Embed Size (px)
Citation preview

lable at ScienceDirect
Neurobiology of Aging 35 (2014) 1065e1073
Contents lists avai
Neurobiology of Aging
journal homepage: www.elsevier .com/locate/neuaging
Age and duration of inflammatory environment differentially affectthe neuroimmune response and catecholaminergic neurons in themidbrain and brainstem
Isabelle Bardou a,b, Roxanne M. Kaercher a,b, Holly M. Brothers a,b, Sarah C. Hopp a,b,Sarah Royer a,b, Gary L. Wenk a,b,*
aDepartment of Psychology, The Ohio State University, Columbus, OH, USAbDepartment of Neuroscience, The Ohio State University, Columbus, OH, USA
a r t i c l e i n f o
Article history:Received 29 May 2013Received in revised form 17 October 2013Accepted 9 November 2013Available online 19 November 2013
Keywords:Alzheimer’s diseaseParkinson’s diseaseNeuroinflammationSubstantia nigraLocus coeruleusMicrogliaRatAgingCytokines
* Corresponding author at: 1835 Neil Avenue, DepOhio State University, Columbus, OH 43210, USA. Tel.:4733.
E-mail address: [email protected] (G.L. Wenk).
0197-4580/$ e see front matter � 2014 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.neurobiolaging.2013.11.006
a b s t r a c t
Neuroinflammation and degeneration of ascending catecholaminergic systems occur early in the neuro-degenerative process. Age and the duration of a pro-inflammatory environment induced by continuousintraventricular lipopolysaccharide (LPS) differentially affect the expression profile of pro- and anti-inflammatory genes and proteins as well as the number of activated microglia (express major histocom-patibility complex II; MHC II) and the integrity and density of ascending catecholaminergic neural systemsoriginating from the locus coeruleus (LC) and substantia nigra pars compacta (SNpc) in rats. LPS infusionincreased gene expression and/or protein levels for both pro- and anti-inflammatory biomarkers. AlthoughLPS infusion stimulated a robust increase in IL-1ß gene and protein expression, this increase was bluntedwith age. LPS infusion also increased the density of activated microglia cells throughout the midbrain andbrainstem. Corresponding to the development of a pro-inflammatory environment, LC and SNpc neuronsimmunopositive for tyrosine-hydroxylase (the rate-limiting synthetic enzyme for dopamine and norepi-nephrine) decreased in number, along with a decrease in tyrosine-hydroxylase gene expression in themidbrain and/or brainstem region. Our data support the concept that continuous exposure to a pro-inflammatory environment drives exaggerated changes in the production and release of inflammatorymediators that interact with age to impair functional capacity of the SNpc and LC.
� 2014 Elsevier Inc. All rights reserved.
1. Introduction
Activation of the brain’s resident microglia occurs during normalaging, is associated with many neurodegenerative diseases such asParkinson’s disease (PD) and Alzheimer’s disease (AD), and maydrive a self-propagating toxic cycle promoted by the release of pro-inflammatoryand loss of protectivemediators (Aarsland et al., 2001;Akiyama et al., 2000; Bartels and Leenders, 2005; Block and Hong,2005; Cribbs et al., 2012; Griffin et al., 1989; Hobson and Meara,2004; Hughes et al., 2000; Swardfager et al., 2010; Whitton, 2007).When these processes are triggeredwithin vulnerable brain regions,theymay lead to the loss of acetylcholinergic neurons in the nucleusbasalis magnocellularis (Whitton, 2007;Willard et al., 1999) as wellas dopaminergic neurons in the substantia nigra pars compacta(SNpc), noradrenergic (NE) neurons in the locus coeruleus (LC) and,
artment of Psychology, The614-688-3404; fax: 614-688-
ll rights reserved.
all regions that show significant early cell loss in the brains of pa-tients with PD and AD (Braak et al., 2003; Grudzien et al., 2007;Halliday et al., 2006; Rudow et al., 2008; Szot et al., 2006).
We and others have speculated that the consequences of neu-roinflammation associated with microglial activation, are carefullyregulated until, because of normal aging or the deposition of toxicproteins, there is a gradual shift to a nonequilibrium state that ispermissive for neurodegenerative processes (Block and Hong,2005; Colton and Wilcock, 2010; Smith et al., 2012; Wenk andHauss-Wegrzyniak, 2001). Microglia can assume various pheno-types that are associated either with the release of potentiallydestructive, pro-inflammatory cytokines, and other toxic moleculesor the expression of a cytokine profile that sustains repair, recovery,and growth. Microglia in various states of activation are detectablemany years before the onset of neuropathological changes (Cagninet al., 2006; Gerhard et al., 2006; Imamura et al., 2003). Becausevulnerable brain regions are likely exposed for many decades to acomplex combination of microglia in various activation states(Bilbo, 2010; Eikelenboom et al., 2010; Heneka et al., 2010; Herrup,2010). The present study investigated the differential influence of

I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e10731066
brain age and the duration of the pro-inflammatory environmentupon the expression of pro- and anti-inflammatory genes andproteins as well as the number of activated microglia and theintegrity and density of ascending catecholaminergic neural sys-tems originating in the LC and SNpc.
2. Methods
2.1. Experimental design
Young (3 months), middle-aged (9 months), and aged (23months) male F-344 rats (Harlan SpragueeDawley) receivedchronic infusion of lipopolysaccharide (LPS) or its vehicle (artificialcerebral spinal fluid; aCSF) into the fourth ventricle for 21 or 56days. We believe that this approach best represents the situationpresent during the early stages of many chronic neurodegenerativediseases. Multiple counter-balanced iterations of this study wereperformed to produce a total of 132 rats; yielding experimentalgroups with a minimum of 11 rats that were divided betweenbiochemical (minimum 6 rats/group) and histologic (minimum 5rats/group) analysis. Midbrain and/or brainstem regions wereevaluated for protein and messenger RNA (mRNA) expression ofinflammatory markers and the LPS receptor (toll-like receptor 4;TLR4), as well as the presence of MHC II-IR microglia, which wasused to define activatedmicroglia. Changes in these immune factorsat 3 ages and after short (21 days) or long (56 days) of continuousLPS infusion were then evaluated with respect to changes inneurotransmitter systems including expression of genes involved inthe regulation of glutamate (glutamate transporter 1; GLT1, and thecystine-glutamate anti-porter; XcT), and gene and histologicexpression of the enzymes responsible for production of dopamine(tyrosine hydroxylase; TH) and norepinephrine (dopamine-ß-hy-droxylase; DBH). DBH immunostaining was examined to determinespecifically the integrity of norepinephrine innervation of thedentate gyrus region of the hippocampus, which does not receive adense dopaminergic input (Gasbarri et al., 1994). TH immuno-staining was used to define specifically the norepinephrine neuronsin the LC and dopaminergic neurons in the SNpc.
2.2. Subjects
Rats were maintained on a 12/12-hours lightedark cycle withlights off at 09:00 in a temperature-controlled room (22 �C) withfree access to food and water. All rats were sacrificed during thedark phase of the diurnal cycle. Body weights and general healthwere closely monitored throughout the study. All rats were allowedat least 1 week to adapt to their new environment before surgery.The experiments were carried out in accordance with the NationalInstitute of Health Guide for the Care and Use of Laboratory Animals(NIH Publications No. 80-23) revised 1996, and formal approval toconduct the experiments was obtained from the animal subjectsreview board from The Ohio State University.
2.3. Surgery
aCSF (140 mM NaCl, 3.0 mM KCl, 2.5 mM CaCl2, 1.0 mM MgCl2,and 1.2 mMNa2HPO4 adjusted to pH 7.4) or LPS (0.25 mg/h, 1.66 mg/mL prepared in aCSF; Escherichia coli, serotype 055:B5, TCAextraction, Sigma-Aldrich, St. Louis, MO, USA) were continuouslyinfused via a cannula implanted into the fourth ventricle (�2.5 mmanterior-posterior and �7.0 mm dorsal-ventral, relative to lambda)and attached (via Tygon tubing, 0.06 O.D.) to an osmotic minipump(Alzet model #2006, to deliver 0.15 ml/h; DURECT Corporation,Cupertino, CA, USA) as previously described (Hauss-Wegrzyniaket al., 1998; Marchalant et al., 2007; Rosi et al., 2004). The average
fill volume and release rates for the pump allows for an infusion upto 56 days. Postoperative care included lidocaine 1% solutionapplied to the exposed skin upon closure, 2 mL of isotonic saline bysubcutaneous injection to prevent dehydration during recovery and2% tylenol in the drinking water for 3 days before and after surgery.
2.4. Tissue collection
Rats used for protein and mRNA analysis were briefly anes-thetized and then rapidly decapitated; their midbrain and/orbrainstem (extending from just rostral to the SNpc and just caudalto the LC) and entire hippocampus were quickly dissected on iceand stored at �80 �C until processed. Blood was collected duringthe rapid decapitation procedure. After centrifugation at 4 �C for 15minutes at 2500�g, serumwas collected and assayed. Rats used forimmunohistochemistry were deeply anesthetized with isofluranefor a transcardiac perfusion with 80 mL of cold 0.9% saline con-taining 50 U/mL heparin, followed by 120 mL of 4% para-formaldehyde in 0.1 M phosphate buffered saline (PBS), pH 7.4, at arate of 10 mL/min. Brains were postfixed overnight in the samefixative and then stored in PBS.
2.5. Protein analysis
Brainstem or serum levels of tumor necrosis factor (TNF)-a,interleukin-1 (IL-1)-a, IL-1b, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 IL-13,IL-18 (interferon-g inducing factor), interferon-g, and granulocyte-macrophage-stimulating factor (GM-CSF) were quantified simulta-neously with a magnetic bead-based immunoassay (Bio-Rad,BioPlex Pro Rat Standard, 171-K1002M), according to the manufac-turer’s protocol. Briefly, total protein was extracted from frozenmidbrain and/or brainstem with a BioPlex Cell Lysis Kit (BioRad,Richmond, CA, USA). A mixture of distinct capture beads (fluo-rescently dyed microspheres) each with a specific spectral addressand conjugated to an antibody against one of the cytokines listedpreviouslyweredispensedacross a 96-well plate andprotected fromlight. Samples and antigen standards were added in duplicate andthe plate was shaken (700 RPM, 1 hour). Then a mixture of bio-tinylated detection antibodies directed against each of the primaryantibodies was added and the plate was shaken (700 RPM, 30 mi-nutes); unbound materials were washed away (3 times) (BioRad,BioPlex Pro wash station). Each well was then incubated with astreptavidin-phycoerythrin conjugate reporter dye that binds to thedetection antibody (700 RPM,10minutes); unboundmaterials werewashed away (3 times). Each well was then suspended in assaybuffer and shaken (1100 RPM for 30 seconds). Finally, the contentswere passed through a dual detection multiplexing machine (Bio-Rad MAGPIX multiplex reader) with a classification laser that dis-tinguishes each of the proteins by color of its bound antigen-specificbead and a reporter laser that quantifies each molecule based uponthe fluorescence of bound antigen-specific streptavidin-phycoery-thrin conjugate reporter dye. Values were standardized to proteincontent of the homogenate obtained with a Bio-Rad protein assay(Bio-Rad), and results are reported as pg/mg protein.
2.6. Real-time polymerase chain reaction mRNA analysis
Brainstems were evaluated for mRNA expression of: TNFa, IL1ß,transforming growth factor-b (TGFß), TLR4, fractalkine receptor(CX3CR1), GLT1, XcT, brain-derived neurotrophic factor (BDNF). THwas evaluated in themidbrain and/or brainstem as a precursor to LCnorepinephrine as well as SNpc dopamine, and DBH was evaluatedin the hippocampus because it is a precursor to norepinephrine thatcan distinguish input from the LC projections. Tissues were ho-mogenized inTrizol (Life Technologies, Carlsbad, CA, USA). Total RNA
I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e1073 1067
was extractedwith a NucleoSpin RNA II kit (Macherey-Nagel, Düren,Germany); 1 mg of total RNA was reverse-transcribed to create acomplementary DNA library using the iScript reverse transcriptionSupermix for quantitative real-time polymerase chain reaction(Bio-Rad). PrimersweredesignedwithPrimerQuest software (version3.4Integrated DNA Technologies, Coralville, IA, USA; Table 1), andspecificity was ensured by examining primer alignments with theBLAST database. Primers and Sso Advanced SYBR Green Supermix(BioRad) were prepared with RNase-free water. For PCR amplifica-tion, mix (19 mL) was added to reverse transcription reaction (1 mL)previously diluted (1:20). Assayswere run in triplicate on the CFX96,C1000 Thermal Cycler (Bio-Rad). Amplification conditions were: 95�C for 30 seconds and 40 cycles of PCR (denaturation: 95 �C for 5seconds, annealing and/or extension: 60 �C for 30 seconds), followedby melting curves to verify the absence of primer dimers. Twonegative controls were performed during each quantitative PCRexperiment: reaction without the reverse transcription (-RT) toconfirm the absence of genomic DNA contamination, and sampleswith no added complementary DNA template (H2O only). The cycle(Ct) at which expression levels crossed threshold was normalized tothe Ct of the reference gene glyceraldehyde-3-phosphate dehydro-genase, producing DCt with arbitrary units of total gene expression.
2.7. Immunocytochemistry
Free-floating coronal sections (40 mm) were obtained with avibratome (Leica, Model VT1000S) from perfused brains that werekept cold but never frozen. All reactions took place on an agitator atroom temperature, except for overnight incubations, and all rinses(3 times 10minutes) were in PBSwith 0.05% tween 20. Tissues wererinsed, quenched of native peroxidase activity with 0.3% H2O2 in50% methanol for 1 hour, rinsed, blocked for nonspecific binding in5% normal goat serum for 1 hour, and incubated in primaryantibody diluted in 5% normal goat serum overnight at 4 �C.Double-immunostaining of all sections was achieved using theseantibodies: anti-class II major histocompatibility complex (MHC II,1: 200, mouse monoclonal, Pharmingen, San Diego, CA, USA#554926) and anti-TH (1:3000 for the LC and 1:1500 for the SNpc,rabbit polyclonal, Millipore, Billerica, MA, USA #AB152). Thereafter,
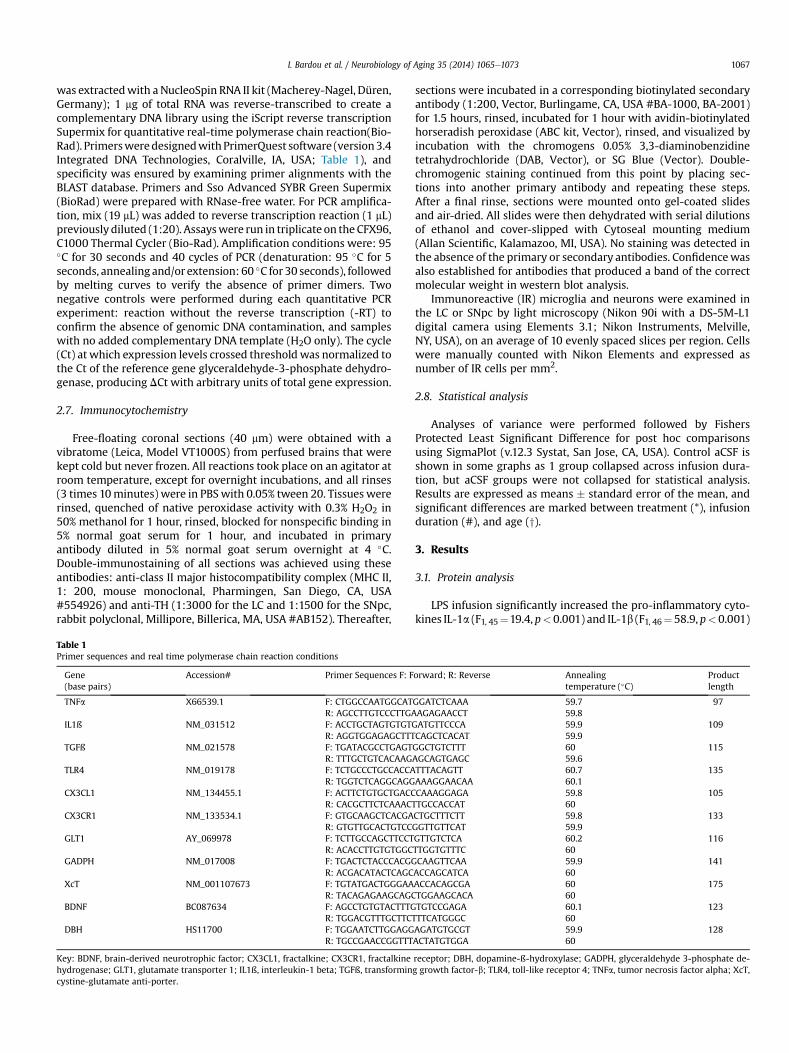
Table 1Primer sequences and real time polymerase chain reaction conditions
Gene(base pairs)
Accession# Primer Sequences F: F
TNFa X66539.1 F: CTGGCCAATGGCATR: AGCCTTGTCCCTTG
IL1ß NM_031512 F: ACCTGCTAGTGTGTR: AGGTGGAGAGCTTT
TGFß NM_021578 F: TGATACGCCTGAGTR: TTTGCTGTCACAAG
TLR4 NM_019178 F: TCTGCCCTGCCACCAR: TGGTCTCAGGCAGG
CX3CL1 NM_134455.1 F: ACTTCTGTGCTGACCR: CACGCTTCTCAAAC
CX3CR1 NM_133534.1 F: GTGCAAGCTCACGAR: GTGTTGCACTGTCC
GLT1 AY_069978 F: TCTTGCCAGCTTCCTR: ACACCTTGTGTGGC
GADPH NM_017008 F: TGACTCTACCCACGGR: ACGACATACTCAGC
XcT NM_001107673 F: TGTATGACTGGGAAR: TACAGAGAAGCAGC
BDNF BC087634 F: AGCCTGTGTACTTTGR: TGGACGTTTGCTTC
DBH HS11700 F: TGGAATCTTGGAGGR: TGCCGAACCGGTTT
Key: BDNF, brain-derived neurotrophic factor; CX3CL1, fractalkine; CX3CR1, fractalkinehydrogenase; GLT1, glutamate transporter 1; IL1ß, interleukin-1 beta; TGFß, transformincystine-glutamate anti-porter.
sections were incubated in a corresponding biotinylated secondaryantibody (1:200, Vector, Burlingame, CA, USA #BA-1000, BA-2001)for 1.5 hours, rinsed, incubated for 1 hour with avidin-biotinylatedhorseradish peroxidase (ABC kit, Vector), rinsed, and visualized byincubation with the chromogens 0.05% 3,3-diaminobenzidinetetrahydrochloride (DAB, Vector), or SG Blue (Vector). Double-chromogenic staining continued from this point by placing sec-tions into another primary antibody and repeating these steps.After a final rinse, sections were mounted onto gel-coated slidesand air-dried. All slides were then dehydrated with serial dilutionsof ethanol and cover-slipped with Cytoseal mounting medium(Allan Scientific, Kalamazoo, MI, USA). No staining was detected inthe absence of the primary or secondary antibodies. Confidencewasalso established for antibodies that produced a band of the correctmolecular weight in western blot analysis.
Immunoreactive (IR) microglia and neurons were examined inthe LC or SNpc by light microscopy (Nikon 90i with a DS-5M-L1digital camera using Elements 3.1; Nikon Instruments, Melville,NY, USA), on an average of 10 evenly spaced slices per region. Cellswere manually counted with Nikon Elements and expressed asnumber of IR cells per mm2.
2.8. Statistical analysis
Analyses of variance were performed followed by FishersProtected Least Significant Difference for post hoc comparisonsusing SigmaPlot (v.12.3 Systat, San Jose, CA, USA). Control aCSF isshown in some graphs as 1 group collapsed across infusion dura-tion, but aCSF groups were not collapsed for statistical analysis.Results are expressed as means � standard error of the mean, andsignificant differences are marked between treatment (*), infusionduration (#), and age (y).
3. Results
3.1. Protein analysis
LPS infusion significantly increased the pro-inflammatory cyto-kines IL-1a (F1, 45¼19.4, p< 0.001) and IL-1b (F1, 46¼ 58.9, p< 0.001)
orward; R: Reverse Annealingtemperature (�C)
Productlength
GGATCTCAAA 59.7 97AAGAGAACCT 59.8GATGTTCCCA 59.9 109CAGCTCACAT 59.9GGCTGTCTTT 60 115AGCAGTGAGC 59.6TTTACAGTT 60.7 135AAAGGAACAA 60.1CAAAGGAGA 59.8 105TTGCCACCAT 60CTGCTTTCTT 59.8 133GGTTGTTCAT 59.9GTTGTCTCA 60.2 116TTGGTGTTTC 60CAAGTTCAA 59.9 141ACCAGCATCA 60ACCACAGCGA 60 175TGGAAGCACA 60TGTCCGAGA 60.1 123
TTTCATGGGC 60AGATGTGCGT 59.9 128ACTATGTGGA 60
receptor; DBH, dopamine-ß-hydroxylase; GADPH, glyceraldehyde 3-phosphate de-g growth factor-b; TLR4, toll-like receptor 4; TNFa, tumor necrosis factor alpha; XcT,
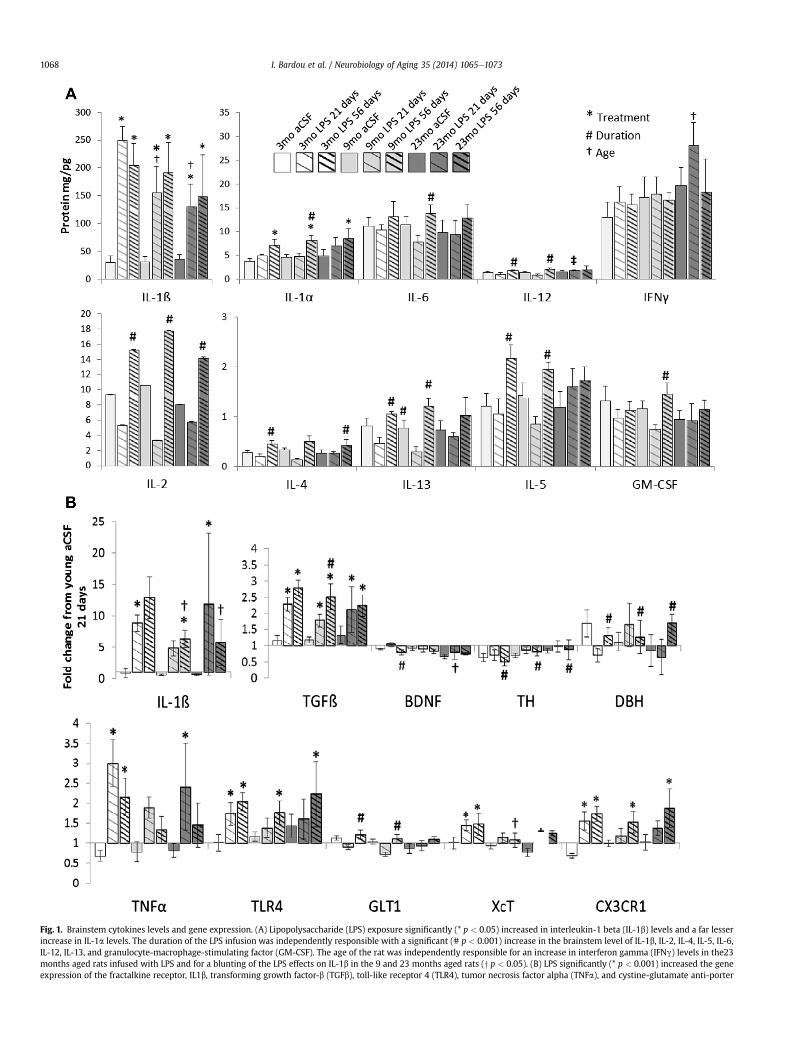
Fig. 1. Brainstem cytokines levels and gene expression. (A) Lipopolysaccharide (LPS) exposure significantly (* p < 0.05) increased in interleukin-1 beta (IL-1b) levels and a far lesserincrease in IL-1a levels. The duration of the LPS infusion was independently responsible with a significant (# p < 0.001) increase in the brainstem level of IL-1b, IL-2, IL-4, IL-5, IL-6,IL-12, IL-13, and granulocyte-macrophage-stimulating factor (GM-CSF). The age of the rat was independently responsible for an increase in interferon gamma (IFNg) levels in the23months aged rats infused with LPS and for a blunting of the LPS effects on IL-1b in the 9 and 23 months aged rats (y p < 0.05). (B) LPS significantly (* p < 0.001) increased the geneexpression of the fractalkine receptor, IL1b, transforming growth factor-b (TGFb), toll-like receptor 4 (TLR4), tumor necrosis factor alpha (TNFa), and cystine-glutamate anti-porter
I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e10731068

I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e1073 1069
levels in the midbrain and/or brainstem regions that included thesubstantia nigra and LC (Fig. 1A). However, the IL-1ß response to 21days of LPS infusion was significantly (p < 0.05) blunted in bothmiddle-aged and aged rats. LPS infusion over 56 days significantly(all F> 24, p< 0.001) increasedmidbrain and/or brainstem levels ofIL-1a, IL-2, IL-4, IL-5, IL-6, IL-12, IL-13, andGM-CSF comparedwith21days of LPS infusion in rats of the same age. Although aged ratsinfusedwith LPS for 21days had a blunted IL-1ß response, this grouphad elevated pro-inflammatory IFNg levels compared with 21 daysof LPS infusion in younger rats (F2, 48 ¼ 3.62, p < 0.05).
Serum levels of IL-1a, IL-1b, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, IL-13, GM-CSF, INFg, and TNFa were unchanged across all age andtreatment groups (data not shown), and may indicate that intra-cranial infusion of LPS did not have a marked response on the pe-ripheral immune response.
3.2. mRNA expression
LPS exposure significantly (all F > 32, p < 0.001) increased thegene expression of pro-inflammatory IL1b and TNFa, the LPS re-ceptor TLR4, anti-inflammatory TGFß and CX3CR1 as well as theglutamate-antiporter XcT (Fig. 1B). Consistent with the resultsobserved in protein analysis, the IL-1ß response to 56 days of LPSinfusion was significantly (p < 0.05) blunted in both middle-agedand aged rats compared with young rats. Gene expression ofTGFß increases more after 56 days of LPS infusion than 21 daysinfusion in middle-aged rats (F1, 48 ¼ 6.39, p < 0.01) and a similarduration-dependent increase in GLT1 is observed in young andmiddle-aged rats (F1, 48 ¼ 12.1, p < 0.001). TGFß is anti-inflammatory and GLT1 serves to sequester extracellular gluta-mate that can be excitotoxic; an increase in these factors afterprolonged LPS exposure could be a protective mechanism that doesnot occur in aged rats. Consistent with this, aged rats infused withLPS express less of the trophic factor BDNF than younger ratsinfused with LPS (F1, 48 ¼ 8.94, p < 0.001) and by middle-aged ratsinfused with LPS express less XcT (F1, 48 ¼ 4.13, p < 0.05) whichcould otherwise be used to acquire cysteine for anti-oxidant pro-duction. Interestingly, across all ages, 56 days of LPS infusion wasresponsible for a significant decrease (all F> 6, p< 0.01) in the geneexpression of TH within the midbrain and/or brainstem and a cor-responding elevation in DBH gene expression within the hippo-campus; suggesting a loss of midbrain and/or brainstemcatecholamine production capacity and a compensatory increase inhippocampal catecholamine production capacity.
3.3. Immunohistochemistry
The density of TH-IR cells in the LC, that is the number of cellprofiles/sq. mm, was significantly decreased by the LPS infusion(F1, 41¼7.44, p< 0.01, Fig. 2A vs. B) andwas not dependent upon theage of the rat or the duration of the LPS exposure (p > 0.1). Thedensity of MHC II-IR microglia (Fig. 2B and C) throughout thebrainstem and particularly within the LC was significantlyincreased by the LPS infusion (F1, 41 ¼ 14.3, p < 0.001) and furtherincreased by the duration of the exposure (F1, 41 ¼ 11.0, p < 0.01).
The density of TH-IR cells in the SNpc (Fig. 3) was significantly(F2, 42 ¼ 5.26, p < 0.01) decreased because of aging. There was asignificant main effect of duration of LPS exposure (F1, 42 ¼ 6.78,
(XcT). The duration of the LPS infusion was responsible with a significant (# p < 0.01) incduration of the LPS infusion was responsible with a significant decrease (# p < 0.01) in thfactor (BDNF) within the brainstem and dopamine-ß-hydroxylase (DBH) gene expression wit(y p < 0.001) decrease in the gene expression of BDNF and XcT. Each biomarker represents a mdopamine-ß-hydroxylase; GLT1, glutamate transporter 1; GM-CSF, granulocyte-macrophagsaccharide; TGFb, transforming growth factor-b; TH, tyrosine hydroxylase; TLR4, toll-like re
p < 0.01); post hoc tests revealed that the number of TH-IR cells inthe SNpc of young andmiddle-aged rats were elevated after 56 daysof LPS infusion compared with 21 days infusion (p < 0.05). Ourprevious stereological analysis did not find evidence for tissueshrinkage (Brothers et al., 2013a). This recovery in the density of TH-IR in the SNpcwas not observed in the aged rats.We have previouslyreported a statistically significant increase in the number of MHC II-IR microglia within the SNpc following LPS exposure that wasassociated with a decline in the number of phosphorylated (i.e.,active) TH-IR SNpc cells (Brothers et al., 2013a).
4. Discussion
We induced a pro-inflammatory environment in the brains ofyoung, middle-aged, and aged male rats, and then systematicallydocumented an evolving series of biochemical and genetic changesin the midbrain and/or brainstem induced by the chronic infusionof picomolar levels of LPS into the fourth ventricle. These changesrepresent a complex interplay between glia and neurons in thepresence of continuous stimulation of TLR4 receptors on microglia,and suggest that a prolonged pro-inflammatory environment in-teracts with age to reduce the capacity of ascending catechol-aminergic nuclei in the midbrain and brainstem. These findingsrelate to the pre-symptomatic inflammatory environment observedin AD and PD and early loss of these ascending systems in diseaseprogression (Aarsland et al., 2001; Akiyama et al., 2000; Bartels andLeenders, 2005; Block and Hong, 2005; Cribbs et al., 2012; Griffinet al., 1989; Hobson and Meara, 2004; Hughes et al., 2000;Swardfager et al., 2010; Whitton, 2007).
LPS was used as a chemical tool to stimulate the TLR4/CD14complex expressed by microglia; administered as a continuous lowdose, LPS was not sufficient to produce any peripheral manifesta-tionsof infectiousdiseaseprocesses (suchaselevated serumlevels ofinflammatory cytokines) or down-regulation in microglial TLR4gene expression in the brain (Bardou et al., 2013; Brothers et al.,2013b). LPS did promote a central pro-inflammatory environment.For example, LPS increasedmidbrain and/or brainstem levels of pro-inflammatory IL-1b and IL-1a. IL-1b gene and protein levels corre-lated significantlywith each other (r¼ 0.74, p< 0.001). This variancein response to LPS is likely because of the differences in post-translational control of IL-1ß protein production (Chen et al., 2006).This distinction may be important, given the role of IL-1b in thedegeneration of SNpc dopaminergic neurons in PD (Koprich et al.,2008). Interestingly, elevations in IL-1ß gene and protein expres-sion because of LPS infusion were blunted in both middle-aged andaged rats. Ordinarily, GM-CSF levels are barely detectable in themiddle-agedbrain (Dameet al.,1999).However, in thepresent study,GM-CSF levels increased within the brainstem in response to pro-longed (56days) LPS exposure, butonly in themiddle-aged rats. GM-CSF is a pro-inflammatory factor released by astrocytes in responseto IL-1bor LPSthatbinds toa specific receptoronmicroglia leading toup-regulation of MHC II expression (Pierson et al., 2012) and TLR4gene expression (Parajuli et al., 2012) similar to that seen in thepresent study. GM-CSF can also enhance the LPS-induced NF-kBnuclear translocation and production of IL-1b (Parajuli et al., 2012),also similar to that seen in the present study. TNFa gene expressionwas increased by the LPS infusion but this increase was not influ-enced by the age of the animal. Typically, with normal aging,
rease in gene expression of glutamate transporter 1 (GLT1) and TGFb. In contrast, thee gene expression of tyrosine-hydroxylase (TH), TNFa, and brain-derived neurotrophichin the hippocampus. The age of the rat was independently responsible for a significantinimum of 6 rats/group. Abbreviations: BDNF, brain-derived neurotrophic factor; DBH,
e-stimulating factor; IFNg, interferon gamma; IL-1b, interleukin-1 beta; LPS, lipopoly-ceptor 4; TNFa, tumor necrosis factor alpha; XcT, cystine-glutamate anti-porter.
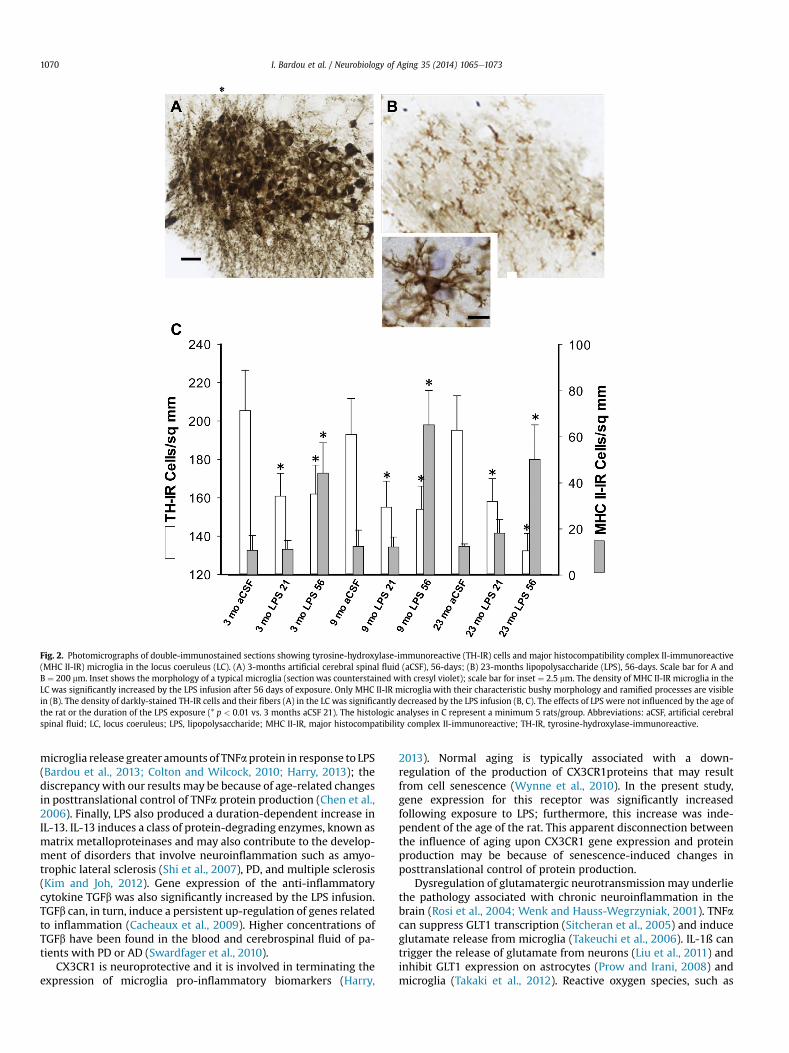
Fig. 2. Photomicrographs of double-immunostained sections showing tyrosine-hydroxylase-immunoreactive (TH-IR) cells and major histocompatibility complex II-immunoreactive(MHC II-IR) microglia in the locus coeruleus (LC). (A) 3-months artificial cerebral spinal fluid (aCSF), 56-days; (B) 23-months lipopolysaccharide (LPS), 56-days. Scale bar for A andB ¼ 200 mm. Inset shows the morphology of a typical microglia (section was counterstained with cresyl violet); scale bar for inset ¼ 2.5 mm. The density of MHC II-IR microglia in theLC was significantly increased by the LPS infusion after 56 days of exposure. Only MHC II-IR microglia with their characteristic bushy morphology and ramified processes are visiblein (B). The density of darkly-stained TH-IR cells and their fibers (A) in the LC was significantly decreased by the LPS infusion (B, C). The effects of LPS were not influenced by the age ofthe rat or the duration of the LPS exposure (* p < 0.01 vs. 3 months aCSF 21). The histologic analyses in C represent a minimum 5 rats/group. Abbreviations: aCSF, artificial cerebralspinal fluid; LC, locus coeruleus; LPS, lipopolysaccharide; MHC II-IR, major histocompatibility complex II-immunoreactive; TH-IR, tyrosine-hydroxylase-immunoreactive.
I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e10731070
microglia release greater amounts of TNFaprotein in response to LPS(Bardou et al., 2013; Colton and Wilcock, 2010; Harry, 2013); thediscrepancywith our results may be because of age-related changesin posttranslational control of TNFa protein production (Chen et al.,2006). Finally, LPS also produced a duration-dependent increase inIL-13. IL-13 induces a class of protein-degrading enzymes, known asmatrix metalloproteinases and may also contribute to the develop-ment of disorders that involve neuroinflammation such as amyo-trophic lateral sclerosis (Shi et al., 2007), PD, and multiple sclerosis(Kim and Joh, 2012). Gene expression of the anti-inflammatorycytokine TGFb was also significantly increased by the LPS infusion.TGFb can, in turn, induce a persistent up-regulation of genes relatedto inflammation (Cacheaux et al., 2009). Higher concentrations ofTGFb have been found in the blood and cerebrospinal fluid of pa-tients with PD or AD (Swardfager et al., 2010).
CX3CR1 is neuroprotective and it is involved in terminating theexpression of microglia pro-inflammatory biomarkers (Harry,
2013). Normal aging is typically associated with a down-regulation of the production of CX3CR1proteins that may resultfrom cell senescence (Wynne et al., 2010). In the present study,gene expression for this receptor was significantly increasedfollowing exposure to LPS; furthermore, this increase was inde-pendent of the age of the rat. This apparent disconnection betweenthe influence of aging upon CX3CR1 gene expression and proteinproduction may be because of senescence-induced changes inposttranslational control of protein production.
Dysregulation of glutamatergic neurotransmission may underliethe pathology associated with chronic neuroinflammation in thebrain (Rosi et al., 2004; Wenk and Hauss-Wegrzyniak, 2001). TNFacan suppress GLT1 transcription (Sitcheran et al., 2005) and induceglutamate release from microglia (Takeuchi et al., 2006). IL-1ß cantrigger the release of glutamate from neurons (Liu et al., 2011) andinhibit GLT1 expression on astrocytes (Prow and Irani, 2008) andmicroglia (Takaki et al., 2012). Reactive oxygen species, such as
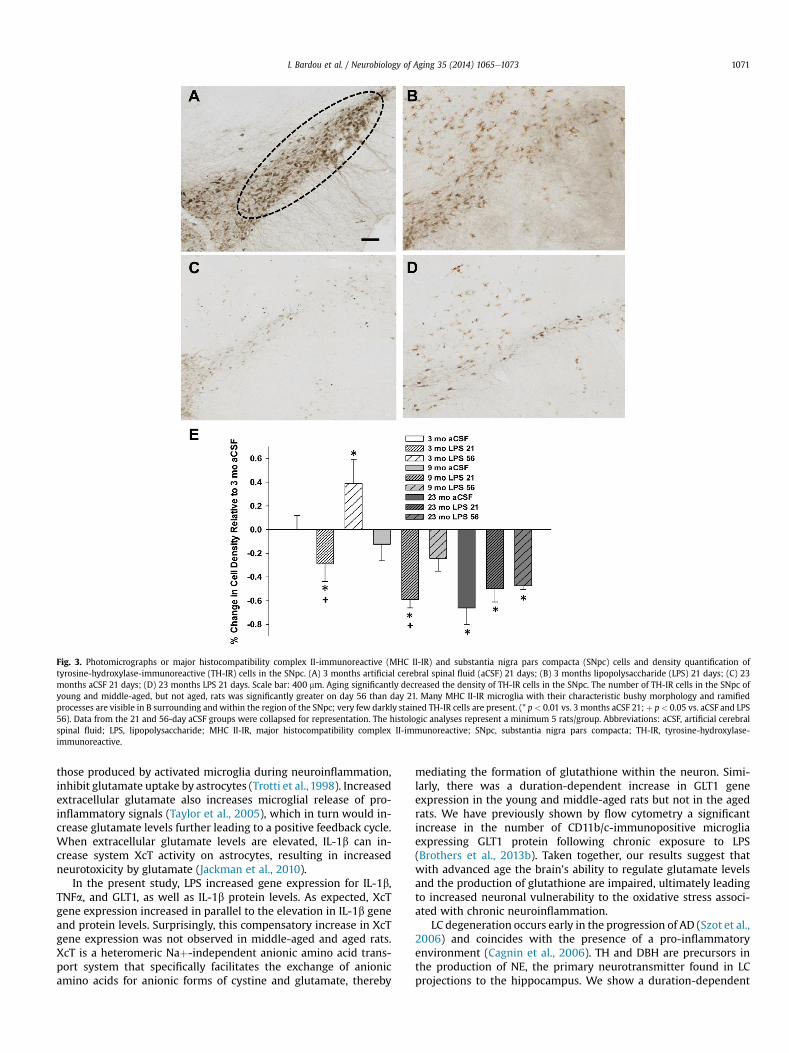
Fig. 3. Photomicrographs or major histocompatibility complex II-immunoreactive (MHC II-IR) and substantia nigra pars compacta (SNpc) cells and density quantification oftyrosine-hydroxylase-immunoreactive (TH-IR) cells in the SNpc. (A) 3 months artificial cerebral spinal fluid (aCSF) 21 days; (B) 3 months lipopolysaccharide (LPS) 21 days; (C) 23months aCSF 21 days; (D) 23 months LPS 21 days. Scale bar: 400 mm. Aging significantly decreased the density of TH-IR cells in the SNpc. The number of TH-IR cells in the SNpc ofyoung and middle-aged, but not aged, rats was significantly greater on day 56 than day 21. Many MHC II-IR microglia with their characteristic bushy morphology and ramifiedprocesses are visible in B surrounding and within the region of the SNpc; very few darkly stained TH-IR cells are present. (* p < 0.01 vs. 3 months aCSF 21; þ p< 0.05 vs. aCSF and LPS56). Data from the 21 and 56-day aCSF groups were collapsed for representation. The histologic analyses represent a minimum 5 rats/group. Abbreviations: aCSF, artificial cerebralspinal fluid; LPS, lipopolysaccharide; MHC II-IR, major histocompatibility complex II-immunoreactive; SNpc, substantia nigra pars compacta; TH-IR, tyrosine-hydroxylase-immunoreactive.
I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e1073 1071
those produced by activated microglia during neuroinflammation,inhibit glutamate uptake by astrocytes (Trotti et al., 1998). Increasedextracellular glutamate also increases microglial release of pro-inflammatory signals (Taylor et al., 2005), which in turn would in-crease glutamate levels further leading to a positive feedback cycle.When extracellular glutamate levels are elevated, IL-1b can in-crease system XcT activity on astrocytes, resulting in increasedneurotoxicity by glutamate (Jackman et al., 2010).
In the present study, LPS increased gene expression for IL-1b,TNFa, and GLT1, as well as IL-1b protein levels. As expected, XcTgene expression increased in parallel to the elevation in IL-1b geneand protein levels. Surprisingly, this compensatory increase in XcTgene expression was not observed in middle-aged and aged rats.XcT is a heteromeric Naþ-independent anionic amino acid trans-port system that specifically facilitates the exchange of anionicamino acids for anionic forms of cystine and glutamate, thereby
mediating the formation of glutathione within the neuron. Simi-larly, there was a duration-dependent increase in GLT1 geneexpression in the young and middle-aged rats but not in the agedrats. We have previously shown by flow cytometry a significantincrease in the number of CD11b/c-immunopositive microgliaexpressing GLT1 protein following chronic exposure to LPS(Brothers et al., 2013b). Taken together, our results suggest thatwith advanced age the brain’s ability to regulate glutamate levelsand the production of glutathione are impaired, ultimately leadingto increased neuronal vulnerability to the oxidative stress associ-ated with chronic neuroinflammation.
LC degeneration occurs early in the progression of AD (Szot et al.,2006) and coincides with the presence of a pro-inflammatoryenvironment (Cagnin et al., 2006). TH and DBH are precursors inthe production of NE, the primary neurotransmitter found in LCprojections to the hippocampus. We show a duration-dependent

I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e10731072
loss in TH gene expression in the midbrain and/or brainstem andreduced number of TH-IR cells in the LC and SNpc with LPS infusionand age. Overall, the oldest rats that experienced the longestduration of LPS infusion demonstrated the greatest decline in TH-IRcells in LC and SNpc, consistent with previous investigations(Mouton et al., 2012). The present studywas designed to investigatefor such an age-dependent increase in vulnerability to chronicneuroinflammation and identify a specific inflammatory biomarkerunderlying the LC cell loss in animal models of AD (Manaye et al.,2013). We anticipated that IL-6 might play a role in the age-related vulnerability (Ye and Johnson, 1999) but IL-6 levels in thepresent study only increased in response to the duration of the LPSinfusion and was not dependent upon the age of the rat. BDNFsignaling via trkB neurotrophin receptors is important for themaintenance of the LC innervation of the hippocampus (von Bohlenund Halbach and Minichiello, 2006); in the present study, aged ratsinfused with LPS expressed significantly less of the gene for BDNFthan the younger rats infused with LPS.
Interestingly, after 56 days of LPS infusion, DBH is significantlyincreased in the hippocampus in all age groups compared with adecrease (although not significant) after 21 days of LPS infusion.This suggests a compensatory up-regulation in the DBH gene,consistent with our previous report (Brothers et al., 2013a). Adecrease in TH enzyme levels in the midbrain and/or brainstemsuggests a decline in catecholamine function that may underlieaspects of the cognitive impairment seen in AD (Grudzien et al.,2007). Cognitive decline may also be related to the fact that NEacts as an anti-inflammatory molecule within the cortex and hip-pocampus (Feinstein et al., 2002; Wenk et al., 2003), stimulatesBDNF production (Mannari et al., 2008) and supports neurogenesis(Masuda et al., 2012); all functions that could be improved by acompensatory increase in hippocampal DBH. Overall, chronicneuroinflammation leads to impaired LC cellular integrity, reducedhippocampal neurogenesis (Marchalant et al., 2009), and reducedBDNF gene expression (Tong et al., 2008).
SNpc cellular degeneration is influenced by the presence ofneuroinflammation early in the progression of PD (Tome et al., 2013;Wanget al., 2013). Previous inflammatorymodels of PD that injectedLPS directly into the substantia nigra significantly reduced thenumber of TH-IR neurons (Herrera et al., 2000; Kim et al., 2000). Incontrast, an acute intra-nigral injection of TNF-a (Castano et al.,2002) or an acute peripheral injection of a high dose of LPS bothfailed to reduce the number of TH SNpc neurons (Mouton et al.,2012). Similarly, we have demonstrated that a single acute injec-tion of LPS into the nucleus basalis magnocellularis failed to reducethe number of acetylcholinergic neurons (Willard et al.,1999). In thepresent study, 3 weeks of continuous LPS infusion produced adecline in the number of TH-IR cells in young and middle-aged rats,but no additional decline in the number of TH-IR cells in the SNpc ofaged rats. After 8 weeks of LPS infusion the number of TH-IR cells inthe SNpc had completely recovered in young and middle-aged rats.In addition, the density of TH-IR cells in the SNpc was decreasedbecauseof normal aging; a decline in SNpc function that is consistentwith previous reports (Gozlan et al., 1990;Miguez et al., 1999). Thus,the consequences of pro-inflammatory environment likely dependupon an interaction between the duration of the exposure to specificcytokines (Bardou et al., 2013), the age of the brain and the specificregion. For example, the vulnerability of SNpc cells was primarilydependent upon the duration of the pro-inflammatory environ-ment, while the vulnerability of LC cells was not changed by longerexposure to LPS. Overall, the infusion of LPS increased the density ofMHC II-IR microglia and expression of pro-inflammatory cytokines(Il-1ß and IL-1a) throughout the midbrain and/or brainstem.
Our data support the concept that continuous exposure to a pro-inflammatory environment drives exaggerated changes in the
production and release of inflammatory mediators, decreasedproduction protective factors, altered glutamate regulation andimpaired cellular function within the SNpc, and LC. Furthermore,our data show that the response to a pro-inflammatory environ-ment changes with age. Overall, these data suggest that early anti-inflammatory intervention is an important therapeutic opportunityin neurodegenerative diseases, which have defining pathology ofneuroinflammation such as PD and AD.
Disclosure statement
The authors declare no conflicts of interest.
Acknowledgements
Supported by U.S. Public Health Service, RO1 AG030331, RO1AG037320 and The Ohio State University Women and PhilanthropyProgram to G.L.W. All authors have contributed to the work, agreewith the presented findings, and that the work has not been pub-lished before nor is being considered for publication in anotherjournal.
References
Aarsland, D., Andersen, K., Larsen, J.P., Lolk, A., Nielsen, H., Kragh-Sorensen, P., 2001.Risk of dementia in Parkinson’s disease: a community based, prospective study.Neurology 56, 730e736.
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cooper, N.R., Eikelenboom, P.,Emmerling, M., Fiebich, B., Finch, C.E., Frautschy, S., Griffin, W.S., Hampel, H.,Landreth, G., McGeer, P.L., Mrak, R., MacKenzie, I., O’Banion, K., Pachter, J.,Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen, Y., Streit, W.,Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F.L., Veerhuis, R., Walker, D.,Webster, S., Wegrzyniak, B., Wenk, G., Wyss-Coray, A., 2000. Inflammation andAlzheimer’s disease. Neurobiol. Aging 21, 383e421.
Bardou, I., Brothers, H.M., Kaercher, R.M., Hopp, S.C., Wenk, G.L., 2013. Differentialeffects of duration and age upon the consequences of neuroinflammation in thehippocampus. Neurobiol. Aging 34, 2293e2301.
Bartels, A.L., Leenders, K.L., 2005. Neuroinflammation in the pathophysiology ofParkinson’s disease: evidence from animal models to human in vivo studieswith [11C] PK11195 PET. Mov. Disord. 22, 1852e1856.
Bilbo, S.D., 2010. Early-life infection is a vulnerability factor for aging-related glialalterations and cognitive decline. Neurobiol. Learn Mem. 94, 57e64.
Block,M.L.,Hong, J.S., 2005.Microglia and inflammation-mediatedneurodegeneration:multiple triggers with a commonmechanism. Prog. Neurobiol. 76, 77e98.
Braak, H., Del Tredici, K., Rub, U., DeVos, R.A., Jansen Steur, E.N., Braak, E., 2003.Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol.Aging 24, 197e211.
Brothers, H.M., Bardou, I., Hopp, S.C., Marchalant, Y., Kaercher, R.M., Turner, S.M.,Mitchem, M.R., Kigerl, K., Wenk, G.L., 2013a, Time-dependent compensatoryresponses to chronic neuroinflammation in hippocampus and brainstem: thepotential role of glutamate neurotransmission. J. Alz. Dis. Parkin, in press.
Brothers, H.M., Bardou, I., Hopp, S.C., Kaercher, R.M., Wynne-Corona, A., Fenn, A.M.,Godbout, J.P., Wenk, G.L., 2013b. Riluzole partially rescues age-associated, butnot LPS-induced, loss of glutamate transporters and spatial memory impair-ment. J. Neuroimmune Pharmacol. 8, 1098e1105.
Cacheaux, L.P., Ivens, S., David, Y., Lakhter, A.J., Bar-Klein, G., Shapira, M.,Heinemann, U., Friedman, A., Kaufer, D., 2009. Transcriptome profiling revealsTGF-beta signaling involvement in epileptogenesis. J. Neurosci. 29, 8927e8935.
Cagnin, A., Kassiou, M., Meikle, S.R., Banati, R.B., 2006. In vivo evidence for micro-glial activation in neurodegenerative dementia. Acta Neurol. Scand. Suppl. 114,107e114.
Castano, A., Herrera, A.J., Cano, J., Machado, A., 2002. The degenerative effect of asingle intranigral injection of LPS on the dopaminergic system is prevented bydexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma.J. Neurochem. 81, 150e157.
Chen, Y.L., Huang, Y.L., Lin, N.Y., Chen, H.C., Chiu, W.C., Change, C.J., 2006. Differentialregulation of ARE-mediated TNFa and IL-1b mRNA stability by lipopolysac-charide in RAW264.7 cells. Biochem. Biophys. Res. Commun. 346, 160e168.
Colton, C.A., Wilcock, D.M., 2010. Assessing activation states in microglia. CNSNeurol. Disord. Drug Targets 9, 174e191.
Cribbs, D.H., Berchtold, N.C., Perreau, V., Coleman, P.D., Rogers, J., Tenner, A.J.,Cotman, C.W., 2012. Extensive innate immune gene activation accompaniesbrain aging, increasing vulnerability to cognitive decline and neuro-degeneration: a microarray study. J. Neuroinflammation 9, 179.
Dame, J.B., Christensen, R.D., Juul, S.E., 1999. The distribution of granulocyte-macrophage colony-stimulating factor and its receptor in the developing hu-man fetus. Pediatr. Res. 46, 358e366.

I. Bardou et al. / Neurobiology of Aging 35 (2014) 1065e1073 1073
Eikelenboom, P., van Exel, E., Hoozemans, J.J.M., Veerhuis, R., Rosemuller, A.J.M., vanGool, W.A., 2010. Neuroinflammation e an early event in both the history andpathogenesis of Alzheimer’s disease. Neurodegener Dis. 7, 38e41.
Feinstein, D.L., Heneka, M.T., Gavrilyuk, V., Dello Russo, C., Weinberg, G., Galea, E.,2002. Noradrenergic regulation of inflammatory gene expression in brain.Neurochem. Int. 41, 357e365.
Gasbarri, A., Packard, M.G., Campana, E., Pacitti, C., 1994. Anterograde and retro-grade tracing of projections from the ventral tegmental area to the hippocampalformation in the rat. Brain Res. Bull. 33, 445e452.
Gerhard, A., Pavese, N., Hotton, G., Turkheimer, F., Es, M., Hammers, A., Eggert, K.,Oertel, W., Banati, R.B., Brooks, D.J., 2006. In vivo imaging of microglial activa-tion with [C-11](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol.Dis. 21, 404e412.
Gozlan, H., Daval, G., Verge, D., Spampinato, U., Fattaccini, C.M., Gallissot, M.C.,Elmestikawy, S., Hamon, M., 1990. Aging associated changes in serotoninergicand dopaminergic pre- and postsynaptic neurochemical markers in the ratbrain. Neurobiol. Aging 11, 437e449.
Griffin, W.S., Stanley, L.C., Ling, C., White, L., MacLeod, V., Perrot, L.J., White III, C.L.,Araoz, C., 1989. Brain interleukin 1 and S-100 immunoreactivity are elevated inDown syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A 86,7611e7615.
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D.C., Mesulam, M.M., 2007.Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impair-ment and early Alzheimer’s Disease. Neurobiol. Aging 28, 327e335.
Halliday, G.M., Del Tredici, K., Braak, H., 2006. Critical appraisal of brain pathologystaging related to presymptomatic and symptomatic cases of sporadic Parkin-son’s disease. J. Neural Trans. Suppl. 70, 99e103.
Harry, G.J., 2013. Microglia during development and aging. Pharmacol. Ther. 139,313e326.
Hauss-Wegrzyniak, B., Dobrzanski, P., Stoehr, J.D., Wenk, G.L., 1998. Chronic neu-roinflammation in rats reproduces components of the neurobiology of Alz-heimer’s disease. Brain Res. 780, 294e303.
Heneka, M.T., O’Banion, M.K., Terwel, D., Kummer, M.P., 2010. Neuroinflammatoryprocesses in Alzheimer’s disease. J. Neural Trans. 117, 919e947.
Herrera, A.J., Castano, A., Venero, J.L., Cano, J., Machado, A., 2000. The singleintranigral injection of LPS as a new model for studying the selective effectsof inflammatory reactions on dopaminergic system. Neurobiol. Dis. 7,429e447.
Herrup, K., 2010. Reimagining Alzheimer’s disease- an age-based hypothesis.J. Neurosci. 30, 16755e16762.
Hobson, P., Meara, J., 2004. Risk and incidence of dementia in a cohort of oldersubjects with Parkinson’s disease in the United Kingdom. Mov. Disord. 19,1043e1049.
Hughes, T.A., Ross, H.F., Musa, S., Bhattacherjee, S., Nathan, R.N., Mindham, R.H.,Spokes, E.G., 2000. A 10-year study of the incidence of and factors predictingdementia in Parkinson’s disease. Neurology 54, 1596e1602.
Imamura, K., Hishikawa, N., Sawada, M., Nagatsu, T., Yoshida, M., Hashizume, Y.,2003. Distribution of major histocompatibility complex class II-positivemicroglia and cytokine profile of Parkinson’s disease brains. Acta Neuro-pathol. 106, 518.
Jackman, N.A., Uliasz, T.F., Hewett, J.A., Hewett, S.J., 2010. Regulation of system x(c)(-) activity and expression in astrocytes by interleukin-1 beta: implications forhypoxic neuronal injury. Glia 58, 1806e1815.
Kim, Y.S., Joh, T.H., 2012. Matrix metalloproteinases, new insights into the under-standing of neurodegenerative disorders. Biomol. Ther. 20, 133e143.
Kim, W.G., Mohney, R.P., Wilson, B., Jeohn, G.H., Liu, B., Hong, J.S., 2000. Regionaldifference in susceptibility to lipopolysaccharide-induced neurotoxicity in therat brain: role of microglia. J. Neurosci. 20, 6309e6316.
Koprich, J.B., Reske-Nielsen, C., Mithal, P., Isacson, O., 2008. Neuroinflammationmediated by IL-1b increases susceptibility of dopamine neurons to degenera-tion in an animal model of Parkinson’s disease. J. Neuroinflammation 5, 8.
Liu, L., Aboud, O., Jones, R.A., Mrak, R.E., Griffin, W.S.T., Barger, S.W., 2011. Apoli-poprotein E expression is elevated by interleukin 1 and other interleukin1-induced factors. J. Neuroinflammation 8, 175.
Manaye, K.F., Mouton, P.R., Xu, G., Drew, A., Lei, D.-L., Sharma, Y., Rebeck, G.W.,Turner, S., 2013. Age-related loss of noradrenergic neurons in the brains of tripletransgenic mice. Age 35, 139e147.
Mannari, C., Origlia, N., Scatena, A., Del Debbio, A., Catena, M., Dell’agnello, G.,Barraco, A., Giovannini, L., Dell’osso, L., Domenici, L., Piccinni, A., 2008. BDNFlevel in the rat prefrontal cortex increases following chronic but not acutetreatment with duloxetine, a dual acting inhibitor of noradrenaline and sero-tonin re-uptake. Cell Mol. Neurobiol. 28, 457e468.
Marchalant, Y., Brothers, H.M., Norman, G.J., Karolina, K., DeVries, C., Wenk, G.L.,2009. Cannabinoids attenuate the effects of aging upon neuroinflammation andneurogenesis. Neurobiol. Dis. 34, 300e307.
Marchalant, Y., Rosi, S., Wenk, G.L., 2007. Anti-inflammatory property of thecannabinoid agonist WIN-55212-2 in a rodent model of chronic brain inflam-mation. Neuroscience 144, 1516e1522.
Masuda, T., Nakagawa, S., Boku, S., Nishikawa, H., Takamura, N., Kato, A., Inoue, T.,Koyama, T., 2012. Noradrenaline increases neural precursor cells derived frommiddle-aged rat dentate gyrus through beta2 receptor. Prog. Neuro-psychopharmacol. Biol. Psychiatary 36, 44e51.
Miguez, J.M., Aldegunde, M., Paz-Valinas, L., Recio, J., Sanchez-Barcelo, E., 1999.Selective changes in the contents of noradrenaline, dopamine and serotonin inrat brain areas during aging. J. Neural. Transm. 106, 1089e1098.
Mouton, P.R., Kelly-Bell, B., Tweedie, D., Spangler, E.L., Perez, E., Carlson, O.D.,Short, R.G., deCabo, R., Chang, J., Ingram, D.K., Li, Y., Greig, N.H., 2012. The effectsof age and lipopolysaccharide (LPS)-mediated peripheral inflammation onnumbers of central catecholaminergic neurons. Neurobiol. Aging 33, 423e427.
Parajuli, B., Sonobe, Y., Kawanokuchi, J., Doi, Y., Noda, M., Takeuchi, H., Mizuno, T.,Suzumura, A., 2012. GM-CSF increases LPS-induced production of proin-flammatory mediators via upregulation of TLR4 and CD14 in murine microglia.J. Neuroinflammation 9, 268.
Pierson, E., Simmons, S.B., Castelli, L., Goverman, J.M., 2012. Mechanisms regulatingregional localization of inflammation during CNS autoimmunity. Immunol. Rev.248, 205e215.
Prow, N.A., Irani, D.N., 2008. The inflammatory cytokine, interleukin-1 beta, medi-ates loss of astroglial glutamate transport and drives excitotoxic motor neuroninjury in the spinal cord during acute viral encephalomyelitis. J. Neurochem.105, 1276e1286.
Rosi, S., Ramirez-Amaya, V., Hauss-Wegrzyniak, B., Wenk, G.L., 2004. Chronic braininflammation leads to a decline in hippocampal NMDA R1 receptors.J. Neuroinflammation 1, 12e18.
Rudow, G., O’Brien, R., Savonenko, A.V., Resnick, S.M., Zonderman, A.B.,Pletnikova, O., Marsh, L., Dawson, T.M., Crain, B.J., West, M.J., Troncoso, J.C.,2008. Morphometry of the human substantia nigra in ageing and Parkinson’sdisease. Acta Neuropathol. 115, 461e470.
Shi, N., Kawano, Y., Tateishi, T., Kikuchi, H., Osoegawa, M., Ohyagi, Y., Kira, J., 2007.Increased IL-13-producing T cells in ALS: positive correlations with diseaseseverity and progression rate. J. Neuroimmunol. 182, 232e235.
Sitcheran, R., Gupta, P., Fisher, P.B., Baldwin, A.S., 2005. Positive and negativeregulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlledrepression. EMBO J. 24, 510e520.
Smith, J.A., Das, A., Ray, S.K., Banik, N.L., 2012. Role of pro-inflammatory cytokinesreleased from microglia in neurodegenerative diseases. Brain Res. Bull. 87,10e20.
Swardfager, W., Lanctôt, K., Rothenburg, L., Wong, A., Cappell, J., Herrmann, N., 2010.A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68,930e941.
Szot, P., White, S.S., Greenup, J.L., Leverenz, J.B., Peskind, E.R., Raskind, M.A., 2006.Compensatory changes in the noradrenergic nervous system in the locus ce-ruleus and hippocampus of postmortem subjects with Alzheimer’s disease anddementia with Lewy bodies. J. Neurosci. 26, 467e478.
Takaki, J., Fujimori, K., Miura, M., Suzuki, T., Sekino, Y., Sato, K., 2012. L-glutamatereleased from activated microglia downregulates astrocytic L-glutamate trans-porter expression in neuroinflammation: the “collusion” hypothesis forincreased extracellular L-glutamate concentration in neuroinflammation.J. Neuroinflammation 9, 275.
Takeuchi, H., Jin, S.J., Wang, J.Y., Zhang, G.Q., Kawanokuchi, J., Kuno, R., Sonobe, Y.,Mizuno, T., Suzumura, A., 2006. Tumor necrosis factor-alpha induces neuro-toxicity via glutamate release from hemichannels of activated microglia in anautocrine manner. J. Biol. Chem. 281, 21362e21368.
Taylor, D.L., Jones, F., Kubota, E.S., Pocock, J.M., 2005. Stimulation of microglialmetabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand.J. Neurosci. 25, 2952e2964.
Tome, C.M.L., Tyson, T., Rey, N.L., Grathwohl, S., Britschgi, M., Brundin, P., 2013.Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease-is there a link? Mol. Neurobiol. 47, 561e574.
Tong, L., Balazs, R., Soiampornkul, R., Thangnipon, W., Cotman, C.W., 2008. Inter-leukin-1 beta impairs brain derived neurotrophic factor-induced signal trans-duction. Neurobiol. Aging 29, 1380e1393.
Trotti, D., Danbolt, N.C., Volterra, A., 1998. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neuro-degeneration? Trends Pharmacol Sci. 19, 328e334.
Von Bohlen und Halbach, O., Minichiello, L., 2006. Neurotrophin receptor hetero-zygosity causes deficits in catecholaminergic innervation of amygdala andhippocampus in aged mice. J. Neural Transm. 113, 1829e1836.
Wang, J., Song, N., Jiang, H., Wang, J., Xie, J.X., 2013. Pro-inflammatory cytokinesmodulate iron regulatory protein 1 expression and iron transportation throughreactive oxygen/nitrogen species production in ventral mesencephalic neurons.Biochim. Biophys. Acta Mol. Basis Dis. 1832, 618e625.
Wenk, G.L., Hauss-Wegrzyniak, B., 2001. Animal Models of Chronic Neuro-inflammation as a Model of Alzheimer’s Disease. In: Bondy, S., Campbell, A.(Eds.), Inflammatory Events in Neurodegeneration. Prominent Press, Scottsdale,AZ, pp. 83e87.
Wenk, G.L., McGann, K., Hauss-Wegrzyniak, B., Rosi, S., 2003. The toxicity of tumornecrosis factor-a upon cholinergic neurons within the nucleus basalis and therole of norepinephrine in the regulation of inflammation: implications forAlzheimer’s disease. Neuroscience 121, 719e729.
Whitton, P.S., 2007. Inflammation as a causative factor in the aetiology of Parkin-son’s disease. Br. J. Pharmacol. 150, 963e976.
Willard, L.B., Hauss-Wegrzyniak, B., Wenk, G.L., 1999. Pathological and biochemicalconsequences of acute and chronic neuroinflammation within the basal fore-brain cholinergic system of rats. Neuroscience 88, 193e200.
Wynne, A.M., Henry, C.J., Huang, Y., Cleland, A., Godbout, J.P., 2010. Protracteddownregulation of CX3CR1 on microglia of aged mice after lipopolysaccharidechallenge. Brain Behav. Immun. 24, 1190e1201.
Ye, S.M., Johnson, R.W., 1999. Increased interleukin-6 expression by microglia frombrain of aged mice. J. Neuroimmunol. 93, 139e148.




























