Embed Size (px)
Citation preview

Electrochimica Acta 45 (1999) 387–397
New developments in cyclic voltammetry at the droppingmercury electrode
Francisco Martınez-Ortiz *, Marıa-Luisa Alcaraz, Isidoro RocaDepartamento de Quımica-Fısica, Uni6ersidad de Murcia, E-30071 Murcia, Spain
Received 27 January 1999; received in revised form 7 June 1999
Abstract
New theoretical and experimental results are presented for the response corresponding to a charge transfer reactionwhen cyclic voltammetry is applied to a dropping mercury electrode (DME). Amalgam formation for both reversibleand slow electrode processes has also been considered. We show that sphericity and convective effects can becontrolled experimentally so that it is easier to detect possible kinetic effects. Working curves for determining the rateconstants of electrode reactions are also given. © 1999 Elsevier Science Ltd. All rights reserved.
Keywords: Cyclic voltammetry; Amalgam formation; Dropping mercury electrode
www.elsevier.nl/locate/electacta
1. Introduction
The dropping mercury electrode (DME) possesses aset of suitable characteristics for use as a powerfulelectroanalytical tool. Among these are its clean andreproducible surface, as well as the absence of mechan-ical parts requiring regular attention. Hence, with littleeffort we can obtain a device useful for years. However,this electrode does have some disadvantages, for exam-ple, the dependence of electrode area on time, theconvective effects caused by growing the drop, thesphericity effects and the inconstancy of the mercuryflow rate in the early drop life, which give rise, undersome experimental conditions, to behaviour differentfrom that predicted by the physical models used todescribe the DME, and also the popularisation of staticelectrodes are responsible for decreasing use of theDME.
The DME has been used in linear sweep voltammetrysince its early application in polarographic studies [1].
Owing to the complexities of time-dependent electrodearea, sphericity and convective effects, the scans werecarried out on mature drops [2,3], with the suppositionthat the electrode then behaves as a stationary one andthe classic Randles–S& evcik [4,5] approach can be ap-plied. However, as we show in this paper, new applica-tions for the DME are possible in cyclic voltammetry.Thus, for example, the problems caused by the time-de-pendent electrode area can be eliminated by calculationof current density (this task is very easy using moderndigital storage systems) and sphericity and convectiveeffects can be controlled by keeping the appropriateexperimental variables constant. Problems associatedwith the inconstancy of mercury flow rate, which isonly relevant when the time scale used is in the range0.1–3 s, can be avoided simply by using a blank periodin the early drop life. The blank period can easily beobtained keeping the electrode either at open circuit orat a potential in which no appreciable faradaic elec-trode process takes place. Thus, during this blank pe-riod no other process, except the growth of the drop,takes place.
In order to show the above advantages, we havedivided this paper into two parts. In the first part, we
* Corresponding author.E-mail addresses: [email protected] (F. Martınez-Ortiz),
[email protected] (M.-L. Alcaraz)
0013-4686/99/$ - see front matter © 1999 Elsevier Science Ltd. All rights reserved.
PII: S 0 0 1 3 -4686 (99 )00279 -0

F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397388
have obtained theoretical cyclic voltammograms for asimple charge transfer reaction (for both electrolytesolution soluble product and amalgamation) takinginto account convective and sphericity effects for re-versible and slow electrode processes. Since it has notbeen possible to find the complete analytical solutionfor this problem, we have obtained a numerical one.There are some previous numerical descriptions for aDME applying other techniques [6,7] but, as far as weknow, there is no other theoretical treatment for theDME using cyclic voltammetry (with the considerationof sphericity and convective effects) in the literature. Inthe approach used in this paper, we have introduced aset of dimensionless variables and, after discretisationof the diffusion equations, we have carried out theimplicit calculation of both reduced and oxidised spe-cies concentration profiles, using the Crank–Nicolsonprocedure. The dependence of the theoretical voltam-mograms on the variables related to sphericity, convec-tive effects and kinetic parameters, is analysed andsome working curves, useful for the determination ofthe rate of the charge transfer reaction, are also given.In the second part of this paper we have obtained someexperimental cyclic voltammograms at a DME. Wediscuss therein both conditions under which convectiveand sphericity effects are important and how theseeffects can be controlled. We also show that in manycases, for identification of a slow electrode process,sphericity effects can be ignored. In addition, we indi-cate how the methodology presented in this paper canbe easily extended to more complex electrode processes.
2. Experimental
A computer driven potentiostat and sweep generatorwere designed and constructed by QUICELTRON(Spain). Data acquisition was performed using aDAS16-330i (ComputerBoards, USA) board. All com-puter programs for data acquisition and data transfor-mation were written in our laboratory. A DME EA1019-2 (METRHOM) was used, with mercury flowrates between 0.71 and 2.40 mg s−1. A saturatedcalomel electrode and a platinum electrode were used asreference and auxiliary electrodes. The blank periodwas obtained holding the electrode at the initial poten-tial, at which, under the experimental conditions givenin this paper, no measurable faradaic electrode processtakes place. Chemicals were analytical grade fromMERCK and were used as received. In all experimentsthe temperature was kept constant at 2590.2°C. Forcalculations, we have used literature values for thediffusion coefficients. Hence, we have used DCd
2+ =6.4×10−6 cm2 s−1 [8], DCd(Hg)=1.6×10−5 cm2 s−1
[8], D zn2+ =8.4×10−6 cm2 s−1 [9] and DZn(Hg)=1.7×
10−5 cm2 s−1 (see Ref. [3] p. 107).
3. Theory
Let us consider a charge transfer reaction
A+ne-?B (1)
where the product B is soluble either in solution or inthe electrode. Under these conditions, the mass trans-port, when cyclic voltammetry is applied to a DME,can be formulated as follows if we take the model of anexpanding sphere (see Ref. [3] p. 92).
#CA
#t=DA
�#2CA
#r2 +2r
#CA
#r�
−a3
3r2
#CA
#r(2a)
#CB
#t=DB
�#2CB
#r2 +2r
#CB
#r�
−a3
3r2
#CB
#r(2b)
where
a= (3mHg/4pdHg)1/3 (3)
In Eqs. (2a) and (2b) the first two terms of the right-hand side describe the diffusion to a stationary sphere;the third term accounts for the change in concentrationcaused by the motion of the medium.
The boundary value problem associated with thisprocess is given by (see nomenclature)
t=0,t\0,
r]r0
r��"
: cA=cA* (4)
t=0,t\0,
r]r0
r��"
: cB=cB* (SOLUBLE) (5a)
t=0,t\0,
r5r0
r�−�"
: cB=cB* (AMALGAM) (5b)
t\0, r=r0:
I(t)nFA(t+ te)
=DA
�#cA
#r�
r 0
=�DB
�#cB
#r�
r 0
(6)
I(t)nFA(t+ te)
=kf(E(t))cA(r0,t)−kb(E(t))cB(r0,t) (7)
te being a blank period previous to the application ofthe potential and r0 is the time dependent electroderadius. Note that during te there is no change in theinitial conditions, only the electrode area changes.
When the product is soluble in solution the condi-tions Eqs. (4), (5a) and (6) (with the upper sign) andEq. (7) are applied, whereas Eqs. (4), (5b) and (6) (withthe lower sign) and Eq. (7) describe the conditionscorresponding to amalgam formation. Note that wehave applied the Koutecky approximation [10] in Eq.(5b).
The heterogeneous rate constants kf and kb are re-lated to the potential applied E through the equations:
kf(E(t))=ksexp�−anF
RT(E(t)−E0)
�(8)

F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397 389
kb(E(t))=ksexp�(1−a)nF
RT(E(t)−E0)
�(9)
where the dependence on time of the potential E is asfollows:
05 t5 tR/2: E=E0−6t (10a)
t] tR/2: E=E0−6tR+6t (10b)
In these equations, E0 is the initial potential, n is thepotential sweep rate, tR is the experimental time-scalecoincident with the duration of the experiment and tR/2is the half period of the symmetric triangular wave.
We introduce the dimensionless variables
Ci=ci
cA*(i=A, B) (11)
V=t
tR
(12)
R=r−r0
(DAtR)(13)
z=a
DA1/2tR
1/6 (14)
G=ItR
1/2
nFA(te+ t)DA1/2cA*
(15)
KS=kStR
1/2
DA1/2 (16)
h=nF
RT(E−E0) (17)
where the normalized variable z is related to the rate offlow and density of mercury through a (see Eqs. (14) and(3)).
Then, the differential equations system correspondingto the diffusion becomes
#CA
#V=#2CA
#R2
+� z
3(Ve+V)2/3+2
R+z(Ve+V)1/3
−z3
3(R+z(Ve+V)1/3)2
n#CA
#R(18)
#CB
#V=
1g2
�#2CB
#R2
�+� z
3(Ve+V)2/3+2
g2(R+z(Ve+V)1/3)
−z3
3(R+z(Ve+V)1/3)2
n #CB
#R(19)
where
Ve=te
tR
(20)
g=�DA
DB
�1/2
(21)
and the normalised boundary conditions are
V=0,V\0,
R]0R��
": CA=1 (22)
V=0,V\0,
R]0R��
": CB=CB* (SOLUBLE) (23a)
V=0,V\0,
R50R�−�
": CB=CB* (AMALGAM)
(23b)
V\0, R=0:
G=�#CA
#R�
0
=�1g2
�#CB
#R�
0
(24)
G=Ks(CAe−ah−CBe (1−a)h) (25)
Under these conditions, the dependence on V of thedimensionless potential h is
05V50.5: h=h0−hMV (26a)
V]0.5: h=h0+hM(V−1) (26b)
where hM is the dimensionless potential sweep amplitude(see nomenclature) and h0 is the dimensionless initialpotential.
In our digital simulation we have considered theCrank–Nicolson procedure [11], with implicit calcula-tions for surface concentrations. By applying this methodwe find a recursive relation (see Appendix) which allowsus to carry out numerical calculations for surface concen-trations and to obtain theoretical curves.
Moreover, for convenience, in the dimensionlessvoltammograms included in this paper we have used theclassical current function c [12], given by
c=G
hM1/2=
I
nFA(te+ t)DA1/2cA*
�RT
nFn
�1/2
(27)
instead of the dimensionless variable G defined by Eq.(15).
4. Results and discussion
4.1. Numerical results
In the theoretical model used in this paper for theDME, we have used the Koutecky approximation [10](Eqs. (5b) and (23b)), which greatly simplifies the calcu-lations at a growing electrode and has been largely usedin the literature (see for example Ref. [13]). To test thisapproximation, we have carried out some calculationswith the stationary spherical electrode when amalgamformation takes place, using both the approximatemethod and the rigorous model [8]. The goodness of theKoucteky approximation depends on electrode spheric-
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397390
ity. Thus, for example, the error committed at a sta-tionary spherical electrode by use of this approximation(for 0.6BgB1.4) is lower than 0.3% in the peak cur-rents if (r0)/(DAtR)\1.5. In regard to a DME, thesignificant parameter is (z(Ve+V)1/3)= (r0)/(DAtR)(see notation and Eqs. (12), (14) and (20)). In order toensure good results, the faradaic process always beginsfor z(Ve+V)1/3\2 in all the calculated current-poten-tial curves included in this paper.
Once the Koutecky approximation is assumed in thetheoretical calculations carried out in cyclic voltamme-try at the DME, we must take into account the follow-ing variables: h0, hM, z, Ve CB* and g. If the chargetransfer reaction is slow, we must also consider a andKS. In all the experiments and calculations carried outin this paper we consider CB*=0 (note that if amalga-mation takes place, the consideration CB*"0 impliesthe use of an amalgam electrode. There is no theoreticallimitation on imposing this condition, but its use is veryinfrequent).
The current function depends on h0 and hM. Thedependence of cathodic peak on h0 is very weak andcan be ignored in most situations. Similarly, with welldeveloped peaks the dependence of the anodic peak onh0 at fixed hM is caused by the decaying cathodiccurrent, which attains different values depending on h0
(note that the potential at which the scan is reversedwill change if h0 varies). Taking this decaying cathodiccurrent as a base line, we find that the dependence onh0 of the current function corresponding to the anodicpeak is also very weak. The dependence on hM ismainly caused by the change in the reference time scale
(tR) on changing the sweep amplitude. However, in anykinetic experimental study it is usual to keep the sweepamplitude constant. Consequently, the influence of hM
on the cyclic voltammograms has no practical rele-vance. Thus, for a reversible process our attention willbe focused on the influence of Ve, z and g besides thebehaviour of the reaction product in relation to eitherthe formation of a solution soluble species oramalgamation.
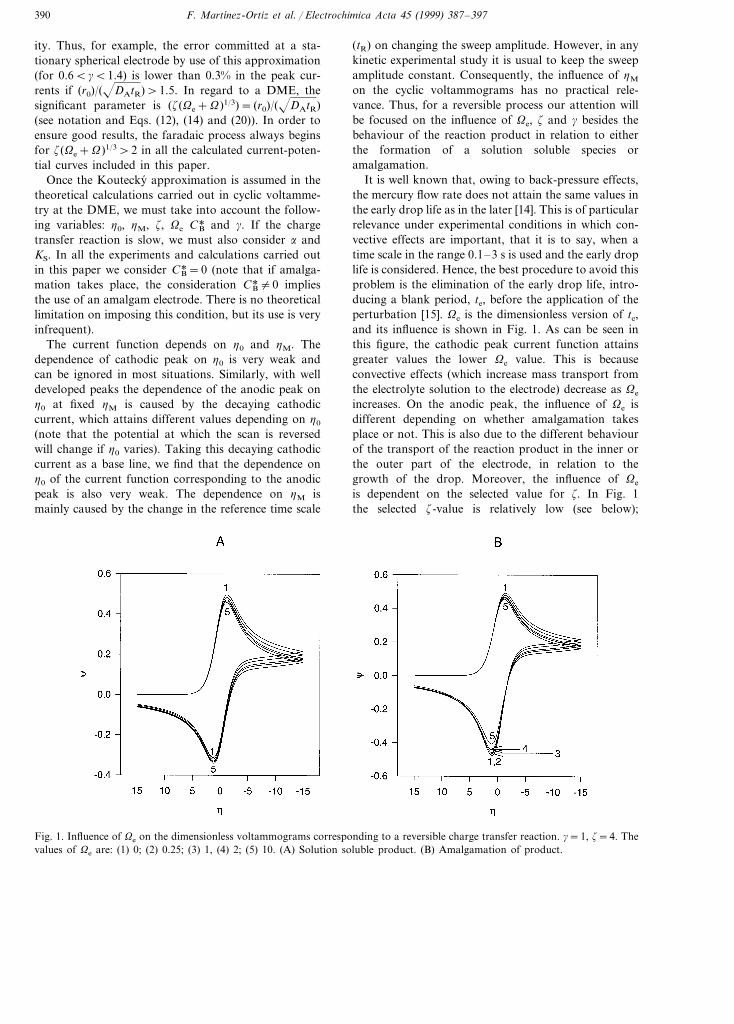
It is well known that, owing to back-pressure effects,the mercury flow rate does not attain the same values inthe early drop life as in the later [14]. This is of particularrelevance under experimental conditions in which con-vective effects are important, that it is to say, when atime scale in the range 0.1–3 s is used and the early droplife is considered. Hence, the best procedure to avoid thisproblem is the elimination of the early drop life, intro-ducing a blank period, te, before the application of theperturbation [15]. Ve is the dimensionless version of te,and its influence is shown in Fig. 1. As can be seen inthis figure, the cathodic peak current function attainsgreater values the lower Ve value. This is becauseconvective effects (which increase mass transport fromthe electrolyte solution to the electrode) decrease as Ve
increases. On the anodic peak, the influence of Ve isdifferent depending on whether amalgamation takesplace or not. This is also due to the different behaviourof the transport of the reaction product in the inner orthe outer part of the electrode, in relation to thegrowth of the drop. Moreover, the influence of Ve
is dependent on the selected value for z. In Fig. 1the selected z-value is relatively low (see below);
Fig. 1. Influence of Ve on the dimensionless voltammograms corresponding to a reversible charge transfer reaction. g=1, z=4. Thevalues of Ve are: (1) 0; (2) 0.25; (3) 1, (4) 2; (5) 10. (A) Solution soluble product. (B) Amalgamation of product.
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397 391
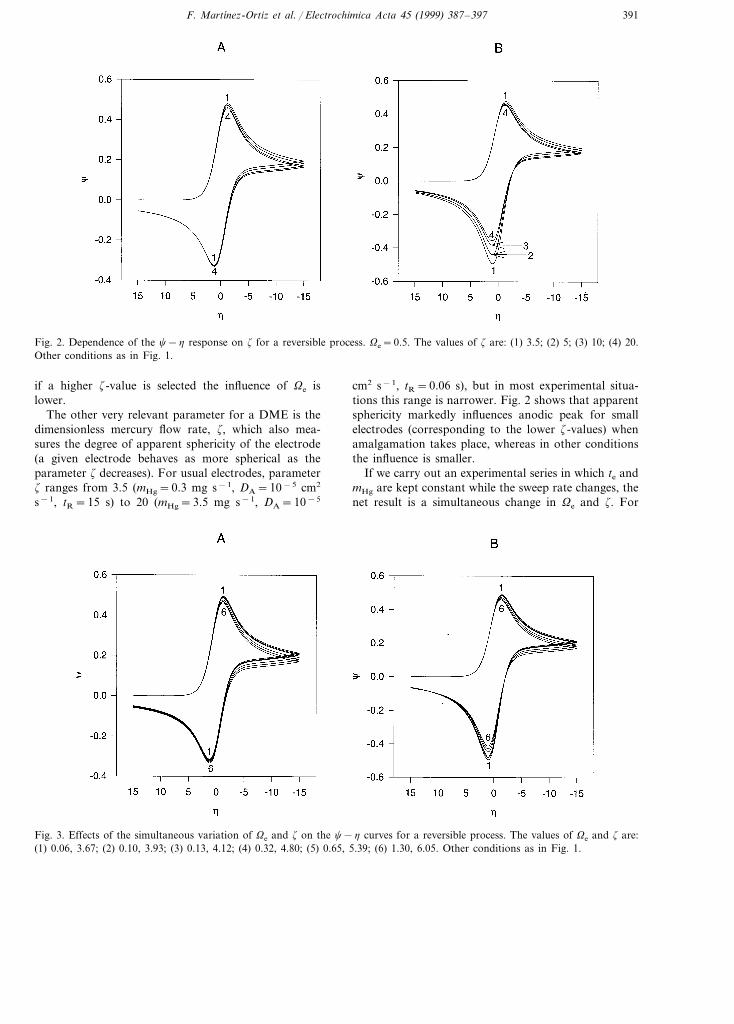
Fig. 2. Dependence of the c−h response on z for a reversible process. Ve=0.5. The values of z are: (1) 3.5; (2) 5; (3) 10; (4) 20.Other conditions as in Fig. 1.
if a higher z-value is selected the influence of Ve islower.
The other very relevant parameter for a DME is thedimensionless mercury flow rate, z, which also mea-sures the degree of apparent sphericity of the electrode(a given electrode behaves as more spherical as theparameter z decreases). For usual electrodes, parameterz ranges from 3.5 (mHg=0.3 mg s−1, DA=10−5 cm2
s−1, tR=15 s) to 20 (mHg=3.5 mg s−1, DA=10−5
cm2 s−1, tR=0.06 s), but in most experimental situa-tions this range is narrower. Fig. 2 shows that apparentsphericity markedly influences anodic peak for smallelectrodes (corresponding to the lower z-values) whenamalgamation takes place, whereas in other conditionsthe influence is smaller.
If we carry out an experimental series in which te andmHg are kept constant while the sweep rate changes, thenet result is a simultaneous change in Ve and z. For
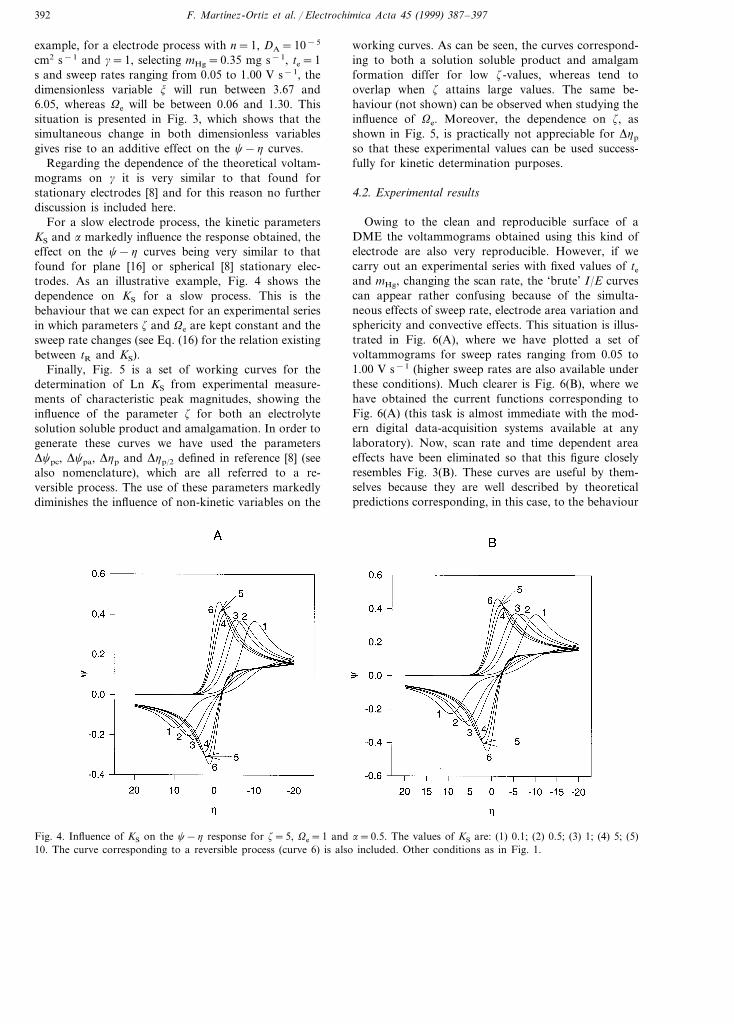
Fig. 3. Effects of the simultaneous variation of Ve and z on the c−h curves for a reversible process. The values of Ve and z are:(1) 0.06, 3.67; (2) 0.10, 3.93; (3) 0.13, 4.12; (4) 0.32, 4.80; (5) 0.65, 5.39; (6) 1.30, 6.05. Other conditions as in Fig. 1.
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397392
example, for a electrode process with n=1, DA=10−5
cm2 s−1 and g=1, selecting mHg=0.35 mg s−1, te=1s and sweep rates ranging from 0.05 to 1.00 V s−1, thedimensionless variable j will run between 3.67 and6.05, whereas Ve will be between 0.06 and 1.30. Thissituation is presented in Fig. 3, which shows that thesimultaneous change in both dimensionless variablesgives rise to an additive effect on the c−h curves.
Regarding the dependence of the theoretical voltam-mograms on g it is very similar to that found forstationary electrodes [8] and for this reason no furtherdiscussion is included here.
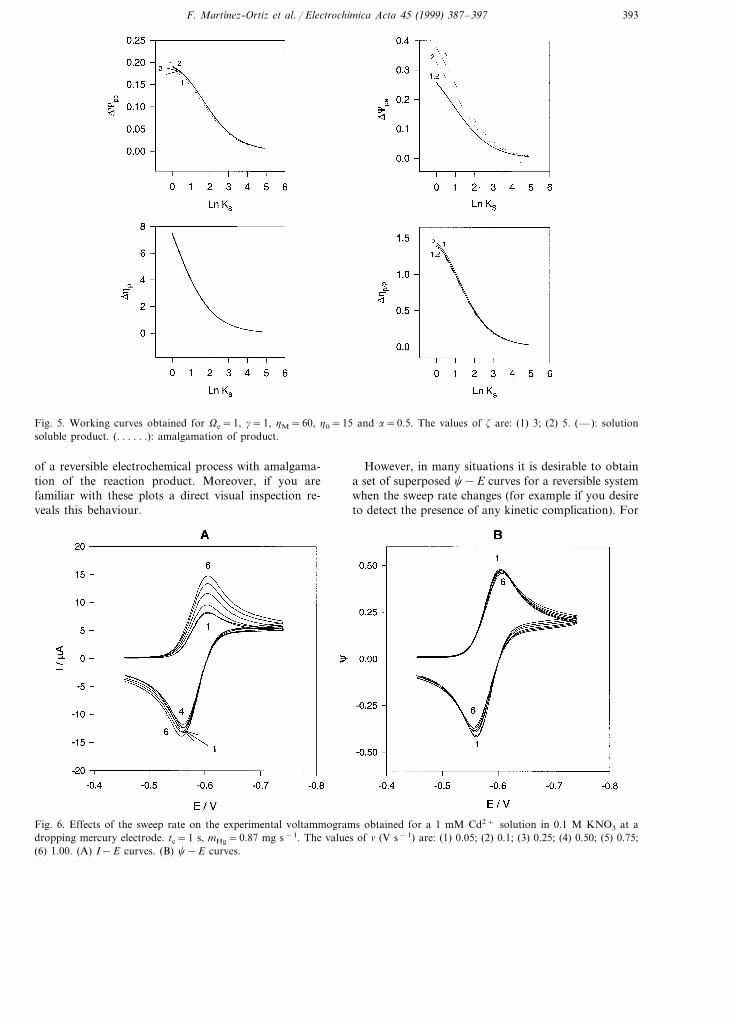
For a slow electrode process, the kinetic parametersKS and a markedly influence the response obtained, theeffect on the c−h curves being very similar to thatfound for plane [16] or spherical [8] stationary elec-trodes. As an illustrative example, Fig. 4 shows thedependence on KS for a slow process. This is thebehaviour that we can expect for an experimental seriesin which parameters z and Ve are kept constant and thesweep rate changes (see Eq. (16) for the relation existingbetween tR and KS).
Finally, Fig. 5 is a set of working curves for thedetermination of Ln KS from experimental measure-ments of characteristic peak magnitudes, showing theinfluence of the parameter z for both an electrolytesolution soluble product and amalgamation. In order togenerate these curves we have used the parametersDcpc, Dcpa, Dhp and Dhp/2 defined in reference [8] (seealso nomenclature), which are all referred to a re-versible process. The use of these parameters markedlydiminishes the influence of non-kinetic variables on the
working curves. As can be seen, the curves correspond-ing to both a solution soluble product and amalgamformation differ for low z-values, whereas tend tooverlap when z attains large values. The same be-haviour (not shown) can be observed when studying theinfluence of Ve. Moreover, the dependence on z, asshown in Fig. 5, is practically not appreciable for Dhp
so that these experimental values can be used success-fully for kinetic determination purposes.
4.2. Experimental results
Owing to the clean and reproducible surface of aDME the voltammograms obtained using this kind ofelectrode are also very reproducible. However, if wecarry out an experimental series with fixed values of te
and mHg, changing the scan rate, the ‘brute’ I/E curvescan appear rather confusing because of the simulta-neous effects of sweep rate, electrode area variation andsphericity and convective effects. This situation is illus-trated in Fig. 6(A), where we have plotted a set ofvoltammograms for sweep rates ranging from 0.05 to1.00 V s−1 (higher sweep rates are also available underthese conditions). Much clearer is Fig. 6(B), where wehave obtained the current functions corresponding toFig. 6(A) (this task is almost immediate with the mod-ern digital data-acquisition systems available at anylaboratory). Now, scan rate and time dependent areaeffects have been eliminated so that this figure closelyresembles Fig. 3(B). These curves are useful by them-selves because they are well described by theoreticalpredictions corresponding, in this case, to the behaviour
Fig. 4. Influence of KS on the c−h response for z=5, Ve=1 and a=0.5. The values of KS are: (1) 0.1; (2) 0.5; (3) 1; (4) 5; (5)10. The curve corresponding to a reversible process (curve 6) is also included. Other conditions as in Fig. 1.
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397 393
Fig. 5. Working curves obtained for Ve=1, g=1, hM=60, h0=15 and a=0.5. The values of z are: (1) 3; (2) 5. (—): solutionsoluble product. (. . . . . .): amalgamation of product.
of a reversible electrochemical process with amalgama-tion of the reaction product. Moreover, if you arefamiliar with these plots a direct visual inspection re-veals this behaviour.
However, in many situations it is desirable to obtaina set of superposed c−E curves for a reversible systemwhen the sweep rate changes (for example if you desireto detect the presence of any kinetic complication). For
Fig. 6. Effects of the sweep rate on the experimental voltammograms obtained for a 1 mM Cd2+ solution in 0.1 M KNO3 at adropping mercury electrode. te=1 s, mHg=0.87 mg s−1. The values of n (V s−1) are: (1) 0.05; (2) 0.1; (3) 0.25; (4) 0.50; (5) 0.75;(6) 1.00. (A) I−E curves. (B) c−E curves.
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397394
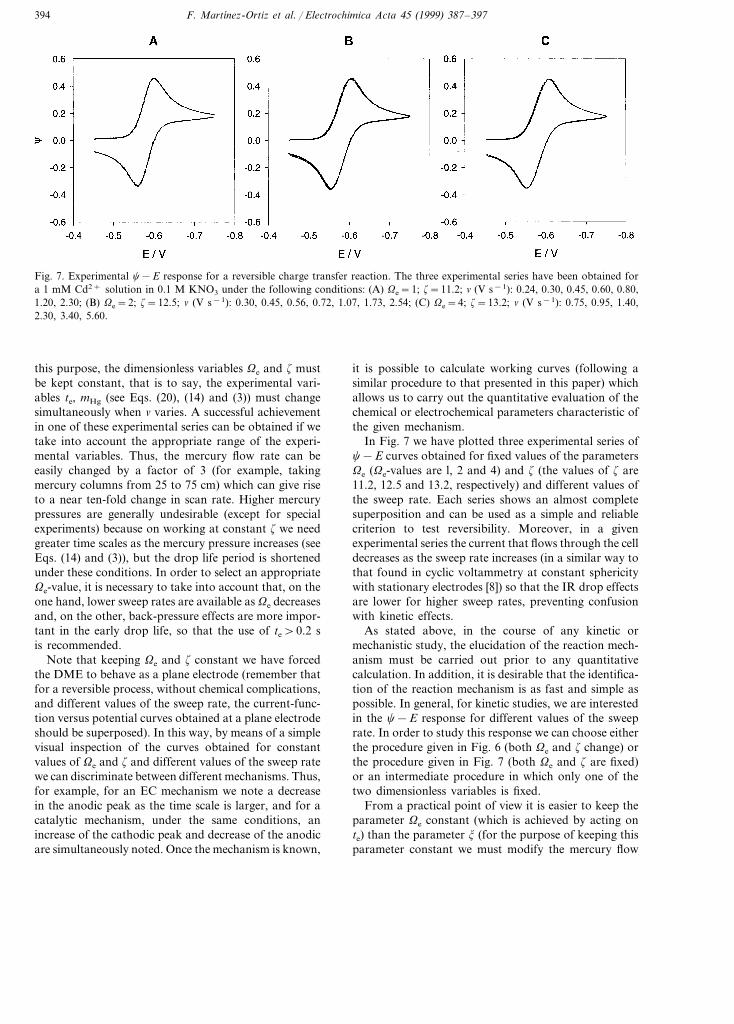
Fig. 7. Experimental c−E response for a reversible charge transfer reaction. The three experimental series have been obtained fora 1 mM Cd2+ solution in 0.1 M KNO3 under the following conditions: (A) Ve=1; z=11.2; n (V s−1): 0.24, 0.30, 0.45, 0.60, 0.80,1.20, 2.30; (B) Ve=2; z=12.5; n (V s−1): 0.30, 0.45, 0.56, 0.72, 1.07, 1.73, 2.54; (C) Ve=4; z=13.2; n (V s−1): 0.75, 0.95, 1.40,2.30, 3.40, 5.60.
this purpose, the dimensionless variables Ve and z mustbe kept constant, that is to say, the experimental vari-ables te, mHg (see Eqs. (20), (14) and (3)) must changesimultaneously when n varies. A successful achievementin one of these experimental series can be obtained if wetake into account the appropriate range of the experi-mental variables. Thus, the mercury flow rate can beeasily changed by a factor of 3 (for example, takingmercury columns from 25 to 75 cm) which can give riseto a near ten-fold change in scan rate. Higher mercurypressures are generally undesirable (except for specialexperiments) because on working at constant z we needgreater time scales as the mercury pressure increases (seeEqs. (14) and (3)), but the drop life period is shortenedunder these conditions. In order to select an appropriateVe-value, it is necessary to take into account that, on theone hand, lower sweep rates are available as Ve decreasesand, on the other, back-pressure effects are more impor-tant in the early drop life, so that the use of te\0.2 sis recommended.
Note that keeping Ve and z constant we have forcedthe DME to behave as a plane electrode (remember thatfor a reversible process, without chemical complications,and different values of the sweep rate, the current-func-tion versus potential curves obtained at a plane electrodeshould be superposed). In this way, by means of a simplevisual inspection of the curves obtained for constantvalues of Ve and z and different values of the sweep ratewe can discriminate between different mechanisms. Thus,for example, for an EC mechanism we note a decreasein the anodic peak as the time scale is larger, and for acatalytic mechanism, under the same conditions, anincrease of the cathodic peak and decrease of the anodicare simultaneously noted. Once the mechanism is known,
it is possible to calculate working curves (following asimilar procedure to that presented in this paper) whichallows us to carry out the quantitative evaluation of thechemical or electrochemical parameters characteristic ofthe given mechanism.
In Fig. 7 we have plotted three experimental series ofc−E curves obtained for fixed values of the parametersVe (Ve-values are l, 2 and 4) and z (the values of z are11.2, 12.5 and 13.2, respectively) and different values ofthe sweep rate. Each series shows an almost completesuperposition and can be used as a simple and reliablecriterion to test reversibility. Moreover, in a givenexperimental series the current that flows through the celldecreases as the sweep rate increases (in a similar way tothat found in cyclic voltammetry at constant sphericitywith stationary electrodes [8]) so that the IR drop effectsare lower for higher sweep rates, preventing confusionwith kinetic effects.
As stated above, in the course of any kinetic ormechanistic study, the elucidation of the reaction mech-anism must be carried out prior to any quantitativecalculation. In addition, it is desirable that the identifica-tion of the reaction mechanism is as fast and simple aspossible. In general, for kinetic studies, we are interestedin the c−E response for different values of the sweeprate. In order to study this response we can choose eitherthe procedure given in Fig. 6 (both Ve and z change) orthe procedure given in Fig. 7 (both Ve and z are fixed)or an intermediate procedure in which only one of thetwo dimensionless variables is fixed.
From a practical point of view it is easier to keep theparameter Ve constant (which is achieved by acting onte) than the parameter j (for the purpose of keeping thisparameter constant we must modify the mercury flow
F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397 395
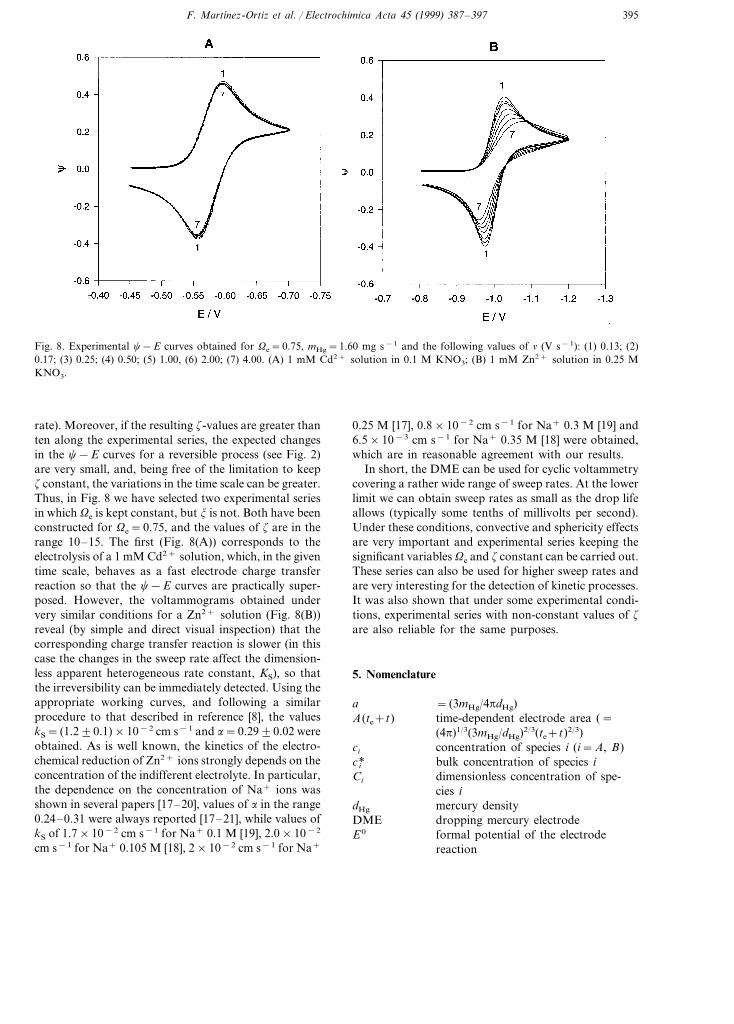
Fig. 8. Experimental c−E curves obtained for Ve=0.75, mHg=1.60 mg s−1 and the following values of n (V s−1): (1) 0.13; (2)0.17; (3) 0.25; (4) 0.50; (5) 1.00, (6) 2.00; (7) 4.00. (A) 1 mM Cd2+ solution in 0.1 M KNO3; (B) 1 mM Zn2+ solution in 0.25 MKNO3.
rate). Moreover, if the resulting z-values are greater thanten along the experimental series, the expected changesin the c−E curves for a reversible process (see Fig. 2)are very small, and, being free of the limitation to keepz constant, the variations in the time scale can be greater.Thus, in Fig. 8 we have selected two experimental seriesin which Ve is kept constant, but j is not. Both have beenconstructed for Ve=0.75, and the values of z are in therange 10–15. The first (Fig. 8(A)) corresponds to theelectrolysis of a 1 mM Cd2+ solution, which, in the giventime scale, behaves as a fast electrode charge transferreaction so that the c−E curves are practically super-posed. However, the voltammograms obtained undervery similar conditions for a Zn2+ solution (Fig. 8(B))reveal (by simple and direct visual inspection) that thecorresponding charge transfer reaction is slower (in thiscase the changes in the sweep rate affect the dimension-less apparent heterogeneous rate constant, KS), so thatthe irreversibility can be immediately detected. Using theappropriate working curves, and following a similarprocedure to that described in reference [8], the valueskS= (1.290.1)×10−2 cm s−1 and a=0.2990.02 wereobtained. As is well known, the kinetics of the electro-chemical reduction of Zn2+ ions strongly depends on theconcentration of the indifferent electrolyte. In particular,the dependence on the concentration of Na+ ions wasshown in several papers [17–20], values of a in the range0.24–0.31 were always reported [17–21], while values ofkS of 1.7×10−2 cm s−1 for Na+ 0.1 M [19], 2.0×10−2
cm s−1 for Na+ 0.105 M [18], 2×10−2 cm s−1 for Na+
0.25 M [17], 0.8×10−2 cm s−1 for Na+ 0.3 M [19] and6.5×10−3 cm s−1 for Na+ 0.35 M [18] were obtained,which are in reasonable agreement with our results.
In short, the DME can be used for cyclic voltammetrycovering a rather wide range of sweep rates. At the lowerlimit we can obtain sweep rates as small as the drop lifeallows (typically some tenths of millivolts per second).Under these conditions, convective and sphericity effectsare very important and experimental series keeping thesignificant variables Ve and z constant can be carried out.These series can also be used for higher sweep rates andare very interesting for the detection of kinetic processes.It was also shown that under some experimental condi-tions, experimental series with non-constant values of z
are also reliable for the same purposes.
5. Nomenclature
a = (3mHg/4pdHg)A(te+t) time-dependent electrode area (=
(4p)1/3(3mHg/dHg)2/3(te+t)2/3)ci concentration of species i (i=A, B)c i* bulk concentration of species iCi dimensionless concentration of spe-
cies idHg mercury densityDME dropping mercury electrode
formal potential of the electrodeE0
reaction

F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397396
E0 initial potential applied to theelectrode
G dimensionless current (=ItR1/2/
(nFA(te+t)DA1/2c*A))
h dimensionless space intervalheterogeneous rate constants of thekf, kb
forward and reverse charge trans-fer reactions
kS standard rate constant of an elec-trode reaction
dimensionless standard rate constantKS
of an electrode reaction (=kStR1/2/
DA1/2)
potential sweep amplitudeMmercury flow ratemHg
distance from the centre of therelectrode
r0 electrode radius at time te+t (=(3mHg/4pdHg)1/3(te+t)1/3)
R dimensionless variable correspondingto the distance from the surfaceof the dropping mercury electrode(= (r−r0)/(DAtR)1/2)
time elapsed between the applicationtof the potential and the currentmeasurement
blank period previous to the appli-te
cation of the potentialexperimental time-scale coincidenttR
with the duration of the experiment(=M/n)potential sweep raten
Greek letters
electron transfer coefficienta
= (DA/DB)1/2g
= (Vj+1−Vj)/(h)2l
dimensionless variable (=a/(DA1/2tR
1/z
6))dimensionless potential (=nF(E−h
E0)/(RT))h0 dimensionless initial potential
dimensionless potential sweep am-hM
plitude (= (nF/RT)M)dimensionless cathodic half-peakhpc/2
potentialdimensionless cathodic half-peakhpc/2(rev)potential corresponding to a re-versible processdimensionless cathodic and anodichpc, hpa
peak potentialsdimensionless cathodic and anodichpc(rev), hpa(rev)peak potentials corresponding to areversible process
�(hpc−hpa)−(hpc(rev)−hpa(rev))�Dhp
�(hpc−hpc/2)−(hpc(rev)−hpc/2(rev))�Dhp/2
(cpc(rev)−cpc)/cpc(rev)Dcpc
(cpa−cpa(rev)/cpc(rev)Dcpa
c current function (=G/hM1/2)
cpc, cpa current functions corresponding tothe cathodic and anodic peakscurrent functions corresponding tocpc(rev), cpa(rev)the cathodic and anodic peaks fora reversible processdimensionless time of application ofV
the potential (= t/tR)Ve dimensionless blank periodOther symbols are conventional
Acknowledgements
The authors greatly appreciate the financial supportfrom the Direccion General de Investigacion Cientıficay Tecnica (Spain), project no PB96-1095.
Appendix A
The discretized forms of the partial differential diffu-sion Eq. (18) and Eq. (19) are
aj+1, ik Cj+1, i−1
k +bkCj+1, ik +dj+1, i
k Cj+1, i+1k =e j,i
k
(k=A,B); 15 i5 imax−1 (A1)
where
aj+1, iA =1−
hz
6(Ve+Vj+1)2/3−h
ih+z(Ve+Vj+1)1/3
+hz3
6(ih+z(Ve+Vj+1)1/3)2 (A2a)
aj+1, iB =
1g2−
hz
6(Ve+Vj+1)2/3
−h
g2(9 ih+z(Ve+Vj+1)1/3)
+hz3
6(9 ih+z(Ve+Vj+1)1/3)2 (A2b)
bA= −2l
(l+1) (A3a)
bB= −2l
� l
g2+1�
(A3b)
dj+1, iA =1+
hz
6(Ve+Vj+1)2/3+h
ih+z(Ve+Vj+1)1/3
−hz3
6(ih+z(Ve+Vj+1)1/3)2 (A4a)

F. Martınez-Ortiz et al. / Electrochimica Acta 45 (1999) 387–397 397
G=−25Cj+1,0
A +48cJ+1,1A −36Cj+1,2
A +16Cj+1,3A −3Cj+1,4
A
12h
= −1g2
�−25Cj+1,0B +48Cj+1,1
B −36Cj+1,2B +16Cj+1,3
B −3Cj+1,4B
12h�
(A13)
dj+1, iB =
1g2+
hz
6(Ve+Vj+1)2/3
+h
g2(9 ih+z(Ve+Vj+1)1/3)
−hz3
6(9 ih+z(Ve+Vj+1)1/3)2 (A4b)
e j,iA = −aj, i
A Cj, i−1A +
2l
(l−1)Cj,iA −dj,i
ACj, i+1A (A5a)
e j,iB = −aj,i
BCj, i−1B +
2l
� l
g2−1�
Cj,iB −dj,i
BCj, i+1B (A5b)
with
l=Vj+1−Vj
h2 (A6)
Cj,i max
k =Cj+1,i max
k =Ck* (k=A,B) (A7)
In these equations the subindex j refers to the differentdimensionless discrete time-values. Moreover, we haveassumed R= ih (for the diffusion of species A) andR=9 ih (for the diffusion of species B), and we haveapplied the Crank–Nicolson procedure. The upper signin R=9 ih, which also appears in Eqs. (A2b) and (A4b),refers to a solution soluble product and the lower onerefers to amalgam formation.
By carrying out a reiterative procedure we transformthe tridiagonal system (A1) into a bidiagonal one. In thisway the following recursive relation is found:
aj+1,ik Cj+1,i−1
k +bj+1,i’k Cj+1,i
k =e j,i’k (k=A, B) ; 15 i
5 imax−1 (A8)
where
bj+1,i’k =bk−
aj+1,i+1k d j+1,i
k
b j+1,i+1’k (k=A, B) ; 15 i
5 imax−2 (A9)
e j+1,i’k =e j,i
k −dj+1,i
k e j,i+1’k
b j+1,i+1’k (k=A, B) ; 15 i5 imax−2
(A10)
bj+1,i max−1
’k =bk (k=A, B) (A11)
e j,i max−1
’k =e j,i max−1
k −dj+1,i max−1
k Ck* (k=A, B)(A12)
By using the five-point approximation for the individualconcentration gradients at the electrode surface weobtain:
where the dimensionless faradic flux at the electrodesurface G is related to the dimensionless potential h
through the equation:
G=KS(Cj+1,0A e−ahj+1−Cj+1,0
B e (1−a)hj+1) (A14)
Finally, if we use the first four equations of the sys-tem (A8) (for k=A and k=B) and the three corre-sponding to Eqs. (A13) and (A14), we can solve asystem of 11 equations and in this way the implicitcalculation of surface concentrations may be carriedout.
References
[1] N. Nichols, Trans. Electrochem. Soc. 73 (1938) 193.[2] J. Vogel, in: P. Zuman, I.M. Kolthoff, Progress in Po-
larography, vol. II, Interscience, New York, 1962, p.429.
[3] J. Heyrovsky, J. Kuta, Principles of Polarography, Aca-demic Press, New York, 1966.
[4] J.E.B. Randles, Trans. Faraday Soc. 44 (1948) 322.[5] A. S& evcik, Collect. Czech. Chem. Commun. 13 (1948)
349.[6] S.W. Feldberg, J. Electroanal. Chem. 109 (1980) 69.[7] S. Pons, B. Speiser, J.F. McAleer, Electrochim. Acta 27
(1982) 1711.[8] F. Martınez-Ortiz, M.L. Alcaraz, I. Roca, M. Lopez-
Tenes, J. Electroanal. Chem. 443 (1998) 243.[9] P. Deenen, M. Lindstrom, G. Sundholm, J. Electroanal.
Chem. 101 (1979) 291.[10] J. Koutecky, Czech. J. Phys. 2 (1953) 50.[11] D. Britz, Digital Simulation in Electrochemistry,
Springer, Berlin, 1988.[12] R.S. Nicholson, I. Shain, Anal. Chem. 36 (1964) 706.[13] A. Molina, F. Martınez-Ortiz, C. Serna, J. Electroanal.
Chem. 336 (1992) 1.[14] G.C. Barker, A.W. Gardner, in: I.S. Longmuir (Ed.),
Advances in Polarography, vol. I, Pergamon, London,1960, p. 330.
[15] A. Molina, F. Martınez-Ortiz, J. Zapata, J. Albaladejo,J. Electroanal. Chem. 227 (1987) 1.
[16] R.S. Nicholson, Anal. Chem. 37 (1965) 1351.[17] J. Koryta, Electrochim. Acta 6 (1962) 67.[18] J.H. Christie, E.P. Parry, R.A. Osteryoung, Elec-
trochim. Acta 11 (1966) 1525.[19] N. Tanaka, Pure Appl. Chem. 44 (1975) 627.[20] B. Workie, T. Solomon, J. Electroanal. Chem. 224
(1987) 49.[21] A. Baars, M. Sluyters-Rehbach, J.H. Sluyters, J. Elec-
troanal. Chem. 364 (1994) 189.













![Cyclic Voltammetry of Zirconyl Chloride (ZrOCl2) in KF ......Solutions of zirconyl chloride reach their maximum hydrolysis in three hours [7-10]. Table 3. Cyclic voltammetry parameters](https://img.pdfslide.us/doc/110x75/611a62ecf0687b2382647c1c/cyclic-voltammetry-of-zirconyl-chloride-zrocl2-in-kf-solutions-of-zirconyl.jpg)















