Embed Size (px)
Citation preview

HOST NUCLEIC ACID SYNTHESIS IN CHICKEMBRYO FIBROBLASTS FOLLOWING
INFECTION BY ROUS SARCOMA VIRUS
Item Type text; Dissertation-Reproduction (electronic)
Authors DeLamarter, John Frederic, 1948-
Publisher The University of Arizona.
Rights Copyright © is held by the author. Digital access to this materialis made possible by the University Libraries, University of Arizona.Further transmission, reproduction or presentation (such aspublic display or performance) of protected items is prohibitedexcept with permission of the author.
Download date 03/03/2021 00:18:09
Link to Item http://hdl.handle.net/10150/290397

INFORMATION TO USERS
This material was produced from a microfilm copy of the original document. While
the most advanced technological means to photograph and reproduce this document
have been used, the quality is heavily dependent upon the quality of the original
submitted.
The following explanation of techniques is provided to help you understand
markings or patterns which may appear on this reproduction.
1. The sign or "target" for pages apparently lacking from the document
photographed is "Missing Paga(s)". If it was possible to obtain the missing
page(s) or section, they are spliced into the film along with adjacent pages.
This may have necessitated cutting thru an image and duplicating adjacent
pages to insure you complete continuity.
2. When an image on the film is obliterated with a large round black mark, it
is an indication that the photographer suspected that the copy may have
moved during exposure and thus cause a blurred image. You will find a
good image of the page in the adjacent frame.
3. When a map, drawing or chart, etc., was part of the material being
photographed the photographer followed a definite method in
"sectioning" the material. It is customary to begin photoing at the upper
left hand corner of a large sheet and to continue photoing from left to
right in equal sections with a small overlap. If necessary, sectioning is
continued again — beginning below the first row and continuing on until
complete.
4. The majority of users indicate that the textual content is of greatest value,
however, a somewhat higher quality reproduction could be made from
"photographs" if essential to the understanding of the dissertation. Silver
prints of "photographs" may be ordered at additional charge by writing
the Order Department, giving the catalog number, title, author and
specific pages you wish reproduced.
5. PLEASE NOTE: Some pages may have indistinct print. Filmed as received.
Xerox University Microfilms 300 North Zeeb Road Ann Arbor, Michigan 48106

76-18,255
DeLAMARTER, John Frederic, 1948-HOST NUCLEIC ACID SYNTHESIS IN CHICK EMBRYO FIBROBLASTS FOLLOWING INFECTION BY ROUS SARCOMA VIRUS.
The University of Arizona, Ph.D., 1976 Biology
Xerox University Microfilms, Ann Arbor, Michigan 48106

HOST NUCLEIC ACID SYNTHESIS IN CHICK
EMBRYO FIBROBLASTS FOLLOWING INFECTION BY
ROUS SARCOMA VIRUS
by
John Frederic DeLamarter
A Dissertation Submitted to the Faculty of the
DEPARTMENT OF MICROBIOLOGY
In Partial Fulfillment of the Requirements For the Degree of
DOCTOR OF PHILOSOPHY WITH A MAJOR IN MOLECULAR BIOLOGY
In the Graduate College
THE UNIVERSITY OF ARIZONA
19 7 6

THE UNIVERSITY OF ARIZONA
GRADUATE COLLEGE
I hereby recommend that this dissertation prepared under my
direction by John Frederic DeLamarter
entitled HOST NUCLEIC ACID SYNTHESIS IN CHICK EMBRYO FIBROBLASTS
FOLLOWING INFECTION BY ROUS SARCOMA VIRUS
be accepted as fulfilling the dissertation requirement of the
degree of Doctor of Philosophy •
1 /-•< .
Dissertation Director Date T 6
After inspection of the final copy of the dissertation, the
following members of the Final Examination Committee concur in
its approval and recommend its acceptance:-
This approval and acceptance is contingent on the candidate's adequate performance and defense of this dissertation at the final oral examination. The inclusion of this sheet bound into the library copy of the dissertation is evidence of satisfactory performance at the final examination.

STATEMENT BY AUTHOR
This dissertation has been submitted in partial fulfillment of requirements for an advanced degree at The University of Arizona and is deposited in the University Library to be made available to borrowers under rules of the Library.
Brief quotations from this dissertation are allowable without special permission, provided that accurate acknowledgment of source is made,, Requests for permission for extended quotation from or reproduction of this manuscript in whole or in part may be granted by the head of the major department or the Dean of the Graduate College when in his judgment the proposed use of the material is in the interests of scholarship* In all other instances, however, permission must be obtained from the author.

ACKNOWLEDGMENTS
I wish to express my sincere appreciation to Dr. Marshall
Dinowitz for his steadfast encouragement, constructive criticisms,
and generous friendship. Dr. Harris Bernstein's critical reading of
this dissertation and the timely encouragements of Drs. Harris Bern
stein, Charles J. Gauntt, and David Mount were most helpful to me. I
also wish to acknowledge the friendship of many graduate students,
particularly that of Eileen Nonn, that greatly enriched my graduate
years.
iii

TABLE OF CONTENTS
Page
LIST OF TABLES vi
LIST OF ILLUSTRATIONS ..... vii
ABSTRACT o © o o ® o o o ® . ® o « o . . . . . . . . . ® . . o I X
INTRODUCTION o o o o o o o o o o o o o o o o o . o . o o o o o I
RSV Life Cycle ooooo.o.eoooo.oooooooo 1 Provirus Synthesis o.o.o.ooooooo.oooo 2 Integration of the Provirus .0.000..0.000 3 Viral RNA Synthesis 00.00000000.000.0 ^
Virus-Host Interactions ................. 5 Cellular RNA Synthesis 5 Cellular DNA Synthesis and Division . 6 Endogenous Viral Sequences . 7 RNA Tumor Virus Origins .... ..... 8
Cellular Aspects ..... ... 9 Cellular DNA Synthesis ....... . o . o . . ® o . 9 Cellular RNA .00.000 O O . O . O O O D ® . . . ® 1 1
This Investigation 00.0.000000.0000.0.0 11
MATERIALS AND METHODS ... .00.00.00000.0.00 13
Cells OO.OO...............OOOOOO 13 Infections with RSV and Labeling ...... 13 Extraction and Preparation of Host Nucleic Acids ..... 1^
Preparation of Radioactive DNA . . . . . o o . . . . . 15 Extraction of Host RNA .................. 16
Nuclear RNA . . . . . . . . . . . ^ . . . . . o o o o 16 Validation of Protocol Modifications ......... 17 Polysomal RNA Extraction . . o o . . . . . . . . . . . 2 0
DNA/DNA Reassociation ...... ... 22 Assay of DNA Annealing ......000.0000.0 23
RNA/DNA Hybridization ..... .... 2b Assay of RNA/DNA Duplexes ......... 26 Modification of RNase Assay ... 26
iv

V
TABLE OF CONTENTS—Continued
Page
RESULTS 29
Cell Transformation . 29 Host Cell DNA Synthesis Following Infection ........ 29
Labeling of Host Cell DNA «>«*««ooo»o«»oo 32 DNA/DNA Reassociation Kinetics .............. 36 Analysis of Host RNA Synthesis Following Infection . . . » bj>
Labeling of Host Cell RNA .000.0.00.00000 bb Hybridization of Pulse-Labeled Host Cell nRNA » . o . o o o bb
RNA/DNA Hybridizations of Control Cell nRNA » . . o 0 «, ^6 InfeCted Cell nRNA ooeeoeoooeo.ooooOQ ^9
Constant DNA/RNA Ratio Comparison Hybridizations . . . . . 5b Polysome-Associated "Repetitive" RNA .......so 59 Labeling and Extraction of pRNA oo.oooo....e 59 Low CQt Hybridizations ....osoeoooooo.. 60
Post-Transcriptional Processing of "Repetitive" RNA . . . « Gb
DISCUSSION 66
DNA/DNA Reassociations . . * 66 Theoretical and Practical Aspects o 66 Observations and Interpretations 69
RNA/DNA Hybridizations . . . . . . . . . . . . . « o S S . 70 Theoretical and Practical Aspects . . . . . . o . o . o 70 Observations and Interpretations ....... . . . o 7 ^ Repetitive Sequence Transcripts ...........s 75
Conclusions ......... 8l
LIST OF REFERENCES Sb

LIST OF TABLES
Table Page
1. Comparison of digestion schemes for radioactive RNA ... 28
2. Summary of pulse-labeled DNA extractions ........ 33
3. Summary of reassociation kinetics for pulse-labeled Cellular DNA o.oe..eoe.oooeeeo.o..oo ^f2
k. Summary of pulse-labeled nuclear RNA extractions .... ^5
5. Summary of pulse-labeled polyribosomal RNA extractions . 62
vi

LIST OF ILLUSTRATIONS
Figure Page
1. Sephadex chromatography of DNase-treated nucleic acids . . 19
2. Sedimentation of the cytoplasmic fraction of chick cells on a sucrose gradient 00.0.0000000000000 21
3» Liability of RNA during RNA/DNA hybridization . » . » o . 25
k. Morphological transformation of normal cells by RSV infection o o o o o o o o o o o o o o o o o o o . o o o o 30
5. Partial synchrony of cellular DNA synthesis following subculturmg o o o o o o o o o o o o o o o o o o o o o o o 35
6. Reassociation analyses of uninfected cell DNA ...«.« 37
7. Composite of control-cell DNA reassociation curves . » . . 38
8. Reassociation analyses of RSV-infected cell DNA . . » o » kO
9. Composite of infected-cell DNA reassociation curves o . » *tl
10. Hybridization of normal CEF nuclear RNA ..o.ooooo ^7
11. Hybridization of RSV-infected cell nRNA with cell DNA ® o 50
12. Hybridization kinetics for unique sequence transcripts from RSV-infected and control cell nRNA ......... 51
13. Hybridization kinetics for repeat sequence transcripts from RSV-infected and control-cell nRNA ......... 53
1^-. Low CQt hybridizations of control cell and RSV-infected cell nRNA 0000.00.00000.000.0000.0 55
15. The fraction of nRNA hybridized by a CQt of 10 for control a n d R S V — i n f e c t e d c e l l s 0 0 0 0 0 0 0 0 . 0 0 . 0 0 0 . 0 0 5 ^
16. Hybridizations of nuclear RNA at a constant DNA/RNA r a t i o . O O O . O . O O . O O O O O 0 . . . O O O . . 0 0 5 8
17. Sedimentation profiles of cytoplasmic fractions from c o n t r o l a n d R S V - i n f e c t e d c e l l s . . . . . . . . . . . . . . 6 l
vii

viii
LIST OF ILLUSTRATIONS—Continued
Figure Page
18. Ratio of infected to control cell polysomal RNA hybridized at a CQt of 15 63
19. The relationship between the polysomal and nuclear "repetitive" RNA fractions as a function of infection . . 65
20. Summary of cellular events after subculturing or infection with RSV 82

ABSTRACT
Host cell nucleic acid synthesis was monitored for alterations
during infection by a transforming RNA tumor virus. Nucleic acids
pulse labeled during the transformation of infected cells were analyzed
by their annealing kinetics in liquid reaction mixtures,. When suscep
tible chick embryo fibroblasts (CEF) were infected with Rous sarcoma
virus (RSV), within 2k hours individual cells showed transformed mor
phology and by 50 hours the entire culture was morphologically trans
formed.
Conditions used for subculturing and infecting CEF cells re
sulted in partial synchrony for DNA synthesis of both normal and
infected cells, VJhen normal cells stopped synthesizing DNA, the addi
tion of fresh serum to the culture medium induced new DNA synthesis®
For DNA samples pulse-labeled at different times during the provirus
phase of RSV replication (3-12 hours after infection), DNA/DNA re-
association kinetics were very similar whether the DNA was derived
from infected or control cells.
Transcription in normal and infected cells was analyzed by
liquid RNA/DNA hybridizations of pulse-labeled RNA. The fraction of
repetitive sequence transcripts in normal cell nuclei decreases
slightly during active cell growth compared to cultures of confluent
monolayers. Similarly the repetitive sequence transcripts found on
the polyribosomes from normal cells represent a smaller fraction in
ix

X
rapidly dividing cells. However, late after infection by RSV (25-50
hours), nuclei of infected cells contained twice the fraction of repeat
sequence transcripts as control cell nuclei® By approximately 15 hours
after infection, repetitive sequence transcripts found on infected-cell
polyribosomes show half the concentration of those from control cells.
The observed changes in representation of repetitive sequence RNA in
the different cellular compartments suggests that the transport of
these transcripts may be inhibited in RSV-infected cells. Very late
(58 hours) after infection — at a time when control cells had reached
confluency and infected cells were all transformed as well as having
reached saturation density — the proportion of repeat sequence tran
scripts were similar for control and RSV infected cells in both the
polyribosomal and nuclear RNA fractions. These data are consistent with
models which have assigned regulatory functions to the transcripts of
repetitive-sequence DMA.

INTRODUCTION
RNA tumor viruses replicate in an intimate relationship with
their host cell (Tooze, 1973)« More than host RNA and protein machinery
is required for the synthesis of their progeny (Temin, 1971a)„ Alter
ations in cellular functions that follov; infection are indications of a
virus-host inter-relationship existing at the level of the gene0 In
fection of susceptible cells with the RNA tumor virus, Rous sarcoma
virus (RSV), causes major inheritable changes in cell morphology (Temin
and Rubin, 1953; Temin, 1962) and metabolism (Bissell, Hatie and Rubin,
1972; Buck, Glick and Warren, 1971) including continued production of
virus. These characteristics of RSV infection of its host, chick
embryo fibroblasts (CEF), make it an appropriate system in which to in
vestigate altered regulation of host-cell nucleic-acid synthesis. In
this study CEF cellular DNA and RNA were analyzed to determine whether
there were differences in DNA replication and transcription following
infection by RSV.
RSV Life Cycle
Replication of RSV genomic single-stranded RNA is a complex
process beginning with virus adsorption to appropriate receptors on the
cell surface (Piraino, 1967), penetration of the cell membrane by
phagocytosis (Crittenden, 1968), and probable uncoating of the virus
in the cytoplasm (Tooze, 1973)• From this point on the fate of the
virion RNA is not certain. In an electron micrographic study, Dales

2
and Hanafusa (1972) showed virion-sized parental RNA in autoradiograms
of the host cell nucleus one hour post infection. Synthesis of a DNA
intermediate, the provirus, is the next major event in RSV replication.
However, evidence establishing its existence conflicts with the nuclear
location for the parental RNA just described,,
Provirus Synthesis
Temin (1962) first postulated a DNA intermediate for RSV repli
cation based on the Mendelian inheritance of viral information in cells
once transformed by RSV. Indirect evidence accumulated to support this
hypothesise Virus production proved to be sensitive to agents inter
acting with DNA such as Actinomycin D and Bromodeoxyuridine (Temin,
1963; Bader, 196^; Boettiger and Temin, 1970; Balduzzi and Morgan, 1970).
Observations yielding indirect support to the DNA provirus hypothesis
culminated in the discovery of the virion associated RNA-dependent DNA
polymerase (reverse transcriptase) by Temin and Mizutani (1970) and
Baltimore (1970). Only very recently has it been possible to directly
isolate the proviral DNA. Varmus and co-workers (Varmus, Guntaka, Deng
and Bishop, 197*0 used RSV infected Duck cells from which they ex
tracted 0o002& of the total DNA representing proviral sequences as
shown by hybridization to viral 70S RNA.
Several studies are pertinent to the provirus synthesis. Again
using the RSV-duck cell system, Varmus, Guntaka, Deng and Bishop (197*0
showed cytoplasmic synthesis of viral DNA beginning at 3 hours after
infection with transfer to the nucleus seen at 6 hours reaching a
maximum at 9«5 hours. This time frame had been indicated

by previous investigations. For example, Bader and Brown (1971) had
induced mutations in RSV (an RNA virus) by use of a DNA analogue
(Bromodeoxyuridine, BrdU) finding that only the first 12 hours follow
ing infection proved to be sensitive to the mutagen,, During neither
the preceding nor the following 12 hour periods was viral replication
susceptible to the presence of the mutagen,. These studies implicated
the time between 3 and 12 hours after infection for synthesis of the
proviral DNA„ Rather than existing as a viral episome, however, RSV
DNA appears to integrate its sequences into the host chromosome (Varmus,
Guntaka, Deng and Bishop, 197*0 in a manner somewhat analogous to the
insertion of lambda phage into the bacterial chromosome (Hershey, 1971)-
Integration of the Provirus
Baluda and Nayak (1970) demonstrated increased viral sequences
in host DNA following transformation of cells by an avian oncornavirus.
They hybridized avian myeloblastosis virus (AMV) labeled RNA to trans
formed myeloblast cell DNA and found an increased hybridization when
compared to control cell DNA. Using similar viral-RNA/cellular-DNA
hybridizations, Neiman (1972) demonstrated that RSV transformed cells
contained many more copies of a part of the transforming genome plus
a more complete copy of the viral sequences. While these studies
documented the integrated state of the viral genome in transformed
cells, the integration step remained unproven. Schincariol and Joklik
(1973) prepared radioactive viral DNA in vitro from the RNA template
by reverse-transcriptase and used this to probe infected cell DNAe
Their data show an increasing number of copies of viral sequences

beginning at 3 hours post infection such that by 10 hours twice as many
copies of viral DNA sequences were present as in uninfected cells<>
Samples taken at later times yielded approximately 3 copies of the
viral sequences present per cello Their work did not rule out possible
plasmids for the increased number of copies seen. Varmus, Vogt, and
Bishop (1973)? however, cleverly demonstrated viral integration into
the host chromosome. Taking advantage of the normal repeated sequences
found in eukaryotic DNA, these investigators demonstrated that network-
made by denaturing and quickly reannealing infected host cell DNA
showed increasing amounts of virus-homologous sequences as infection
proceededo Subsequently, they presented data which placed viral DNA in
the host nucleus 6 hours after infection (Varmus, Guntaka, Warner et al,
1974). The average copy number/nucleus in infected cells increased
to 0.8 by 9„5 hours after infection with 90$ of the RSV genome repre
sented,, Taken together these investigations demonstrate that the first
12 hours after infection are the critical period for proviral integra
tion into the host chromosome. Evidence indicates that synthesis of
viral RNA follows this step.
Viral RNA Synthesis
Studies with Actinomycin D had indicated that viral RNA was
transcribed from a DNA template. Near the end of the proviral phase
(10 hours post infection) the first detectable increases in viral RNA
following infection sire found (Schincariol and Joklik, 1973). The con
centration of viral RNA continued to increase until late after infec
tion (96 hours). Infectious virus production was not detected until

5
18 hours after the infection and leveled off after 72 hours. This se
quence is consistent with the timing of synthesis and integration of
the provirus and the fact that viral transcription requires the host
nuclear RNA polymerase form II (Jaquet et al„, 197^5 Rymo et al„, 197*M
Dinowitz, 1975b)o
Virus-Host Interactions
Information concerning host-virus interdependence is limited.
The topics investigated to date have of necessity been restricted by
our understanding of virus specific and normal cell functions.
Cellular RNA Synthesis
RSV infection has been shown to increase the rate of cellular
RNA synthesis in general and of one class (ribosomal RNA, rRNA) in par
ticular (Colby and Rubin, 1969). The synthesis of RNA in transformed
cells is also more resistant to low concentrations of BrdU and to
a-amanitan than normal cells (Levinson et al., 1970; Dinowitz, 1975a).
Recently the activation of embryonic globin genes in CEF cells by RSV
infection has been reported (Groudine and Weintraub, 1975). These in
vestigators found 100-500 transcripts/cell of the embryonic globin gene
sequence, but none of the adult globin sequence. From these data it
can be inferred that RSV infection and transformation of CEF cells
results in a difference in the general regulation of transcription as
well as the expression of specific genes.

6
Cellular DNA Synthesis and Division
The requirements for cellular division or DNA synthesis in the
life cycle of RSV remains unresolved,, The necessity of these host
functions has been investigated both in transformed cells continuously
releasing virus and in newly infected cells becoming transformed by
RSV «
Leong, Levinson and Bishop (1972) studied the effect of the
host cell cycle on virus production in cells transformed by RSV„ When
fresh serum was added to transformed cells grown in serum-free medium,
the cells left the early "Gi" phase and progressed through the cell
cycle in a synchronous fashion0 Using the virion-associated reverse-
transcriptase product for hybridizations with cellular RNA, the authors
showed maximum viral RNA synthesis occurring in cells from early "S"
phase® Virion protein synthesis followed during the early "G^" phase.
Similarily, Paskind, Weinberg and Baltimore (1975) grew Moloney Murine
Leukemia virus transformed mouse cells in medium with reduced serum to
induce cell cycle synchrony,. Release of the cells from serum depri
vation resulted in DNA synthesis and virus release as measured by
assaying reverse-transcriptase activity in the culture fluids® These
studies link viral production to the host cell cycle®
Temin (1967a) investigated the requirement for cellular DNA
synthesis and division in transformation of the host„ These experi
ments produced evidence in favor of host cell division as a requirement
for RSV transformation of CEF cells® Conversely, data derived from
mitotic inhibitor studies were interpreted by Bader (1972; 1973) to
show virus-induced cell transformation without cell division® More

7
recently, Humphries and Terain (197*0 used chick cells synchronized for
cell growth by serum deprivation to study this relationship„ In these
experiments cells were infected 12 hours prior to the addition of fresh
serum which had previously been shown to induce cell DNA synthesis0
Cells treated in this manner did not synthesize detectable viral RNA,
as assayed by hybridization of cellular RNA to viral DNA until 18 hours
after the addition of serum* Furthermore, if these cells were stimu
lated with serum in the presence of colchicine, the detection of viral
RNA was delayed 12-2^ hours over colchicine-minus controls,, The
authors interpret these results as supportive of a requirement for cell
DNA synthesis and division before infection by RSV can result in virus
replication®
Endogenous Viral Sequences
The presence of avian leukosis and sarcoma viral sequences in
most chick cells is now well documented (Rosenthal et al., 1971; Baluda,
1972; Neiman, 1972; Neiman, 1973)» Expression of these sequences in
the form of a viral protein (gs) and a helper virus activity (chf) has
been shown to be determined by inherited genes which are not coordi-
nately controlled (Hanafusa et alc, 197*0° Whether these regulatory
genes are cellular or viral in origin is not clear (Tooze, 1973)» Some
form of transcriptional control appears to exist for these nearly
ubiquitous endogenous viral sequences» Hayward and Hanafusa (1975)
have recently isolated a new Rous associated virus (RAV 60) which they
believe to be recombinant between the host's endogenous virus and the
infecting exogenous virus. These authors used reverse-transcriptase

viral DNA from RAV 60 to show partial homology with the exogenous viral
RNA, host cell RNA, and the endogenous viral DNA. Their data indicate
that infecting viruses can interact v/ith endogenous viral sequences to
yield recombinant genomes- The exchange of genetic information ob
served shows a close relationship between some host and viral DNA®
RNA Tumor Virus Origins
Two theories attributing cellular origins to oncornaviruses are
described here as possible links between cellular and viral functions0
Temin (1970) published his protovirus theory following the discovery of
the RNA dependent DNA polymerase0 Simply put, the theory suggests that
RNA tumor viruses are one of the current evolutionary products of an
original "protovirusThe protovirus was a piece of the cellular
genome capable of being copied into either RNA or DNA and replicating
in the later form (Temin, 1971b)«, Cellular information was transferred
from cell to cell by infrequent infection v/ith this uncoated nucleic
acido Protoviruses then would be potential sources for single-
generation information transfer in a stable form,, Their evolution to
an enveloped virion with associated reverse-transcriptase can readily
be imagined in terms of the selective advantage accrued,,
Recently, Gillespie and Gallo (1975) have also argued that RNA
tumor viruses originated from normal cell transcripts via a mechanism
they term "paraprocessing«M They have integrated evidence which sup
ports the idea that virion RITA resembles normal cell nuclear RNA which
has not undergone the usual processing to messenger RNA (mRNA), hence
paraprocessingo This process, v/hen coupled with cytoplasmic transport,

acquisition of a cellular reverse-transcriptase, and budding from the
cell membrane could account for the origins of many of the character
istics currently associated with RNA tumor viruses,, Paraprocessing as
a possible mechanism for oncornavirus evolution is an attractive theory
for two reasons that are also valid for the protovirus theory,, Both
these hypotheses give plausable reasons for the _a priori existence of
these viruses plus the special relationship that exists between their
host and viral genomes.
Cellular Aspects
The analyses of cellular nucleic acid synthesis undertaken in
this study were done to identify virus-induced changes in normal cell
metabolism,, To provide the framework for these investigations, normal
cell functions including information about synchronous synthesis of
DNA, transcriptional control, and the nature of cellular RNA are dis
cussed below0
Cellular DNA Synthesis
Eukaryotic DNA replication involves discontinuous synthesis
initiated at many sites (Callan, 1973). Since chromosomal replication
does not begin at a unique origin and progress in a linear manner to
completion, distinct classes of DNA sequences may be synthesized at
different times. Virus infections might then influence the timing of
synthesis for these classes. Linear replication of eukaryotic chromo
somal DNA during a single period has been challenged by a number of
investigators (Brown and Blackler, 1972; Tobia, Schildkraut and Maio,

10
1970; Gall, 1969; Dickson, Boyd and Laird, 1971). Their evidence indi
cates several alternative modes for replication; including over-
synthesis followed by degradation, extra-chromosomal synthesis, and/or
amplification of genetic information., Klevecz, Kapp and Remington
(197^) synchronized several different mammalian cell lines by mitotic
selection and then monitored their DNA synthesis by long (30 minute)
3 and short (2 minute) pulses with H-thymidine. Long pulses showed a
single peak of DNA synthesis, while shorter ones showed discontinuous
synthesis throughout "S" phase. These data suggest the transient syn
thesis and catabolism of undefined classes of the cell's DNAo
Balazs, Brown and Schildkraut (1973) investigated the temporal
replication of different cellular cistrons. They labeled the DNA
replicated at different times during "S" phase of a synchronously
growing SV4o transformed mouse cell line. These investigators deter
mined that integrated SVkO sequences were maximally replicated in
middle "S" phase at approximately tv/ice the rate of early and late "S"
phaseo In the same study they report an alteration in the time of
satellite DNA replication during the induced DNA synthesis that fol
lows SVkO infection of permissive cells* In uninfected synchronously
dividing mouse cells, satellite DNA replicates very late in the "S"
phase, but infection by SV^O causes the replication of satellite se
quences to be shifted to the period of bulk DNA synthesis. These data
imply that specific cistrons replicate at different times during the
"S" phase and that viral infections may cause temporal alterations in
the order in which some repetitive DNA is replicated.

11
Cellular RNA
In the nucleus of eukaryotes, large heterogeneously-sized mole
cules of RNA (HnRNA) are rapidly synthesized which contain both repeti
tive and unique DNA sequence transcripts including sequences homologous
to protein-specific mRNA (Lewin, 1975a)» Poly-adenylate tracts are
added post transcriptionally to the 3' end of the HnRNA and the mole
cule is probably then cleaved to a single protein messenger size in a
manner analogous to the processing of ribosomal RNA (Greenberg, 1975;
Lewin, 1975b)» In growing normal cells only about bC#> of the unique
sequence diversity present in nuclear RNA (nRNA) is found on polyribo
somes (Grady and Campbell, 1975a)„ On the average this nRNA is rapidly
turning over (half-life of approximately 23 minutes) compared to some
classes of mRNA which have half-lives of 2b hours (Brandhorst and
McConkey, 197^5 Perry and Kelley, 1968; Singer and Penman, 1973)«. The
existence of repeat sequence transcripts in mRNA similar to those in
nRNA has been uncertain,, Recent evidence supports the contention that
two distinct classes of mRNA exist — the major fraction being tran
scribed only from unique sequences and a minor fraction transcribed
solely from repeat DNA (Campo and Bishop, 197^5 Klein et al., 197*0 •
This Investigation
Host cell nucleic acid synthesis was studied during the conver
sion of normal CEF to RSV transformed cells. Secondly, alterations of
a relatively transient nature were identified by pulse labeling with
radioactive precursors® In this manner, temporal changes as a function

12
of RSV infection in both cellular DNA synthesis and transcription were
examined.
Host cell DNA pulse-labeled during the synthesis and integra
tion of the RSV provirus was analyzed by denaturation and reassociation
of the labeled DNA. Results of this technique indicate the relation
ship between different reiteration classes of DNA synthesized at the
time of the pulse (Britten and Kohne, 1968)0 Analogous changes to the
shift in replication of the highly repeated mouse satellite DNA by SVbO
infection were sought throughout the period of host and viral DNA
interactionso
Host cell RNA was pulse labeled during the conversion of the
RSV-infected culture from normal to transformed morphology. Cells were
fractionated into nuclear and cytoplasmic components in order to sepa
rately investigate RNA from these two cellular compartments. The RNA
found in the nucleus and that subsequently appearing on the cytoplasmic
polyribosomes was examined by RNA/DNA hybridizations. Similar to DNA
reassociation, the RNA hybridizations carried out in this study deter
mined the fractions of RNA transcribed from DNA templates that occur
in repeat or single copy frequency in the genome (Wilt, 1971)®
The period after infection that was investigated corresponded
to the time of probable maximum alterations for both cellular DNA and
RNA synthesis due to the virus infection. Since repetitive sequences
have been hypothesized to play a role in the regulation of growth and
differentiation in eukaryotic cells (Britten and Davidson, 19&9)»
changes in the relationship between repetitive and unique sequence
elements were sought during these periods®

MATERIALS AND METHODS
Cells
Ten to 11 day old chick embryos (Kimber Farms; Freemont, Ca„)
were decapitated, evicerated, minced, and trypsinized (0„05$> trypsin
in TBS /tris(Tris hydroxy-methyl-aminomethan)-buffered saline;(Vogt,
1969)/, 39°C) for 5 minutes, three times., The cell suspension was
plated on 100 mm plactic petri culture dishes (Falcon Plastics) and
passaged at least once before use. As a source of Rous sarcoma virus
(RSV), a second cell-type culture was maintained,, These were the con
tinuous line of RSV transformed and virus-producing cells, RTAZ-1,
established by Dr. Marshall Dinowitz (1976).
Cell cultures were grown in standard medium (SM) consisting of
medium 199 (GIBCO) supplemented with 10^ tryptose phosphate broth, 8^
calf serum and 2% fetal calf serum,, To retard microbial growth 100
units/ml penicillin, 100 /xg/ml streptomycin, and 2.5 /ig/ml amphotericin
B (Fungizone, Squibb) were added to the medium. Cells were incubated
in a humidified CO^ incubator under an atmosphere of 95$ air and 5%
C02 at 39°C.
Infections with RSV and Labeling
Infections of CEF were carried out with tissue culture fluids
from confluent RTAZ-1 cultures. The culture medium was clarified by
centrifugation to remove cells and cellular debris (5 minutes, 2,000xg).
This supernatant routinely contained 2-k x 10^ FFU/ml (Dinowitz, 1975c)
13

when assayed for focus formation on CEF (Temin and Rubin, 1958)e Cells
were infected in suspension with a mixture of fresh medium and the
clarified tissue culture fluid in equal volumes.. Control cells were
mock infected as above by substituting conditioned medium from normal
cell cultures, ioe„, normal cell tissue culture fluids clarified and
mixed with an equal volume of fresh medium. Cells were infected at a
multiplicity of infection (moi) of ~3 focus forming units (FFU).
For experiments where DNA was to be examined, the cells were
exposed to 5 HCi/m± ^H-thymidine (TdR) (k0-60 Ci/mM; NEN) during the
indicated 3 hours of incubation. Cells in which RNA was to be examined
were pulse-labeled with 50 /iCi/ml ^H-(5?6) uridine C+0-50 Ci/mM; MEN)
for 2 hours. Incorporation of radioactive precursors were terminated
by removing the cells from the culture dish and placing them in ice-cold
TBS."
Extraction and Preparation of Host Nucleic Acids
Chick embryo fibroblast DNA was prepared from 10-11 day old
chick embryos decapitated, eviscerated, and minced in TBS. Nuclear
cell DNA was extracted from this tissue by a procedure modified from
that of Marmur's (1961)= Cells were placed in reticulocyte standard
buffer (RSB: 0.01 M tris, pH 7®^; 0.01 M NaCl; 105 mM MgC^) and
homogenized in a dounce homogenizer. The disrupted cells were centri-
fuged at 1500 x g, 5 minutes and the nuclear pellet was resuspended in
a NaCl-EDTA buffer (0.15 M, 0.1 M) which was made 1% in sodium dodecyl-
sulfate (SDS) and then 1 M in sodium perchlorate. The suspension was

15
extracted three times with an equal volume of chloroform-isopentyl
alcohol (v/v, 2^:1) by shaking for 15 minutes in a wrist-action shaker
and separating the phases by centrifugation (1600 x g for 10 minutes).
The DNA was spooled from the final aqueous phase by adding two volumes
of 9% ethanol, resuspended in IX SSC(0„15 M NaCl, 0.015M sodium
citrate), treated with bovine pancreatic RNase A (50 fxg/ml, previously
boiled for 10 minutes to eliminate DNases) and a-amalyse (50 /ig/ml) for
30 minutes at 37° C followed by pronase (250 g/ml, 15-18 hours, 37° C)0
The DNA was then extracted two more times as above, spooled, resuspended
in IX SSC, re-extracted once, spooled again, and finally resuspended in
3 mM EDTAo Normal CEF DNA prepared in this manner exhibited absorbance
ratios of 260nm/230nm of >2. and at 260nm/280nm of >1.8. For all DNA
samples the DNA concentration was determined by absorbance at a wave
length of 260nm (21 O.D. units=lmg/ml). This DNA was then sheared by
sonication in an ice bath (10, 1 minute pulses separated by 0.5 minute
cooling periods) using a Heat Systems-Ultrasonic, sonifier (Heat Sys
tems Inc., Plainview, N.Y.).
Preparation of Radioactive DNA
Pulse-labeled cellular DNA was extracted and prepared in the
same manner as unlabeled cell DNA except that total cellular DNA was
isolated. The concentration of DNA was too low to allow spooling of
the nucleic acid; therefore, for each spooling step in the previous
procedure pelleting of the DNA by centrifugation (27*000 x g, 30 min
utes) was substituted. Radioactivity was monitored during the extrac
tion by counting trichloroacetic acid (TCA) precipitatable DNA.

Aliquots were spotted onto Whatman 3MM filter dies, dried and treated
twice with cold 5^ TCA„ The filter dies were then rinsed in cold 9%
ethanol, dried and counted in a liquid scintillation fluid gm BBOT/
L of toluene) used for the liquid scintillation, spectrophotometer.
Extraction of Host RNA
Nuclear RNA
Labeled cells were fractionated into cytoplasmic and nuclear
fractions using the Non-idet NP^O procedure (Borun, Scharff and Robbins,
1967) as modified to substitute Triton X-100 (Dinowitz, 1975b) «> Cells
were washed with cold TBS, then treated with cold 0e5/e Triton X-100 in
RSB. After 5 minutes incubation at *f°C the lysed cells were removed
with the aid of a rubber policeman, placed in a tube, vortexed vigor
ously, and centrifuged to pellet the nuclei (1500 x g, 5 minutes)» The
supernatant was retained as the cytoplasmic fraction for immediate pro
cessing as described below. Further cleaning of the nuclear pellet was
accomplished by resuspension in RSB; making the solution 0.5^ in sodium
deoxycholate, vortexing, then 1% in Tween 0, and mixing again (Penman,
1966). Nuclei were pelleted from the wash by centrifugation as before
and the supernatant discarded. The clean nuclei were then extracted by
a modified protocol of Penman (1966). The extraction was carried out
by resuspending the nuclei in a lysis buffer (lOmM tris, 1 mM EDTA,
lOOmM NaCl, 0o5% SDS, pH 7°*0 and the suspension was extracted with an
equal volume of phenol (redistilled), chloroform, isopental alcohol
(50:^8:2, v/v/v) saturated with the lysis buffer shaken for 5 minutes

on a wrist-action shaker. The extraction mixture was centrifuged at
10,000 x g for 5 minutes and the solvent removed from the bottom, then
replaced by an equal volume of fresh solvent» After repeating the ex
traction three more times, the aqueous phase was removed, made up to
0.15 M NaCl, 2 volumes of cold 95^ ethanol were added to precipitate
the RNA and the mixture was stored at -20°C for three or more hours®
Precipitated nucleic acids were pelleted by centrifugation at 27,000
x g for 30 minutes at it-°C0 The pellet was resuspended in a DNA diges
tion buffer (50 mM tris, 25 mM NaCl, 10 mM MgC^, 2o5 niM CaCl^, pH 7®2),
100 /ig/ml bovine pancreatic DNase I was added and the samples were in
cubated at 37°C for 30 minutes- These samples were then chromatographed
through a G-50 Sephadex column using 0,15 Mo NaCl as the eluent buffer.
The column had been previously washed with 2 column volumes of buffer,
brought to pH 11 by passing a 0.1 M NaOH solution through it in order
to denature RNase which may have been present, then returned to neutral
pH by flushing with 5-10 more column volumes of buffer,. Void volume
fractions were collected from the loaded column and the RNA was pre
cipitated by ethanol and pelleted as described above.
Validation of Protocol Modifications
Coupling the DNase with sephadex chromatography is a modifi
cation of the Penman technique suggested by Birnie et al.,(197if). In
further modifications, extractions were initially carried out with
CaCl^ present in a modified lysis buffer to allow for an earlier DNase
step as described by Penman (1966). This addition caused nearly 90$>
of the labeled nucleic acid to remain either in the solvent phase or

18
at the solvent-aqueous interface. By using the lysis buffer described
above, extractions resulted in the retention of greater than 8($ of the
total input radioactivity..
To test whether the DNase-sephadex step was to be an effective
method for removing the small DNA fragments remaining after DNase
treatment, eluent fractions were examined to determine where DNase-
treated DNA was eluted from the column,, Purified native DNA eluted
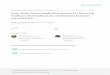
exclusively in the void volume of the G-50 Sephadex column (Fig. 1A).
The DNase-treated material eluted at about one half column volume
•z latere Nucleic acids labeled for 2k hours with H-adenosine, extracted,
DNased, and chromatographed as before resulted in radioactivity eluting
in both the void volume and the position characteristic of DNA frag
ments (Fig® IB). Since this nucleotide is a precursor to both DNA and
RNA, an elution profile consistent with the presence of both nucleic
acids was expected,, In contrast, nucleic acids pulse-labeled for 2
3 hours with H-uridine, DNased, and chromatographed as above eluted
exclusively in the void volume (Fig. IB).
Nuclear RNA samples were resuspended in 2x glass distilled water
and the RNA concentrations measured in a spectrophotometer by absorbance
at a wavelength of 260 nm (2k 0D„ units = 1 mg/ml). Recovery of RNA
was monitored during the extraction procedure by measuring TCA-
precipitable radioactivity as described for the labeled DNA® . For use
in hybridization reaction mixtures, nRNA samples were concentrated by
precipitation with ethanol pelleting and resuspending in the required
amount of 10X SSC.

19
a o
8
7
6
5
4
3
2
I
O
4
A
o—°—. os *0
—gz §/> —©—o—o—®-
B to
o
5 CL o
t
l.Q-0-
Ul
t i
f * °X /
A---oy * '•r\ CXzzS—A—A.-- A--- ""A- DV Oy
I I I I I I I 10 20 30 40 50 60 70
FRACTION NUMBER
Figure 1. Sephadex chromatography of DNase-treated nucleic acids. — Nucleic acids were digested with DNase I, chromatographed on a G50 Sephadex column, and fractions collected as described in Materials and Methods. The radioactivity in eluent fractions was determined by counting in a scintillation spectrophotometer. Panel A shows the elution profile for purified, native DNA (s o) and for the same DNA after DNase treatment (o- - -o)«, Panel B depicts elution profiles for purified DNase-treated cellular nucleic acids, which were either labeled with 3H-adenosine for 2k hours ( • •) or 3jl-uridine for 120 minutes (A A).

Polysomal RNA Extraction
Cytoplasmic fractions isolated as described above were immedi
ately layered onto a sucrose sedimentation gradients Linear 15-30^
(w/v) sucrose gradients with RNase free sucrose in RSB were centrifuged
for 90 minutes at 200,000xg in a Spinco SW *f0 rotor at k°Ca Fractions
were collected from the bottom of the gradient through a continuous
flow cell in the light path of a spectrophotometer set at a wavelength
of 260 nm. Figure 2 shows typical tracing from one of these gradients
demonstrating typical polysome, monosome (80S) and ribosomal subunit
(60S and hOS) peakso Fractions representing the polysomes (up to, but
not including the monosome peak) were pooled, mixed with an equal vol
ume of 2X SDS buffer (SDS buffer is 0.1 M NaCl; 0.025 M tris, pH 7.^;
Q.OJfa SDS), precipitated with cold ethanol, and pelleted as described
before« The pellets were resuspended in a tris-EDTA-NaCl-SDS buffer
(0.01 M tris, pH lnM EDTA, 0.1 M NaCl, and 1% SDS) and extracted
by a method designed to minimize the loss from the polysomal fraction
of RNA's containing poly-adenosine tracts (Perry et alo, 1972). The
resuspended polysomes were extracted 3 times with an equal volume of
phenol-chloroform (1:1, v/v) saturated with 0.01 M Acetate, pH 6.0,
containing 0.1 M NaCl, and IrriM EDTA. Extractions included 5 minutes
of shaking on a wrist-action shaker followed by centrifugation at
10,000 x g for 5 minutes to separate the phases,, After the final ex
traction the aqueous phase was precipitated at -20°C with cold 9
ethanol and the precipitate was pelleted as before. The pellets were
resuspended in 2X distilled water, the concentration of RNA was

Figure 2. Sedimentation of the cytoplasmic fraction of chick cells on a sucrose gradient.
The cytoplasmic fraction of CEF cells, prepared as described in the text, was centrifuged on a linear 15-3^ sucrose sedimentation gradient at 200,000 x g for 90 minutes at k°C. The gradient was collected from the bottom through a continuous flow cell placed in the optical path of a spectrophotometer set at a wavelength of 260 nm. The tracing shown is a record of the absorbance at 260 nm exhibiting polysomal peaks as seen in a typical gradient.

21
80s
0.2
E c O CD CM
I-<
LLI O ;2: < CD CC O CO CD <
0
0.0
Pooled Polysomal Fractions
Bottom «s5- Top Direction of sedimentation
Figure 2. Sedimentation of the cytoplasmic fraction of chick cells on a sucrose gradient.

determined by optical density and the radioactivity measured by TCA-
precipitable counts. The RNA in these samples was concentrated by
ethanol precipitation and resuspension in 10X SSC. Radioactivity in
the samples was again determined by TCA precipitable counts.
DNA/DNA Reassociation
Reassociation analysis of denatured, pulse-labeled, host cell
DNA was performed by a modification of the original Britten and Kohne
(1967; 1968) procedure,, Reaction mixtures contained sheared, labeled
DNA and normal CEF DNA, phosphate buffer (PB: equal volumes of mono and
dibasic sodium phosphate, pH 6.8), and 0.2% SDS. The concentrations
of the excess, unlabeled DNA and the PB were of two values chosen to
readily reach low or high Cot values in short incubation times where
CQt is the initial concentration of the DNA (moles/Liter) multiplied
by the time of incubation (seconds) (Britten and Kohne, 1968). For low
2 CDt reactions (CQt <10 ) the excess, unlabeled DNA concentration was
p 0.5 mg/ml and PB was 0.12 M. High CQt reactions (CDt >10 ) on the
other hand, contained 5 mg/ml excess, unlabeled DNA and 0.'+ M PB. Mix
tures of the appropriate concentrations of PB and unlabeled DNA plus
labeled DNA (800-5,000 cpm/reaction mixture) were sealed in microcapil-
lary tubes (2.5-25 ML), placed in a boiling water bath for ten minutes
to denature the DNA strands, plunged into an ice-water bath, and incu
bated for the appropriate times at 60°Co Triplicate reaction mixtures
were removed from the 60°C water bath, terminated by rapid immersion in
an ice-water bath, and then stored at -20°C until assayed. The

resultant equivalent CQ s were calculated from the time of incubation
and the final concentration of total DNA present, corrected for rate
acceleration due to salt concentrations (Britten and Smith, 1970)•
Assay of DNA Annealing
The extent of reassociation was assayed by the single stranded
specific nuclease, SI (Ando, 1966) purchased from Sigma Chemical Co.
(St. Louis, Missouri)o This enzyme will digest single-stranded nucleic
acids and portions of duplexes that are not complementary (Sutton,
1971)« Before using the SI preparation, the enzyme was diluted 1:1
with a solution of 50/6 glycerol in SI digestion buffer (0.03 M Sodium
Acetate buffer, pH 5; 0.01 M ZnSO/^. and 10 /ig/ml denatured, calf
thymus DNA) and the activity was titrated with labeled single stranded
DNA. The concentration needed to completely digest ten times the DNA
present was determined and used in subsequent assays. Aliquots of the
enzyme were sorted frozen at -70°C until used.
Reassociation reaction mixtures were thawed and diluted into
1 ml of Sl-digestion buffer. The final concentration of PB present did
not exceed 2-h mM as greater concentrations inhibited the enzyme ac
tivity (Dinowitz, 1975c). Reaction mixtures were divided equally into
two 0.5 ml aliquots. One tube received an appropriate amount of SI
nuclease and the other served as a control. After digestion at 50°
for 2 hours, 0.05 mg/ml of calf thymus DNA carrier was added to each
tube followed by k ml of TCA. After 20-60 minutes incubation of the
mixture at ^°C, the contents of the tubes were filtered through Whatman

2b
glass filter fiber dies (GF/C), washed twice with cold 5% TCA, dried
and placed in 5 ml of organic scintillation fluid and counted in a
liquid scintillation spectrophometer. The percentage of double-stranded,
labeled DNA present in the various samples was calculated by dividing
the precipitable counts in the SI digested tube by those in the un
treated tube. Digestion of known, sheared, single stranded and native
DNA standards were included to monitor the SI nuclease activity.
RNA/DNA Hybridization
All hybridizations performed followed the essentials of a pro
cedure described by Neiman et al„ (197*0» These reaction conditions,
termed liquid hybridization in modest DNA excess, were chosen for their
ability to show nearly complete hybridization of a single-copy sequence
RNA molecule. Reaction conditions were designed to reduce loss of RNA
by degredation over long periods of incubation at elevated temperatures.
Neiman, Purchase and Okazaki (1975) found no loss of precipi
table CPM after 672 hours of incubation, but a halfing of the size of
the RNA every 168 hours. In this laboratory greater than 959^ of the
input radioactive RNA remained acid precipitable for 200 hours incuba
tion under these conditions. A trend in the loss of precipitable
counts was observed following this time (Fig. 3)» Extrapolation of
this curve indicates that 1/2 the input RNA would be acid soluble
3 after 1.9 x 10 hours of incubation. However, hybridization incuba
tions did not exceed 900 hours in any of these experiments.
Pulse-labeled RNA in 10X SSC was mixed with an equal volume of
normal CEF DNA which had been resuspended in formamide. Reaction

(3 2 2 < 5 UJ a:
1.0
q: UJ _i m <
S0.5 o LU a: a a o < 2 o i— a <0.0
.90 x I03hrs.
0 LOGj0TIMEOF INCUBATION (HOURS)
Figure 3. Lability of RNA during KNA/DNA hybridization. — Hybridization reaction mixtures made to a final concentration of 50^ in formamide and 5X SSC, contained equal volumes of cellular DNA (25.5 nag/ml) and radioactive RNA (60-300 fig/ml). The fraction of the initial labeled RNA remaining acid-precipitable is shown as a function of incubation time at 50°C.
ro VJ1

26
mixtures were drawn into micropapillary pipettes (2.5-5 /*L/pippet),
sealed, and denatured in a boiling water bath for 10 minuteso These
reaction mixtures were then incubated at 50°C for up to four weeks to
5 reach a Cct of 10 (Neiman et al., 197*0. Following the appropriate
incubations, replicate samples were removed from the 50° water bath,
immersed in an ice-water bath and stored frozen at ~20°C until a com
plete set was ready to assay.
Assay of RNA/DNA Duplexes
Hybrid formation was assayed by a modification of the method
described by Klein et al. (197*0 • Reaction mixtures were diluted into
1 ml of 2X SSC, split into equal aliquots with one tube receiving 10
pig/ml bovine pancreatic Ribonuclease A and the other serving as an un
digested control. The digestion mixtures were incubated at 22°C for
one hour. The method for precipitation and counting of the undigested
RNA hybrids was the same as that described for DNA/DNA reassociations.
As was previously reported (Neiman et al„, 1975; Klein et al., 197*0?
a fraction of input RNA is resistant to digestion under these con
ditions before hybridization. To account for this resistant fraction,
duplicate hybridization reaction mixtures were frozen unincubated fol
lowing the denaturation step. The percentage of labeled RNA remaining
in these samples after RNase digestion was subtracted from the values
obtained for the incubated samples.
Modification of RNase Assay
To obtain maximum digestion of RNA which had not hybridized
with DNA, several digestion conditions were tested. Various

27
combinations of RNase A and RNase T1 at 25°C or 37°C incubation for 1
hour were examined. The maximum digestion of non-hybridized cytoplas
mic RNA in solution was obtained using RNase A (10 fj.g/ml) plus T1 (10
units/ml) with incubation for 1 hour at 37°C (Table 1)» Hence, RNA/
DNA hybridizations in later studies were assayed using both RNases A
and T1 under the conditions just described. Unincubated hybridization
reaction mixtures digested by this scheme averaged 3-^9° resistant
counts which were subtracted from the percent hybrid found in incubated
samples.

28
Table 1, Comparison of digestion schemes for radioactive RNA.a
RNase A (10 jig/ml)
RNase T1 (10 u./ml)
RNase A (10 g/ml)+ RNase T1 (10 u./ml)
25°C/60 min. 9.1(3)b 91.5(*0 8.1(4)
k.k(k)
37°C/60 min. 6.5CO 5^.6CO 3.25(5)
a. Approximately 3i000 cpm/sample of cellular H-RNA in 2X SSC was treated with RNase(s) as shown. The average fraction of radioactivity remaining acid-precipitable after the treatments listed is expressed as a percent of the controlT incubated without RNase present.
b. The number of samples digested.

RESULTS
Cell Transformation
The time course for conversion of susceptible cells to tumor
cells by RSV infection was established for the conditions used in sub
sequent experimentso V/hen CEF cells were infected with RSV as de
scribed under Methods, individual rounded cells overlaying neighboring
cells were noted within 2b hours (Fig* *fA and B) . During the next 2b
hours these characteristic morphological changes became apparent for
all cells of the culture (Fig. and D). At this time control cells
had grown to confluent monolayers and the transformed cells had reached
high population density. The analyses of nucleic acids isolated from
infected cells reported below were done with cultures that were consis
tently transformed over the time course shown in Figure b.
Host Cell DNA Synthesis Following Infection
Host cell DNA, pulse-labeled after infection, was examined by
DNA/DNA reassociation analysis for differences in the relationship be
tween reiterated and unique sequences. If the proportion of these
classes of DNA synthesized were altered by infection, this change should
be evident in the reassociation kinetics of the cell DNA. Experiments
designed to explore the effect that RSV infection has on the relation
ship of newly synthesized repeat to single copy DNA are presented.
29

,{
30
ms-.: •
5 '• -V ••"•:;•••' - ; •
5 r v • ' . • " ; ; • ••
.v '• .*•
. \ \ • '• • • • o* »• , V V'V
•v; s:;
• • • :
W.
v-
-V T
\
. v ' Si-
'"v
. ;•
• " " ,\ . v,t, v -' v
: -• - • X. , A. . • *..,,y
\ ' •
•a ' ':v '•••}'•'
• ; :v;
• ' '• ''
"
Figure k. Morphological transformation of normal cells by RSV infection. — Confluent monolayer cultures of CEF cells were infected as described in Materials and Methods. Equivalent cultures 2*f hours after plating of uninfected cells (Panel A) and infected cells (Panel B) are shown. The same cultures are shown 50 hours after subculturing in Panel C, and infection in Panel D (mag. = V76X).

V.
31
ifOj/p : :V.
'• **' ' J • ' •
y J» .-..4 . tf , • > t . S • i . ' - - :
V 5 >"• • ' ' 'r W-.'t ; • :
• '>„• •' /'v y. • V - V.
D ' " r t ' ' ^ ~ yyyy'• L/ • .'• . •'••• "U'.~ : V---' '• - '-•••
• . -.'4 ? : '' ° y ,
> '•SsV :'>-v'. y ~. . ' •-'•'• i -
f; M.' .. -,•> •.-••; .: -6 ?>;.'• .-•••• - ••.•:•..•• w / ° . , ' ' • " • ;•'••••. ••••.' •••,,. ;>.i< '. •' .< J-1 ; •• ."•• v.'- ' 1 '"• '
-:.v. t f • ... '. - '••' • •
, v -
' / r - ' t ' . C , . V C I • >7, * !r," • '
-,-y •^'•ys^y.
;'' • !. '"-V* •••' '•,• .i'i "ft "r'V; . o...• • ' -"V/ • • -V y !
, •• ^ ••• : -t-. ^ J"" , >'%•«)* •
- • •• ' •--/ . r>- _ ~ ;>_ • .
Figure continued. Morphological transformation of normal cells by RSV infection.

Labeling of Host Cell DNA
The period of proviral synthesis and integration (3-12 hours
after infection; Varmus, Guntaka, Deng and Bishop (197*0 was chosen for
this study since it is during this period that the DNA copy of the
virus is synthesized and processed,, The possibility that a transient
alteration in host DNA synthesis may result from these interactions was
examined by pulse labeling the DNA«> Methods used for subculturing
cells resulted in partial synchrony in cellular DNA synthesis.. Since
expression of viral functions is host cell cycle dependent in infected
(Humphries and Temin, 197*0 and transformed cells (Leong, Levinson and
Bishop, 1972), processing and integration of the viral genome may also
be cell cycle dependent„ In such a case the partial synchrony induced
in the host cell cycle would be an advantage for isolating viral in
fluences on cellular DNA synthesis* To specifically identify host DNA
synthesized during short periods of the proviral phase, cells were
pulse-labeled with ^H-thymidine for 3 hour intervals beginning 3 hours
after infection, then harvested and pelleted for DNA extraction as
previously outlined- The preparations are all shown to be relatively
free of contaminating protein by their 260nm/280nm absorbance ratios
exceeding 1„8,, Results of these extractions demonstrate consistent
i s o l a t i o n o f p u r i f i e d D N A ( T a b l e 2 ) „
The partial synchrony introduced into DNA synthesis by sub
culturing CEF cells is a consistent finding under the conditions used
in these experiments® Partial synchrony in cellular DNA synthesis is
shown by the peak in specific activity seen for DNA labeled 6-9 hours

33
Table 2. Summary of pulse-labeled DNA extractions.
Time of Labeling (hrs.)
Control Cells Infected Cells Time of Labeling (hrs.)
^260nm A280nm
Specific Activity (cpm)
(fig )
^260nm A230nm
Specific Activity (cpm)
( (iS )
3-6 N.D.b NeDo 1.91 1.8%103
6-9 1.84 k
2.3^x10 1.88 1.05x10^
9-12 1.85 k
1.30x10 1.91 if.92xl05
12-15 1.86 6.11xl03 1.91 2.69xl05
15-18 1.82 5.70xlo5 N.D. N.D.
n a. RSV-infected and mock-infected (control) cells (1-2x10 cells/sample)
were pulse-labeled for 3 hour periods with 5^Ci/ml 3H-thymidine. Cellular 3H-DNA was isolated and extracted as detailed in Materials and Methods.
b. Not determined.

after plating (Table 2). These data indicate that the rate of DNA
synthesis increases then declines over the period examined,, A separate
experiment was performed to test the conditions which resulted in par-
3 tial synchrony0 Normal CEF cells were subcultured with H-thymidine
present in the media from passage until the cultures were harvested.
The rate of DNA synthesis for duplicate cultures was calculated from
the amount of DNA synthesized by the indicated harvest times (Fig. 5A).
3 Incorporation of H-thymidine into whole cell DNA increases until
approximately 10 hours post plating then declines to very low levels
by ifO hours. Under the conditions used here, the addition of fresh
serum induced new DNA synthesis in cells in which DNA synthesis had
stopped (Fig. 5A). The addition of fresh serum to cultures which were
no longer dividing has been previously shown to induce new rounds of
DNA synthesis and division (Temin, 1971b). Other experiments similar
to those reported in Table 2 and Figure 5A resulted in nearly identical
observations- These experiments were designed to further examine the
early period of DNA synthesis (i.e., the first 15 hours) following pas
sage of the cells. For cells pulse labeled with ^H-thymidine, both
whole-cell acid-precipitable radioactivity and the specific activity
of DNA isolated from these cells showed a peak in the rate of DNA syn
thesis 6-9 hours after plating (Fig. 5B and C). Whether or not the
cells were infected with RSV, the rate of DNA synthesis in the culture
was similar,. These results (Table 2, Fig. 5) demonstrate that in both
infected and control cells, partial synchrony in cellular DNA synthesis
occurs under the conditions used here to infect cells. This

Figure 5» Partial synchrony subculturing.
of cellular DNA synthesis following
Acid-precipitable radioactivity was measured in cells labeled with 3H-thymidine (^H-TdR) after passage and plating of the cells. Panel A shows the rate of DNA synthesis calculated from duplicate cultures of normal CEF cells where label (0.1 MCi/ml, ^H-TdR) was present continuously up to the time when cells were harvested. Panel B shows the rate of DNA synthesis for duplicate cultures of normal CEF cells (o o) and PSV-infected cells (e o), pulse-labeled with 3H-TCLR (5 Ci/ml) for the 3 hours prior to harvest of the cells. Panel C shows the specific activity of DNA from normal cells (A~ A) and RSV-infected cells (A A) isolated from the cultures described in Panel B.

3.0 - A
2.0
M I o
£ a. u
1.0 Fresh serum added
3.0 - B
0 2 0 - 4 0 6 0
Hours after plating cells
80 0 5 .10 15
.Hours after plating cells
0 5 10 .15
Hours after plating cells
< 2 O
U z a
o
t > o t- v
X < E y s-U_ w u lU CL <0
Figure 5. Partial synchrony of cellular DNA synthesis follov/ing subculturing.
VJ1

36
characteristic of subcultured CEF cells along with their response to
serum deprivation may contribute to some of the effects noted for host
nucleic acid synthesis.
DNA/DNA Reassociation Kinetics
The relationship between repeat and unique sequence cell DNA
synthesized following infection by RSV was examined by reassociation of
pulse-labeled DMA. If sequences of a particular reiteration were pref~
erentially synthesized at a given time after infection, this might be
seen in reassociation analyses of pulse-labeled DNAo The possibility
that infection might alter replication of DNA sequences occurring at
different copy numbers/genome was investigated in this manner,
Reassociation of the individual pulse-labeled control cell DNA
is shown in Figure 6. These reassociation curves demonstrate charac
teristic second order reaction kinetics in the high CQt (unique
sequence) region (Cot>200); completing the reannealing approximately
two decades after it begins. The unique DNA C0t^ (the CQt at which the
reassociation of unique sequences is half completed) varies between
•z 1.6-2.0 x 10 as calculated from the reassociation curves. In the low
CDt (repeat sequence) region (CQt of 1 to 50) reassociation is essen
tially completed before CQt < 1. The repetitive DNA which reassociated
before a CGt of 1 represented 9-1% of the labeled cell DNA. The dif
ferences noted between the reassociation kinetics of the DNA samples
pulse labeled at different times after mock infection are small as can
be seen in Figure 7•

Figure 6. Reassociation analyses of uninfected cell DNA.
Pulse-labeled ^H-DNA (55-120 jig/ml) was mixed with an excess of unlabeled normal CEF DNA (0.5 mg/ml for low CQt and 5 mg/ml for high C0t analyses) in liquid associ ation reaction mixtures. After denaturation, the 3h-DNA was allowed to reanneal at 60°C until the desired C0t values were obtained. The extent of reassociation was assayed by the single strand specific nuclease, SI, as described in Materials and Methods. Panel A: reassociation curves for DNA labeled 6-9 hours post-plating. Panel B: 9-12 hours post-plating. Panel C: 12-15 hours post-plating. Panel D: 15-18 hours post-plating.

—d-°-d—•-D DVd
X cl
6 - 9 H r s . p o s t p l a t i n g ( p . p . )
Log(0equivalent CQt
—Q.
B
9 - 1 2 H r s . p . p .
\
2 3 Logloequivalent CQt
a—
2 O
0
20
40
60
80 < o 0 100 CO , tn ' < UJ cc
1 0 a
z 20 UJ 0 £ 40 01
60
80
100
—D-0-
• u\
\ 12 -15 Hrs. p. p.
Log equivalent C.t 10 o
D " a
\
15 -18 Hrs.p.p.
Log|()equivalent CQt
Figure 6. Reassociation analyses of uninfected cell DNA.

50 CONTROL CELLS
6 - 9 H r s . p . i . 9 - 1 2 H r s . p . i .
12 -15 Hrs. p.i. 15 -18 Hrs. p.i.
100
LOG|0 EQUIVALENT CQt
Figure 7* Composite of control-cell DNA reassociation curves. — Curves from Figure 6 are shown together on a single graph for comparison.
oo

39
Reassociations of the DNA derived from cells infected with RSV
are shown in Figure 8. As noted for the control cell DNA associations,
all the infected cell DNA reassociation curves show second order reac
tion kinetics for single copy sequences,, CDty2 values for unique DNA
3 lie between lol-2o0 x 10 for the various pulse-labeled DNA's,, Similar
to the reannealing of control cell DNA, infected cell DNA demonstrates
very little additional reassociation between a Cot of 1 and 50o Rapidly
reassociating DNA increased from 6o5 to approximately 10„5 percent
from the earliest to latest pulse-labeling times. Data from Figure 8
have been combined in Figure 9 in order to show that as with the con
trol cell DNA, the differences observed between curves for each pulse-
labeling time are small„
Table 3 summarizes values for the kinetics of reassociation
from the curves of Figures 6 and 8C These reactions were kinetically
complete as demonstrated by the 75 to 85^ of the labeled DNA that re-
if association by sin equivalent CDt > 10 0 Small differences m unique
DNA C0ti£ and the fraction of repeat DNA reassociating were noted be
tween labeling timesa The percent reassociating as repeat DNA generally
increased with time, while the values for unique DNA Coty2 were about
3 1»5-1<>9 x 10 o These fluctuations in kinetics may be due to the
preferential labeling of specific sequences during the partial syn
chrony observed for DNA synthesis0 Differences in the temporal repli
cation of sequences represented at reiterated frequencies in the genome
might then be responsible for these variations,.

Figure 8. Reassociation analyses of RSV-infected cell DNA.
Pulse-labeled cell DNA(60-90 jzg/ml) extracted from CEF cells infected with RSV was mixed with an excess of unlabeled normal CEF DNA (0.5 mg/ml for low C0t analyses) in liquid reassociation reaction mixtures., After de-naturation, the 3H-DNA was allowed to reanneal at 60°C until the desired CQt values were achieved. The extent of reassociation was assayed by the single strand specific nuclease, SI, as described in Materials and Methods. Panel A: reassociation kinetics for infected cell DNA labeled 3-6 hours post infection. Panel B: 6-9 hours post infection. Panel C: 9-12 hours post infection. Panel D: 12-15 hours post infection.

2 O
0
20
40
60
80 •— < 0 8 100 cn i < UJ cr. <
1 0
2 20 UJ o £ 40 a
60
80
100
3 - 6 h r s . p o s t i n f e c t i o n ( p . i )
I i I
Log,, equivolent C t
_Q_
B
6 - 9 h r s . p . i .
Log|oequivolent CQt
o-~-o—
0
20
40
z 60 0 < 80 u to 100 CO { < UJ or <
1 0
z 20 UJ o £ 40 CL 60
80
100
9-12 hrs.p.i.
i
\ V
Log equivolent C.t 10 0
—O-O—O-
D
12-15 hrs.p.i.
V
\ On«o
Log|0equivalent CQt
Figure 8. Reassociation analyses of RSV-infected cell DNA.
s

0
50 INFECTED CELLS
3 - 6 H r s . p . i . 6 - 9 H r s . p . i . 9 - 1 2 H r s . p . i .
12 -15 Hrs. p. i.
100 _L
Figure 9«
2 LOG |0 EQUIVALENT CQt
Composite of infected-cell DNA reassociation curves. — Curves from Figure 8 are plotted together for comparison.

k2
Table 3« Summary of reassociation kinetics for pulse-labeled cellular DNA.a
Time of Labeling Unique DNA C 3^ oti/2 x 10 Percent Repeat DNAC
(hrs.) Control Cells Infected Cells Control Cells Infected Cells
3-6 N.D.d 1.15 N.D. 6.5
6-9 1.97 1.92 9.0 8.0
9-12 1.87 1.93 10.0 9.5
12-15 1.56 1.57 9.0 10.5
15-18 1.76 N.D. 15.5 N.D.
a. Parameters reported here for the reassociation kinetics of denatured cell DNA were determined from data in Figures 6 and 8.
b. Got at which 5Q?o of the unique sequences are reassociated. c. Fraction of DNA reannealed at C0t = 10. d. Not determined.

k3
The unique DNA C0ty2 changes less than 5^ in value from infected
to control cells for comparable labeling periods. While variations in
the percent of repeat DNA are greater during the same labeling times,
the absolute differences are quite small» Hence, both the single copy
DNA C0ty2 and the percent repeat DNA annealed change very little as a
result of RSV infection* These data suggest that synthesis and inte
gration of the RSV DNA provirus do not perturb host DNA synthesis
sufficiently to be measured by reassociation kinetics under the con
ditions employed here»
Analysis of Host RNA Synthesis Following Infection
Alterations in RNA synthesis seemed likely after infection,
since the synthesis of the RSV genome and new or altered proteins are
found in transformed cells* Possible viral effects on host RNA syn
thesis seemed most probable during the host's transition from a normal
to a transformed cello An understanding of the effects of virus in
fection on cellular RNA synthesis might yield information indicative
of the mechanism(s) by which cells are transformed. Since budding RSV
is first detected 18 hours after infection of chick cells and the cul
ture morphology is completely transformed by 30 hours after infection,
the period examined for transcriptional alterations was extended to
over 50 hourso During this period infected and control cells were
"T pulse labeled with H-uridine and the isolated RNA analyzed by RNA/DNA
hybridization. The relationship between repeat and unique sequence

kk
transcripts was examined in this manner to identify virus induced
changes in cell transcription.,
Labeling of Host Cell RNA
Transient differences in host cell RNA synthesis during infec
tion could be studied by pulse labeling RNA which would allow examina
tion of RNA synthesized only during the labeling period. Secondary CEF
3 were either mock infected or RSV infected and pulse labeled with H-
uridine for two hours at various times after replating the cells« These
pulse-labeled cells were harvested, fractionated into cytoplasmic and
nuclear samples, and nRNA was extracted as detailed in Materials and
Methods*
Results for two separate experiments of the extractions and
treatments described are shown in Table Approximately kO-5C$> of the
acid-precipitable whole-cell radioactivity was recovered after this ex
tensive purification.. The specific activity of RNA from the control
and infected cells of the same labeling period were comparable indi
cating that the rate of RNA synthesis in control and infected cells is
similar.. These data further demonstrate that the isolation and re
covery of nRNA from both control and RSV infected cells is consistent.
Overall, extraction yielded uniform nRNA that had a molecular weight
greater than 50,000.
Hybridization of Pulse-Labeled Host Cell nRNA
In order to investigate differences in the populations of RNA
synthesized in RSV-infected cells, the RNA was analyzed by hybridization

Table 4. Summary of pulse-labeled nuclear RNA extractions.a
Experiment 1 J Time Time j at Percent CPM Concentration Specific Activity at Percent CPIs
Pulse Recovered (fig/ml) (cpm x 10~b/ug) Pulse Recovered] (hrs) Control Infect,, Control Infect, Control Infecto (hrs) Control Infec
5 1+8 bz 15d 21.5 2.11 1o36 12 39 U
15 52 bz 29=5 17.3 1.02 1.56 2b 39 5<
25 53 3b *fl.5 29°7 lo28 lobo b6 39 55
50 67 50 b3ol 68.0 2.03 l.6l 58 b3 4C
n a. RSV-infected and control cells (1-2 x 10 cells/sample) were pulse-labeled
with H-uridine (50 Ci/ml) for 120 minuteso Nuclear fractions were isolated and the RNA extracted from them as detailed in Materials and Methods.
b. Limited recovery of RNA in void volume of sephadex column.

if5
ed nuclear RNA extractions.3
Experiment 2
tration /ml)
Specific Activity (cpm x 10-Vug)
Time at
Pulse Percent CPM Recovered
Concentration (ug/ml)
Specific Activity (cpm x 10 /j/g)
Infect. Control Infect. (hrs) Control Infect. Control Infect. Control Infect.
21 o5 2 oil 1«. 36 12 39 l^b 10.2 7 ok 9O60 ka66
17 °3 1.02 1o56 2k 39 50 16.3 1^.1 7«>78 10A
29<»7 1O28 lo^O k6 39 55 35«8 37®3 9<>22 llo9
68o0 2o03 I06I 58 ^3 ko ^3o2 59cl 3=91 3-72
7 s (1-2 x 10 cells/sample) were pulse-labeled or 120 minutes„ Nuclear fractions were d from them as detailed in Materials and
id volume of sephadex column.

46
with cell DNA® The kinetics of these RNA/DNA hybridizations defined
the classes of unique and repeat sequence transcripts and indicated the
relative concentration of repeat sequence transcripts,. Differences in
these parameters were sought between nENA from RSV-infected and control
cellSo
Several broad classifications are used here for RNA hybridizing
in these experiments., RNA which is presumed to be transcribed from the
unique DNA sequences (termed "unique" RNA) hybridizes with apparent
second order reaction kinetics at high Coto The RNA transcribed from
reiterated DNA sequences (called "repetitive" RNA) forms duplexes at
low Cot's (C0t = 1-100) and RNA which hybridizes below a CQt of 1 is
transcribed from highly repetitive DNA templates and will also be re
ferred to as "highly repetitive" RNA.
RNA/DNA Hybridizations of Control Cell nRNA
For hybridizations of pulse-labeled nRNA from control cells,
the representation of the three classes of RNA varies with the time at
which the nRNA was labeled (Fig® 10). "Highly repetitive" RNA is de
tected for nRNA labeled 5 and 25 hours after plating but very little
"repetitive" RNA is seen to hybridize from these samples (Fig® 10A and
10C). nRNA labeled 15 hours after plating shows "repetitive" RNA hybrid
ization but no "highly repetitive" RNA hybridization (Figo 10B)« The
RNA/DNA hybridization of nRNA labeled at 50 hours demonstrates hybrid
ization of both "highly repetitive" and "repetitive" RNA (Fig 10D)«
From a low of almost no detectable hybridization for "highly repetitive"

Figure 10. Hybridization of normal CEF nuclear RNA.
Nuclear RNA (75.5 to 215*5 ug/ml) from cells pulse-labeled 120 minutes with 50 MCi/ml H-uridine was mixed in equal volume with normal CEF DNA (25.5 mg/ml for high C0t reactions and 0.212 mg/ml for low CQt analyses) in liquid reaction mixtures containing a final concentration of 50/6 formamide and 5X SSC. The DNA/RNA ratio ranged between ' approximately 1-^ x 102 for high CQt reactions and between 3:1 to less than 1:1 for low 0ot samples. Reaction mixtures contained 2,000 to 10,000 cpm of labeled RNA. Following incubation at 50°C for times appropriate to reach the desired Cot values, duplicate reaction mixtures were assayed for RNase A-resistent hybrids. Hybridization curves for nRNA labeled 5 hours post-plating (Panel A); 15 hours post-plating (Panel B); 25 hours post-plating (Panel C); 50 hours post-plating (Panel D) are shown.

0
5
10
1 5
20
2 5
3 0
0
5
10
1 5
20
2 5
3 0
" °oo
- A \ o
5 hrs. post plating (p.p.) X
1 1 1 1 1 1
_ B \
ay
15 hrs. p.p. 9
o*-| I | 1 I |° | 1 1 1 1 1 1 1
o-O-Q-o—o—o^o 1 I 1 1 1 1 1 1
°\ \ _ c \ _ D o.
°\ \ 25 hrs. p.p. \o 50 hrs. p.p. \
\ J QyO
V -1 1 1 1 1 1
O^Q.
1 1 1 1 I I 1 I 0 I 2 3 4 5 - 1 0 1 2 3 4 5 6
log C t 10 0
Figure 10. Hybridization of normal CEF nuclear RNA.
•P--n3

^8
nRNA labeled at 15 hours, a maximum of 2 percent of the input RNA
hybridizes for RNA labeled at 50 hours- These extremes are closely
coincident with the rates of maximum and minimum DNA synthesis observed
with increasing time after plating of the cells (Fig. 5)° "Repetitive"
RNA hybridizing by a CQt of 10 shows similar temporal differences
varying between 1 and 3 percent of the input RNA labeled at these
time So
The "unique" RNA hybridization varies very little from one
labeling time to the next hybridizing with apparent second order reac
tion kinetics for each sample„ Estimates of the C0ty2 values for the
3 RNA annealings (approximately 2.5 x 10 ) are consistent with kinetics
for unique sequence transcripts. The total fraction of "unique" nRNA
hybridized was between 18 and 23 percent for the different RNA samples.
These differences may represent small changes in the fraction of the
genome transcribed as discussed below.
Data presented for control cell nRNA hybridizations indicate
two points about transcription in these cells. The "unique" RNA for
hybridizations of nRNA labeled at various times after passage are rela
tively uniform and consistent. Both "highly repetitive" and "repeti
tive" nRNA synthesized during different periods show a small increase
with time after subculturing. During active cell growth these frac
tions of the RNA account for a smaller portion of the newly transcribed
nRNA than during the late, stationary-growth phase.

k9
Infected Cell nRNA
RSV-infected cells pulse-labeled with "^H-uridine as described
in Materials and Methods, were fractionated into cytoplasmic and
nuclear fractions and the nRNA was isolated,, Labeled nRNA was allowed
to hybridize to cell DNA in liquid hybridization reaction mixtures
identical in reactants and conditions to those of the control cell nRNA
annealings. Results of these hybridizations are shown in Figure llo
The data demonstrate a pattern similar in several ways to that seen for
control cell nRNA hybridization., The "unique" RNA hybridization kinet
ics Eire very much alike for all time periods examined® Estimated Cotyz
3 for "unique" RNA hybridizations were approximately 2„5 x 10 « The
portion of total nRNA hybridized in the unique sequence region of these
curves ranged from approximately 20 to 2b percent0
The fraction of the nRNA which hybridized as "repetitive" RNA
increased with time after infection, as did the nRNA from control cells.
For the first two time points (5 and 15 hours post-infection) the same
fraction of nRNA represented by "highly repetitive" and "repetitive"
RNA are seen to hybridize as with control cell nRNA« Repeated sequence
RNA found in RSV infected cells with increasing time after infection
while following the trend of control cell nRNA hybridization — in
creased beyond that observed in the uninfected controls during the
period 25 to 50 hours after infection,,
The data for "unique" RNA are compared in Figure 12 with those
of the control cell nRNA hybridizations in order to demonstrate their
similarities. Hybridizations of unique sequence transcripts for each

Figure 11. Hybridization of BSV-infected cell nRNA with cell DNA.
Pulse-labeled nRNA (76.5 to 3^0 fig/ml) was mixed with equal volumes of normal CEF DNA (25-5 mg/ml for high C0t reactions and 0.212 mg/ml for low CQt analyses) in liquid reaction mixtures. These reaction mixtures, the hybridization condition, and the assay procedure were all identical to those described in the legend to Figure 10. Hybridization curves are shown for nRNA labeled 5 hours post infection (Panel A); 15 hours post infection (Panel B); 25 hours post infection (Panel C); 50 hours post infection (Panel D).

0
5
10
15
20
25
30
0
5
10
15
20
25
30
Q-o- -o—o—O-
5 hrs. post infection (p.i.)
CT
15 hrs. p.i.
-QJ
25 hrs. p. i 50 hrs. p. I.
J L J I L 12 3 4 5
'°5,(P0t
0 I 2 3 4 5
'OflKpO*
re 11. Hybridization of RSV-infected cell nRNA with cell DNA.

CD 10
5 hrs. p.i 25 hrs.p.i
m
15 hrs. p.i. 50 hrs
l°9,oCot
Figure 12. Hybridization kinetics for unique sequence transcripts from RSV-infected and control cell nRNA„ — The unique sequence regions (between a CQt of 102 and 105) of the hybridization curves for control cell nRNA (o——o) from Figure 10 and those of RSV-infected cell nRNA (o- - -©) from Figure 11 are shown together for comparison of similar pulse-labeling times.

of the times assayed are not readily distinguishable0 The small dif
ferences in the total amount of RNA hybridized noted earlier can also
be identified, but no consistent differences between normal and in
fected cells have been observed,,
To demonstrate the differences between control and infected
cell "repetitive" nRNA hybridizations, the results from control cell
hybridizations (Fig. 10) and those from infected cells (Fig. 11) are
combined in Figure 13° These differences are seen at late times after
infection when transformed morphology becomes recognizable for infected
cellso Nuclear nRNA labeled 5 or 15 hours after infection or plating
hybridized with similar kinetics» Hybridizations of nRNA synthesized
25 or 50 hours after infection, however, diverge from the results
obtained with normal cells. During this period when the transformed
character of the culture is established, the fraction of "repetitive"
RNA in infected cells increases over that observed in uninfected cells
by more than twofold.
In order to verify the differences between repeat sequence RNA
transcribed in control and infected cells, a second, independent ex
periment in which cells were mock or RSV infected and cellular RNA
pulse-labeled was carried out. The method of infection, RNA labeling,
cell fractionation, nRNA extraction, and RNA/DNA hybridization were all
done as before. In this second experiment the times of pulse-labeling
were chosen to examine the periods shortly after infection when no dif
ferences in control and infected cell nRNA hybridizations were seen,
late after infection when differences were observed, and very late

© O O ' O Q
25 hrs.p.i 5 hrs. p. i.
O-Q-O
15 hrs. p. i. 50 hrs. p.i.
'° ioCo*
Figure 13® Hybridization kinetics for repeat sequence transcripts from RSV-infected and control-cell nRNA. — The repeat sequence region (between a Cot of 10"- and 10 of the curves for control cell nRNA hybridizations (o o) from Figure 10 and those of RSV-infected cell nRNA hybridizations (©_ - -o) from Figure 11 are shown together for comparison of similar pulse-labeling times.
vn

5*+
after infection when both infected and control cells were rarely divid
ing. nRNA hybridizations of infected and control cells from the same
labeling times are shown together in Figure l*fo As before, nRNA from
the earliest time period examined (12 hours) demonstrated similar
kinetics for low CQt hybridizations, while during the 2k~b6 hour inter
val nuclei from infected cells again exhibited a larger fraction of
newly-synthesized "repetitive" RNA than did control cells. By ^6 hours
after infection a maximum difference in the hybridization curves for
infected and control cell RNA is evident. Analysis of nRNA synthesized
at 58 hours after infection indicated that RNA from infected cells con
tained the same fraction of repetitive sequence transcripts as controls,
although the sequences transcribed may not be the same as those synthe
sized in control cells.
This independent repeat of the earlier experiment confirmed the
original observations,, Data from these experiments are summarized in
Figure 15 where the fraction of nRNA hybridized by a CQt of 10 for each
of the RNA annealing curves (Figs. 13 and 14) are shown. Between 2b
and 50 hours after infection the concentration of "repetitive" RNA in
infected cells is greater than controls. The difference in the frac
tion hybridized by C0t = 10 reaches a maximum by 50 hours and then
declines by 58 hours after infection.
Constant DNA/RNA Ratio Comparison Hybridizations
Since the repeat DNA sequences occur at different frequencies
and their transcripts occur at unknown frequencies, specifying the mass

Figure lA- Low CQt hybridizations of control cell and RSV-infected cell nRNA.
Pulse-labeled nRNA (70.5 to 275 from a second, independent experiment (see the legend to Table k) was mixed with an equal volume of normal CEF DNA for liquid hybridization, and assayed under conditions described in the legend to Figure 10. Low CQt hybrids formed with nRNA from control cells (o o) and from RSV-infected cells (©- - -©) from the same labeling times are shown.

5 o cr 00 >
5 io < 2 cc
< z Q
1-z UJ o cc UJ Q.
0
10
o S o © ° _o 0
• •
12 hrs.p.i.
A
• < t
X cr^Q. *© ®***«».
© «°
46 hrs p.i. ®
c
1 ! 1 0J 1 I ©X 1 ' '
Gl>—? o ® 'O-® S ° ©•» _ ©
©®^8
24 hrs.p.i. 58 hrs.p.i.
B
i i i
D
i i i
Figure l*t. Low C0t hybridizations of control cell and ESV-infected cell nRNA.

56
O LLJ N
Q
CT CD >-
or
h-"Z. LiJ O £T LJ CL
7.5
5.0
2.5
0
/
/
/A / /
/4 I / \
\ \
o JA
03
o
0 20 40 60 TIME AT LABELING (HOURS)
Figure 15. The fraction of nRNA hybridized by a CDt of 10 for control and RSV-infected cells. — The percent of nRNA found in hybrids at a CQt of 10 in the curves of Figures 13 (circles) and Ik (triangles) are shown for both control (open symbols) and RSV-infected cells (closed symbols).

57
ratios of DNA to RNA does not indicate the sequence ratio for particu
lar DNA sequences and their transcripts,, If in general the "repeat"
RNA hybridizing is less concentrated than the repeat sequences from
which they were transcribed, all the RNA should hybridize. However,
it may be possible that the RNA copies exceed in number their templates
even at high DNA/RNA mass ratioso Hence, it is necessary to specify
some constant DNA/RNA ratio for comparison of hybridizations<> To rule
out the possibility that a variable DNA/RNA ratio might account for
some of the differences observed, hybridizations in which the DNA/RNA
ratios were held constant were performed.. Although the actual sequence-
to-transcript ratio was indeterminant, a constant DNA/RNA mass ratio
implied that the extent of DNA saturation by RNA is consistent for each
hybridization,, nRNA was diluted to a constant concentration and
hybridized to normal CEF DNA to a CQt of 14-16. The hybrids formed
were assayed using the complete digestion conditions provided by the
combination of RNases A and T1 as described in Materials and Methods®
Results of constant DNA/RNA ratio hybridizations are shown in
Figure l6A. These findings confirm the pattern previously seen,, Con
trol cell nRNA labeled at different times varies slightly in the frac
tion of "repetitive" RNA hybridized. Hybridization of pulse-labeled
nRNA from infected cells increases with time and this increase again
surpasses the normal cells in the period 25-50 hours after infection.
Arithmetic means for the percent of infected cell nRNA hybridized for
both the k6 and 50 hour time points are statistically different from
control cell values at the 99% confidence level (t = -5.56 and -7-56

Figure 16. Hybridizations of nuclear RNA at a constant DNA/RNA ratio.
Nuclear RNA was diluted to a constant concentration (50 /ig/ml) and hybridized with an excess of normal cell DNA to a CQt of 1^ or 16 as described in Materials and Methods. Quadruplicate samples were assayed by digestion with both RNases A and T1 and the mean values for remaining hybrids plotted. The RNA used in this experiment was the same as used for the experiments described in Figure 13 (square symbols) and Figure 1^ (circles). Panel A shows the percent of control cell nRNA (open symbols) hybridized and that of RSV-infected cell nRNA (closed symbols) hybridized for the labeling times indicated. Panel B shows the ratio of hybridization of infected cell nRNA to control cell nRNA.

58
< Zio cr —
< •*-<> 2: O O •}—
S s o »-
_Q CD >>
Q_ X
5.0 - A
2.5
0
*o <D N
-Q >» X
or c
CD O
T3 CD
O a>
"O CD M
-g
-Q >* X
cc c
"a5 o
c o o
3.0
0 20 40 60 Time After Infection (hrs.)
- B
2.0
1.0
0
- •
0 20 40 60 Time After Infection (hrs.)
Figure 16. Hybridizations of nuclear RNA at a constant DNA/RNA ratio.

respectively)0 The ratios of the fraction of RNA hybridized from in
fected cell RNA to control cell RNA are shown in Figure l6B« A two
fold increase is observed for the fraction of "repetitive" RNA found
in the nuclei of cells infected with RSV during the 25-50 hour period
after infection — a time when the majority of the cell population is
becoming morphologically transformed.
Polysome-Associated "Repetitive" RNA
Alterations observed in the newly-synthesized "repetitive" nRNA
of infected cells raised an immediate question: could changes also be
seen in mRNA, a putative product of nRNA processing (Greenberg, 1975)?
Since polyribosomal associated RNA (pRNA) is representative of the
mRNA population actively being translated, this RNA was examined for
differences in the representation of "repetitive" transcripts,, Cyto
plasmic fractions from infected and control cells were centrifuged on
sedimentation gradients to isolate polyribosomes <> RNA extracted from
the pooled polysomes was then analyzed by means of low CQt RNA/DNA
hybridization.
Labeling and Extraction of pRNA
During the period when infected cells are converted from normal
3 to transformed characteristics, cells were pulse-labeled with H-
uridine for 120 minutes and the cytoplasms were prepared and layered
onto sucrose sedimentation gradients. The sedimentation of the cyto
plasmic fractions routinely yielded absorbance (260 nm) profiles

6o
demonstrating intact polyribosomes (Fig. 17). Separation of polyribo
somes allowed isolation of RNA sedimenting with diribosome complexes
or faster., Approximately 20 percent of the total acid-precipitable
radioactivity present in the cytoplasm was isolated from the polyribo
some region of the gradients (Table 5)° Nearly complete recovery of
this pRNA followed the phenol-chloroform extraction designed to retain
polyadenylated RNA molecules by minimizing their loss in the organic
solvent (Perry et al0, 1972)„ Table 5 shows that the resulting specif
ic activity for infected and control cell pRNA from comparable label
ing periods are similar.
Low Opt Hybridizations
pRNA samples were adjusted to a constant concentration and
hybridized with excess normal cell DNA to a CQt of 1^-16. The results
from separate experiments show that early control cell pRNA hybridizes
to about Zfoo For later times this percentage drops to 1-1.5% hybrid
ized. The infected cell pRNA follows and amplifies this trend.
Hybridization of "repetitive" pRNA initially represents 2% of the pRNA,
then falls to approximately 0.5% for the next kO hours. Fifty-eight
hours after infection this trend is reversed and control pRNA hybrid
ized by a CQt of l*f-l6 surpasses that of infected cells0
The relationship between the fraction of control and infected
cell pRNA hybridizing is shown in Figure 18. At the earliest time
examined (5 hours), control and infected cells hybridize very similar
fractions of their pRNA. By 12 hours after infection hybridization of

Figure 17. Sedimentation profiles of cytoplasmic fractions from control and RSV-infected cells.
Cytoplasm isolated from infected and control cells from 12 to 58 hours after infection were centrifuged on 15-30^ sucrose gradients as described in the legend to Figure 2. The gradients were collected from the bottom through a continuous flow cell placed in the optical path of a spectrophotometer set at a wavelength of 260nm. Tracings at A260 are shown for the different samples. Panel At sedimentation profile of control cell cytoplasmic fraction labeled 12 hours post plating. Panel B: sedimentation profile of infected cell cytoplasmic fraction labeled 12 hours post infection. Panel C: sedimentation profile of control cell cytoplasmic fraction labeled ^6 hours post plating. Panel D: sedimentation profile of infected cell cytoplasmic fraction labeled k6 hours post infection. Panel E: sedimentation profile of control cell cytoplasmic fraction labeled 58 hours post plating. Panel F: sedimentation profile of infected cell cytoplasmic fraction labeled 58 hours post infection.

DIRECTION OF SEDIMENTATION
Figure 17. Sedimentation profiles of cytoplasmic fractions from control and RSV-infected cells.

Table 5* Summary of pulse-labeled polyribosomal RNA extractions,
Experiment 1 Time at Pulse (hrs) Control Infect,
Percent CPM Recovered
Time Concentration Specific Activity at
(fig/ml) (cpm x 10-3//jg) Pulse Control Infect. Control Infect. (hrs)
Percent d Recovertj
Control lnf(
5 12 16 93 78 0o4l o.6l 12 7
25 16 16 121 100 0.76 1.04 46 22
46.5 18 21 110 114 l044 1.55 58 21
n RSV-infected and control cells (1-2x10 cells/sample) were pulse labeled with 3H-uridine (50 MCi/ml) for 1-20 minutes. Cytoplasmic fractions were prepared and the polyribosomal RNA was isolated on sedimentation gradients (see legend to Fig. 17) and extracted as described in Materials and Methods.

9 62
beled polyribosomal RNA extractions»a
L 1
Experiment 2 Time
;entration Specific Activity at Percent CPM Concentration Specific Activity Ifig/ml) (cpm x 10-3/ur) Pulse Recovered (/ig/ml) (cpm x ! 10-3/ug) :ol Infect. Control Infect. (hrs) Control Infect. Control Infect, Control Infect.
5 78 O.kl 0.61 12 7 13 170 102 1=07 3*72
: 100 0.76 1.0*f ke 22 23 215 297 2.16 2.19
) Ilk lokk 1.55 58 21 25 168 269 2.89 2.55
h ells (1-2x10 cells/sample) were pulse labeled ) for 120 minutes„ Cytoplasmic fractions were lomal RNA was isolated on sedimentation gradients .d extracted as described in Materials and
I $

63
"D <D M
jg
-Q > n
< ;z or Q_
<D CJ
~o CD
*+— O <D
"O <D N
jg
_Q
X
1.5
.0
a: CL
3 0.5 "o c o o
0
0 20 40 60
TIME AFTER INFECTION (HRS.)
Figure 18. Ratio of infected to control cell polysomal RNA hybridized at a CDt of 15® — Pulse-labeled pRNA isolated from the polyribosomal fractions of RSV-infected and control cells was diluted to a constant concentration (250 fig/ml), hybridized with excess cell DNA to a Cot = 15, and assayed under stringent conditions as described in the legend to Figure 16. The ratio of the percent hybrid for infected-cell pRNA to that of control-cell pRNA is shown for RNA from two separate experiments (Experiment 1 - open symbols; Experiment 2 - closed symbols).

6k
pRNA from infected cells is reduced to less than half that of controls.
This difference continues during the period of transformation of in
fected cells from 12-^8 hours® Late after infection when all the cells
are transformed and their population density is very high this differ
ence diminishes.
Post-Transcriptional Processing of "Repetitive" RNA
Data presented so far indicate several interesting points about
newly synthesized "repetitive" RNAo While the fraction of repeat se
quence transcripts from control cell nuclei increases slightly with
time after plating, the polysomal associated "repetitive" RNA decreases.
RSV infected cells also follow but accentuate this trendo Infected
cell nuclei have a larger fraction of "repetitive" RNA than controls
while the "repetitive" RNA from polyribosomes represent a smaller frac
tion than controls,, These results from Figures 16B and 18 have been
combined in Figure 19 to show the virus induced alterationso The in
crease compared to controls in "repetitive" RNA from the nuclei of
infected cells has been divided by the fraction found on infected-cell
polysomes to yield the ratios shown* This index suggests an alteration
in the post-transcriptional processing of these repeat sequence tran
scripts due to the infection by RSV„ During the cells' conversion from
normal to transformed the appearance of the expected proportions of
"repetitive" RNA in the infected cell cytoplasm was inhibited.

Figure 19. The relationship between the polysomal and nuclear "repetitive" RNA fractions as a function of infection.
RSV-induced changes in the representation of repetitive sequence transcripts on the polyribosomes compared with that in the nucleus of infected cells is shown. The ratio of infected to control pRNA concentration of repetitive sequence transcripts (from Fig. 18) has been divided by the same ratio for nuclear RNA (from Fig. 16B) to yield these values plotted against the time at which the RNA was labeled.

65
o> D> c c *N N
"O jo "l— X3 JD >» >»
JZ sz
< < ;z
(Z £T CL c
"<D "<D > >
•4— cu 0) CL CL <D CD
%-
"o3 ~c5 o o o o i— c c o o o o o o •H
•O "O <D CD
-J— o o CD CD 4— «+-c c H— o o o o
'•*—
a D tr CL
2.0
1.0
0.5
0.0 0 20 40 60
Time after infection (hours)
Figure 19. The relationship between the polysomal and nuclear "repetitive" RNA fractions as a function of infection.

DISCUSSION
The results presented in this study indicate that some tran
scriptional controls in CEF cells may be influenced by both the growth
state and RSV infection,. Transcription of repetitive sequence DNA
increased twofold in cells as a result of infection by RSV. Changes in
cellular DNA synthesized after infection were not identified when
measured by DNA/DNA reassociation techniques- As shown here, these
observations correlate with the transformed state of the cells.
DNA/DNA Reassociations
Theoretical and Practical Aspects
DNA/DNA reassociation has proved a powerful tool for investi
gating the organization of the eukaryotic genome. Data generated by
this technique have demonstrated a high degree of complexity for the
structure of the genome of higher organisms. The assay consists of
shearing chromosomal DNA to small fragments and melting the double
helices into their component single strands. These molecules are then
incubated to allow reannealing through complementary base pairing be
tween the two strands. The extent of reassociation is most accurately
measured by exhaustively digesting single stranded molecules or por
tions thereof and determining the resistent double stranded fraction.
In the ideal case reassociation of DNA is a second order reaction
dependent on the concentration of the two reactants (the single
66

stranded molecules) and the time of incubation.. Britten and Kohne
(1967) used these values to create standard measurements of the extent
of the reaction by multiplying the initial concentration of the total
DNA (Co) by the incubation time (t) C0t0 The unexpected result of
Britten and Kohne®s (1968) experiments was the demonstration of multiple
copies of some DNA sequences present in the genome of eukaryotes® They
g found repeat sequences occur from a few to 10 copies per genome, repre
senting families of similar but not necessarily identical base compo
sitions The occurrence of multiple copies of nearly identical sequences
has also been shown for ribosomal DNA (Moore and McCarthy, 1968) and
some RNA tumor-virus integrated genes (Gillespie and Gallo, 1975)°
Investigating reassociations of repetitious DNA lead to the demonstra
tion of the interspersion of these sequences amongst the unique se
quences of the chromosome (Davidson and Britten, 1973)° This arrange
ment of repeat DNA has also been shown by Kleinschmidt electron
microscopy for reassociation fragments of DNA (Davidson et alo, 1973) <•
The presence of repeated sequences in the DNA raised questions
about their function.. Very highly reiterated DNA (satellite DNA) from
mouse cells is composed of tandem repeats of 300 nucleotides which are
not transcribed (Flamm, Walker and McCallum, 1969)0 These sequences
have been hypothesized to have a "housekeeping" function in maintain
ing the structure of the chromosome (Waiker, Flamm and McLaren, 1969)®
Other repetitive sequences, whose functions remain unknown, are tran°
scribed, though a smaller fraction of these transcripts are found in
the cytoplasm than in the nucleus (Greenberg, 1975)• Britten and

Davidson (1969) first proposed that repeat sequences represented
address sites which were differentially activated for control of growth
and differentiation,, This theory postulates that either sequence-
specific proteins or RNA binds to the address sites (repetitive DNA)
resulting in transcription of the associated unique sequences (David
son and Britten, 1973)o Their model allows for elaborate coordinate
and integrated transcription of physically-separated sequences through
these moleculeso The possibility of regulatory repeat sequence tran
scripts is particularly interesting in light of the finding of in
creased "repeat" RNA in infected cells reported in this investigation.
The presence of repeat sequences in animal cell genomes is seen
in the DNA renaturation assay at low Cots. Before the unique sequences
have had an opportunity to reanneal, repetitive sequences reassociate
due to their relatively higher concentration.. The fraction of repeat
sequence DNA varies from one eukaryotic genome to another (see Britten
and Kohne, 1968), amounting to about 25-30% of the total for chick DNA
(Rosen, Liarakos and O'Malley, 1973; Schincariol and Joklik, 1973;
Neiman, 1972). DNA/DNA reassociation assays are useful for character
izing both the fraction and redundancy of these repeat sequences. This
technique can also measure the kinetic complexity of the unique se
quences (Davidson and Britten, 1973)»
A practical consideration germane to this study is the method
of observing these different sequences. If hyper-chromicity of DNA
or the duplex formation of uniformly labeled DNA are followed during
renaturation, then the entire polynucleotide population will be

69
monitored. However, pulse-labeling during DNA synthesis results in
incorporation of radiolabeled precursors only into DNA sequences repli
cated during the labeling period,, For a population of cells, only
those cells synthesizing DNA and of these only the specific sequences
replicated will be labeled with a radioactive isotope0 Hence, in a
DNA/DNA reassociation where pulse-labeled DNA is assayed, only the re-
association of radioactive sequences will be monitored,, Such assays
measure the relationship between repeat and unique DNA that is synthe
sized during the time of pulse labeling.
Observations and Interpretations
The reassociations of pulse-labeled cellular DNA from both in
fected and control cells show very similar kinetics (Figs. 7 and 9)*
For those samples where both normal and RSV infected cell DNA was
examined, the fraction of repeat DNA reassociation varied no more than
lo% (Table 3)° Unique sequence DNA reassociated with comparable
kinetics varying no more than 3^ in their CQtyz values for similar
labeling times. Differences noted between the various labeling pulses
for these measures are relatively small and were not further investi
gated. It may be that they are a function of both the induced partial
synchrony in cellular DNA synthesis and the differential labeling of
sequences during the pulse. These results imply that the cellular DNA
synthesized in RSV infected and control cells are very similar when
assayed by DNA/DNA reassociation techniques.

70
RNA/DNA Hybridizations
Theoretical and Practical Aspects
In order to interpret data presented in this study and the
findings derived by different means by other investigators, a general
discussion of hybridization techniques illustrating the power and
limitations of each of the various methods will be useful«,
Gillespie and Spiegelman (1965) developed the filter-bound DNA
hybridization technique which was widely used prior to the advent of
the liquid hybridization procedure,, Denatured DNA was bound to nitro
cellulose filters which were then immersed in solutions of radioactive
RNA. Following incubations of 18-2^ hours or longer, these filters
were washed, treated with RNase and counted for radioactivity remain
ing in RNase-resistent RNA/DNA hybridso When increasing amounts of RNA
were present in the hybridization solutions, the number of counts
hybridized to the DNA reached a maximum that did not increase with
greater quantities of RNA. The plateau which resulted was interpreted
to mean that all available DNA sites were saturated„ By knowing the
specific activity of the input RNA, the amount of RNA bound can be cal
culated and from that the amount of DNA saturated by RNA is calculable0
(The percent DNA saturated is also defined when the amount of filter-
bound DNA remaining is known*) Although not appreciated at the time,
these plateau values reflect the complexity of the DNA transcribed;
i.e., the total number of DNA sequences which were used as template for
RNA synthesis. However, early experiments suffered from two central

problems; a reduced specificity of hybridization and an inability to
measure unique sequence transcripts (McCarthy and Church, 1970). The
low temperature and high salt concentrations used resulted in less
stringent homology requirements for hybridization. The relatively
short incubation times and low RNA concentrations employed were gener
ally sufficient to allow only repeated sequences to hybridize, thereby
exacerbating the problem of mismatch. Early plateaus were probably
achieved due to the kinetic separation between hybridization of repeat
sequence and that of unique sequence transcripts,. For example, calf
thymus DNA repeat sequences have completely reassociated three decades
of Cot before unique sequences begin to anneal (Britten and Kohne, 1968).
Still, this technique is effectively used today when the limitations
just described are carefully considered in designing and interpreting
the experimentSo
A more sensitive method has been developed for analyzing total
genomic transcription.. High specific-activity, denatured DNA is mixed
with a vast excess of unlabeled cellular RNA in a liquid hybridization
mixture (Kohne, 1968). DNA can hybridize with the RNA much more readily
than it can reanneal with itself because the RNA is present in higher
concentration than the DNA. When the reaction is complete, i.e., when
no more DNA will hybridize, the percent of DNA in DNA/RNA hybrids is a
measure of the total portion of the genome transcribed. While results
from this type of procedure are easier to interpret than the filter
method described above, they also present problems. First, the method
is not very sensitive to small changes. A differential transcription

72
of a percent or less of the genome would be difficult to monitor by
this method, although it may represent a major alteration in the RNA
present in the cello This is true for two reasons* First, a one per
cent difference in the genomic transcription represents a large number
of sequenceso For the chicken chromosomes this fraction would involve
expression of approximately 1„3 x 10"^ daltons out of the total 1®3 x
12 10 daltons of genetic information (Mirsky and Ris, 1950)„ Secondly,
the technique is relatively insensitive to the copy number of a tran
script since only a few copies are necessary to form hybrids with the
7 limited amount of DNA» For example, if 10 copies of these sequences
2 were present in one cell and 10 in another, both cases would likely
result in no differences in the final percent DNA hybridized,, Both of
these limitations are found in filter-bound DNA saturation experiments
where RNA complexity is also assayed,.
An additional theoretical limitation of DNA/RNA hybridization
using excess unlabeled RNA and labeled DNA is pertinent to this study0
The use of unlabeled, whole cell RNA combined with the fact that in
some cells 1/3 of mRNA's have an average half-life of 24 hours (Singer
and Penman, 1973) reduces many results to steady-state (or time average)
considerations,. This is a direct outcome of the inability to specify
at what point the RNA was synthesized., When all the above information
is weighed, total cellular RNA isolated from cells differing 24-48
hours in age may show a similar percent of labeled DNA hybridized
though the instantaneous transcription may be very dissimilar in these
cells of differing ages.

73
RNA/DNA hybridization, where the RNA is labeled and the DNA is
in excess, is a technique that has been used recently to examine the
hybridization kinetics of RNA present in eukaryotic cells (Gelderman,
Rake and Britten, 1971; Melli et al», 1971)o Employing model systems
for this type of hybridization, Bishop has shown that the reaction
kinetics of the labeled RNA faithfully reflect the kinetic complexity
of the DNA from which it is transcribed (Bishop, 1972)o Since he ob
served a slower rate for RNA hybridization than DNA reassociation, how
ever, the C0ti/2 for hybridization of RNA transcribed from a particular
DNA is greater than the C0ty2 for the reassociation of that DNA. Hence,
a known standard must exist to relate any given CDty2 to the absolute
complexity of the DNA from which the RNA is transcribed.
The most outstanding drawback to this technique is the limit
to the total amount of RNA that will hybridize. While Bishop's model
systems show 10C$ hybridization, in hybridizations of eukaryote cellu
lar RNA to DNA, 25-50?o of the total RNA hybridizes, depending on the
method of assay and the cell system used (Goldberg et al., 1973; Klein
et al«, 197^; Campo and Bishop, 197*0• Evidence reported by Goldberg
et ale (1973) and Smith et al. (197*+) supports the hypothesis that the
extent of hybridization is limited by the extremely high number of
copies of some unique sequence transcripts. Their data indicate that
a large portion of the total RNA is composed of greater than 10^ copies
of these unique sequence transcripts.

7^
Observations and Interpretations
In the study reported here, unique sequence transcripts showed
similar hybridization kinetics for both control and infected cell nRNA
(Figo 12)o Using a different technique, Grady and Campbell (1975a)
assayed transcription in both normal and polyoma transformed 3T3 cells.
By hybridizing labeled DNA to unlabeled nRNA in excess, they found a
small difference (1%) in normal cell transcription as a function of the
growth stateo They deemed this insignificant when compared to the 50%
increase over controls of transformed cell transcription regardless of
the growth state or cell population densityD It is possible that the
differences noted by Grady and Campbell are a function of the cell-line
clones that they examined,, Dissimilarities may then be a function of
the original clones, AL/N and PY AL/N clone 3» as much as they could be
due to transformation by polyoma,. Unlike the system examined by Grady
and Campbell, RSV infected and control cells used in this study were
derived from the same chick embryo cultures which were either infected
or mock infected at the same time0 Furthermore, Grady and Campbell
assayed the extent of unique sequence transcription, whereas the hybrid
ization kinetics of unique sequence transcripts were examined in this
investigation.. Results reported here support the conclusion that dif
ferences in "unique" RNA hybridization kinetics between normal and RSV
infected cells are small. However, changes noted in the "repeat" RNA
fraction require that differences in the "unique" RNA fraction must
exist, although they are likely to be only a few percent.

75
Repetitive Sequence Transcripts
Birnie et al„ (197*0 analyzed nuclear RNA which was labeled
during growth or non-growth states of several mouse cell systems.
Using filter bound DNA for labeled RNA hybridizations, they found that
a greater number of repeat sequences were transcribed when cells were
not growing than when they were dividingo On the other hand, Grady
and Campbell (1975b) were unable to detect any differences in the
extent of repeat sequence transcription between growing and confluent
cultures of a mouse cell line0 These investigators hybridized labeled
DNA to excess whole-cell RNA to arrive at their conclusion,. The dif
ference in results may be due to the undetermined time of synthesis for
the hybridizing RNA used in the experiments of Grady and Campbells
Since the RNA species hybridizing were not restricted to those tran
scribed during a particular time period, they may represent an average
of the RNA populations present at different times in the cells' growth
cycle»
The low CDt hybridizations reported in this study address the
question of the concentration of repeat sequence transcripts present,
rather than the fraction of the DNA transcribed,. Since these reactions
are driven by DNA, the kinetics are determined by the DNA concentra-
tionB Thus, it is not possible by this technique to determine the
number of different repeat sequences which were transcribed* The
values seen for these hybridizations yield the portion of the total
labeled RNA which was transcribed from repeat sequence DNA during the
pulse-labeling period.

76
Since "repeat" RNA may hybridize to DNA families of similar but
not identical base sequences, the extent of mismatch of the hybrids
formed is higher than that observed with unique sequences„ Hybridi
zation conditions used here are quite stringent due to the combination
of high formamide concentration and high temperature plus the use of
extensive RNase digestion (see McConaughy, Laird and McCarthy, 1969;
Hayward and Hanafusa, 1975)• The potential for similar but hetero
logous sequences hybridizing is reduced by elevated temperatures which
select against duplexes with lower "stacking-free energy" (Kennel,
1971)o The RNase assay used for hybrid formation minimizes the pos
sibility of mismatched sequences being scored as hybrids since both
free ends and internal unhybridized loops are digestedo Where both
RNase A and T1 at 37°C are used, the amount of non-specific hybridiza
tion can be presumed to be very small due to the complementary substrate-
specificities of these enzymes (Barman, 1969) and the higher incubation
temperature,, It should be noted that all the differences observed in
this study are derived from comparisons between identically treated
infected cell and normal cell RNA samples®
Examining "repeat" RNA hybridization in this investigation
generated two observations — one a function of the time after sub-
culturing cells (growth state) and the other a function of infection
by RSV. In separate experiments where pulse-labeled nRNA was hybrid
ized to cell DNA, the data show comparable results for the indepen
dently prepared sets of nRNA. A slightly larger fraction of nRNA
isolated from uninfected control cells early after plating hybridizes

to repeat DNA than that isolated during the period 15-25 hours after
plating (see Fig„ 10), nRNA isolated even later after subculturing,
hybridizes a greater portion of its total to repeat sequences (Figs®
10 and 16). Hence, it is concluded that non-growing CEF cells tran
scribe a slightly greater fraction of their total nRNA from repeated
DNA sequences than do dividing cells« The second observation with
pulse-labeled nRNA was the presence of proportionately greater amounts
of repeat sequence transcripts within the nuclei of infected cells late
after infection,. Between 25 and 50 hours after infection by RSV cellu
lar nRNA is demonstratably richer in repeat sequence transcripts®
Grady and Campbell (1975b), using labeled DNA in hybridization
with excess RNA, found no difference in the number of repeat sequences
used as templates for RNA synthesis between normal and polyoma trans
formed 3T3 cells* Earlier, Neiman and Henry (1969) hybridized labeled
RNA to filter-bound DNA and showed that normal lymphocyte nuclei con
tain "repetitive" RNA of greater complexity than leukemic cells0 Con
versely, the same technique demonstrated nRNA from hepatoma cells had
greater complexity than the control liver cell nBNA (Church, Luther
and McCarthy, 1969)0 These filter hybridization studies contain all
the inherent problems discussed above that limit their sensitivity to
the overall RNA synthesis occurring* Without contradicting these
findings, the data presented in this study demonstrate that a greater
portion of the nRNA synthesized late after infection is transcribed
from repeated DNA than is transcribed in uninfected cells, regardless
of the number of different sequences expressed.

78
The dissimilarities seen between infected and control cell nBNA
hybridizations diminished at 58 hours after infection (Figs„ 15 and 16).
It would appear that the increase in the fraction of repeat sequence
transcripts observed during the transformation of CEF cells does not
continue indefinitely.. Infection of normal cells by RSV may be causing
a transient alteration of repeat sequence transcription that recedes
after transformation. In fact, morphological transformation of cells
begins about 2k hours after infection (see Figo k) at approximately the
same time that differences are first noted in nBNA synthesis® These
observations argue for the possibility that the transient alteration in
••repeat" RNA synthesis marks the passage of cells from normal to trans
formed states and is in some way related to this phenomena. A second
explanation would be that transformed-cell transcription responded to
the culture conditions late after passage<, According to this hypothe
sis, the differences between normal cells and transformed cells would
be detected when both these cell types are actively gro\ving, but not
seen when they are rarely dividing.. Data presented in this study plus
evidence cited from other investigations indicate that normal cells do
respond to this form of regulation,. It was shown here that normal
cells stopped synthesizing DNA after kO hours in culture and that fresh
serum could induce new DNA synthesis at this time* The effect of serum
on the multiplication and saturation density of RSV transformed cells
is also well documented (Temin, 1967b).. It seems plausible that the
return to control values for the amount of repeated sequence nRNA
hybridized from transformed cells could be a function of growth limiting

79
factors in the culture. This is not to say that the "repetitive" nRNA
found late after infection in control and infected cells represents the
same transcripts but only that the fraction of nRNA that is "repeti
tive" RNA is similar for both cell types0 In fact, it seems much more
likely that these transcripts are different but that that difference
is not measured by the technique usedo
The examination of polyribosomal RNA for the amount of "repeat"
RNA showed that, while normal cells had a reduced concentration of
"repetitive" RNA associated with the polysomes, RSV infected cells
exhibit a further decrease in this proportion (Figo 18)<> This finding
is consistent with the fact that representation of total nRNA in the
polysomal fraction is known to be very limited (see Greenberg, 1975)®
Early hybridization studies with filter bound DNA demonstrated greater
complexity for repeat sequence transcripts found in nuclear compared
to that found in cytoplasmic fractions of tumor cells (Drews, Brawerman
and Morris, 1968).
One possible contribution to the observed increased concentra
tion of repetitive sequences in infected cell nuclei would be an in
creased synthesis of ribosomal RNA (rRNA)„ Colby and Rubin (1969) have
noted that RSV transformed chick cells do synthesize more rRNA than
normal confluent cells. Other investigators have established that
normal cells — both CEF and the 3T3 cell line — synthesize more rRNA
while growing than when contact inhibited (Emerson, 1971; Johnson et al.,
197*0• However, it is not likely that increased rRNA synthesis could
account for the higher fraction of repeat sequence transcripts observed

in this study. The labeling pulse used here was sufficiently long to
allow synthesis and transport of rRNA to the polysomes (Perry, 1967;
Dinowitz, 1975b)o If the "repeat" RNA were rRNA, one would predict
that greater concentrations of "repeat" RNA in the nucleus would also
be seen in pRNA from infected cells and this was not observed (Figo l8)o
In addition, the copy number of ribosomal cistrons/genome is incon
sistent with the CDt at which differences in "repetitive" RNA was
observed,, Since chick ribosomal sequences occur at 100 times the fre
quency of unique sequences (Attardi and Amaldi, 1970), the C0ti/2 for
rRNA should be 100 times faster than the "unique" RNA (0oty2 = 2„5 x 10 )
or 25° All of the differences noted for repetitive sequence transcript
hybridizations were completed before this CQt (see Figs0 13 and 1*0«
The relative distribution of "repetitive" RNA in nuclear and
polysomal fractions appears to be greatly altered by virus infection
(Fig0 19)<= It is tempting to speculate that the increased "repetitive"
RNA seen in the nucleus and the decreased "repetitive" RNA found on the
polysomes results from a reduction in processing or transport of the
RNA from nucleus to the cytoplasm — as has been observed in another
virus-infection system (Dinowitz and Green, 1972)» This hypothesis
would specify that similar proportions of "repetitive" RNA are tran
scribed in RSV infected and control cells but that some event following
synthesis is altered in transforming cells. Late after RSV infection
both the nuclear and polysomal differences in concentration of "repeti
tive" RNA diminish, suggesting that the degree of altered transcription
or processing reaches a peak and then is reduced. However, different

81
gene sequences may be transcribed before and after the time of altered
RNA synthesis.
Conclusions
Evidence presented in this investigation suggests that a change
occurs in RSV infected cells in the concentration of RNA transcribed
from reiterated DNA» This difference is sufficiently large to be
measured by the procedures used here beginning about 25 hours after in
fection and reaches a maximum ^5-50 hours after infection., "Repetitive?'
RNA found on the polysomes of infected cells decreases in concentration
over controls during this same period,, These findings have been sum
marized in Figo 20 which shows the relationship between cellular events
and the hours in culture* Observations characteristic of normal and
RSV-infected cells — as documented in the literature •— are listed
immediately above and below the time line showing the order of their
occurrenceo Moving further away from the time line, observations from
this study are shown- It is interesting to note the coincidence of
several changes in growth and morphological characteristics with bio
chemical alterations., The decreased concentration of "repetitive" RNA
associated with infected cell polysomes occurs at approximately the end
of the provirus integration phase. The concentration of newly synthe
sized "repetitive" RNA from infected cell nuclei increases at about the
time the culture first contains cells appearing morphologically trans
formed. These differences all occur during the period in which RSV-
infected chick cells are converted from the normal to transformed
morphology®

Figure 20. Summary of cellular events after subculturing or infection with RSV.
Selected events occurring in normal and RSV-infected cells in culture are compared with emphasis on growth conditions, nucleic acid synthesis, and transformation to tumor cells. Immediately above and below the time line are observations of changes related to cell density and RSV infection reported in the literature (/—* \). Moving away from the time line are observations supported by data from this study (y—™x) concerning DNA synthesis and transcription.

CO o h-cn cc UJ h* O < cc < x o
UJ o
or o
CO u h-CO
or UJ t— o < cc < X o
UJ o Q UJ I— o UJ UL
Newly synthesized "repetitive" nRNAand pRNA decrease in concentration
Cellular DNA" synthesis reaches
a maximum
Newly synthesized "repetitive"nRNA and pRNA
increase in concentration
Cellular DNA synthesis is no longer detectable
/"
Growing cells A
10 Hrs. p. plating
./•
Confluent monolayer a _
30 50
0
\_
Hrs. p. i 20 40 60
Provirus phase Budding RSV detectable
N ..
TJ Q) O a>
0) O
Culture morphologically transformed
Newly synthesized "repetitive" nRNA increases in concentration over controls
Newly synthesized "repetitive" pRNA decreases in concentration over controls
Figure 20. Summary of cellular events after subculturing or infection with RSV.
oo

83
An attractive hypothesis to account for these observations is
that some repeat sequences or repeat sequence families are involved in
the regulation of changes in cell metabolism related to growth and
differentiation (Britten and Davidson, 1969; Davidson and Britten,
1973)i and that infection with RSV affects these controlling sequences.
The data presented in this investigation are consistent with such a
possibilityc, Further characterization of specific repeat-sequence
transcripts and their functional products (or primary effects) may pro
vide some insight into the changes induced in cellular transcription
by RSV infection. However, until the role of repetitive sequence DNA
and its transcripts are better understood, the fundamental significance
of these observed alterations will remain a matter for speculation.

LIST OF REFERENCES
Ando, T. 1966. A nuclease specific for heat-denatured DNA isolated from a product of Aspergillus oryzae. Biochim. Biophys. Acta 11^:158.
Attardi, G. and F. Amaldi. 1970.. Structure and synthesis of ribo-somal RNAo Ann. Rev, Biochemo 39tl83<>
Bader, J„ P. 196ka The role of DNA in the synthesis of Rous sarcoma virus. Virology 22:k620
Bader, J. P. 1972., Metabolic requirements for infection by Rous sarcoma virus IV. Virus reproduction and cellular transformation without cellular division,, Virology k8:k^ha
Bader, J. P. 1973° Virus induced transformation without cell division. Science l80:1069.
Bader, J. P. and N. R. Brown. 1971= Induction of mutations in an RNA tumor virus by an analogue of a DNA precursor. Nature (New Biology) 23^:11.
Balazs, I., E. H. Brown and C. L. Schildkraut. 1973° The temporal order of replication of some DNA cistrons. Cold Spring Harbor Symp. Quant. Biol. 38:239°
Balduzzi, P. and H. R. Morgan. 1970. Mechanisms of oncogenic transformation by Rous sarcoma virus. II. Intracellular inactiva-tion of cell transforming ability of Rous sarcoma virus by 5-bromodeoxy uridine and light. J. Virol. 5^70°
Baltimore, D. 1970. RNA-dependent DNA polymerase in virons of RNA tumor viruses. Nature (London) 226:1209.
Baluda, M. A. 1972. Widespread presence, in chickens, of DNA complementary to the RNA genome of avian leukosis viruses. Proc. Nat. Acad. Sci. (U.S.A.) 69:576.
Baluda, M. A. and D. P. Nayak. 1970. DNA complementary to viral RNA in leukemic cells induced by avian myeloblastosis. Proc. Nat. Acad. Sci. (U.S.A.) 66:329.
Barman, T. E. (Ed.). 1969. Enzyme Handbook. Springer-verlag, New York, pp. 76, 48^.
8^

85
Birnie, G. D., J. Delcour, D. Angus, G. Threlfell, and J. Paul. 197^« Distribution of nuclear RNAs in growing and nongrowing cells. In Control of Proliferation in Animal Cells, edited by Clarkson Baserga. Cold Spring Harbor Laboratory, New York.
Bishop, J. 0. 1972. Molecular hybridization of ribonucleic acid with a large excess of deoxyribonucleic acid. Biochem. J. 126:171.
Bissell, M. Jo, Co Hatie', and H. Rubin, 1972. Patterns of glucose metabolism in normal and virus-transformed chick cells in tissue culture. J. Nat. Cancer Inst. 9:555°
Boettiger, D. and H. M. Temin. 1970. Light inactivation of focus formation by chicken embryo fibroblasts infected with avian sarcoma virus in the presence of 5-bromodeoxyuridine0 Nature (London) 228;621.
Borun, T. W., M. D. Scharff and E. Robbins. 1967. Preparation of mammalian polyribosomes with detergent Nonidet 9-^0. Biochim. Biophys. Acta 1^9:3020
Brandhorst, B. P. and E. H. McConkey. 197^+. Stability of nuclear RNA in mammalian cells. J. Mol. Biol. 85:^51.
Britten, R. J. and E. H. Davidson. 1969. Gene regulation for higher cells: a theory. Science 165:3^9°
Britten, R. J. and D. E. Kohne. 1967= Repeated nucleotide sequences. Carnegie Inst. Wash. Year Book 66:73.
Britten, R. J. and D. E. Kohne. 1968. Repeated sequences in DNA. Science l6l:529.
Britten, R. J. and J. Smith. 1970. A bovine genome. Carnegie Inst, of V/ash. Year Book 68:378.
Brown, D. D. and A. W. Blackler. 1972. Gene amplification proceeds by a chromosome copy mechanism. J. Mol. Biol. 63:75»
Buck, C. A., M. C. Glick, and L. Warren* 1971. Glycopeptides from the surface of control and virus-transformed cells. Science 172:169.
Callan, H. G. 1973. DNA replication in the chromosomes of eukaryotes. Cold Spring Harbor Symp. 38:195.
Campo, M. S. and J. 0. Bishop. 197^° Two classes of messenger RNA in cultured rat cells: repetitive sequence transcripts and unique sequence transcripts. J. Mol. Biol. 90:6^9®

86
Church, R. B., S. W. Luther, and B. J. McCarthy. 1969. RNA synthesis in taper hepatoma and mouse-liver cells. Biochim. Biophys. Acta 190;30.
Colby, Co and H. Rubin. 1969. Growth and nucleic acid synthesis in normal cells and cells infected with Rous sarcoma virus. J. Nat. Cancer Inst. bj>: bj>7.
Crittenden, L. B. 1968. Observations on the nature of genetic cellular resistance to avian tumor viruses. J. Nat. Cancer Inst. jOt1^5.
Dales, S. and H. Hanafusa. 1972. Penetration and intracellular release of the genomes of avian RNA tumor viruses. Virology 30?bk0o
Davidson, E. H. and R. J. Britten. 1973. Organization transcription, and regulation in the animal genome. Quarterly Rev. Biol.
^•8; 565.
Davidson, E, H., B. Hough, C. Amenson and R. J. Britten. 1973. General interspersion of repetitive with non-repetitive sequence elements in the DNA of Xenopus. J. Mol. Biol. 77:1«
Dickson, E., J. B. Boyd and C. E. Laird. 1971. Sequence diversity of polytene chromosome DNA from Orosophilia hydei. J. Mol. Biol. 61:615.
Dinowitz, M. 1975a- Resistence of Rous sarcoma virus transformed cells toa-amanitin inhibition of RNA synthesis. Unpublished paper, Department of Microbiology, University of Arizona, Tucson.
Dinowitz, M. 1975b. Inhibition of Rous sarcoma virus by a-amanitin: possible role of cell DNA-dependent RNA polymerase form II. Virology 66:1.
Dinowitz, M. 1975c. Unpublished observations. Department of Microbiology, University of Arizona, Tucson
Dinowitz, M. 1976. Establishment of a continuous line of Rous sarcoma virus transformed chick embryo cells. Unpublished paper, Department of Microbiology, University of Arizona, Tucson.
Dinowitz, M. and M. Green. 1972. The effect of infection with adenovirus 2 on the transcription of cellular RNA. Virology 50:619.
Drews, J., G. Brawerman and H. P. Morris. 1968. Nucleotide sequence homologies in nuclear and cytoplasmic ribonucleic acid from rat liver and hepatomas. European J. Biochem. 3:28^.

87
Emerson, C. P. 1971« Regulation of the synthesis and the stability of ribosomal RNA during contact inhibition of growths. Nature (New Biology) 232:101'.
Flamm, W. G., P., Mo B. Walker, and Mo McCallunio 1969® Some properties of the single strands from the DNA of the nuclear satellite of the mouse,, J. Molo Biol. 40:423.
Gall, Jo G0 1969® The genes for ribosomal RNA during oogenesis. Genetics (supplement) 6l;1210
Gelderman, A. Ho, Ao V. Rake and Re Jo Britten. 1971o Transcription of nonrepeated DNA in neonatal and fetal mice» Proco Nat. Acad. Scio (U.S.Ao) 68;172.
Gillespie, Do and R. C= Gallo. 1975o RNA processing and RNA tumor virus origin and evolution. Science l88;802»
Gillespie, D. and S. Spiegelman. 1965= A quantitative assay for DNA-RNA hybrids with DNA immobilized on a membrane. J. Mol. Biol. 12:829.
Goldberg, R. B., G. A. Galau, R. J. Britten, and E. H. Davidson. 1973. Non-repetitive DNA sequence representation in sea urchin embryo messenger RNA. Proc. Nat. Acad. Sci. (U.S.A.) 70:3516.
Grady, L. J. and W. P. Campbell. 1975a. Non-repetitive DNA transcripts in nuclei and polysomes of polyoma-transformed and non-trans formed mouse cells. Nature (London) 254: 356.
Grady, L. J. and V/o P. Campbello 1975bo Transcription of the repetitive DNA sequences in polyoma-transformed and nontransformed mouse cells in culture. Cancer Res. 35:1559»
Greenberg, J. R. 1975. Messenger RNA metabolism of animal cells. Possible involvement of untranslated sequences and in RNA-associated proteins. J. Cell Biol. 64:269.
Groudine, M. and H. Weintraub. 1975. Rous sarcoma virus activates embryonic globin genes in chicken fibroblasts. Proc. Nat. Acad. Sci. (U.S.A.) 72:4464.
Hanafusa, H., W. S. Hayward, J„ H. Chen, and T. Hanafusa. 1974. Control of expression of tumor virus genes in uninfected chicken cells. Cold Spring Harbor Symp. Quant. Biol. 39: 1139.
Hayward, W. S. and H. Hanafusa. 1975° Recombination between endogenous and exogenous RNA tumor virus genes as analyzed by nucleic acid hybridization. J. Virol. 15:1367®

88
Hershey, Ao D. (Ed.). 1971. The Bacteriophage Lambda. Cold Spring Harbor Laboratory, New York.,
Humphries, E. H. and Ho Mo Temin» 197^. Requirement for cell division for initiation of transcription of the Rous sarcoma virus RNA. Jo Virole lA:531.
Jaquet, Mo, Y. Groner, G» Monvoy and J. Hurwitz. 197^<> The in vitro synthesis of avian myeloblastosis virus nuclear acid in cells producing Rous sarcoma virus: Detection and characterization. Jo Virole 891o
Johnson, L. F., H. T„ Abelson, H„ Green, and S. Penman., 197^° Changes in RNA in relationship to growth of the fibroblast Io Amounts of mRNA, rRNA, and tRNA in resting and growing cells. Cell _1: 12 o
Kennel, D, E. 1971= Principles and practices of nucleic acid hybridization Progo in Nuco Acid Res, and Molo Biolo 11:259°
Klein, Wo H., W. Murphy, G. Attardi, R, J. Britten, and E. Ho Davidson. 197^o Distribution of repetitive and non-repetitive sequence transcripts in Hela mRNA. Proc. Nat. Acad. Scio (U.S.A.) 71t 1785.
Klevecz, R. R., L. N. Kapp, and J. A. Remington. 197^. Intermittent amplification and catabolism of DNA and its correlation with gene expression. In Control of Proliferation in Animal Cells, edited by Clarkson/Baserga, New York.
Kohne, D. E. 1968. Isolation and characterization of bacterial ribo-somal RNA cistronso Biophysical J. 8:110^o
Leong, J. A., W. Levinson and J. M. Bishop. 1972. Synchronization of Rous sarcoma virus production in chick embryo cells. Virology k7}133 °
Levinson, V/., Jo M. Bishop, No Quintrell, J. Jakson, L. Franshier. 1970. Synthesis of RNA in normal and RSV-infected cells: effect of bromodeoxyuridine. Virology k2:221„
Lewin, B. 1975a. Units of transcription and translation: the relationship between heterogeneous nuclear RNA and messenger RNA Cell jf:ll„
Lewin, B. 1975b. Units of transcription and translation: sequence components of heterogeneous nuclear RNA and messenger RNA. Cell k:77.

89
Marmur, J. 196lo A procedure for the isolation of deoxy. ribonucleic acid from micro-organisms. J. Mol. Biol. 3s208.
McCarthy, B. Jo and R» Bo Church. 1970® The specificity of molecular hybridization reactions., Ann„ Rev. Biocheme 39:131q
McConaughy, B. L., Co D. Laird, B„ J. McCarthy0 1969- Nucleic acid reassociation in formamide. Biochem. 8:3289.
Melli, M., Co Whitfield, K, V. Rao, M. Richardson, and J. 0. Bishop. 1971» DNA-RNA hybridizations in vast DNA excess. Nature (New Biology) 251:8.
Mirsky, A. E. and H. Ris. 1950. The desoxyribonucleic acid content of animal cells and its evolutionary significance. J. Gen. P h y s i o l . 3 ^ : .
Moore, R. L. and B. J. McCarthy. 1968. Related base sequences in the DNA of simple and complex organisms III. Variability in the base sequence of the reduplicated genes for ribosomal RNA in the rabbit. Biochem. Genetics 2:75.
Neiman, P. E. 1972. Rous sarcoma virus nucleotide sequences in cellular DNA: measurement by RNA:DNA hybridization. Science 178:750.
Neiman, P. E. 1973. Measurement of endogenous leukosis virus nucleotide sequences in the DNA of normal avian embryos by RNA-DNA hybridization. Virology 53:196.
Neiman, P. E. and P. H. Henry. 19^9. Ribonucleic acid-deoxyribonucleic acid hybridization and hybridization-competition studies of the rapidly labeled ribonucleic acid from normal and chronic lymphocytic leukemia lymphocytes. Biochem. _8: 275.
Neiman, P. E., H. Graham Purchase and W. Okazaki. 1975. Chicken leukosis virus genome sequences in DNA from normal chick cells and virus-induced bursal lymphonas. Cell
Neiman, P. E., S. E. Wright, C. McMillin, and D. MacDonnell. 197^. Nucleotide sequence relationships of avian RNA tumor viruses: measurement of the deletion in a transformation-defective mutant of Rous sarcoma virus. J. Virol. 13s837°
Paskind, M. P., R. A. Weinberg and D. Baltimore. 1975. Dependence of Moloney murine leukemia virus production on cell growth. Virology 67:2^2.

90
Penman, S. 1966. RNA metabolism in the Hela cell nucleus. J. Mol. Biol. 17s117 o
Perry, R. P. 1967° The nucleolus and the synthesis of ribosomes. Prop;, Nuclo Acid Res. Mol= Biolo 6_:219 °
Perry, K. P. and D. E. Kelley. 1968, Messenger RNA-protein complexes and newly synthesized ribosomal subunite: analysis of free particles and components of polyribosomes,, J. Mol. Biol„ 35s
37.
Perry, R. P., Jo LaTorre, D. E. Kelley, and J. R. Greenberg. 1972. On the lability of poly(A) sequences during extraction of messenger SNA from polyribosomes,. Biochim. Biophys<> Acta 262;220o
Piraino, F. 1967. The mechanism of genetic resistance of chick embryo cells to infection by Rous sarcoma virus-Bryan strain. Virology 32:700„
Rosen, J. H., C. D. Liarakos and B. W. O'Malley. 1973. Effect of estrogen on gene expression in the chick oviduct I. Deoxyribonucleic acid-deoxribonucleic acid renaturation studies« Biochem. 12:2803-
Rosenthal, P. No, H. L. Robinson, W. S. Robinson, T. Hanafusa and Ho Hanafusa. 1971. DNA in uninfected and virus-infected cells complementary to avian tumor virus RNA. Proc. Nat. Acad. Sci. (U.S.A.) 68:2336.
Rymo, L«, J. T. Parsons, J. M. Coffin, and C. Weissmann. 197^. In vitro synthesis of Rous sarcoma virus-specific RNA is catalyzed by a DNA-dependent RNA polymerase. Proc. Nat. Acad. Sci. (U.S.A.) 71:2782.
Schincariol, A. L. and W. J. Joklik. 1973. Early synthesis of virus-specific RNA and DNA in cells rapidly transformed with Rous sarcoma virus. Virology 56:532.
Singer, R. H. and S. Penman. 1973. Messenger RNA in HeLa cells: kinetics of formation and decay. J. Mol. Biol. 78:321.
Smith, M. J., B. R. Hough, M» E. Chamberlin and E. Davidson. 197^® Repetitive and non-repetitive sequence in sea urchin heterogeneous nuclear RNA. J. Mol. Biol. 85:103<>
Sutton, W. D. 1971. A crude nuclease preparation suitable for use in DNA reassociation experiments. Biochim. Biophys. Acta 240:522.

Temin, H„ M. 1962. Separation of morphological conversion and virus production in Rous sarcoma virus infection. Cold Spring Harbor Symp. Quant. Biol. 27:^07«
Temin, H. M. 1963. The effects of actinomycin D on growth of Rous sarcoma virus in vitro. Virology 20:377°
Temin, H. M« 1967a. Studies on carcinogenesis by avian sarcoma viruses, V. Requirement for new DNA synthesis and for cell division. J. Cell Physiol. 69:33°
Temin, H. M. 1967b. Studies on carcinogenesis by avian sarcoma viruses III. The differential effect of serum and polyanions on multiplication of uninfected and converted cells. J. Nat. Cane. Inst. 37:167°
Temin, H. M. 1970. The role of the DNA provirus in carcinogenesis by RNA tumor viruses. In The Biology of Oncogenic Viruses, edited by L. G. Silvestri, New York, Proceedings of the 2nd Lepetit Colloquium.
Temin, H. M. 1971a. Mechanism of cell transformation by RNA tumor viruses. Ann. Rev. Micro. 23:609.
Temin, H. M. 1971b. Stimulation by serum multiplication of stationary chick cells. J. Cell Physiol. 70:l6l.
Temin, II. M. and S= Mizutani. 1970. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature (London) 226:1211.
Temin, H. M. and H. Rubin. 1958. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology _6:669.
Tobia, A. M., C. L. Schildkraut, and J. J. Maio. 1970. Deoxyribonucleic acid replication in synchronized cultured mammalian cells I. Time of synthesis of molecules of different average guanine + cytosine content. J. Mol. Biol. 3^:^-99°
Tooze, J. (Ed.) 1973. The Molecular Biology of Tumor Viruses. Cold Spring Harbor Laboratory, New York.
Varmus, H. E., R. V. Guntaka, C. T. Deng and J. M. Bishop. 197^. Synthesis, structure and function of avian sarcoma virus-specific DNA in permissive and nonpermissive cells. Cold Spring Harbor Symp. Quant. Biol. 39:987•

92
Varmus, H. E., R. V. Guntaka, F. Warner, So Heasley and J. M. Bishop. 197^. Synthesis of viral DNA in the cytoplasm of duck embryo fibroblasts and in enucleated cells after by avian sarcoma virus. Proc. Nat. Acad. Sci. (U.S.A.) 71:387°
Varmus, H. E., P. K. Vogt and J. M. Bishop. 1973. Integration of deoxyribonucleic acid specific for Rous sarcoma virus after infection of permissive and nonpermissive hosts. Proc. Nat. Acad. Sci. (U.S.A.) 7O:3067»
Vogt, P. K. 1969« Focus assay of Rous sarcoma virus. In Fundamental Techniques in Virology, edited by K. Habel and N. P. Salzman. Academic Press, Inc., New York.
Walker, P. M. B., W. G. Flamm and A. McLaren. 1969. The specificity of molecular hybridization in relation to studies on higher organisms. Prog. Nucl. Acid Res. Mol. Biol. 9:301»
Witt, F. H. 1971° Advances in nucleic acid hybridization. Develop. Bio. 26:358.