Embed Size (px)
Citation preview

Neuroprotection by Chlorpromazine and Promethazine in SevereTransient and Permanent Ischemic Stroke
Xiaokun Geng1,2 & Fengwu Li1 & James Yip2& Changya Peng2 & Omar Elmadhoun2
&
Jiamei Shen1& Xunming Ji1,3 & Yuchuan Ding1,2
Received: 29 June 2016 /Accepted: 31 October 2016 /Published online: 28 November 2016# Springer Science+Business Media New York 2016
Abstract Previous studies have demonstrated depressive orhibernation-like roles of phenothiazine neuroleptics [com-bined chlorpromazine and promethazine (C + P)] in brainactivity. This ischemic stroke study aimed to establish neuro-protection by reducing oxidative stress and improving brainmetabolism with post-ischemic C + P administration.Sprague-Dawley rats were subjected to transient (2 or 4 h)middle cerebral artery occlusion (MCAO) followed by 6 or24 h reperfusion, or permanent (28 h) MCAO without reper-fusion. At 2 h after ischemia onset, rats received either anintraperitoneal (IP) injection of saline or two doses of C + P.Body temperatures, brain infarct volumes, and neurologicaldeficits were examined. Oxidative metabolism and stress weredetermined by levels of ATP, NADH, and reactive oxygenspecies (ROS). Protein kinase C-δ (PKC-δ) and Akt expres-sion were determined by Western blotting. C + P administra-tion induced a neuroprotection in both transient and perma-nent ischemia models evidenced by significant reduction ininfarct volumes and neurological deficits post-stroke. C + Pinduced a dose-dependent reduction in body temperature asearly as 5 min post-ischemia and lasted up to 12 h. However,reduction in body temperature either only slightly or did not
enhance C + P-induced neuroprotection. C + P therapy im-proved brain metabolism as determined by increased ATPlevels and NADH activity, as well as decreased ROS produc-tion. These therapeutic effects were associated with alterationsin PKC-δ and Akt protein expression. C + P treatments con-ferred neuroprotection in severe stroke models by suppressingthe damaging cascade of metabolic events, most likely inde-pendent of drug-induced hypothermia. These findings furtherprove the clinical potential for C + P treatment and may directus closer towards the development of an efficacious neuropro-tective therapy.
Keywords Hibernation-like therapeutic effect . Ischemia/reperfusion . Brain metabolism . ROS
Introduction
Stroke is one of the most debilitating vascular diseases world-wide with accompanying health care costs as high as $38.6billion each year in the USA [1]. Reperfusion strategies suchas systemic thrombolysis with intravenous (IV) tissue plas-minogen activator (tPA) and in situ clot retrieval, but not neu-roprotection strategies, remain the major therapy for strokepatients. Given the several limitations and potential complica-tions of reperfusion therapy, however, the vast majority ofpatients with acute ischemic stroke have not benefited fromthis strategy. Although a small portion of patients (17%) ex-perience spontaneous thrombolysis by 6–8 h [2], many pa-tients suffer from permanent arterial occlusion [3]. Even ifrecanalization is successful, outcome is sometimes poor dueto reperfusion injury [4].
Experimental animals have been shown to be protectedfrom the adverse effects of blood loss and oxygen deprivationwhen they are maintained in a suspended [5] or hibernation
* Xunming [email protected]
* Yuchuan [email protected]
1 China-America Institute of Neuroscience, Beijing Luhe Hospital,Capital Medical University, Beijing 101100, China
2 Department of Neurosurgery, Wayne State University School ofMedicine, 550 E Canfield, Detroit, MI 48201, USA
3 Department of Neurosurgery, Xuanwu Hospital, Capital MedicalUniversity, Beijing, China
Mol Neurobiol (2017) 54:8140–8150DOI 10.1007/s12035-016-0280-x

state, a result of the downregulation of energy metabolism [6,7]. Thus, inducing a “hibernation-like” state with depressedenergy utilization through the use of anesthetics has drawnmuch interest as a potential neuroprotective strategy.However, limitations such as the need to apply anestheticsbefore the onset of ischemia as well as several toxicity issueshave narrowed their clinical potential [8]. Alternatively, hypo-thermia has been recognized as a robust “hibernation-like”neuroprotectant because of its profoundly depressive effecton metabolism [9]. The application of hypothermia in ische-mic stroke patients is largely limited by the delayed coolingonset, prolonged duration, extensive medical and nursing ef-forts, and secondary complications [10]. Instead, phenothia-zine drugs, in addition to their antipsychotic and sedative ef-fects, which have been demonstrated to induce “artificial hi-bernation” [11] and neuroprotection by previous experimentalwork in ischemia [12–15], are therefore the focus of this study.
Since an early reperfusion strategy may not be viable formost stroke patients, and since the ischemic regions of thebrain have patent collateral circulations for effective drug de-livery [16–18], we determined whether chlorpromazine andpromethazine (C + P) therapy confers neuroprotection in se-vere stroke by reducing brain metabolism. We aimed to applyC + P therapy in more clinically relevant stroke models byinducing either longer ischemia periods (4 h) or permanent(28 h) ischemia without reperfusion. As protein kinase C-δ(PKC-δ) and Akt/PKB are thought to play key roles in reper-fusion injury [19], changes in both PKC-δ and Akt were alsoinvestigated to understand the mechanisms underlying neuro-protection induced by C + P. Results from this study couldprovide the basis for a potential stroke therapy that is relativelyeasy to implement.
Materials and Methods
Subjects All experimental procedures were approved by theInstitutional Animal Investigation Committee of CapitalMedical University in accordance with the NationalInstitutes of Health (USA) guidelines for care and use of lab-oratory animals. A total of 272 adult male Sprague-Dawleyrats (280–300 g, Vital River Laboratory Animal TechnologyCo., Ltd., Beijing, China) were randomly divided into thefollowing groups: (1) a sham-operated group without middlecerebral artery occlusion (MCAO) (n = 8), (2) 2 h MCAO(n = 8 × 13), (3) 4 h MCAO (n = 8 × 12), and (4) permanent(28 h) MCAO (n = 8 × 9). The 2 h MCAO groups wererandomly assigned to 13 subgroups, receiving either saline(sham treatment) or an intraperitoneal (IP) injection of twodoses C + P (1:1, 2 mg/kg + 2 mg/kg or 4 mg/kg + 4 mg/kg);the first dose at 2 h after the onset of ischemia followed by asecond dose after another hour at 1/3 of the initial amount.Two sets of animals were used: one with body temperature
maintained at physiological levels (rectal temperature at 36.5–37.5 °C) and one without body temperature control. Becauseof the close correlation between body and brain temperatures[20] and increased risk of intracranial hemorrhage, rectal tem-perature was monitored instead of brain temperature. Duringthe recovery period (24–28 h), in all temperature-controlledgroups, rats were placed on a 37 °C insulation blanket as wellas under a warm light to maintain their temperature. In groupswithout temperature control, rats were placed in a 25 °C en-vironment. For the 4 h MCAO, rats in a total of 12 groupswere randomly assigned to receive one of 3 different treat-ments: (1) saline, (2) 8 mg/kg C + P at 2 h after the onset ofMCAO followed by 2.6 mg/kg (1/3 of the initial amount) 2 hlater ± body temperature preservation, or (3) 12 mg/kg C + Pat 2 h after the onset of MCAO followed by 4 mg/kg 2 h laterwithout temperature control. Animals with transient MCAOwere analyzed at 24 h of reperfusion for infarct volume, and at6 or 24 h of reperfusion for protein and biochemical measure-ments. Similarly, for permanent stroke, rats in 8 groups wererandomly assigned to receive one of 3 treatments: (1) saline;(2) 12 mg/kg C + P at 2 h after the onset of MCAO, followedby 4 mg/kg 2 h later; or (3) 24 mg/kg C + P at 2 h after theonset of MCAO, followed by 8 mg/kg 2 h later. At 28 h afterischemia onset, animals from each group were examined forneurological deficits and processed for infarct volumes or bio-chemical analyses. The mortality rate was low (less than 10%)and was about equal between paired groups (with or withouttreatment). The death of ischemic rats in the present study wascaused by the operative skills and skull base hemorrhage dueto arterial rupture during filament insertion, rather than theischemic time. All data were analyzed in a blind manner.
MCA Occlusion Animals were fasted 12 h prior to the pro-cedures. Animals were anesthetized in a chamber with 1–3%isoflurane along with a mixture of 70% nitrous oxide and 30%oxygen. The rats were intubated, and anesthesia was main-tained with 1% isoflurane delivered from a calibrated preci-sion vaporizer. Rats were subjected to a right side MCAO foreither 2, 4, or 28 h using the intraluminal filament model [21].Reperfusion was achieved by the withdrawal of the filament at2 or 4 h of MCAO. Blood pCO2, pO2, mean arterial pressure(MAP), and blood glucose were monitored throughout theprocedure. Heating lamps and pads were utilized to maintainrectal temperature at 36.5–37.5 °C.
Chlorpromazine and Promethazine Administration In allischemia models with 2, 4, and 28 hMCAO ± reperfusion, thecombination of chlorpromazine and promethazine (1:1) atdoses of 4, 8, 12, or 24 mg/kg in 3 mL saline (as determinedby a preliminary study to induce significant neuroprotection)were injected IP at 2 h after the onset of ischemia. A secondinjection with one third of the original dose was added 1–2 hlater to enhance the drugs’ effects.
Mol Neurobiol (2017) 54:8140–8150 8141

Neurological Deficit The modified scoring systems (5 and 12scores) proposed by Zea Longa [21] and Belayev et al. [22]were used to examine the severity of neurological deficits inrats before surgery, duringMCAO, after 24 h reperfusion, andafter 28 h MCAO without reperfusion. A 28-h time point waschosen in MCAO without reperfusion to coincide with the24-h reperfusion group, accounting for the 4-h reperfusiontime. The severity of brain damage and consistency in eachgroup are highly important in this study. After MCAO, themodified scoring systems (five scores) for neurological defi-cits were used to confirm brain injury. If the scores were 1 orbelow, theMCA occlusions were considered unsuccessful andthe rats were excluded from further studies. In our study, about10% of animals with MCAO were discarded for this reason.
Cerebral Infarct VolumeAfter 24 h of reperfusion in 2 or 4 hMCAO, or after 28 h MCAO without reperfusion, the brainswere resected from ischemic rats and cut into 2-mm-thicks l i ces (bra in mat r ix ) and t rea ted wi th 2 , 3 , 5 -triphenyltetrazolium chloride (TTC, Sigma, USA) for stain-ing. An indirect method for calculating infarct volume wasused to minimize error caused by edema [23]. We also mea-sured and compared infarct size of cortex and striatum at threedifferent levels from anterior +1.00 mm to posterior −4.8 mmto the bregma of the brain.
ATP Production The BioVision Apo SENSOR Assay Kit(Biovision, CA) was used. Brain tissue samples containingthe frontoparietal cortex and dorsolateral striatum, which areMCA-supplied territories, were processed as described previ-ously by us [24].
NADH Assay The quantification kit (Biovision, CA) wasused to determine brain NADH levels as described previouslyby us [24].
Reactive Oxygen Species Production Reactive oxygen spe-cies (ROS) levels in the frontoparietal cortex and dorsolateralstriatum were detected as described previously by us [25].
Protein Expression Western blot was used to detect proteinlevels of PKC-δ and phosphorylated Akt (pAkt). Brain tissuescontaining the frontoparietal cortex and dorsolateral striatumwere processed as described previously by us [24, 25] andincubated with primary antibodies (polyclonal rabbit anti-PKC-δ at 1:5000, Santa Cruz Biotechnology, Inc.; and poly-clonal rabbit anti-phospho-Akt 1:1000, Cell SignalingTechnology, Inc) at 4 °C.
Statistical Analysis (SPSS Software, Version 17, SPSS Inc)All data were described as mean ± SE. Differences amongmultiple groups were assessed using both one-way and two-way analysis of variance (ANOVA) with a significance level
at p < 0.05. Post-hoc comparison between groups wasachieved using the least significant difference (LSD) method.
Results
Physiological Parameters There were no significant differ-ences in blood pH, pO2, pCO2, MAP, or blood glucose be-tween the groups.
Body Temperature In 2 h MCAO rats, body temperatureswere significantly reduced by 1 °C after C + P administration,with both 4 and 8 mg/kg doses achieving this value within asearly as 5 min (Fig. 1). At about 2 h after administration, bodytemperatures reached 35.7 and 34 °C with 4 and 8 mg/kg C +P, respectively. These temperatures remained significantly lowfor up to 6 h and subsequently returned to normal levels. In 4 hMCAO rats, 3 different doses of C + P (8, 12, and 24 mg/kgplus an additional one-third dose) all resulted in dose-dependent reductions in body temperatures by 1–2 °C withinas early as 5 min after administration (Fig. 1). At about 2 hafter administration, body temperatures reached their lowestlevels, with the three different doses yielding 35.7 °C(8 mg/kg), 32.3 °C (12 mg/kg), and 30.5 °C (24 mg/kg).These temperatures remained significantly depressed for upto 12 h and returned to normal levels thereafter. In the perma-nent (28 h) MCAO without reperfusion model, again, 3 dif-ferent doses of C + P (8, 12, and 24 mg/kg plus an additionalone-third dose) resulted in a dose-dependent decrease bodytemperatures by 1–2 °C (p < 0.05) within as early as 5 minof C + P administration (Fig. 1). Body temperatures reachedtheir lowest levels at 32.0 °C (24 mg/kg) after 2 h, 34.0 °C(12 mg/kg) after 3 h, and 35.5 °C (8 mg/kg) after 6 h post-C +P administration. These temperatures remained significantlyreduced for up to 12 h before it returned to normal levels.
Infarct VolumeWith 2 hMCAO, when body temperatures inischemic rats were maintained at 37.0 °C, both the low(4 mg/kg) (39.4%) and high (8 mg/kg) (35.7%) doses ofC + P significantly (p < 0.05) decreased infarct volumes ascompared to non-treatment groups (50.3 %) (Fig. 2a, d), witha greater infarct volume reduction seen in the higher dose. Ifbody temperature was not controlled and allowed to reach ahypothermic state after C + P administration, brain infarctvolumes were not significantly (albeit slightly) reduced anyfurther with either the 4 mg/kg dose (37.9 vs. 39.4%) or the8 mg/kg dose (29.8 vs. 35.7%). In the 4 h MCAO group, the8 mg/kg dose with temperature controlled at 37 °C induced amild decrease in infarct volume (41.6 vs. 50.2%, p < 0.05,Fig. 2b, e). A greater reduction (35.4%) was obtained withouttemperature control, but it did not reach a significant level.However, an additional reduction (p < 0.05) in brain infarctvolume was further seen with higher doses at 12 (31.6%) and
8142 Mol Neurobiol (2017) 54:8140–8150
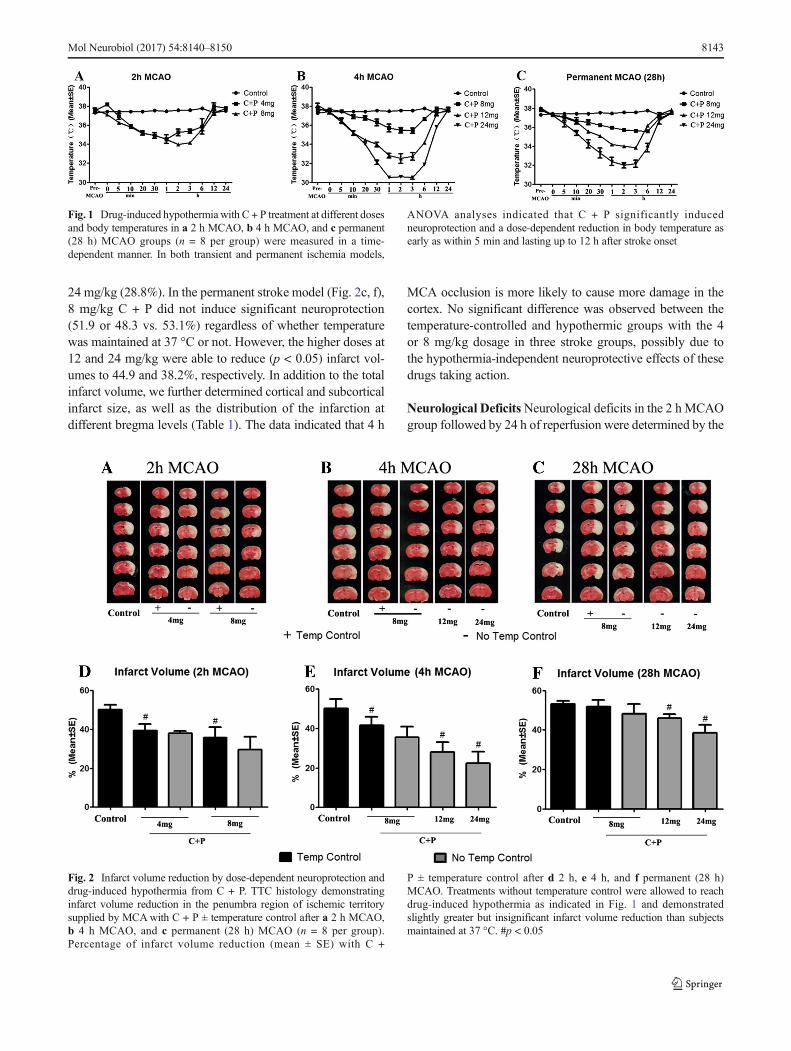
24 mg/kg (28.8%). In the permanent stroke model (Fig. 2c, f),8 mg/kg C + P did not induce significant neuroprotection(51.9 or 48.3 vs. 53.1%) regardless of whether temperaturewas maintained at 37 °C or not. However, the higher doses at12 and 24 mg/kg were able to reduce (p < 0.05) infarct vol-umes to 44.9 and 38.2%, respectively. In addition to the totalinfarct volume, we further determined cortical and subcorticalinfarct size, as well as the distribution of the infarction atdifferent bregma levels (Table 1). The data indicated that 4 h
MCA occlusion is more likely to cause more damage in thecortex. No significant difference was observed between thetemperature-controlled and hypothermic groups with the 4or 8 mg/kg dosage in three stroke groups, possibly due tothe hypothermia-independent neuroprotective effects of thesedrugs taking action.
Neurological DeficitsNeurological deficits in the 2 h MCAOgroup followed by 24 h of reperfusion were determined by the
Fig. 2 Infarct volume reduction by dose-dependent neuroprotection anddrug-induced hypothermia from C + P. TTC histology demonstratinginfarct volume reduction in the penumbra region of ischemic territorysupplied by MCAwith C + P ± temperature control after a 2 h MCAO,b 4 h MCAO, and c permanent (28 h) MCAO (n = 8 per group).Percentage of infarct volume reduction (mean ± SE) with C +
P ± temperature control after d 2 h, e 4 h, and f permanent (28 h)MCAO. Treatments without temperature control were allowed to reachdrug-induced hypothermia as indicated in Fig. 1 and demonstratedslightly greater but insignificant infarct volume reduction than subjectsmaintained at 37 °C. #p < 0.05
Fig. 1 Drug-induced hypothermia with C + P treatment at different dosesand body temperatures in a 2 h MCAO, b 4 h MCAO, and c permanent(28 h) MCAO groups (n = 8 per group) were measured in a time-dependent manner. In both transient and permanent ischemia models,
ANOVA analyses indicated that C + P significantly inducedneuroprotection and a dose-dependent reduction in body temperature asearly as within 5 min and lasting up to 12 h after stroke onset
Mol Neurobiol (2017) 54:8140–8150 8143
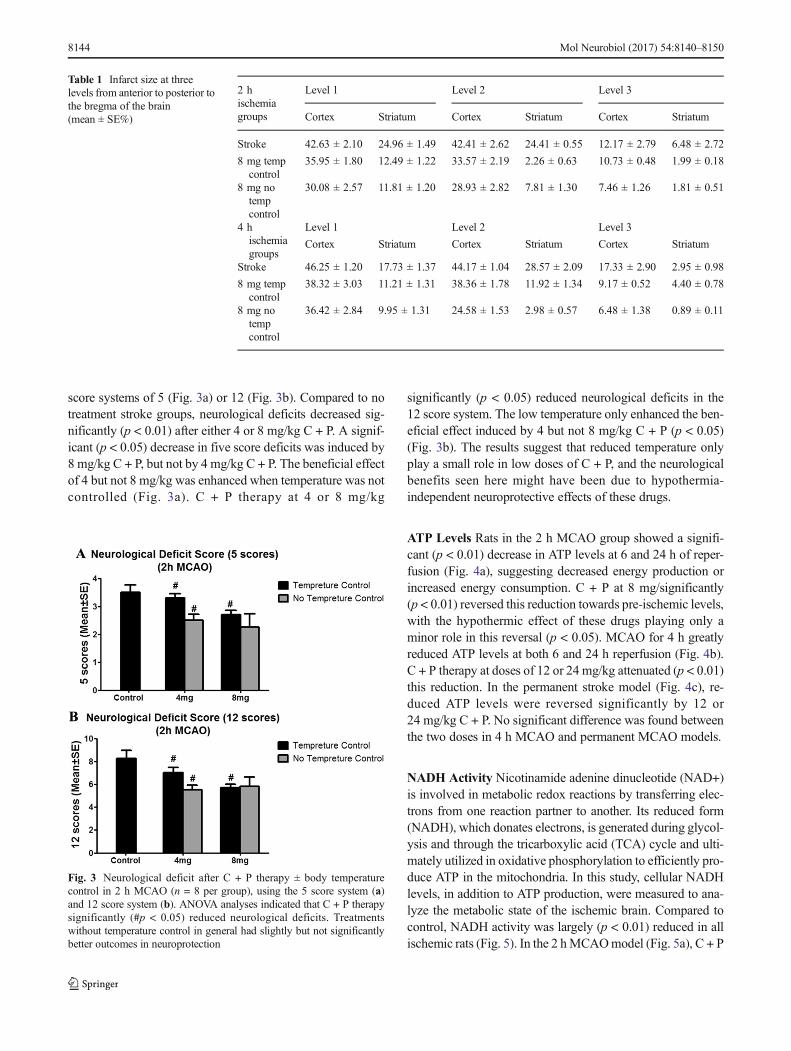
score systems of 5 (Fig. 3a) or 12 (Fig. 3b). Compared to notreatment stroke groups, neurological deficits decreased sig-nificantly (p < 0.01) after either 4 or 8 mg/kg C + P. A signif-icant (p < 0.05) decrease in five score deficits was induced by8 mg/kg C + P, but not by 4 mg/kg C + P. The beneficial effectof 4 but not 8 mg/kg was enhanced when temperature was notcontrolled (Fig. 3a). C + P therapy at 4 or 8 mg/kg
significantly (p < 0.05) reduced neurological deficits in the12 score system. The low temperature only enhanced the ben-eficial effect induced by 4 but not 8 mg/kg C + P (p < 0.05)(Fig. 3b). The results suggest that reduced temperature onlyplay a small role in low doses of C + P, and the neurologicalbenefits seen here might have been due to hypothermia-independent neuroprotective effects of these drugs.
ATP Levels Rats in the 2 h MCAO group showed a signifi-cant (p < 0.01) decrease in ATP levels at 6 and 24 h of reper-fusion (Fig. 4a), suggesting decreased energy production orincreased energy consumption. C + P at 8 mg/significantly(p < 0.01) reversed this reduction towards pre-ischemic levels,with the hypothermic effect of these drugs playing only aminor role in this reversal (p < 0.05). MCAO for 4 h greatlyreduced ATP levels at both 6 and 24 h reperfusion (Fig. 4b).C + P therapy at doses of 12 or 24 mg/kg attenuated (p < 0.01)this reduction. In the permanent stroke model (Fig. 4c), re-duced ATP levels were reversed significantly by 12 or24 mg/kg C + P. No significant difference was found betweenthe two doses in 4 h MCAO and permanent MCAO models.
NADH Activity Nicotinamide adenine dinucleotide (NAD+)is involved in metabolic redox reactions by transferring elec-trons from one reaction partner to another. Its reduced form(NADH), which donates electrons, is generated during glycol-ysis and through the tricarboxylic acid (TCA) cycle and ulti-mately utilized in oxidative phosphorylation to efficiently pro-duce ATP in the mitochondria. In this study, cellular NADHlevels, in addition to ATP production, were measured to ana-lyze the metabolic state of the ischemic brain. Compared tocontrol, NADH activity was largely (p < 0.01) reduced in allischemic rats (Fig. 5). In the 2 hMCAOmodel (Fig. 5a), C + P
Table 1 Infarct size at threelevels from anterior to posterior tothe bregma of the brain(mean ± SE%)
2 hischemiagroups
Level 1 Level 2 Level 3
Cortex Striatum Cortex Striatum Cortex Striatum
Stroke 42.63 ± 2.10 24.96 ± 1.49 42.41 ± 2.62 24.41 ± 0.55 12.17 ± 2.79 6.48 ± 2.72
8 mg tempcontrol
35.95 ± 1.80 12.49 ± 1.22 33.57 ± 2.19 2.26 ± 0.63 10.73 ± 0.48 1.99 ± 0.18
8 mg notempcontrol
30.08 ± 2.57 11.81 ± 1.20 28.93 ± 2.82 7.81 ± 1.30 7.46 ± 1.26 1.81 ± 0.51
4 hischemiagroups
Level 1 Level 2 Level 3
Cortex Striatum Cortex Striatum Cortex Striatum
Stroke 46.25 ± 1.20 17.73 ± 1.37 44.17 ± 1.04 28.57 ± 2.09 17.33 ± 2.90 2.95 ± 0.98
8 mg tempcontrol
38.32 ± 3.03 11.21 ± 1.31 38.36 ± 1.78 11.92 ± 1.34 9.17 ± 0.52 4.40 ± 0.78
8 mg notempcontrol
36.42 ± 2.84 9.95 ± 1.31 24.58 ± 1.53 2.98 ± 0.57 6.48 ± 1.38 0.89 ± 0.11
Fig. 3 Neurological deficit after C + P therapy ± body temperaturecontrol in 2 h MCAO (n = 8 per group), using the 5 score system (a)and 12 score system (b). ANOVA analyses indicated that C + P therapysignificantly (#p < 0.05) reduced neurological deficits. Treatmentswithout temperature control in general had slightly but not significantlybetter outcomes in neuroprotection
8144 Mol Neurobiol (2017) 54:8140–8150
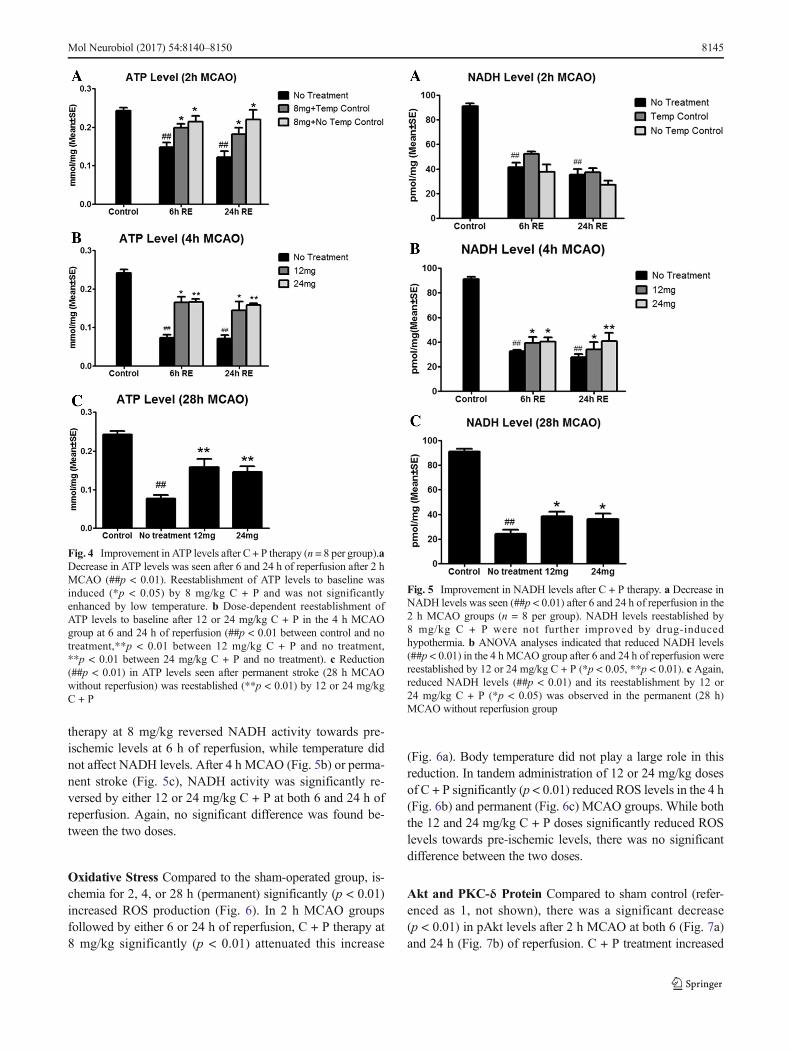
therapy at 8 mg/kg reversed NADH activity towards pre-ischemic levels at 6 h of reperfusion, while temperature didnot affect NADH levels. After 4 h MCAO (Fig. 5b) or perma-nent stroke (Fig. 5c), NADH activity was significantly re-versed by either 12 or 24 mg/kg C + P at both 6 and 24 h ofreperfusion. Again, no significant difference was found be-tween the two doses.
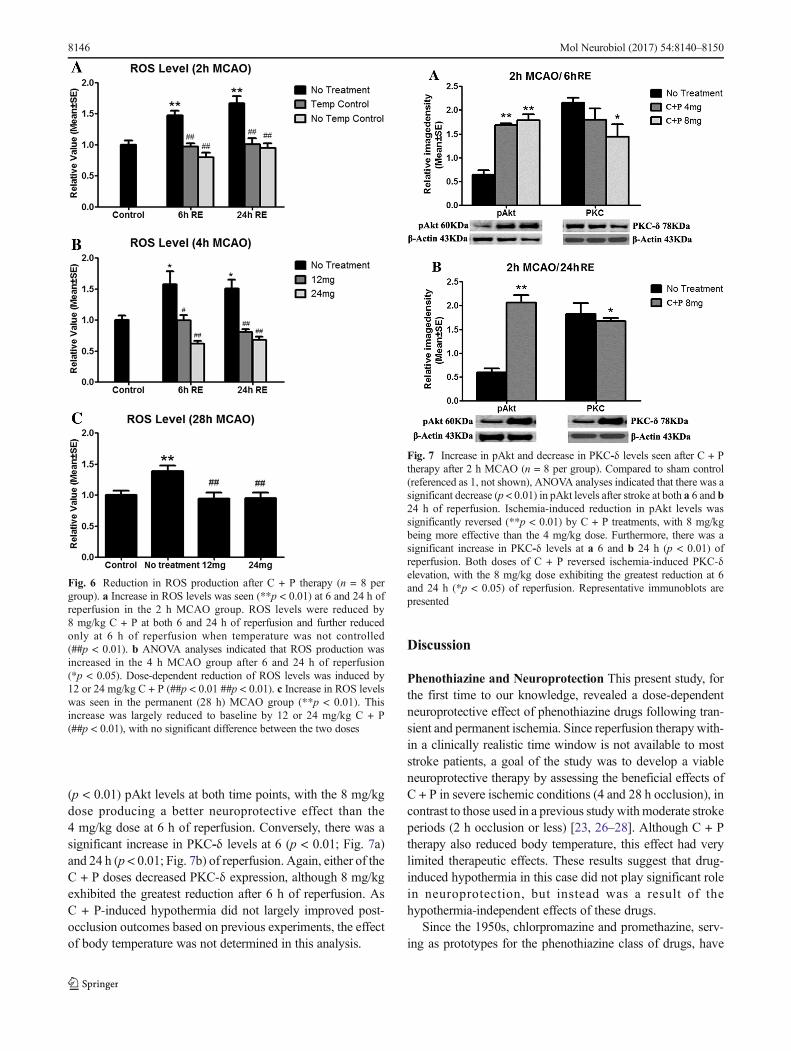
Oxidative Stress Compared to the sham-operated group, is-chemia for 2, 4, or 28 h (permanent) significantly (p < 0.01)increased ROS production (Fig. 6). In 2 h MCAO groupsfollowed by either 6 or 24 h of reperfusion, C + P therapy at8 mg/kg significantly (p < 0.01) attenuated this increase
(Fig. 6a). Body temperature did not play a large role in thisreduction. In tandem administration of 12 or 24 mg/kg dosesof C + P significantly (p < 0.01) reduced ROS levels in the 4 h(Fig. 6b) and permanent (Fig. 6c) MCAO groups. While boththe 12 and 24 mg/kg C + P doses significantly reduced ROSlevels towards pre-ischemic levels, there was no significantdifference between the two doses.
Akt and PKC-δ Protein Compared to sham control (refer-enced as 1, not shown), there was a significant decrease(p < 0.01) in pAkt levels after 2 h MCAO at both 6 (Fig. 7a)and 24 h (Fig. 7b) of reperfusion. C + P treatment increased
Fig. 5 Improvement in NADH levels after C + P therapy. a Decrease inNADH levels was seen (##p < 0.01) after 6 and 24 h of reperfusion in the2 h MCAO groups (n = 8 per group). NADH levels reestablished by8 mg/kg C + P were not further improved by drug-inducedhypothermia. b ANOVA analyses indicated that reduced NADH levels(##p < 0.01) in the 4 h MCAO group after 6 and 24 h of reperfusion werereestablished by 12 or 24 mg/kg C + P (*p < 0.05, **p < 0.01). c Again,reduced NADH levels (##p < 0.01) and its reestablishment by 12 or24 mg/kg C + P (*p < 0.05) was observed in the permanent (28 h)MCAO without reperfusion group
Fig. 4 Improvement in ATP levels after C + P therapy (n = 8 per group).aDecrease in ATP levels was seen after 6 and 24 h of reperfusion after 2 hMCAO (##p < 0.01). Reestablishment of ATP levels to baseline wasinduced (*p < 0.05) by 8 mg/kg C + P and was not significantlyenhanced by low temperature. b Dose-dependent reestablishment ofATP levels to baseline after 12 or 24 mg/kg C + P in the 4 h MCAOgroup at 6 and 24 h of reperfusion (##p < 0.01 between control and notreatment,**p < 0.01 between 12 mg/kg C + P and no treatment,**p < 0.01 between 24 mg/kg C + P and no treatment). c Reduction(##p < 0.01) in ATP levels seen after permanent stroke (28 h MCAOwithout reperfusion) was reestablished (**p < 0.01) by 12 or 24 mg/kgC + P
Mol Neurobiol (2017) 54:8140–8150 8145
(p < 0.01) pAkt levels at both time points, with the 8 mg/kgdose producing a better neuroprotective effect than the4 mg/kg dose at 6 h of reperfusion. Conversely, there was asignificant increase in PKC-δ levels at 6 (p < 0.01; Fig. 7a)and 24 h (p < 0.01; Fig. 7b) of reperfusion. Again, either of theC + P doses decreased PKC-δ expression, although 8 mg/kgexhibited the greatest reduction after 6 h of reperfusion. AsC + P-induced hypothermia did not largely improved post-occlusion outcomes based on previous experiments, the effectof body temperature was not determined in this analysis.
Discussion
Phenothiazine and Neuroprotection This present study, forthe first time to our knowledge, revealed a dose-dependentneuroprotective effect of phenothiazine drugs following tran-sient and permanent ischemia. Since reperfusion therapy with-in a clinically realistic time window is not available to moststroke patients, a goal of the study was to develop a viableneuroprotective therapy by assessing the beneficial effects ofC + P in severe ischemic conditions (4 and 28 h occlusion), incontrast to those used in a previous study withmoderate strokeperiods (2 h occlusion or less) [23, 26–28]. Although C + Ptherapy also reduced body temperature, this effect had verylimited therapeutic effects. These results suggest that drug-induced hypothermia in this case did not play significant rolein neuroprotection, but instead was a result of thehypothermia-independent effects of these drugs.
Since the 1950s, chlorpromazine and promethazine, serv-ing as prototypes for the phenothiazine class of drugs, have
Fig. 6 Reduction in ROS production after C + P therapy (n = 8 pergroup). a Increase in ROS levels was seen (**p < 0.01) at 6 and 24 h ofreperfusion in the 2 h MCAO group. ROS levels were reduced by8 mg/kg C + P at both 6 and 24 h of reperfusion and further reducedonly at 6 h of reperfusion when temperature was not controlled(##p < 0.01). b ANOVA analyses indicated that ROS production wasincreased in the 4 h MCAO group after 6 and 24 h of reperfusion(*p < 0.05). Dose-dependent reduction of ROS levels was induced by12 or 24 mg/kg C + P (##p < 0.01 ##p < 0.01). c Increase in ROS levelswas seen in the permanent (28 h) MCAO group (**p < 0.01). Thisincrease was largely reduced to baseline by 12 or 24 mg/kg C + P(##p < 0.01), with no significant difference between the two doses
Fig. 7 Increase in pAkt and decrease in PKC-δ levels seen after C + Ptherapy after 2 h MCAO (n = 8 per group). Compared to sham control(referenced as 1, not shown), ANOVA analyses indicated that there was asignificant decrease (p < 0.01) in pAkt levels after stroke at both a 6 and b24 h of reperfusion. Ischemia-induced reduction in pAkt levels wassignificantly reversed (**p < 0.01) by C + P treatments, with 8 mg/kgbeing more effective than the 4 mg/kg dose. Furthermore, there was asignificant increase in PKC-δ levels at a 6 and b 24 h (p < 0.01) ofreperfusion. Both doses of C + P reversed ischemia-induced PKC-δelevation, with the 8 mg/kg dose exhibiting the greatest reduction at 6and 24 h (*p < 0.05) of reperfusion. Representative immunoblots arepresented
8146 Mol Neurobiol (2017) 54:8140–8150

been widely used as neuroleptics because of their antipsychot-ic and sedative effects [29, 30]. These two drugs, which havebeen commonly used in combination [30], are two of theoldest and well-studied drugs from the phenothiazine class.Due to their high lipophilicity, C + P easily pass the blood-brain barrier (BBB) to exert depressive metabolic effects onthe central nervous system. Chlorpromazine is a low-potencytypical antipsychotic with primarily anticholinergic andantiadrenergic side effects. It has been reported to interferewith a number of receptors and ion channels, including sero-tonin receptors, Ca2+channels, K+ channels, and Cl-channels[31]. Chlorpromazine has been reported to block excitatoryglutamatergic signal transmission by inhibiting NMDA recep-tors [32]. It also confers neuroprotection against brain ische-mia by activating Ca2+-associated potassium channels by lim-iting Ca2+ entry, thus reducing excessive release of excitatoryneurot ransmi t te rs and energy expendi ture [31] .Chlorpromazine has been shown to exert neuroprotective ef-fect on the spinal cord ischemia [33], glutamate-induced neu-rotoxicity [34], ethanol-induced neuronal apoptosis [35], andin vitro hippocampal neuronal cell death [36]. Promethazine,on the other hand, is a first-generation antihistamine clinicallyused for its strong sedative, antiemetic, and weak antipsychot-ic effects. Promethazine has been shown to inhibit NMDAreceptors, Na+/K+-ATPase, and the mitochondrial permeabil-ity transition pore, all of which may contribute to its sedativeand neuroprotective effects [37]. Mitochondrial permeabilitytransition pores cause leakage in the mitochondrial innermembrane and subsequently result in cell death. By this mech-anism, promethazine may reduce ischemic neuronal injurysuch as those found in stroke pat ients [37–41].Phenothiazine derivatives have been reported to protect hu-man cells against oxidative stress [42].
“Hibernation-Like” Effect on Brain Metabolism Energyfailure and oxidative stress with ROS generation, secondaryto impaired oxidative phosphorylation and increased anaero-bic glycolysis (hyperglycolysis), are well-documented patho-physiologies of ischemic neuronal injury [43]. Recent re-search, however, has failed to develop targeted therapies thatmay confer neuroprotection acutely after stroke by addressingthe above metabolic dysfunctions [43]. Current study withC + P in transient or permanent ischemia demonstrated a tem-poral relationship between early improvement in ATP andNADH production and reduced ROS generation with subse-quent reduction in infarct volume and neurological deficits.Although the measures here cannot conclusively explain thebetter outcomes, our findings support a role of C + P in stabi-lizing the dysfunctional metabolic pathways mediated by thePKC and Akt signaling in ischemia.
C + P have been shown to alter glucose metabolism [44]and inhibit glucose uptake [12, 45–47], suggesting that theirdepressing effects may involve inhibiting carbohydrate
oxidation on the nervous system. This impaired carbohydrateutilization may occur even under normal oxygen consumption[48].Moreover, the induced-hyperglycemic response could beblocked by promethazine [49, 50]. Our parallel studies indi-cated a “hibernation-like” effect of these phenothiazine agentson suppressing glucose utilization and brain metabolism (datanot shown), which are similar to that of anesthetic drugs, suchas pentobarbitone [51, 52]. Not only does administration ofphenothiazines cause a reduction in blood flow and oxygenconsumption [53], but they also reduce energy metabolism[12, 44].
We have previously reported a suppressive effect of alcoholon brain metabolismwhich leads to neuroprotection followingthe onset of ischemic stroke [23–25]. However, in comparisonto phenothiazine drugs, there are many societal implicationsand significant side effects associated with alcohol which maylimit its clinical translation. In addition to their clinical use fortheir antipsychotic and sedative effects, the present experi-mental studies along with previous studies [12–14] support anew application of phenothiazine drugs as a neuroprotectantin stroke for their ability to induce an “artificial hibernation”status [11].
PKC-δ is upregulated in ischemic stroke and is potentiatedby the accumulation of ROS [19, 54, 55]. There are severalpathways in cerebral glucose metabolism that may affectPKC. In hyperglycemic states, glucose can elevate diacylglyc-erol (DAG), a second messenger lipid essential for PKC acti-vation, or activate microvascular PKC isoforms to result invascular complications [56]. Fibroblast growth factors (FGF)such as FGF-1 and FGF-2 may also be involved in neuropro-tection against insults such as ischemia, injury, or stress [57,58]. Studies have shown FGF to be mediated by Src tyrosinekinase phosphorylation and phospholipase Cγ, leading to in-creased DAG production and PKC activation [59]. FGFs werealso shown to activate PKC through SNT/FRS2 [59].Although the entire mechanism is still unclear, here, we haveshown for the first time that C + P downregulates PKC-δ, thusmaking these drugs possible therapeutic strategies for futureischemic stroke studies.
Akt, also known as PKB, is a protein kinase involved inmultiple pathways such as in the cell cycle and insulin signal-ing pathways. Studies have elucidated in detail that Akt carriesout the downstream effects controlled by a class of kinasesknown as phosphoinositide 3-kinases (PI 3-kinases). Akt’sinvolvement in insulin signaling could be another method bywhich Akt prevents neuronal damage following stroke bypromoting normal metabolic process and recovery [60].Although the exact mechanism of Akt-associated neuropro-tection still needs to be elucidated, our results show that post-ischemic Akt levels increased with C + P administration,supporting their potential in this stroke therapy. While theprimary focus here is centered on the treatment effects in is-chemic brain tissues, additional mechanistic studies are
Mol Neurobiol (2017) 54:8140–8150 8147

needed to assess the correlation between functional outcomesand the regulatory roles of the PKC-Akt signaling. At a morefundamental level, future studies will aim to determine thecause-and-effect relationships between C + P outcomes andits regulators in ischemic, as well as non-ischemic, braintissues.
Clinical Perspectives of C + P Therapy This study demon-strated the therapeutic possibilities of C + P in a severe ische-mic stroke model. There may be a concern that a large infarctin our model may compromise the hypothalamic arteries,leading to hypothalamic infarction and consequently temper-ature dysregulation. However, the arterial supply of the hypo-thalamus is derived from multiple perforating vessels fromvarious parts of the circle of Willis and the superior hypophy-sial arteries arising from the internal carotid arteries [61].Furthermore, an average of 10 anastomoses was found amongthe hypothalamic arteries [62]. Therefore, it is unlikely thatblood supply to the hypothalamus was further reduced by themodel. In this study, as the first step, we chose the 24 and 28 htime points for neurological assessment and final infarct vol-ume after transient and permanent MCAO because these timepoints were used in previous studies conducted by us andothers, which allows better comparison for the therapeuticeffect of C + P treatment. Furthermore, the average time ofprogression from stroke onset has been shown to be between22 and 48 h [63]. Since the infarcted lesion may still be evolv-ing and the drugs may be slowing down ischemic progressionrather than providing neuroprotection, especially with largerinfarcts, a long-term time point will be used in our futurestudy. In the present study, rectal temperature was monitoredcontinuously to reflect the systemic temperature whichreaches its lowest point at 2 h of drug administration. Wedid not directly measure the temperature of brain tissue sincewe considered the increased risk of intracranial hemorrhage inpatients with acute ischemia, who usually receive antithrom-botic treatment. Nevertheless, our previous study [20] andparallel studies (data not shown) have indicated a good corre-lation between brain and rectal temperatures.
Collateral perfusion has been widely recognized to re-main functional after stroke [16–18, 64]. This may exert adramatic effect on the time course of ischemic injury,stroke severity, imaging findings, as well as therapeuticopportunities and subsequent neurological outcomes afterstroke. In addition, phenothiazine drugs readily cross theBBB and easily diffuse through the collateral circulationinto ischemic regions before reperfusion is evenestablished. The clinical potential of this widely availableFDA-approved drug is apparent as stroke therapeutics.Future studies of C + P in a clinical setting may moveus closer towards the development of an efficacious neu-roprotective therapy.
Compliance with Ethical Standards All experimental procedureswere approved by the Institutional Animal Investigation Committee ofCapital Medical University in accordance with the National Institutes ofHealth (USA) guidelines for care and use of laboratory animals.
Sources of Funding This work was partially supported by AmericanHeart Association Grant-in-Aid (14GRNT20460246), Merit ReviewAward (I01RX-001,964-01) from the US Department of VeteransAffairs Rehabilitation R&D Service, National Natural ScienceFoundation of China (81501141), and Beijing NOVA program(xx2016061) as well as National Outstanding Youth Science Fund ofChina (no. 81325007).
Disclosures None.
References
1. Go AS, Mozaffarian D, Roger VL et al (2014) Heart disease andstroke statistics—2014 update: a report from the American HeartAssociation. Circulation 129(3):e28–e292
2. Kassem-Moussa H, Graffagnino C (2002) Nonocclusion and spon-taneous recanalization rates in acute ischemic stroke: a review ofcerebral angiography studies. Arch Neurol 59(12):1870–1873
3. Michalski D, Hartig W, Schneider D, Hobohm C (2011) Use ofnormobaric and hyperbaric oxygen in acute focal cerebral ische-mia—a preclinical and clinical review. Acta Neurol Scand 123(2):85–97
4. Kent TA, Mandava P (2007) Recanalization rates can be mislead-ing. Stroke 38(10):e103 author reply e104
5. Roth MB, Nystul T (2005) Buying time in suspended animation.Sci Am 292(6):48–55
6. Nathaniel TI (2008) Brain-regulated metabolic suppression duringhibernation: a neuroprotective mechanism for perinatal hypoxia-ischemia. Int J Stroke 3(2):98–104
7. Dave KR, Christian SL, Perez-Pinzon MA, Drew KL (2012)Neuroprotection: lessons from hibernators. Comp BiochemPhysiol B Biochem Mol Biol 162(1–3):1–9
8. Seyfried FJ, Adachi N, Arai T (2005) Suppression of energy re-quirement by lidocaine in the ischemic mouse brain. J NeurosurgAnesthesiol 17(2):75–81
9. Yenari M, Kitagawa K, Lyden P, Perez-PinzonM (2008) Metabolicdownregulation: a key to successful neuroprotection? Stroke39(10):2910–2917
10. Kim J, Yenari M (2015) Hypothermia for treatment of stroke. BrainCirculation 1(1):14–25. doi:10.4103/2394-8108.164997
11. Lopez-Munoz F, Alamo C, Cuenca E, Shen WW, Clervoy P, RubioG (2005) History of the discovery and clinical introduction of chlor-promazine. Ann Clin Psychiatry 17(3):113–135
12. MacMillan V (1982) Effects of promethazine on the energy metab-olism of normoxic and hypoxic rat brain. Stroke 13(4):464–469
13. Narayanan MV, Zhang W, Stavrovskaya IG, Kristal BS,Friedlander RM (2004) Promethazine: a novel application as aneuroprotectant that reduces ischemia-mediated injury byinhibiting mitochondrial dysfunction. Clin Neurosurg 51:102–107
14. Chien KR, Abrams J, Pfau RG, Farber JL (1977) Prevention bychlorpromazine of ischemic liver cell death. Am J Pathol 88(3):539–557
15. Liu S, Geng X, Forreider B et al (2015) Enhanced beneficial effectsof mild hypothermia by phenothiazine drugs in stroke therapy.Neurol Res 37(5):454–460
16. Liebeskind DS (2003) Collateral circulation. Stroke 34(9):2279–2284
8148 Mol Neurobiol (2017) 54:8140–8150

17. Liebeskind DS (2014) Collateral lessons from recent acute ischemicstroke trials. Neurol Res 36(5):397–402
18. Liebeskind D (2015) The collaterome: a novel conceptual frame-work of systems biology in cerebrovascular disorders. BrainCirculation 1(1):3–8. doi:10.4103/2394-8108.162411
19. Chou WH, Messing RO (2005) Protein kinase C isozymes instroke. Trends Cardiovasc Med 15(2):47–51
20. Wang F, Luo Y, Ling F et al (2010) Comparison of neuroprotectiveeffects in ischemic rats with different hypothermia procedures.Neurol Res 32(4):378–383
21. Longa EZ,Weinstein PR, Carlson S, Cummins R (1989) Reversiblemiddle cerebral artery occlusion without craniectomy in rats. Stroke20(1):84–91
22. Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD (1996)Middle cerebral artery occlusion in the rat by intraluminal suture.Neurological and pathological evaluation of an improved model.Stroke 27(9):1616–1622 discussion 1623
23. Wang F, Wang Y, Geng X et al (2012) Neuroprotective effect ofacute ethanol administration in a rat with transient cerebral ische-mia. Stroke 43(1):205–210
24. Kochanski R, Peng C, Higashida T et al (2013) Neuroprotectionconferred by post-ischemia ethanol therapy in experimental stroke:an inhibitory effect on hyperglycolysis and NADPH oxidase acti-vation. J Neurochem 126(1):113–121
25. Geng X, Fu P, Ji X et al (2013) Synergetic neuroprotection ofnormobaric oxygenation and ethanol in ischemic stroke throughimproved oxidative mechanism. Stroke 44(5):1418–1425
26. Garcia JH, Liu KF, Ho KL (1995) Neuronal necrosis after middlecerebral artery occlusion in Wistar rats progresses at different timeintervals in the caudoputamen and the cortex. Stroke 26(4):636–642 discussion 643
27. Fisher M, Takano K (1995) The penumbra, therapeutic time win-dow and acute ischaemic stroke. Baillieres Clin Neurol 4(2):279–295
28. Ginsberg MD, Pulsinelli WA (1994) The ischemic penumbra, inju-ry thresholds, and the therapeutic window for acute stroke. AnnNeurol 36(4):553–554
29. Cote CJ, Karl HW, Notterman DA, Weinberg JA, McCloskey C(2000) Adverse sedation events in pediatrics: analysis of medica-tions used for sedation. Pediatrics 106(4):633–644
30. Burn JH (1954) The pharmacology of chlorpromazine andpromethazine. Proc R Soc Med 47(8):617–621
31. Li HJ, Zhang YJ, Zhou L et al (2014) Chlorpromazine confersneuroprotection against brain ischemia by activating BKCa chan-nel. Eur J Pharmacol 735:38–43
32. Zarnowska ED,Mozrzymas JW (2001) Differential effects of chlor-promazine on ionotropic glutamate receptors in cultured rat hippo-campal neurons. Neurosci Lett 305(1):53–56
33. Sader AA, Barbieri-Neto J, Sader SL, Mazzetto SA, Alves P Jr,Vanni JC (2002) The protective action of chlorpromazine on thespinal cord of rabbits submitted to ischemia and reperfusion is dose-dependent. J Cardiovasc Surg 43(6):827–831
34. Stone JM, Pilowsky LS (2007) Novel targets for drugs in schizo-phrenia. CNS Neurol Disord Drug Targets 6(4):265–272
35. Wu J, SongR, SongWet al (2011) Chlorpromazine protects againstapoptosis induced by exogenous stimuli in the developing rat brain.PLoS One 6(7):e21966
36. Bastianetto S, Danik M, Mennicken F, Williams S, Quirion R(2006) Prototypical antipsychotic drugs protect hippocampal neu-ronal cultures against cell death induced by growth medium depri-vation. BMC Neurosci 7:28
37. Adolph O, Koster S, Georgieff M, Georgieff EM, Moulig W, FohrKJ (2012) Promethazine inhibits NMDA-induced currents—newpharmacological aspects of an old drug. Neuropharmacology63(2):280–291
38. Sharma A, Hamelin BA (2003) Classic histamine H1 receptor an-tagonists: a critical review of their metabolic and pharmacokineticfate from a bird’s eye view. Curr Drug Metab 4(2):105–129
39. Morota S, Mansson R, Hansson MJ et al (2009) Evaluation ofputative inhibitors of mitochondrial permeability transition forbrain disorders—specificity vs. toxicity. Exp Neurol 218(2):353–362
40. Cleren C, Starkov AA, Calingasan NY, Lorenzo BJ, Chen J, BealMF (2005) Promethazine protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Neurobiol Dis 20(3):701–708
41. Cleren C, Calingasan NY, Starkov A et al (2010) Promethazineprotects against 3-nitropropionic acid-induced neurotoxicity.Neurochem Int 56(2):208–212
42. Gonzalez-Munoz GC, Arce MP, Lopez B et al (2010) Old pheno-thiazine and dibenzothiadiazepine derivatives for tomorrow’s neu-roprotective therapies against neurodegenerative diseases. Eur JMed Chem 45(12):6152–6158
43. Forreider B, Pozivilko D, Kawaji Q, Geng X, Ding Y (2016)Hibernation-like neuroprotection in stroke by attenuating brainmetabolic dysfunction. Prog Neurobiol. doi:10.1016/j.pneurobio.2016.03.002
44. Zager EL, Ames A 3rd (1988) Reduction of cellular energy require-ments. Screening for agents that may protect against CNS ischemia.J Neurosurg 69(4):568–579
45. Krausz Y, Eylon L, Cerasi E (1987) Calcium-binding proteins andinsulin release. Differential effects of phenothiazines on first- andsecond-phase secretion and on islet cAMP response to glucose.Acta Endocrinol 116(2):241–246
46. Larsson S (1961) The effect of chlorpromazine on the glucose me-tabolism in different parts of the goat brain. Acta Physiol Scand 53:68–74
47. Dwyer DS, Liu Y, Bradley RJ (1999) Dopamine receptor antago-nists modulate glucose uptake in rat pheochromocytoma (PC12)cells. Neurosci Lett 274(3):151–154
48. Skinner A, Spector RG (1968) The effect of chlorpromazine on 14-C-glucose metabolism in mouse liver and brain. Br J PharmacolChemother 33(1):129–135
49. Nishibori M, Itoh Y, Oishi R, Saeki K (1987) Mechanism of thecentral hyperglycemic action of histamine in mice. J Pharmacol ExpTher 241(2):582–586
50. Nagai K, Frohman LA (1978) Neurotensin hyperglycemia: evi-dence for histamine mediation and the assessment of a possiblephysiologic role. Diabetes 27(5):577–582
51. Bachelard HS, Lindsay JR (1966) Effects of neurotropic drugs onglucose metabolism in rat brain in vivo. Biochem Pharmacol 15(8):1053–1058
52. Bachelard HS, Gaitonde MK, Vrba R (1966) The effect of psycho-tropic drugs on the utilization of glucose carbon atoms in the brain,heart and liver of the rat. Biochem Pharmacol 15(8):1039–1043
53. Berntman L, Carlsson C (1978) Influence of “lytic cocktail” onblood flow and oxygen consumption in the rat brain. ActaAnaesthesiol Scand 22(5):515–518
54. Gao X, Zhang H, Takahashi T et al (2008) The Akt signaling path-way contributes to postconditioning’s protection against stroke; theprotection is associated with the MAPK and PKC pathways. JNeurochem 105(3):943–955
55. DaveKR, Bhattacharya SK, Saul I et al (2011) Activation of proteinkinase C delta following cerebral ischemia leads to release of cyto-chrome C from the mitochondria via bad pathway. PLoS One 6(7):e22057
56. Miele C, Paturzo F, Teperino R et al (2007) Glucose regulatesdiacylglycerol intracellular levels and protein kinase C activity bymodulating diacylglycerol kinase subcellular localization. J BiolChem 282(44):31835–31843
57. Li AJ, Oomura Y, Sasaki K, Suzuki K, Hori T (1999) Protectiveeffect of acidic fibroblast growth factor against ischemia-induced
Mol Neurobiol (2017) 54:8140–8150 8149

learning and memory deficits in two tasks in gerbils. Physiol Behav66(4):577–583
58. Bland ST, Tamlyn JP, Barrientos RM et al (2007) Expression offibroblast growth factor-2 and brain-derived neurotrophic factormRNA in the medial prefrontal cortex and hippocampus after un-controllable or controllable stress. Neuroscience 144(4):1219–1228
59. Reuss B, von Bohlen und Halbach O (2003) Fibroblast growthfactors and their receptors in the central nervous system. CellTissue Res 313(2):139–157
60. Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cellproliferation, survival and insulin responses? J Cell Sci 114(Pt 16):2903–2910
61. Daniel PM (1976) Anatomy of the hypothalamus and pituitarygland. J Clin Pathol Suppl (Assoc Clin Pathol) 7:1–7
62. Marinkovic SV, Milisavljevic MM, Marinkovic ZD (1989)Microanatomy and possible clinical significance of anastomosesamong hypothalamic arteries. Stroke 20(10):1341–1352
63. DeGraba TJ, Hallenbeck JM, Pettigrew KD, Dutka AJ, Kelly BJ(1999) Progression in acute stroke: value of the initial NIH strokescale score on patient stratification in future trials. Stroke 30(6):1208–1212
64. Bang OY, Saver JL, Kim SJ et al (2011) Collateral flow avertshemorrhagic transformation after endovascular therapy for acuteischemic stroke. Stroke 42(8):2235–2239
8150 Mol Neurobiol (2017) 54:8140–8150





























