Embed Size (px)
Citation preview

Published: September 08, 2011
r 2011 American Chemical Society 10550 dx.doi.org/10.1021/jp201067r | J. Phys. Chem. A 2011, 115, 10550–10555
ARTICLE
pubs.acs.org/JPCA
New Generation of Dialkylsilylenes with Stabilities Comparable toDiaminosilylenes: A Theoretical StudyMohammad R. Momeni, Farnaz A. Shakib,* and Zahra Azizi
Department of Chemistry, Shahr_e Qods Branch, Islamic Azad University, Tehran, 37541-13115, Iran
Department of Chemistry, Karaj Branch, Islamic Azad University, Karaj, 31485-313, Iran
bS Supporting Information
1. INTRODUCTION
The development of N-heterocyclic silylenes is a classicprogression started from transient intermediates which turnedto matrix-isolated molecules in 1980s.1 Although no one ex-pected to observe these compounds at room temperature,Arduengo’s isolation of the stable carbene I in 19912 encouragedthe endeavors toward the synthesis of stable silylenes (Figure 1).In 1994, Denk and his co-workers synthesized II as the first stablesilylene,3 which benefits from the similar stabilizing effectsenjoyed by I, that is, electronic stabilization by the amino groups,steric protection by the bulky alkyl groups, and additionalstabilization of aromatic π-electron delocalization over the ring.Subsequently, several experimental and theoretical investigationsappeared on similar silylenes4,5 until Veszpr�emi et al. found 1H-2-silapyridine-2-ylidene (III) more stable than its full valence2-silapyridine isomer.6 They also established the kinetic stabilityof III through theprohibitive high energybarrier (about 60 kcal/mol)required for its intramolecular isomerization to 2-silapyridine. Ina further step, in 1999, Kira et al.7,8 reported the synthesis of thefirst isolable dialkylsilylene IV, prepared by reductive debromi-nation of the precursor dibromosilane. Interestingly, the stabilityof this species is derived entirely from steric protection ofthe divalent silicon atom by two SiMe3 groups attached toeach α-carbon.
Herein, we are pleased to introduce a new family of dialkylsi-lylenes which benefits from the stabilizing interaction of theoccupied Walsh orbital with the vacant p-orbital of the silylenecenter. Indeed, this interaction was the basis of the spectro-scopic characterization of dicyclopropylcarbene V, generatedfrom the irradiation of dicyclopropyldiazirine in a matrix of N2
at 6 K, by Ammann et al. in 1992.9 Our work is aimed to shedsome light on the relative stabilities and reactivities of acyclicand saturated/unsaturated cyclic dicyclopropylsilylenes 1�3(Figure 2).
2. COMPUTATIONAL DETAILS
Full geometry optimizations are accomplished using Møller�Plesset MP210 and hybrid functional B3LYP11�13 methods andthe 6-31G(d)14,15 and 6-31+G(d)16,17 basis sets, respectively,using the Gaussian 98 code.18 To obtain more accurate energeticdata, single point calculations are performed at MP2/6-311++G(d,p) and B3LYP/AUG-cc-pVTZ19 levels of theory based on
Figure 1
Figure 2. Schematic representation of the scrutinized silylenes.
Received: February 1, 2011Revised: August 8, 2011
ABSTRACT: A new family of dialkylsilylenes is introduced which enjoys the stabilizingeffect of α-cyclopropyl substituents. The singlet and triplet states of acyclic and saturated/unsaturated cyclic dicyclopropylsilylenes are fully optimized using MP2/6-31G(d) andB3LYP/6-31+G(d) levels. Their higher ΔES-T values compared to the correspondinganalogues which possess isopropyl groups instead of cyclopropyl represents the stabilizinginteraction of the occupiedWalsh orbital of the cyclopropyls with the vacant p-orbital of thesilylene center. Appropriate isodesmic reactions clearly show that the stabilizing effect of this interaction on the singlet state is muchmore considerable than the corresponding triplet state. Cyclopropyls can serve as good sites for bulky substitution and, hence,provide steric protection for silylene.

10551 dx.doi.org/10.1021/jp201067r |J. Phys. Chem. A 2011, 115, 10550–10555
The Journal of Physical Chemistry A ARTICLE
the corresponding optimized structures. Vibrational frequenciesare calculated to establish the nature of stationary points as trueminima.20 The NBO21 population analysis on optimized struc-tures is accomplished at B3LYP/AUG-cc-pVTZ level.
The nucleophilicity index, N, which was introduced byDomingo et al.,22 is calculated asN = EHOMO(Nu)� EHOMO(TCNE),where tetracyanoethylene (TCNE) is chosen as the reference.The global electrophilicity, ω,23 is also calculated followingthe expression, ω = (μ2/2η), where μ is the chemical potential(μ ≈ (EHOMO + ELUMO)/2) and η is the chemical hardness(η = ELUMO � EHOMO).
24
3. RESULTS AND DISCUSSION
The MP2/6-31G(d) and B3LYP/6-31+G(d) (in parentheses)levels are applied to fully optimize acyclic and saturated/unsaturatedcyclic dicyclopropylsilylenes (1�3; Figure 2).3.1. Stabilizing Effects of W f p Interactions through
Geometrical Parameters and NBO Analysis. The optimizedstructure of singlet (s) 1 adopts C2v symmetry with an axiscrossing divalent center and two perpendicular planes bisectingthe molecule. The carbon�silicon bond is 1.873 (1.887) Å,which is shorter than a typical single H3C�SiH3 bond of 1.883(1.890) Å, optimized at the same levels. Replacing two cyclo-propyl groups of 1 with isopropyl substituents (Figure 2) resultsin1a(s) with theC�Si bond of 1.921 (1.939) Å that is considerably
longer than a typical bond. Hence, shortening of C�Si bondfrom 1.921 Å (1.939 Å) in 1a(s) to 1.873 Å (1.887 Å) in 1(s) isthe geometrical result of the cyclopropyl stabilizing effect. Thiseffect originates from the electronic configuration of cyclopropylring which was rationalized by the theory of Walsh orbitals interms of two symmetric (WS) and antisymmetric components(WA; Figure 3).
25 The interaction of the empty silylenic p-orbitalwithWA of the set of Walsh orbitals (Wf p) favors the singletstate and enlarges the corresponding ΔES-T.
26
The NBO analysis can shed more light on this effect from anelectronic viewpoint. The silylene center of 1a(s), which isadjacent to two isopropyl, carries a positive charge of 1.081(Table 1). Replacing the isopropyl substituents with cyclopropylgroups decrease this charge to 1.014 in 1(s) exhibiting theelectron density transfer from cyclopropyls to the empty p-orbitalof the silylene. Based on NBO calculations, one can find W f pinteraction in terms of four σC�Cf pSi electron density transferin 1(s) creating 10.0 kcal/mol stabilizing energy (2.5 kcal/mol ofeach interaction). Such an interaction is absent both in 1(t) andin 1a(s). Hyperconjugation of one of the C�C bonds of eachisopropyl with the empty p-orbital in 1a(s) creates stabilizationenergy of 5.4 kcal/mol (less than that of cyclopropyls). Theoverall energetic result is the increase of the singlet�tripletenergy gap (ΔES-T = ET � ES) from 20.5 (23.2) kcal/mol of1a to 28.3 (31.2) kcal/mol of 1 in favor of the singlet state.To avoid eclipsed hydrogens of the ring, 2 deforms from
planarity leading toC2 symmetry of themolecule. In the case of 3,the presence of a double bond in the ring imposes the planarityandC2v symmetry to the structure. TheWf p interaction can beestimated from the shortening of the C�Si bonds from 1.931(1.958) Å in 2a(s) to 1.883 (1.899) Å in 2(s) and from 1.941(1.951) Å in 3a(s) to 1.888 (1.905) Å in 3(s). Similar to 1, thesilylene center of both 2 and 3 carries a less positive chargecompared to 2a and 3a, respectively (Table 1). While thecompletely parallel WA orbitals of 3 and 1 with the emptyp-orbital create four σC�C f pSi interactions (causing 10.1 and
Table 1. Principal Geometrical Parameters of the Singlet (s) and Triplet (t) Silylene Species at MP2/6-31G(d) and B3LYP/6-31+G(d) (in Parentheses) along with Their NBO Atomic Charges at B3LYP/AUG-cc-pVTZ//B3LYP/6-31+G(d) and ΔES-Ts atMP2/6-311++G(d,p)//MP2/6-31G(d) and B3LYP/AUG-cc-pVTZ//B3LYP/6-31+G(d) (in Parentheses)
species point group Ccyc�Si (Å) Ciso�Si (Å) C�Si� C (�) qSi ΔES-T (kcal/mol)
1(s) C2v (C2v) 1.873 (1.887) 100.39 (100.63) 1.014 28.3 (31.2)
1(t) C2 (C2) 1.876 (1.888) 118.90 (119.57) 0.894
1a(s) C2 (C2) 1.921 (1.939) 98.64 (99.43) 1.082 20.5 (23.2)
1a(t) C2 (C2) 1.921 (1.944) 117.28 (118.50) 0.854
1b(s) C1 (C1) 1.871 (1.884) 1.923 (1.942) 99.09 (99.75) 1.043 24.3 (26.8)
1b(t) C1 (C1) 1.877 (1.889) 1.922 (1.946) 116.82 (118.35) 0.868
2(s) C2 (C2) 1.883 (1.899) 87.50 (88.49) 1.051 35.9 (36.8)
2(t) C2 (C2) 1.892 (1.909) 96.75 (97.58) 0.906
2a(s) C2 (C2) 1.931 (1.951) 90.31 (91.25) 1.098 25.0 (26.7)
2a(t) C2 (C2) 1.938 (1.964) 100.34 (100.58) 0.853
2b(s) C1 (C1) 1.871 (1.889) 1.943 (1.961) 88.70 (89.72) 1.062 31.1 (32.1)
2b(t) C1 (C1) 1.891 (1.907) 1.939 (1.969) 98.50 (98.97) 0.875
3(s) C2v (C2v) 1.888 (1.905) 87.95 (88.42) 1.048 36.4 (37.9)
3(t) C2v (Cs) 1.892 (1.908) 96.64 (96.99) 0.922
3a(s) C2v (C2v) 1.941 (1.959) 91.87 (91.97) 1.116 24.5 (27.0)
3a(t) C2v (C2v) 1.936 (1.960) 100.08 (99.91) 0.862
3b(s) Cs (Cs) 1.881 (1.897) 1.947 (1.966) 89.82 (90.12) 1.076 30.9 (32.5)
3b(t) Cs (Cs) 1.897 (1.906) 1.935 (1.966) 98.06 (98.33) 0.886
Figure 3. Symmetric (WS) and asymmetric (WA) sets ofWalsh orbitals.

10552 dx.doi.org/10.1021/jp201067r |J. Phys. Chem. A 2011, 115, 10550–10555
The Journal of Physical Chemistry A ARTICLE
7.7 kcal/mol stabilization energy), the deformation of 2 permitsjust two interactions. However, the stabilization energy of each ofthese two interactions is more considerable than the individualinteractions in 1 and 3 (4.5 vs 2.5 and 1.9 kcal/mol). Thisphenomenon is geometrically observable via the unequal bondlengths of each cyclopropyls in 2, 1.549 (1.555) and 1.501(1.510) Å, while in the case of 3 and 1, both bonds of eachcyclopropyl are in a same length. Overall, cyclization increasesthe ΔES-T to 35.9 (36.8 kcal/mol), while unsaturation has asmaller effect and increases it to 36.4 (37.9 kcal/mol).Applying one cyclopropyl substituent on one side of the
silylene and an isopropyl on the other side in 1b�3b (Figure 2)create such structures with NBO atomic charges and ΔES-Tvalues between those of 1�3 and 1a�3a (Table 1). Hence, fromtwo isopropyl substituted silylenes to one and then two cyclopropylsubstituted ones, the stabilizing effect of W f p interaction isapparent from geometrical, energetic, and electronic viewpoints.To have a practical view, the ΔES-T of Kira’s synthesized
dialkylsilylene IV is calculated at the level of B3LYP/AUG-cc-pVTZ. TheΔES-T values of both 2 and 3, 36.8 and 37.9 kcal/mol,are close to that of IV with the ΔES-T of 33.1 kcal/mol. Even theΔES-T of acyclic 1, 31.2 kcal/mol, is just 1.9 kcal/mol less thanthe synthesized IV.3.2. Estimating W f p Interactions through Appropriate
Isodesmic Reactions. The stabilizing effects of W f p interac-tions on both singlet and triplet states of the studied silylenes canbe estimated through isodesmic reactions 1�3 (Scheme 1).Negative ΔES and ΔET values emphasize the stabilizing effectsofWf p interactions on both singlet and triplet states withmorepronounced effects on the formers. This is similar to thestabilizing effect of alkyl groups on both singlet and tripletstates of carbenes. Specifically, the isodesmic reaction appliedby Nemirowski and Schreiner showed that two methyl substit-uents stabilize singlet carbene by 27.8 kcal/mol. The samesubstituents have a less stabilizing effect on the triplet state by15.3 kcal/mol.27
The triplet state of 2 and the singlet state of 3 are the least andthe most stabilized species, respectively. It seems that the pre-sence of a double bond in 3 (s) induces some electron delocal-ization in the ring which accounts for 2.4 (2.4) kcal/mol higherstabilization than that of 2 (s), 15.8 (18.8) vs 13.4 (16.4) kcal/mol, respectively. Similarly, the isodesmic reactions 4�6 are
employed to find the stabilizing effect of only one cyclopropyl.The ΔE values obtained for these reactions are about half ofthose of the corresponding reactions 1�3. This shows that theaddition of the second cyclopropyl not only does not interruptthe stabilizing effect of the first one but also reinforces it.Effects of cyclization and unsaturation on the ΔES-Ts can be
differentiated for the singlet and triplet states using isodesmicreactions 7�9 (Scheme 2). Reaction 7 clearly shows that theincrease of ΔES-T during cyclization of 1 to 2 is related to 6.9(5.8) kcal/mol stabilization of the singlet, while the triplet state isnot considerably affected! Unsaturation of 2 shows a very slightdestabilizing effect on both singlet and triplet states by 0.2 and 0.6(0.2 and 1.3) kcal/mol, respectively (reaction 8). Based onreaction 9, simultaneous cyclization and unsaturation shows aconsiderable stabilizing effect on the singlet state but slightlydestabilizes the triplet state.3.3. Electronic Stabilization: Substituent Effect. Because
the presence of the double bond in 3 makes C3 and C4 reactive,we consider attachment of both electron-releasing and attractinggroups on these two sites. Through substitution on the double
Scheme 1 Scheme 2
Figure 4. MP2/6-31G(d) optimized structures of the singlet and triplet(in parentheses) substituted cyclic unsaturated dicyclopropylsilylenesalong with their MP2/6-311++G(d,p) and B3LYP/AUG-cc-pVTZ(in parentheses) calculated ΔES-Ts in kcal/mol.
10553 dx.doi.org/10.1021/jp201067r |J. Phys. Chem. A 2011, 115, 10550–10555
The Journal of Physical Chemistry A ARTICLE
bond of 3, it is found that electron-releasing NH2 does notsignificantly alter theΔES-T at MP2 level (36.4 vs 36.7 kcal/mol)but decreases it at B3LYP from 37.9 to 32.2 kcal/mol. On theother hand, electron-attracting CN increases it to 38.2 (41.6)kcal/mol (Figure 4). To confirm the favorable effect of electron-attracting groups, CF3 andNO2 are also introduced to the doublebond. The resulting ΔES-T values of 3NO2
and 3CF3, 39.9 (48.4)and 41.4 (42.9) kcal/mol, respectively, are increased comparedto that of 3H 36.4 (37.9) kcal/mol. Thus, electron-attractingsubstitutions on the double bond enlarge theΔES-T value of 3 infavor of the singlet state.3.4. Steric Stabilization: Dimerization. The stability of the
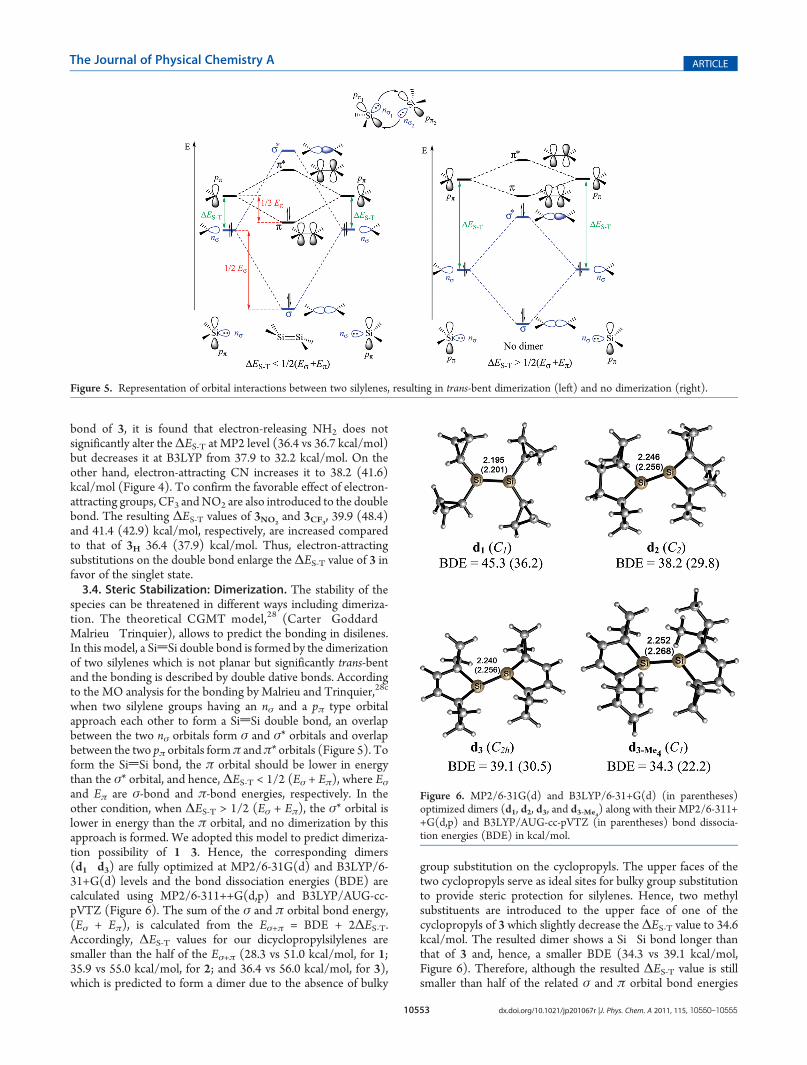
species can be threatened in different ways including dimeriza-tion. The theoretical CGMT model,28 (Carter�Goddard�Malrieu�Trinquier), allows to predict the bonding in disilenes.In this model, a SidSi double bond is formed by the dimerizationof two silylenes which is not planar but significantly trans-bentand the bonding is described by double dative bonds. Accordingto the MO analysis for the bonding by Malrieu and Trinquier,28c
when two silylene groups having an nσ and a pπ type orbitalapproach each other to form a SidSi double bond, an overlapbetween the two nσ orbitals form σ and σ* orbitals and overlapbetween the two pπ orbitals formπ andπ* orbitals (Figure 5). Toform the SidSi bond, the π orbital should be lower in energythan the σ* orbital, and hence, ΔES-T < 1/2 (Eσ + Eπ), where Eσand Eπ are σ-bond and π-bond energies, respectively. In theother condition, when ΔES-T > 1/2 (Eσ + Eπ), the σ* orbital islower in energy than the π orbital, and no dimerization by thisapproach is formed. We adopted this model to predict dimeriza-tion possibility of 1�3. Hence, the corresponding dimers(d1�d3) are fully optimized at MP2/6-31G(d) and B3LYP/6-31+G(d) levels and the bond dissociation energies (BDE) arecalculated using MP2/6-311++G(d,p) and B3LYP/AUG-cc-pVTZ (Figure 6). The sum of the σ and π orbital bond energy,(Eσ + Eπ), is calculated from the Eσ+π = BDE + 2ΔES-T.Accordingly, ΔES-T values for our dicyclopropylsilylenes aresmaller than the half of the Eσ+π (28.3 vs 51.0 kcal/mol, for 1;35.9 vs 55.0 kcal/mol, for 2; and 36.4 vs 56.0 kcal/mol, for 3),which is predicted to form a dimer due to the absence of bulky
group substitution on the cyclopropyls. The upper faces of thetwo cyclopropyls serve as ideal sites for bulky group substitutionto provide steric protection for silylenes. Hence, two methylsubstituents are introduced to the upper face of one of thecyclopropyls of 3 which slightly decrease theΔES-T value to 34.6kcal/mol. The resulted dimer shows a Si�Si bond longer thanthat of 3 and, hence, a smaller BDE (34.3 vs 39.1 kcal/mol,Figure 6). Therefore, although the resulted ΔES-T value is stillsmaller than half of the related σ and π orbital bond energies
Figure 5. Representation of orbital interactions between two silylenes, resulting in trans-bent dimerization (left) and no dimerization (right).
Figure 6. MP2/6-31G(d) and B3LYP/6-31+G(d) (in parentheses)optimized dimers (d1, d2, d3, and d3-Me4) along with their MP2/6-311++G(d,p) and B3LYP/AUG-cc-pVTZ (in parentheses) bond dissocia-tion energies (BDE) in kcal/mol.

10554 dx.doi.org/10.1021/jp201067r |J. Phys. Chem. A 2011, 115, 10550–10555
The Journal of Physical Chemistry A ARTICLE
(34.6 vs 51.8 kcal/mol), but their difference is decreasedcompared to that of 3, indicating the lower tendency fordimerization. In the next step, two methyl substituents areintroduced on each cyclopropyl, which completely rebuffs theprobability of dimerization because no stable dimer is found inthis case.3.5. HOMO�LUMO Gaps: Nucleophilicity and Electrophi-
licity. HOMO�LUMO gaps are traditionally associated withchemical stability against electronic excitations, with larger gapscorresponding to greater stability. Hence, the gaps are calculatedfor 1�3 and compared to those of II and IV at B3LYP/6-311++G(d,p)//B3LYP/6-31+G(d) (Table 2). The HOMO�LUMOgaps of our dicyclopropylsilylenes all are greater than Kira’ssynthesized dialkylsilylene (87.0, 90.4, 88.6 kcal/mol for 1�3,respectively, vs 84.0 kcal/mol for IV). Additionally, they are closeor even higher than the HOMO�LUMO gap of Denk’s synthe-sized diaminosilylene IV (88.1 kcal/mol). The HOMO andLUMO states are naturally attached to the concepts of nucleo-philicity and electrophilicity, respectively. The HOMO states of1�3 with the relative energies of about �5.90 eV are consider-ablymore stabilized than those of II and IV (�4.66 and�5.55 eV).As a result, one expects less nucleophilicities for the formerscompared to the latters. Indeed, the calculated nucleophilicityindices for dicyclopropylsilylenes are about 3.56 eV while thoseof II and IV are 4.80 and 3.91 eV, respectively. On the other hand,more stabilized LUMO states of 1�3 compared to those of IIand IVmake themmore electrophilic. This is consistent with theidea of theWf p interaction, which should stabilize the silyleneLUMO levels.
4. CONCLUSION
The geometrical, electronic, and energetic aspects of acyclicand saturated/unsaturated cyclic dicyclopropylsilylenes 1�3 arecompared and contrasted at the levels of MP2/6-311++G(d,p)//MP2/6-31G(d) andB3LYP/AUG-cc-pVTZ//B3LYP/6-31+G(d).The stabilizing interaction of the occupied Walsh orbitals ofthe cyclopropyls with the vacant p-orbital of the silylene center(W f p interaction) results in the ΔES-T values of 28.3 (31.2),35.9 (36.8), and 36.4 (37.9) kcal/mol for 1�3, respectively. Thisis while the ΔES-T values of the corresponding analogues thatpossess isopropyl groups instead of cyclopropyl are 20.5 (23.2),25.0 (26.7), and 24.5 (27.0) kcal/mol. Applying electron-attract-ing substitutions such as CN, CF3, and NO2 on the double bondincreases the ΔES-T of 3 while electron-releasing NH2 does notalter the gap. CGMT model predicts that bulky substituents onthe upper faces of cyclopropyls diminish the tendency fordimerization since they provide steric protection around the
silylene center. Hence, applying electron-attracting substituentson the double bond and suitable bulky substituents on cyclo-propyls, 3 will generate a new class of dialkylsilylenes.
’ASSOCIATED CONTENT
bS Supporting Information. Full ref 13 and Cartesian co-ordinates for all calculated structures. This material is availablefree of charge via the Internet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*Tel./Fax: +98(261)4463481. E-mail: [email protected].
’ACKNOWLEDGMENT
The authors wish to thank Dr. Mehdi Ghambarian for manystimulating and helpful discussions.
’REFERENCES
(1) See, for example: Drahnak, T. J.; Michl, J.; West, R. J. Am. Chem.Soc. 1979, 101, 5427.
(2) Arduengo, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991,113, 361.
(3) Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Haaland, A.;Belyakov, H.; Verne, P.; Wagner, M.; Metzler, N. J. Am. Chem. Soc.1994, 116, 2691.
(4) (a) Arduengo, A. J.; Bock, H.; Chen, H.; Denk, M.; Dixon, D. A.;Green, J. C.; Herrmann, W. A.; Jones, N. L.; Wagner, M.; West, R. J. Am.Chem. Soc. 1994, 116, 6641.(b) Schmedake, T. A.; Haaf, M.; West, R.Acc. Chem. Res. 2000, 33, 704 and references therein.
(5) Hill, N. J.; West, R. J. Organomet. Chem. 2004, 689, 4165 andreferences therein.
(6) Veszpr�emi, T.; Nyul�aszi, L.; K�arp�ati, T. J. Phys. Chem. A 1996,100, 6262.
(7) Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. J. Am. Chem. Soc.1999, 121, 9722.
(8) Kira, M.; Ishida, S.; Iwamoto, T.; Ichinohe, M.; Kabuto, C.;Ignatovich, L.; Sakurai, H. Chem. Lett. 1999, 263.
(9) Ammann, J. R.; Subramanian, R.; Sheridan, R. S. J. Am. Chem. Soc.1992, 114, 7592.
(10) (a) Møller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618.(b) Krishan, R.; Frisch, M. J.; Pople, J. A. J. Chem. Phys. 1980, 72, 4244.
(11) Becke, A. D. Phys. Rev. A 1988, 38, 3098.(12) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.(13) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.(14) Hariharan, P. C.; Pople, J. A. Mol. Phys. 1974, 27, 209.(15) Francl, M.M.; Pietro,W. J.; Hehre,W. J.; Binkley, J. S.; Gordon,
M. S.; DeFrees, D. J.; Pople, J. A. J. Chem. Phys. 1982, 77, 3654.(16) Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer,
P. v. R. J. Comput. Chem. 1983, 4, 294.(17) Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys. 1984,
80, 3265.(18) Frisch, M. J. et al. Gaussian 98; Gaussian, Inc.: Pittsburgh, PA,
1998. See the Supporting Information for the full reference.(19) Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys.
1992, 96, 6796.(20) Hehre, W. J.; Radom, L.; Schleyer, P. v. R. Pople, J. A. Ab Initio
Molecular Orbital Theory; John Wiley and Sons: New York, 1986.(21) Glendening, E. D.; Reed, A. E.; Carpenter, J. E.; Weinhold, F.
NBO, Version 3.1.(22) Domingo, L. R.; Chamorro, E.; P�erez, P. J. Org. Chem. 2008,
73, 4615.(23) Parr, R. G.; Szentpaly, L.; Liu, S. J. Am. Chem. Soc. 1999,
121, 1922.
Table 2. Nucleophilicity (N) and Global Electrophilicity (ω)Indices for the Singlet Ground States of All the ScrutinizedSilylenes Compared to the Synthesized Silylenes (II and IV)
structure
HOMO
(eV)
LUMO
(eV)
ΔEHOMO�LUMO
(kcal/mol)
N
(eV)
ω
(eV)
1 �5.78 �2.01 87.0 3.68 2.01
2 �5.89 �1.97 90.4 3.57 1.97
3 �6.03 �2.19 88.6 3.43 2.19
II �4.66 �0.84 88.1 4.80 0.99
IV �5.55 �1.91 84.0 3.91 1.91
TCNE �9.46 �5.31 95.7 0.00 6.57

10555 dx.doi.org/10.1021/jp201067r |J. Phys. Chem. A 2011, 115, 10550–10555
The Journal of Physical Chemistry A ARTICLE
(24) (a) Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512.(b) Parr, R. G.; Yang, W. Density Functional Theory of Atoms andMolecules; Oxford University Press: New York, 1989.(25) (a) Walsh, A. D. Nature (London) 1947, 159, 712. Trans.
Faraday SOC 1949, 45, 179. (b) Sugden, T. M. Nature (London) 1947,160, 367.(26) Schoeller, W. W. J. Org. Chem. 1980, 45, 2161.(27) Nemirowski, A.; Schreiner, P. R. J. Org. Chem. 2007, 72, 9533.(28) Thismodelwas suggested independently byCarter andGoddard
and by Trinquier and Malrieu: (a) Carter, A. E.; Goddard, W. A.,III. J. Phys. Chem. 1986, 90, 998. (b) Trinquier, G.; Malrieu, J.-P. J. Am.Chem. Soc. 1987, 109, 5303. (c) Malrieu, J.-P.; Trinquier, G. J. Am. Chem.Soc. 1989, 111, 5916. (d) Trinquier, G. J. Am. Chem. Soc. 1990, 112,1039.





























