. OtterbeinBarbara Wegiel, David J. Gallo, Kathleen G. Raman, Jenny M. Karlsson, Brett Ozanich, Beek
Enhances Endothelial Repair After Vascular InjuryDependent Bone Marrow Progenitor Mobilization by Carbon Monoxide−Nitric Oxide
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2010 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation published online January 18, 2010;Circulation.
http://circ.ahajournals.org/content/early/2010/01/18/CIRCULATIONAHA.109.887695.citationWorld Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2010/01/13/CIRCULATIONAHA.109.887695.DC1Data Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 18, 2018http://circ.ahajournals.org/
Dow
nloaded from

SUPPLEMENTAL MATERIAL
Supplementary Figure Legends
Supplementary Figure 1. The effect of CORM ALF 421 and hypoxia on the
proliferation of RAEC. A. BrdU incorporation was used to assess the proliferation rate
of CORM ALF 421 (1-20 µM) or vehicle treated RAEC cells measured at 24 hours. Data
are representative of 2 independent experiments performed in triplicate. Two-way
ANOVA, p<0.0001. CORM vs control. Tukey post-hoc; Control vs 1µΜ, p=0.505; vs
5µΜ, p=0.079; vs 10µΜ, ∗∗p<0.001; vs 20µΜ, ∗∗p<0.0001 B. BrdU incorporation
testing proliferation of RAEC cells exposed to normoxia (21%O2) and hypoxia (1% O2).
The data are representative of 2 independent experiments in triplicate. One-way ANOVA
*p<0.001 vs normoxia.
Supplementary Figure 2. Densitometric analysis of CO-treated RAEC at 4 hr for (A) P-
Rb; one-way ANOVA, *p=0.003. and (B) P-Akt, one-way ANOVA, *p=0.04 and 10
min for (C) P-eNOS, one-way ANOVA, *p=0.001. Data represent mean ± SD of values
obtained from 3 separate blots comparing CO vs Air.
Supplementary Figure 3. CO blocks the migration of macrophages in vitro and in
vivo. A. Immunostaining for the macrophage marker ED-1 of carotid arteries from air
and CO-treated animals 4 days after injury. Note that CO blocks inflammatory cell influx
into the injured vessel. Images are representative of 6-8 fields from 4-5 animals/group.
Magnification is 40x. Arrows indicate positive staining. Scale bar=50 µm. B. Transwell

- - 2
migration of U937 monocytes treated with Air or CO for 24 hours. Data are
representative of 2 independent experiments preformed in triplicate. One-way ANOVA
**p<0.001. C. Effect of CORM ALF 421 (10-50 µM) treatment on U937 macrophage
migration in Boyden chambers. Cells were treated for 24 hours and the amount of cells
that migrated to the lower chambers were counted. Data represent mean ± SD from 3
independent experiments (n=3/group). Two-way ANOVA; **p<0.001 CORM vs Control
(C). Tukey post-hoc; Air vs 10µM, *p=0.04; vs 20µM, **p=0.009; vs 50µM,
**p<0.0001.
Supplementary Figure 4. Carbon monoxide induced sca-1+ progenitor recruitment
to the injured vessel in mice. Mice were treated with air or CO as described previously.
Carotid artery segments were harvested 12 hrs after injury and sectioned and stained for
the presence of sca1+ and CD31. Vessels from naïve, untreated mice are shown as
control. Note that CO increased the number of sca1+ cells in the artery at 12 hr, which
was not present in air-treated mice. Representative images are from 4-6 animals/group.
Arrows indicate positive staining. Magnification is 20x, Scale bar=50 µm.
Supplementary Figure 5. Carbon monoxide induces recruitment of bone marrow
cells to the circulation as measured by the colony outgrowth assay. A. Representative
images of colony outgrowth (black arrows) 10 days after isolation of blood mononuclear
cells from Air or CO (1h, 250ppm;) treated animals cultured in EC medium (n=4
mice/group). Upper Panels: Air-treated animals, Lower Panels: CO-treated animals. B.
Quantitation of the outgrowth data represented as the number of colonies per field of

- - 3
view (FOV) at 40x magnification. Data represent mean ± SD of 8-10 FOV from each
animal/well in duplicate. One way ANOVA *p=0.007 vs air; Wilcoxon *p=0.031. Note
bottom right panel showing a representative FOV showing tubule-like formation (white
arrows) in a few FOV from cells harvested from animals treated with CO, which was not
observed in air-treated animals.
Supplementary Materials and Methods
Immunoblotting
Cells were lysed by a freeze-thaw cycle in ice-cold lysis buffer (0.5% NP-40, 0.1%
NaDOC, 0.1% SDS, 50mM Tris-HCl pH=7.5, 150mM NaCl, 1mM EDTA pH=8.0, 1mM
NaF, in the presence of a protease inhibitor cocktail. Samples were centrifuged for 30
min at 14000g at 4°C and the supernatants were harvested. 20-40 µg of each protein
sample were electrophoresed on NuPAGE 4-12% Bis-Tris Gel (Invitrogen, CA) followed
by transfer to PVDF membrane. The membranes were then blocked with 5% non-fat dry
milk, probed with appropriate primary antibodies, followed by HRP-conjugated
secondary antibodies at a dilution of 1:5000. Bands were visualized using Super signal
chemiluminescent substrate (Pierce, Rockford, IL) exposed to ECL Film (ISC
BioExpress, Kaysville, UT).
Source of antibodies
The following antibodies were used: rabbit anti-P(Ser473)-Akt (Cell Signaling), rabbit
anti-total Akt (Cell Signaling), rabbit anti-P (Ser780)-Rb (Cell Signaling), mouse anti-
GAPDH (Calbiochem), rabbit anti-P-eNOS (Ser1177) and rabbit anti-total eNOS
(Millipore Upstate), rabbit anti-Histone H3 (Cell Signaling), rat anti-mouse and mouse

- - 4
anti-rat CD31 (BD Biosciences), anti-ICAM (BD Biosciences), mouse anti-rat
mononuclear phagocyte (ED-1) (BD Pharmingen), rat anti-Ly-6A/E (Sca1) (BD
Biosciences) and rabbit anti-GFP (Invitrogen, Molecular Probes).
Cell motility assay
RAEC were grown to confluency on gelatin-coated coverslips. Prior to imaging, a
diametric scratch was made exposing a cell-free region approximately 25 to 30 µm wide.
The coverslips were loaded into 37oC incubated closed chambers (FCS2®, Bioptechs, Inc,
Butler, PA), aligning the scratch longitudinally with the flow of medium at 0.5 ml/hr
delivered by a KDS 100 syringe pump, (KD Scientific). The coverslips were exposed to
buffered medium (CO-saturated and non-saturated containing 5% CO2/air). The medium
was prepared by bubbling with 250 ppm CO continuously for 30 min and then loaded
into an airtight Hamilton syringe. The system was designed and tested to be gas
impermeable). Differential interference contrast (DIC) images were taken every 20 min at
12 different positions along the scratch over 24 hr with a Nikon TE300 with 20X
objectives; the experiments were done simultaneously. Cellular motility was analyzed
with MetaMorph 6.2 (Universal Imaging Corp.), and measured as the average speed of
the cells’ nuclei (n=120 cells) along their migratory pathway.
Monocyte migration. U937 monocytes were seeded in the upper chamber of migration
Boyden chambers (8 µm, Transwell, Costar) in serum free medium. Serum containing
medium was added to the lower chamber and served as the chemoattractant. Cells were
treated with CO or 10-50 µM CORM ALF421 for 24 hours. The amount of cells, which
migrated to the lower chambers were counted using a Neubauer hemocytometer.

- - 5
Phalloidin staining
RAEC were cultured in MCD-B131 complete media (VEC Technology) on 22 mm x 22
mm coverslips in 6 well plates. 1 mm wide scratches of uniform size were then created,
after which the cells were either placed in air or CO for 24 hr. Staining for actin using
Alexa Fluor 488 phalloidin (Molecular Probes, Eugene, OR) was performed according to
manufacturer’s directions. Briefly, the coverslips were fixed in 2% paraformaldehyde for
15 min at room temperature, washed with PBS and incubated with phalloidin for 15 min.
Images were captured of randomized fields using a Zeiss Apotome fluorescent
microscope.


































