Embed Size (px)
Citation preview

Indian Journal of Chemistry Vol. 41A, January 2002, pp. 46-53
New insights into lignin peroxidase
Klaus Piontek
Institute of Biochemistry, Swiss Federal Institute of Technology (ETH), ZUrich, UniversiUitstrasse 16, CH-8092 ZUrich, Switzerland
(E-mail: [email protected])
Received 25 July 2001
Lignin peroxidase an extracellular haem containing glycoprotein is able to oxidise non-phenolic aromatic compounds with redox potentials exceeding 1.4 V. This enzyme is employed by ligninolytic fungi to degrade the recalcitrant biopolymer lignin, a cell wall constituent of woody plants. Due to its enlarged substrate range in the presence of specific mediators and due to its high redox potential this enzyme has the potentiality for the application in various industrial processes. But, until recently information concerning the binding mode of the substrate with the enzyme was lacking, therefore hampering progress in the elucidation of the catalytic mechanism. The finding of an unprecedented amino acid modification at a surface tryptophan initiated several investigations during the past few years on the role of this residue, which resulted in the identification of two distinct substrate interaction sites in lignin peroxidase. This mini-review summarises the major outcome of these investigations and describes the underlying structural factors that govern substrate interaction and electron transfer in lignin peroxidase.
Introduction Haem-containing peroxidases are a Ubiquitous
group of redox enzymes that catalyse the hydrogen peroxide-dependent oxidation of a wide range of organic and inorganic compounds, often with the concomitant production of radical intermediates 1-3.
Structurally, haem peroxidases are globular proteins of about 30-40 kDa. The co-factor haem consists of an iron protoporhyrin IX, being embedded between a proximal and distal domain. The haem iron is coordinated by the four nitrogen atoms of the porphyrin rings, and in the axial directions by a nitrogen of a proximal histidine and by a water or peroxide substrate molecule on the distal side of the haem. The latter, being the sixth ligand, interacts with another histidine, the distal one. The structural motif of the prosthetic group and its surrounding protein matrix are somewhat reminiscent of the haem and its environment in globins. But, while haemoglobin and myoglobin have a positive redox potential and favour the reduced state of the central iron (Fe2+), peroxidases stabilise higher oxidised states. It is assumed that the stronger anionic character of the axial histidine ligand in peroxidases, as compared to the globins, is the major factor, which determines their negative redox potential and enables the stabilisation of high oxidation states4
. But, so far nothing concrete can be said on how the different peroxidases attain their specific redox potential.
The mostly accepted general catalytic cycle of haem peroxidases, as first formulated for horse radish peroxidase5
.6
, starts with the oxidation of the ferric enzyme by H20 2 to compound I (comp-I), a twoelectron-oxidised intermediate with a formal oxidation state of +5 (reaction 1). In a second step (reaction 2) comp-I is then reduced by one substrate molecule to the second intermediate compound II (comp-II) . Further reduction back to the resting enzyme can be accomplished either by the same substrate molecule or by a second one (reaction 3). One oxidising equivalent in comp-I is stored as an oxoferryl species [Fe(IV)=O]2+ and the second either as a porphyrin 1t-radical or a protein based radical cation. In the past, considerable research effort has been invested to identify and localise the radical site in peroxidases7
•9
. However, cytochrome c peroxidase (CcP) was the only example where a protein based (Trp191) radical species could be identified unambigously until lately 10. More recently, by work conducted in our laboratory, which was originally initiated by the finding of a unique amino acid modification in a crystal structure of lignin peroxidase (LiP) II, a transient radical on a surface tryptophan of this peroxidase was identified 12, In the following, other authors have also provided evidence for proteinbased radical formation I3
.15
.
FelIl-PX+H20r~comp-I + H20 (1)
comp-I + AH2 ---t comp-II + AH' (2)

PIONTEK: LIGNIN PEROXIDASE 47
. . . (3)
Based on primary structure similarity and biological origin, the haem peroxidase superfamily can be subdivided into three classes 16. Class I comprises intracellular peroxidases such as yeast CcP, chloroplast and cytosol ascorbate peroxidase, and gene duplicated bacterial peroxidase. Secretory fungal peroxidases like lignin peroxidases (LiP), manganese dependent peroxidase (MnP), Coprinus cinereus peroxidase (CiP) or Arthromyces ramosus peroxidase (ARP) belong to class II, whereas the classical secretory plant peroxidases like horseradish peroxidase (HRP), tobacco peroxidase, or peanut peroxidase form class III. Glycosylation, the existence of two structural Ca-cations, and the formation of four conserved disulphide bridges and their location are examples for structural differences between classes IVIII and class I peroxidases.
Peroxidases have found applications in biosensors and clinical and biochemical assays since a long time. Since a few years, haem peroxidases are used in oxidation catalysis l7-19 and more recently as selective catalysts in oxygen transfer reactions2o.
Since its discovery in 1983, LiP has received extensive attention due to its abi lity to degrade the recalcitrant plant cell wall constituent lignin21. This rather heterogeneous and complex bio-polymer, consists of phenyl propanoid units, linked by various non-hydrolysable C-C- and C-O- bonds22. Several LiP isozymes are secreted by wood-degrading fungi, such as the white-rot fungus Phanerochaete chrysosporium, enabling this micro-organism to attain access to the cellulose of woody material23. On account of its role in lignin biodegradation and its ability to oxidise compounds with reduction potentials exceeding 1.4 V (versus normal hydrogen e\ectrode)24 LiP is considered to become a potential bio-catalyst in biotechnological as well as environmental processes. Despite the availability of high resolution crystal structures and a wealth of kinetic and spectroscopic data the exact catalytic mechanism of LiP, even with small molecular substrates, is still poorly understood. In particular, the exact interaction site(s) of LiP with its substrate(s) are still elusive. During the past three years, investigations based on crystal structure analysis, protein chemistry, site-directed mutagenesis, spectroscopy, and spintrapping provided some new aspects on the functionality of this enzyme. The principal object of this mini-review is to summarise the results of these recent findings and to describe the underlying structural factors that govern substrate interaction and electron transfer in lignin peroxidase.
Overall structure of LiP The first two LiP crystal structures of the same
fungal isozyme were solved independently by two groups and reported in 199325.26 . Albeit these structures were elucidated at somewhat low resolution, their correctness was later confirmed by the determination of this LiP isozyme at high resolution II and even at atomic resolution (Blodig & Piontek, unpublished data) . Furthermore, since then the crystal structures of another fungal isozyme27 and that of recombinant LiPs were determined28
. Before the first structural reports on LiP, CcP was the only available 3-D structure of a haem peroxidase29 on which all structural/functional considerations of related peroxidases were based on at that time. In fact, despite low sequence homology, the fold of the core protein (residues 15-275) of LiP is basically the same as in CcP. It was also found in the crystal structures of other haem peroxidases like Mnp30, HRp31, barely grain peroxidase32, and two other (non-ligninolytic) fungal peroxidases Cip33 and ARP34. The monomeric LiP molecule contains 343-344 amino acids (depending on the isozyme) and forms a globular protein with a size of about 50 x 40 x 40 A3. The folding motif consists of eight major and eight minor helices, and of three short antiparallel B-sheets (Fig. I). Compared to CcP, LiP has an addition of about 50 amino acids, comprised in a C-terminal peptide. This extension has only little secondary structures with few contact sites to the rest of the core protein. The haem divides the protein in a proximal and a distal domain. It is sandwiched between the two domains and buried in the interior of the protein with relatively little access to it from the outer medium via a small channel. In total there are four conserved disulphide bridges, one of them stabilises the C-terminal peptide by connecting it to the core protein. According to the assignment of LiP to the class II peroxidases 16, two integral calcium ions were found in the crystal structures of the fungal enzymes (Fig. 1). One is situated in the distal and one in the proximal domain . Both of them appear to provide rigidity to the protein and possibly an exact geometry for the coordination of the proximal histidine to the heam iron, which could be a determinant for the regulation of the redox potential (see below). Several N- and O-gylcosylation sites could be identified in the crystal structures from fungal sources35,25,27,11. They are clustered in the
proximal domain mostly at the C-terminal portion of the enzyme, where they seem to function as to protect
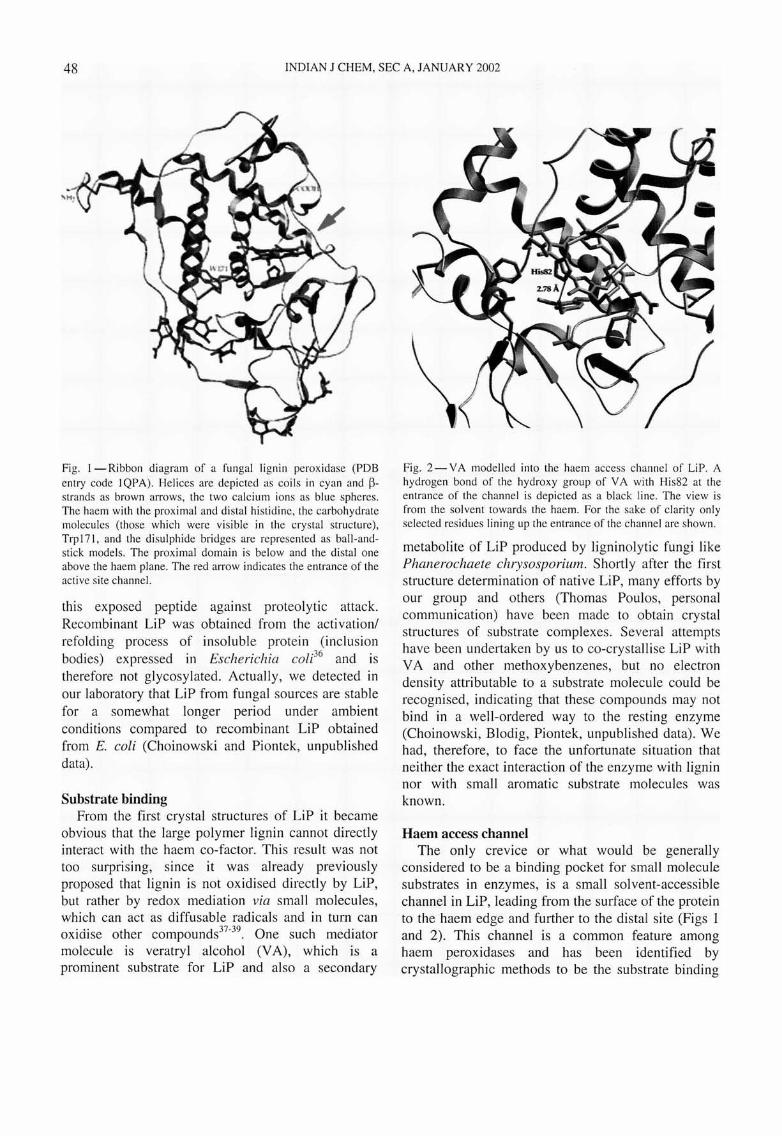
48 INDIAN J CHEM, SEC A, JANUARY 2002
Fig. I - Ribbon diagram of a fungal lign in peroxidase (PDB entry code IQPA). Helices are depicted as coils in cyan and ~strands as brown arrows, the two calcium ions as blue spheres. The haem with the proximal and distal histidine, the carbohydrate molecules (those which were visible in the cry stal structure), Trp 171 , and the disulphide bridges are represented as ball-andstick models. The proximal domain is below and the distal one above the haem plane. The red arrow indicates the entrance of the active site channel.
this exposed peptide against proteolytic attack. Recombinant LiP was obtained from the activation/ refolding process of insoluble protein (inclusion bodies) expressed in Escherichia COli
36 and is therefore not glycosylated. Actually, we detected in our laboratory that LiP from fungal sources are stable for a somewhat longer period under ambient conditions compared to recombinant LiP obtained from E. coli (Choinowski and Piontek, unpublished data).
Substrate binding From the first crystal structures of LiP it became
obvious that the large polymer lignin cannot directly interact with the haem co-factor. This result was not too surprising, since it was already previously proposed that lignin is not oxidised directly by LiP, but rather by redox mediation via small molecules, which can act as diffusable radicals and in turn can oxidise other compounds37
-39
. One such mediator molecule is veratryl alcohol (VA), which is a prominent substrate for LiP and also a secondary
Fig. 2 - V A modelled into the haem access channel of LiP. A hydrogen bond of the hydroxy group of V A with His82 at the entrance of the channel is depicted as a black line. The view is from the solvent towards the haem. For the sake of clarity on ly selected residues lining up the entrance of the channel are shown.
metabolite of LiP produced by ligninolytic fungi like Phanerochaete chrysosporium. Shortly after the first structure determination of native LiP, many efforts by our group and others (Thomas Poulos, personal communication) have been made to obtain crystal structures of substrate complexes. Several attempts have been undertaken by us to co-crystallise LiP with V A and other methoxybenzenes, but no electron density attributable to a substrate molecule could be recognised, indicating that these compounds may not bind in a well-ordered way to the resting enzyme (Choinowski, B1odig, Piontek, unpublished data) . We had, therefore, to face the unfortunate situation that neither the exact interaction of the enzyme with lignin nor with small aromatic substrate molecules was known.
Haem access channel The only crevice or what would be generally
considered to be a binding pocket for small molecule substrates in enzymes, is a small solvent-accessible channel in LiP, leading from the surface of the protein to the haem edge and further to the distal site (Figs 1 and 2)_ This channel is a common feature among haem peroxidases and has been identified by crystallographic methods to be the substrate binding

PIONTEK: LIGNIN PEROXIDASE 49
site in ARp40 and HRp41-42
. The entrance to this site was found to be relatively narrow in LiP as compared to the situation in other peroxidases for which
h· d '1 bl 4331.44,33 ,34 h' h . crystallograp IC ata are aVaI a e ' , w IC IS
somewhat curious in view of the much larger substrate (lignin) of LiP. The channels in HRP and ARP have a net positive charge distribution, whereas in LiP it is more negative, aiding in the stabilisation of a V A cation radical. Due to lack of a substrate complex structure this region in LiP was probed by modelling studies35
,45,46 which supported its suitability for the binding of V A. While these studies are different in detail they share common features, the formation of a hydrogen bond of the OH-group of VA with His82 (Fig. 2) and Van der Waals contacts of the aromatic moiety with the haem edge. In one of these studies the modelled V A molecule is placed not far from the side chains of Ile8S and Asp 18346
. A putative meta-methoxy substituent in the S-position would have steric conflicts with lle8S and Asp183 . Therefore, 3,4,S-trimethoxybenzyl alcohol could be predicted to be a poor substrate for LiP, as observed experimentall/7
. Site-directed mutagenesis of LiP with a charge neutralisation in the haem access channel, in which Glu146 was replaced by a Gly, showed still substantial activity towards VA oxidation, but also a significant increase in pKa for the oxidation of the negatively charged artificial dye substrate 4- [(3 ,S-difluoro-4-hydrox ypheny I )azo ]benzenesulphonic acid48
. These results do not support the proposition that the heam access channel is indeed a substrate binding site for V A, but rather identifies it as a binding site for negatively charged dye substrates.
Post-translational modification of Trp171 and a second substrate interaction site in LiP
During the crystal structure determination of fungal LiP in our laboratory, a significant residual electron density close to the surface residue Trp171 was recognised at a resolution of about 2 A (Fig. 3). The only sensible interpretation of this density was that it corresponds to a hydroxy group covalently bound to the C~-atom of Trp17111. Trp171 is completely conserved in all known lignin peroxidase sequences. To verify the nature of this unprecedented amino acid modification further and to probe the possible role of Trp171 and of its substituent for the catalytic mechanism various protein chemical experiments were performed49
. The results of these investigations had far reaching consequences for the understanding
on LiP substrate interactions and on its redox behaviour.
Tryptic digestion of the enzyme was performed and the Trp 171 containing peptide was isolated using HPLC. While this peptide displays a normal trypthophan absorbance spectrum under ambient conditions, elevated temperature and slightly acid conditions result in an absorbance spectrum with Amax=333 nm. This unusual absorbance could be explained by the occurrence of an extended conjugated system of a, ~-didehydrotrypthophan
resulting from water elimination from ~-hydroxy
tryptophan. The striking similar spectrum of the model compound N-acetyl- a , ~-didehydrotryptho
phanamide is in accordance with this assumption . Experiments with recombinant and refolded lignin peroxidase36 performed in the same way as described above do not yield an absorbance at Amax=333 nm. It was concluded that this enzyme does not have such a modification at Trp17l. But, treatment of two equivalents of hydrogen peroxide resulted in ~
hydroxy tryptophan formation, a process that competes with reducing substrates and does not depend on the presence of 0 2
49. These experiments
strongly suggest that the reaction leading to this unique modification in LiP is an auto-catalytic selfoxidation, which occurs readily in the presence of H20 2• Since such conditions exist in ligninolytic cultures of the fungus , due to the presence of an oxidase-based H20 r generating system, this explains why the modification was found in fungal LiP only .
Even in the absence of reducing substrates LiP comp-I turns quite rapidly into comp-lI5o
. Considering the above finding, it appeares likely that Trp 171 is the endogenous electron donor of LiP comp-I reduction . and implies the formation of a Trp radical cation, and the involvement of Trp 171 in the catalytic cycle. Furthermore, inspection of the LiP crystal structures shows that the local environment of Trp171 is highly acidic (Fig. 3) and could, therefore, contribute to the stabilisation of a cation radical. The assumption that Trp171 participates in the redox cycle of LiP was tested by treating the enzyme with the tryptophan specifc agent N-bromosuccinimide. This procedure led to a drastically reduced activity with respect to VA oxidation49 supporting the above conjecture. Since the neighbouring residue 172 is within Van der Waals contact with its C~-atom to the haem, an electron pathway (with a distance of about 11 A) between the haem and the surface residue Trp 171 can exist via this route (Fig. 3). The residue in position

50 INDIAN J CHEM, SEC A, JANUARY 2002
Fig. 3 - Difference omit map of the hydroxy group at Trp 171 in LiP. The map was contoured at a 40 level. Leu 172, next to the surface residue Trp171, is wi th its C~-atom within Van del' Waals distance to the haem edge (red line), forming the proposed electron pathway between the substrate interaction site at Trp171 and the co-factor. Glu168, Glu250, and Asp264 are labeled to highli ght the ac idic microenvironment in the vici ni ty of Trp 171 .
172 is either a Leu or a Met only , in LiP, which should enable the contact of a C~-atom to the haem edge in either case.
A mechani sm of the hydroxylation at Trp171 was proposed, which starts with the formation of the Trp radical cation49
. Using spin-trapping with methyl nitroso propane (MNP) in combination with peptide mapping and crystallography, evidence for a Trp171 radical intermediate was obtained 12. In the presence of H20 2, MNP forms a covalent adduct with Trp17l. A characteristic visible absorbance spectrum, which is similar to the one obtained from the model compound
N-acetyltryptophanamide, MNP, and a -one-electron oxidation system can be observed from a peptide containing this reaction product. In a crystal structure of LiP, which was treated in the way described above, the precise site, the spin-trap is attached to could be located, which is the C6 atom of the indole ringl2 (Fig. 4) .
Using site-directed mutagenesis the role of Trp 171 in LiP was further investigated48
. In recombinant enzyme, where Trp 171 was replaced by Phe or Ser, VA oxidation activity is completely eliminated48
.
Surprisingly, these two mutants maintain essentially

PIONTEK: LIGNIN PEROXIDASE 51
Fig. 4 - Electron density map from a crystal structure of MNP/H20 r treated LiP at P-hydroxy tryptophan 171. The density in bold represents only one atom of the covalently bound stable adduct, possibly a 6-nitroso or 6-nitro derivative l2
. The electron density of the remaining atom(s) of the nitro or nitroso moiety are disordered and, therefore, not visible in the crystal structure.
unaltered activity with two artificial, negatively charged dye substrates. On the other hand, as mentioned above, a charge neutralisation mutation E146G exhibits an increased pH optimum for the oxidation of a negatively charged difluoroazo dye and a doubling of catalytic activity compared to the wild type enzyme48
. These mutation experiments not only accentuate the importance of Trpl71 for the catalytic mechanism, but are also clear evidence for the existence of two distinct substrate interaction sites in LiP. One site is situated in the "classical" haem access channel and a second, novel site, at Trpl71 which is essential for V A oxidation. Other groups recently supported the significance of Trp171 for VA oxidation. One group introduced oxidation activity for non-phenolic aromatic compounds into a MnP by replacing the Ser by a Trp in a position equivalent to 171 in Lip51
. Wild-type MnP cannot oxidise these substrates. Two other groups have cloned a new type of haem peroxidase, which is able to oxidise Mn2
+ as well as V A and dimethoxybenzene52
,53. This third type of ligninolytic peroxidase, named versatile peroxidase, has a conserved tryptophan in a position equivalent to residue 171 in LiP.
The kinetic work was complemented by the determination of high resolution crystal structures of recombinant LiP in the pristine state (enzyme which has not been reacted with H20 2 and, therefore, is not hydroxylated at Trp17l), of the oxidatively processed enzyme (hydroxylated at Trp171) and of the Trp171Phe mutant28
• Wild-type LiP isolated from fungal cultures is always oxidatively modified due to the presence of hydrogen peroxide-producing oxidases secreted by the fungus. LiP expressed in E.
coli has to be refolded in vitro and therefore allows its preparation in a pristine state. The structures of pristine H20 r treated LiP and the fungal LiP structures are very similar, having small main chain root mean square displacements of about 0.7 A. Significant structural differences of a small number of surface residues can be solely ascribed to crystal packing effects. Comparison of the structure of the pristine enzyme with that of the recombinant, C~ -hydroxylated form reveals that they are virtually identical, besides the absence/presence of the OHgroup. A water molecule found in both structures in a small cavity near the C~-atom of Trp 171 is ideally positioned to attack the C~-atom sterospecifically, leading to an S-configured C~-atom only, as was observed in the crystal structures II ,28 . A Trp 171 Phe mutant structure was determined in order to exclude the possibility that the complete lack of activity in this variant with the natural substrate V A was due to structural changes. Small structural differences can be recognised only in the close vicinity of the side chain of residue 171 . It can be, therefore, concluded that the absence of the redox active indole side-chain is solely responsible for the complete lack of V A oxidation activity of the Trpl71Phe mutant.
Concerning the function of the hydroxy group at the C~-atom of Trp 171 not much specific can be said, besides, that the hydroxylation is not a pre-requisite for V A oxidation by LiP, since this substrate competes with the self-oxidation leading to Trp 171 hydroxylation (see above). Based on simple chemical considerations on substituent effects, it would be expected that the OH-group could have an electron withdrawing effect on the indole ring possibly influencing the stability of a radical cation at the indole ring and subsequently its reactivity, With the stopped-flow technique the kinetics of spontaneous conversion of comp-I to comp-II of recombinant LiP in its pristine and in its oxidatively processed state were examined. (Blodig & Piontek, unpublished data). A 1.5-fold faster decay was found, indicating that an electron withdrawing effect is operative. Possibly, yet another function of the hydroxylation can be envisaged. Since Trp radicals are a reactive species they can cause destructive reactions on the indole ring. LiP, however, relies on a functional Trp, and should protect itself against such reactions. Therefore, the hydroxylation in LiP can be considered as a destructive step, but one that leaves the indole ring intact and functional. Because the OH-group reduces the accessibility to the indole ring and

52 INDIAN J CHEM, SEC A, JANUARY 2002
reduces conformational freedom of the side-chain, subsequent degrading steps could then be prevented.
High redox potential of LiP Regarding the reactivity of LiP, another important
issue is the high redox potential of this enzyme. It was noticed previously that the bonding distance of the proximal His to the iron in the structure of LiP is significantly longer than that in the structure of CCp25
,11. The latter has a lower redox potential than LiP. Based on this observation a relation of the redox potential with the Fe-N (proximal His) distance among haem peroxidases was proposed 1 1,25. Since a longer bond length makes the iron more electron deficient, which in turn destabilises higher oxidation states, this could be the cause for the higher redox potential of LiP. The longer bond length of the iron to the proximal His176 in LiP, on the other hand, is the result of a short hydrogen bond to an adjacent peptide and of a strong interaction of the proximal calcium and Serl77, a completely conserved residue in LiP. By these interactions the proximal His containing helix is forced to move further away from the haem, which results in an increase in the bond length. The previously determined bonding distances of the proximal His to the haem iron in the fungal LiP structures were nicely reproduced in the structures of the recombinant enzymes. Further confirmation of the correctness of these values was recently obtained from the structure of the fungal enzyme, which was meanwhile refined to a resolution of 1.46 A (Blodig & Piontek, unpublished results).
Conclusion Until recently, no information on the interaction
site(s) of LiP with its substrates was available. The crystal structure of the fungal enzyme revealed an unprecedented unique post-translational modification on a surface tryptophan, which is formed autocatalytically during the first few turnover cycles of the enzyme. This finding has highlighted Trp 171 as an essential residue in the catalytic redox cycle for small molecule substrates like V A. Site-directed mutatgenesis experiments showed that V A binds at or close to this redox active tryptophan, but also that another substrate interaction site must exist in LiP, at the "classical" haem access channel of heam peroxidases. The question if and possibly how these two regions communicate with each other is an intriguing one. Also the elucidation of the exact substrate binding mode by means of structural biology is probably a challenging task.
Remark Several structures of LiP, including various
isozymes and variants, are deposited with the Protein Data Bank, under accession codes IB80, 1B82, 1B85, lLGA, lLLP, and lQPA.
Acknowledgement Dr. Thomas Choinowski is thanked for his help in
the preparation of the figures.
Abbreviations used: ARP, Arthromyces ramosus peroxidase; comp-I and comp-II, compound I and II respectively; CcP, cytochrome c peroxidase; CiP, Coprinus cinereus peroxidase; HRP, horse radish peroxidase; LiP, lignin peroxidase; MnP, manganese peroxidase; MNP, methyl nitroso propane; PX, peroxidase; VA, veratryl alcohol (3,4-dimethoxy benzyl alcohol).
References I Saunders B C, Holmes-Siedle A G & Stark B P, Peroxidase
(Butterworths, Washington, DC) 1964. 2 Dunford H B & Stillmann J S, Coord Chem Rev, 19 (1976)
187. 3 Everse J & Everse KE, Peroxidases in chemistry and biology,
Vols I and II, (CRC Press, Boca Raton, FL) 1991. 4 Falk J E, Porphyrin and metailoporphyrins, (Elsevier, New
York) 1964. 5 Chance B, Arch Biochem Biophys, 41 (1952) 416. 6 Dunford H B, Adv /norg Biochem, 4 (1982) 41. 7 Erman J E, Vitello L B, Mauro J M & Kraut J, Biochemistry,
28 (1989) 7992. 8 Erman J E, J Biochem Mol Bioi, 31 (1989) 307. 9 Yonetani T , Schleyer H & Ehrenberg, A, J bioi Chem, 241
(1966) 3240.
10 Sivaraja M, Goodin D B, Smith M, & Hoffmann B M, Science, 245 (1989) 738.
II Choinowski T, Blodig W, Winterhalter K W & Piontek K, J
molec Bioi, 286 (1999) 809. 12 Blodig W, Smith A T, Winterhalter K & Piontek K, Arch
Biochem Biophys, 370 (1999) 86. 13 Mirza 0, Henriksen A, Ostergaard L, Welinder K G, &
Gajhede M, Acta Crystallogr D, 56 (2000) 372. 14 Hiner A N, Martinez J I, Arnao M B, Acosta M, Turner D D,
Lloyd Raven E & Rodriguez-Lopez J N, Eur J Biochem, 268 (2001) 3091.
15 Converso D A & Fernandez M E, Arch Biochem Biophys, 357 (1998) 22.
16 Welinder K G, Curr Opin Struct Bio, 2 (1992) 388. 17 Van Deurzen M P J, Van Rantwijk F & Sheldon R A,
Tetrahedron, 53 (1997) 13183. 18 Colonna S, Gaggero N, Richelmi C & Pasta P, Trends
Biotechnol17 (1999) 163. 19 Adam W, Lazarus M, Saha-Moller C R, Weichold 0, Hoch U,
Haring D & Schreier P, Biotrans!ormations (Springer, Berlin) 1999.

PIONTEK: LIGNIN PEROXIDASE 53
20 Van Rantwijk F, Sheldon R A, Curr Opin Biotechnol, 11 (2000) 554.
21 Tien M & Kirk T K, Science, 221 (1983) 661. 22 Adler E, Wood Sc Technol, 11 (1977) 169. 23 Cai D, Tien M, J Biotechnol, 30 (1993) 70. 24 Steenken S, J phys Chem A, 102 (1998) 7337. 25 Piontek K, Glumoff T & Winterhalter K, FEBS Lett, 315
(1993) 119. 26 Edwards S L, Raag R, Wariishi H, Gold M H & Poulos T L,
Proc Natl Acad Sci USA, 90 (1993) 750.
27 Choinowski T, PhD Thesis, no. 11859, ETH-ZUrich, Switzerland, 1996.
28 Blodig W, Smith A T, Doyle W A & Piontek K, J molec Bioi, 305 (2001) 851.
29 Finzel B C, Poulos T L & Kraut J, J bioi Chem, 259 (1984) 13027.
30 Sundaramoorthy M, Kishi K, Gold M H & Poulos T L, J bioi Chem, 269 (1994) 32759.
31 Gaijhede M, Schuller D J, Henricksen A, Smith AT & Poulos T L, Nat Struc Bioi, 4 (1997) 1032.
32 Henriksen A, Welinder K G, Gajhede M, J bioi Chem, 273 (1998) 2241.
33 Petersen J F W, Kadziola A & Larsen S, FEBS Lett, 339 (1994) 291.
34 Kunishima N, Fukuyama K, Matsubara H, Hatanaka H, Shibano Y & Amachi T, J molec Bioi, 235 (1994) 331.
35 Poulos T L, Edwards S L, Wariishi H & Gold M H, J bioi Chem, 268 (1993) 4429.
36 Doyle W A & Smith A T, Biochem J, 315 (1996) 15. 37 Harvey P, Schoemaker H E, Bowen R M & Palmer J M,
FEBS Lett, 183 (1985) 13.
38 Harvey P, Schoemaker H E & Palmer J M, FEBS Lett, 195 (1986) 242.
39 Goodwin D G, Aust S D & Grover T A, Biochemistry, 34 (1995) 5060.
40 Itakura H, Oda Y & Fukuyama K, FEBS Lett, 412 (1997) 107. 41 Henriksen A, Schuller D J, Meno K, Welinder K G, Smith A
T & Gaijhede M, Biochemistry, 37 (1998) 8054. 42 Henriksen A, Smith A T & Gaijhede M, J bioi Chem, 274
(1999) 35005. 43 Schuller D J, Ban N, Van Huysteen R B, McPherson A
. &Pbulos T L, Structure, 4 (1996) 311. 44 Patterson W R & Poulos T L, J bioi Chem, 296 (1994) 17020. 45 Piontek K, Glumoff T, Winterhalter K H & Schoemaker H E,
Plant peroxidases. Biochemistry and physiology, Geneva, (1993)9.
46 Schoemaker H E, Lundell T K, Floris R, Glumoff T, Winterhalter K H & Piontek K, Biorg med Chem, 2 (1994)
509. 47 Ageorges A, Pelter A & Ward R S, Phytochemistry, 30 (1991)
121. 48 Doyle W A, Blodig W, Veitch N C, Piontek K & Smith A T,
Biochemistry, 37 (1998) 15097. 49 Blodig W, Doyle W A, Smith A T, Winterhal ter K.
Choinowski T & Piontek K, Biochemistry, 37 (1998), 8832. 50 Harvey P J, Palmer J M, Schoemaker H E, Dekker H L &
Wever R, Biochem Biophys Acta, 994 (1989) 59. 51 Timofeevski S L, Nie G, Reading N S & Aust S D, Arch
Biochem Biophys, 373 (2000) 147. 52 Camarero S, Sarkar S, Ruiz-Duenas F J, Martinez M J &
Martinez A, J bioi Chem, 274 (1999) 10324. 53 Mester T & Field J A, J bioi Chem, 273 (1998) 15412.





























