Embed Size (px)
Citation preview

Next Generation SequencingMolecular Methods
Sylvain Foret
March 2010
http://dayhoff.anu.edu.au/~sf/next_gen_seq
1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary

The Genomic Age
Recent landmarks in genomics
1995 First bacterial genome (1.8 Mb)
1996 First eukaryotic genome (12 Mb)
1998 First animal genome (100 Mb)
2000 First human genome (3 Gb)

The Post-Genomic Age
Two big questions
How can we continue sequencing ever faster?
What can be done with all these sequences?
The Archon X Prize
To win the prize purse, the registered group must build adevice and use it to sequence 100 human genomes within10 days or less, with an accuracy of no more than oneerror in every 100,000 bases sequenced for no more than$10,000 per genome.
Other challenges and projects
The $1,000 human genome
The 1,000 genomes project (NHGRI, BGI, . . . )

Course Outline
Next generation (massively parallel) sequencing
Molecular methods (course 1)
Applications (course 2, course 3)
1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary

Sanger Method
Template
Synthesis
DNApolymerase
Primer
T
T
G
C
CAG
CGA AT CG
Electrophoresis
TT
A
A
C
CG
T
T A
T
Chromatogram
Quality score

Sanger Method Summary
Main characteristics
Sequencing by synthesis
Dye terminator method
Input Material: any DNA in sufficient quantity
PCR productsMolecular clones. . .
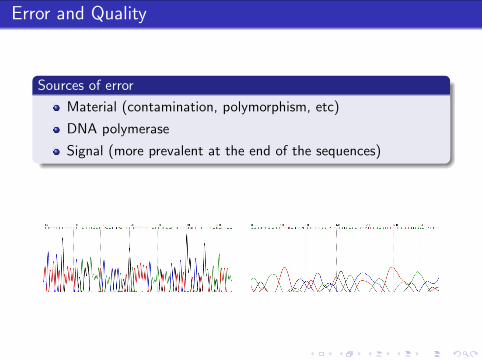
Error and Quality
Sources of error
Material (contamination, polymorphism, etc)
DNA polymerase
Signal (more prevalent at the end of the sequences)
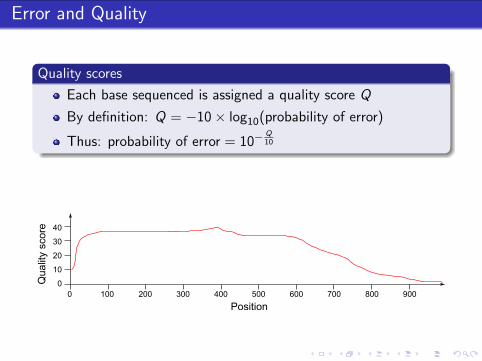
Error and Quality
Quality scores
Each base sequenced is assigned a quality score Q
By definition: Q = −10 × log10(probability of error)
Thus: probability of error = 10−Q10
0 100 200 300 400 500 600 700 800 900
0
10
20
30
40
Position
Qualit
y s
core
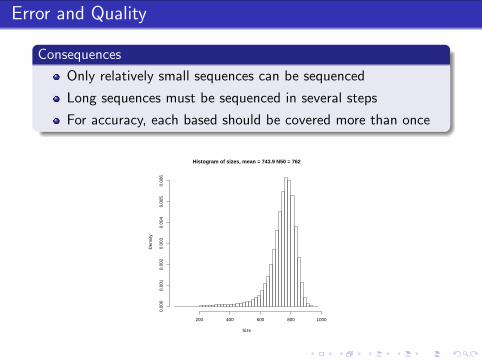
Error and Quality
Consequences
Only relatively small sequences can be sequenced
Long sequences must be sequenced in several steps
For accuracy, each based should be covered more than once
Histogram of sizes, mean = 743.9 N50 = 762
Size
Den
sity
200 400 600 800 1000
0.00
00.
001
0.00
20.
003
0.00
40.
005
0.00
6
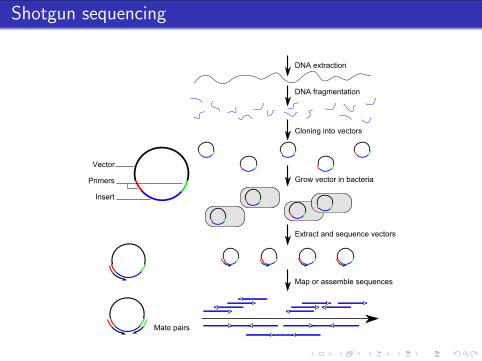
Shotgun sequencing
DNA fragmentation
Cloning into vectors
Grow vector in bacteria
Extract and sequence vectors
Map or assemble sequences
Vector
Primers
Insert
DNA extraction
Mate pairs

1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary
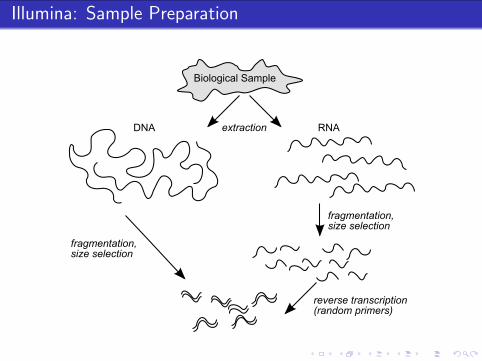
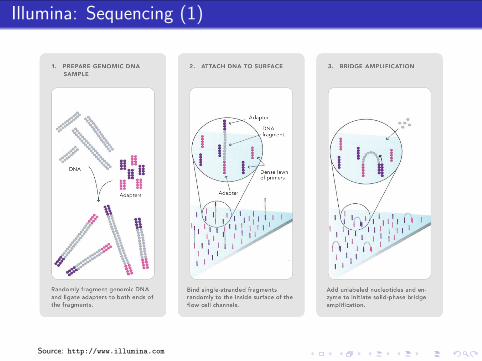
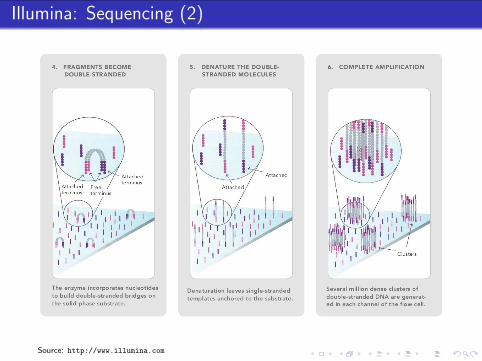
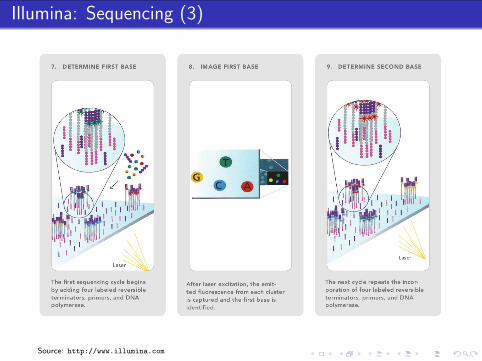
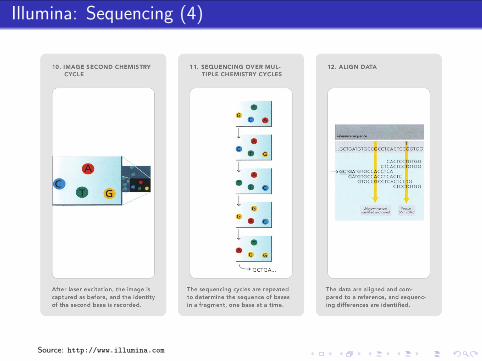
Illumina: Sample Preparation
Biological Sample
extraction
fragmentation,size selection
reverse transcription(random primers)
RNADNA
fragmentation,size selection
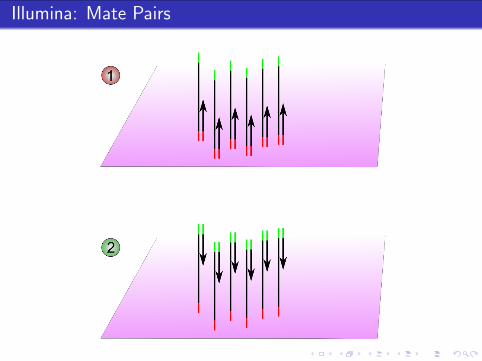
Illumina: Mate Pairs
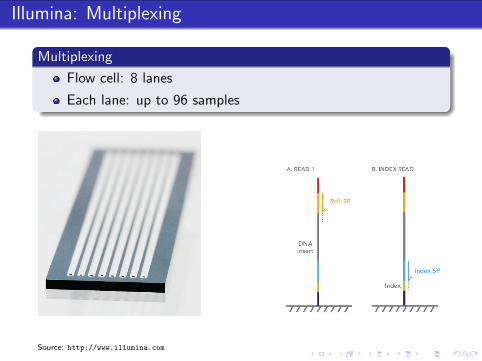
Illumina: Multiplexing
Multiplexing
Flow cell: 8 lanes
Each lane: up to 96 samples
Source: http://www.illumina.com

Illumina Summary
Main characteristics
Sequencing by synthesis
Reversible terminator method
Current size: 100bp
1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary
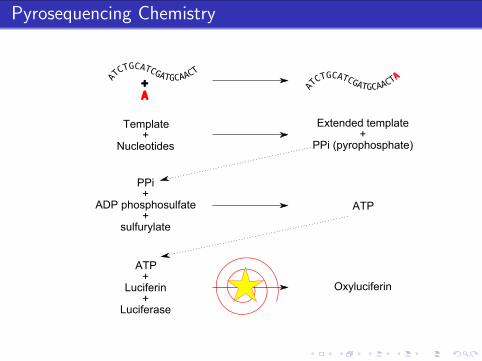
Pyrosequencing Chemistry
A+
Template+
Nucleotides
Extended template+
PPi (pyrophosphate)
PPi+
ADP phosphosulfate+
sulfurylate
ATP
ATP+
Luciferin+
Luciferase
Oxyluciferin
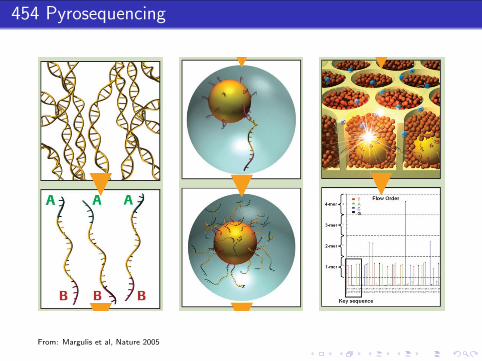
454 Pyrosequencing
From: Margulis et al, Nature 2005

454: Poly-A Tails
AAAAAAAAAAA
TTGTTTCTTTT
AAAAAAAAAAA
TTTTTTTTTTT
AAAAAAAAAAA
TTTTTTTTTTT
RE RE

454: Mate Pairs
Internal adapter Internal adapterInsert (3kb−20kb)
Circularize
Cut150−180 bp 150−180 bp
Sequence
Add sequencing adapters

454: Multiplexing
Multiplexing
Each plate: 1, 2, 4, 8 or 16 regions separated by gaskets
Each region: up to 12 samples
TemplateAdapter
TemplateAdapter + MID(Multiplex Identifier)
Source: http://www.454.com

454 Summary
Main characteristics
Sequencing by synthesis
Pyrosequencing method
Current size: 400bp

1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary
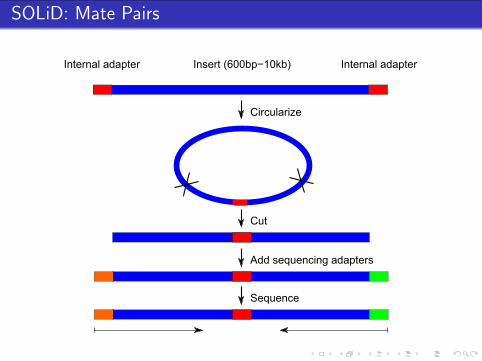
SOLiD: Mate Pairs
Internal adapter Internal adapterInsert (600bp−10kb)
Circularize
Cut
Add sequencing adapters
Sequence

SOLiD: Multiplexing
Multiplexing
Each run: 2 slides
Each slides: 1, 2, 4, 8 regions
Each region: up to 16 samples
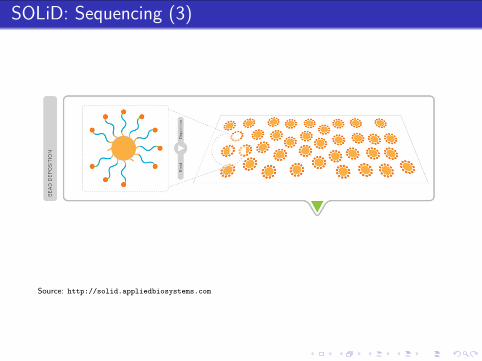
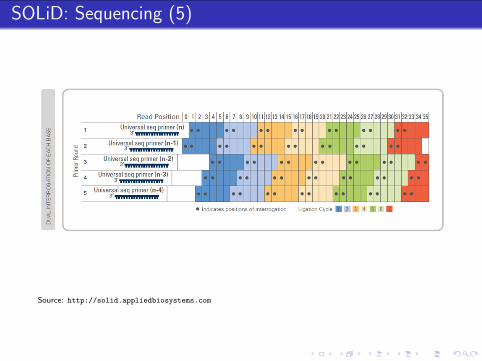
Source: http://solid.appliedbiosystems.com

SOLiD Summary
Main characteristics
Sequencing by ligation
Current size: 50bp
1 Introduction
2 Sanger
3 Illumina
4 454
5 SOLiD
6 Summary

Summary
Numbers, as of March 2010
454 Illumina SOLiD
(Titanium) (Genome Analyser IIx ) (SOLiD 3)
Mean read size 400bp 100bp 50 bp
Reads per run 106 200 × 106 500 × 106
Run time 10 hours 4 days 1 week
Insert size 3kb–20kb 200bp–5kb 600bp–10kb

Summary
Conclusions
Fast moving field
Other players: Helicos, Pacific Biosciences, Nano Pores, ...
A $1,000 human genome seems possible within a few years
Many applications
Genome (re)sequencingTranscriptome sequencingChIP-seqMetagenomics...