Embed Size (px)
Citation preview

Neoceptors: reengineering GPCRs torecognize tailored ligandsKenneth A. Jacobson1, Zhan-Guo Gao1 and Bruce T. Liang2
1 Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, Digestive and Kidney Diseases
(NIDDK), National Institutes of Health (NIH), Bethesda, MD 20892, USA2 Pat and Jim Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, CT 06030, USA
Opinion TRENDS in Pharmacological Sciences Vol.28 No.3
Glossary
Ligand docking: a process of computational identification of an energetically
favorable binding mode of a small molecule in its receptor site.
Efforts to model and reengineer the putative bindingsites of G-protein-coupled receptors (GPCRs) have led toan approach that combines small-molecule ’classical’medicinal chemistry and gene therapy. In this approach,complementary structural changes (e.g. based on novelionic or H-bonds) are made in the receptor and ligand forthe selective enhancement of affinity. Thus, a modifiedreceptor (neoceptor) is designed for activation by tailor-made agonists that do not interact with the nativereceptor. The neoceptor is no longer activated by thenative agonist, but rather functions as a scaffold forthe docking of novel small molecules (neoligands). Intheory, the approach could verify the accuracy of GPCRmolecular modeling, the investigation of signaling, thedesign of small molecules to rescue disease-relatedmutations, and small-molecule-directed gene therapy.The neoceptor–neoligand pairing could offer spatialspecificity by delivering the neoceptor to a target site,and temporal specificity by administering neoligandwhen needed.
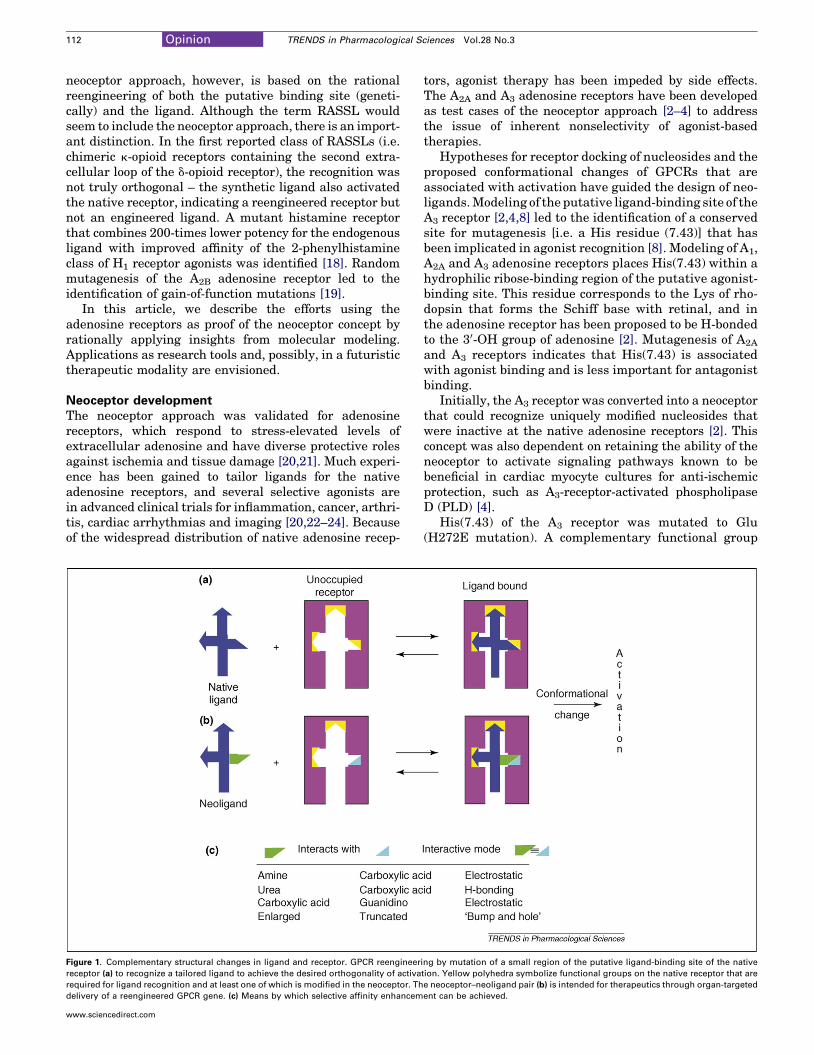
IntroductionThe use of G-protein-coupled receptor (GPCR) agonists fortherapyhas inherent limitations [1] becausedesensitizationand widespread receptor distribution can lead to undesiredside-effects. We are developing an alternative approach tobenefit from GPCR activation in a more spatially andtemporally selective manner than the systemic adminis-tration of agonists to the native GPCR. This ‘neoceptor’approach [2–4] combines small-molecule ‘classical’ medic-inal chemistry and gene or cell therapy. Using this rationaldesign approach, complementary structural changes aremade in the receptor and ligand for the selective enhance-ment of affinity (Figure 1). The activation of a neoceptor in aspatially selective manner would be achieved by cell- ororgan-targeted delivery of the gene, given the developmentof an appropriate delivery method.
Molecular modeling based on homology to thebest-studied GPCR, rhodopsin [5,6], has been used toarrive at hypotheses for ligand docking (see Glossary),which are ideally validated using site-directed mutagen-esis [7,8]. With this knowledge and the ability to tailor-make new analogs of a native agonist, one can design amatched neoceptor and neoligand (i.e. the binding site of a
Corresponding author: Jacobson, K.A. ([email protected]).Available online 5 February 2007.
www.sciencedirect.com 0165-6147/$ – see front matter . Published by Elsevier Ltd. doi:10.101
given GPCR can be engineered to recognize syntheticagonist ligands that do not activate the native receptor).Unlike de novo receptor design [9], this approach uses thenative receptor as a scaffold for the docking of novelmolecules. This reengineered GPCR (neoceptor) ideallyretains its capacity to activate a particular second-messenger pathway, causing beneficial effects identicalto those induced by the native receptor. The uniquelymatched ligands (neoligands) are synthesized based onmolecular complementarity with the neoceptor. The struc-ture–activity relationship (SAR) profile of such modifiedreceptorsneednot correspondtothatof theparent receptors.
It is envisioned that the neoceptor DNA would bedelivered by an appropriate organ-targeted gene or celltherapy. In the absence of the neoligand, the neoceptorwould be ‘silent’ – not subject to activation by the nativeagonist. The side effects normally associated with agonisttherapy would not be expected because the native receptorwould not be activated by the tailored ligand. Thus, bydesign, the interacting pairs of receptor and ligand wouldhave to be orthogonal with respect to the native pair.
The reengineering of enzymes, nuclear receptors andother proteins is practiced in many contexts [10–16]. Var-ious kinases have been reengineered to recognize modifiedATP analogs, for example, by the creation of a ‘bump andhole’ [10]. The microscopic complementarity of the b-adre-noceptor and its chemically modified ligands has beenstudied [12]. Other GPCRs have been engineered for regu-lation by metal ions, leading to insights into the activationmechanism [17], and reengineered receptors have beenproposed for rescue from genetic diseases [4,15].
Although not intended for therapeutic application,receptors activated solely by synthetic ligands (RASSLs)have been introduced by Conklin and colleagues formechanistic probing through conditional expression intransgenicmice [13]. RASSLs beginwith a GPCR for whicha synthetic high-affinity agonist probe is known and thenmutation reduces the affinity of the endogenous ligands,with retention of affinity for the synthetic agonist. The
Orthogonal: multiple systems in which individual elements interact only within
a system and do not cross-react: from the Greek ‘ortho’, meaning ‘right’, and
‘gonia’, meaning ‘angle’.
6/j.tips.2007.01.006
112 Opinion TRENDS in Pharmacological Sciences Vol.28 No.3
neoceptor approach, however, is based on the rationalreengineering of both the putative binding site (geneti-cally) and the ligand. Although the term RASSL wouldseem to include the neoceptor approach, there is an import-ant distinction. In the first reported class of RASSLs (i.e.chimeric k-opioid receptors containing the second extra-cellular loop of the d-opioid receptor), the recognition wasnot truly orthogonal – the synthetic ligand also activatedthe native receptor, indicating a reengineered receptor butnot an engineered ligand. A mutant histamine receptorthat combines 200-times lower potency for the endogenousligand with improved affinity of the 2-phenylhistamineclass of H1 receptor agonists was identified [18]. Randommutagenesis of the A2B adenosine receptor led to theidentification of gain-of-function mutations [19].
In this article, we describe the efforts using theadenosine receptors as proof of the neoceptor concept byrationally applying insights from molecular modeling.Applications as research tools and, possibly, in a futuristictherapeutic modality are envisioned.
Neoceptor developmentThe neoceptor approach was validated for adenosinereceptors, which respond to stress-elevated levels ofextracellular adenosine and have diverse protective rolesagainst ischemia and tissue damage [20,21]. Much experi-ence has been gained to tailor ligands for the nativeadenosine receptors, and several selective agonists arein advanced clinical trials for inflammation, cancer, arthri-tis, cardiac arrhythmias and imaging [20,22–24]. Becauseof the widespread distribution of native adenosine recep-
Figure 1. Complementary structural changes in ligand and receptor. GPCR reengineer
receptor (a) to recognize a tailored ligand to achieve the desired orthogonality of activa
required for ligand recognition and at least one of which is modified in the neoceptor. Th
delivery of a reengineered GPCR gene. (c) Means by which selective affinity enhancem
www.sciencedirect.com
tors, agonist therapy has been impeded by side effects.The A2A and A3 adenosine receptors have been developedas test cases of the neoceptor approach [2–4] to addressthe issue of inherent nonselectivity of agonist-basedtherapies.
Hypotheses for receptor docking of nucleosides and theproposed conformational changes of GPCRs that areassociated with activation have guided the design of neo-ligands.Modeling of the putative ligand-binding site of theA3 receptor [2,4,8] led to the identification of a conservedsite for mutagenesis [i.e. a His residue (7.43)] that hasbeen implicated in agonist recognition [8]. Modeling of A1,A2A and A3 adenosine receptors places His(7.43) within ahydrophilic ribose-binding region of the putative agonist-binding site. This residue corresponds to the Lys of rho-dopsin that forms the Schiff base with retinal, and inthe adenosine receptor has been proposed to be H-bondedto the 30-OH group of adenosine [2]. Mutagenesis of A2A
and A3 receptors indicates that His(7.43) is associatedwith agonist binding and is less important for antagonistbinding.
Initially, the A3 receptor was converted into a neoceptorthat could recognize uniquely modified nucleosides thatwere inactive at the native adenosine receptors [2]. Thisconcept was also dependent on retaining the ability of theneoceptor to activate signaling pathways known to bebeneficial in cardiac myocyte cultures for anti-ischemicprotection, such as A3-receptor-activated phospholipaseD (PLD) [4].
His(7.43) of the A3 receptor was mutated to Glu(H272E mutation). A complementary functional group
ing by mutation of a small region of the putative ligand-binding site of the native
tion. Yellow polyhedra symbolize functional groups on the native receptor that are
e neoceptor–neoligand pair (b) is intended for therapeutics through organ-targeted
ent can be achieved.
Opinion TRENDS in Pharmacological Sciences Vol.28 No.3 113
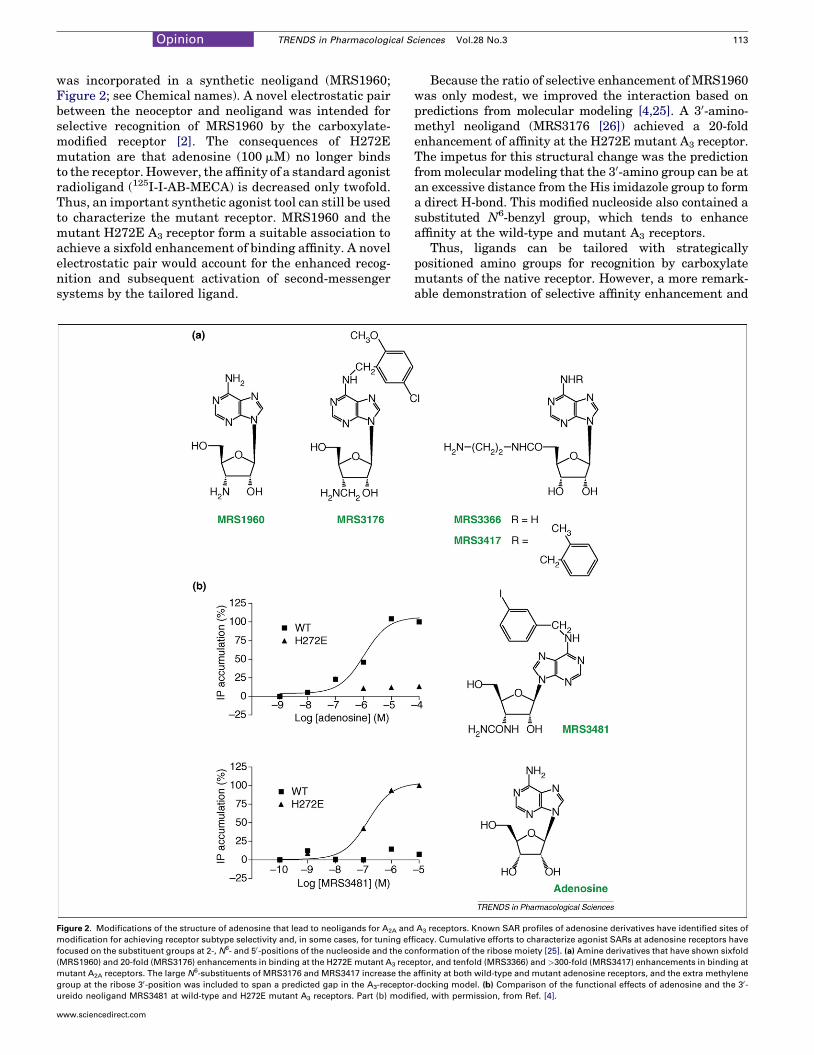
was incorporated in a synthetic neoligand (MRS1960;Figure 2; see Chemical names). A novel electrostatic pairbetween the neoceptor and neoligand was intended forselective recognition of MRS1960 by the carboxylate-modified receptor [2]. The consequences of H272Emutation are that adenosine (100 mM) no longer bindsto the receptor. However, the affinity of a standard agonistradioligand (125I-I-AB-MECA) is decreased only twofold.Thus, an important synthetic agonist tool can still be usedto characterize the mutant receptor. MRS1960 and themutant H272E A3 receptor form a suitable association toachieve a sixfold enhancement of binding affinity. A novelelectrostatic pair would account for the enhanced recog-nition and subsequent activation of second-messengersystems by the tailored ligand.
Figure 2. Modifications of the structure of adenosine that lead to neoligands for A2A and
modification for achieving receptor subtype selectivity and, in some cases, for tuning eff
focused on the substituent groups at 2-, N6- and 50-positions of the nucleoside and the co
(MRS1960) and 20-fold (MRS3176) enhancements in binding at the H272E mutant A3 rece
mutant A2A receptors. The large N6-substituents of MRS3176 and MRS3417 increase the
group at the ribose 30-position was included to span a predicted gap in the A3-receptor
ureido neoligand MRS3481 at wild-type and H272E mutant A3 receptors. Part (b) modif
www.sciencedirect.com
Because the ratio of selective enhancement of MRS1960was only modest, we improved the interaction based onpredictions from molecular modeling [4,25]. A 30-amino-methyl neoligand (MRS3176 [26]) achieved a 20-foldenhancement of affinity at the H272E mutant A3 receptor.The impetus for this structural change was the predictionfrommolecular modeling that the 30-amino group can be atan excessive distance from the His imidazole group to forma direct H-bond. This modified nucleoside also contained asubstituted N6-benzyl group, which tends to enhanceaffinity at the wild-type and mutant A3 receptors.
Thus, ligands can be tailored with strategicallypositioned amino groups for recognition by carboxylatemutants of the native receptor. However, a more remark-able demonstration of selective affinity enhancement and
A3 receptors. Known SAR profiles of adenosine derivatives have identified sites of
icacy. Cumulative efforts to characterize agonist SARs at adenosine receptors have
nformation of the ribose moiety [25]. (a) Amine derivatives that have shown sixfold
ptor, and tenfold (MRS3366) and >300-fold (MRS3417) enhancements in binding at
affinity at both wild-type and mutant adenosine receptors, and the extra methylene
-docking model. (b) Comparison of the functional effects of adenosine and the 30-
ied, with permission, from Ref. [4].

114 Opinion TRENDS in Pharmacological Sciences Vol.28 No.3
orthogonality was desired. The substituent at the 30-pos-ition of ribose was varied in charge, size and H-bondingability, and each analog was examined for binding affinityat several neoceptor variations. The optimal affinityenhancement and orthogonality followed the incorporationof a urea group in place of the 30-OH group of adenosineanalogs, as in MRS3481 (Figure 2). The urea group canform multiple H-bonds with the neoceptor and seems topreclude binding at native adenosine receptors in a stericmanner [27]. A model of this analog docked in the H272Ereceptor showed a bidentate coordination between thecarboxylate group and the urea moiety (Figure 3).
It was necessary to probe the coupling pattern of theneoceptor [4]; although the coupling specificity of GPCRsis principally a function of the second and third intra-cellular loops [28], and ligand specificity is governed byfunctionality within the upper third of the transmem-brane (TM) regions, the preservation of the typical A3
receptor second messengers could not be assumed.Coupling to one known effector pathway of the A3 receptor[i.e. the stimulation of phospholipase C (PLC) through theGb,g-subunits] is preserved upon activation of the H272Emutant receptor by MRS3481. Thus, at least part of thedownstream signaling of this cytoprotective receptor ismaintained. The stimulation of PLC by MRS3481occurred with an EC50 of �100 nM at the neoceptor,whereas MRS3481 was inactive at 10 mM at the wild-typereceptor similarly expressed in COS-7 cells. By compari-son, the EC50 for adenosine was �1 mM at the wild-typereceptor and >100 mM at the neoceptor. The effects ofMRS3481 acting through the H272E neoceptor on othersignaling systems (e.g. cAMP, ion channels and arrestin)remain to be determined.
In a chick cardiac myocyte culture, which is anestablished model for cardioprotection [29], the neoligandMRS3481 induced a potent anti-ischemic protection incells expressing the H272E neoceptor [4]. Also, this protec-
Figure 3. Putative binding-site model of A3 receptor constructs and their nucleoside lig
the neoligand MRS3481 in the H272E neoceptor (b). The homology models were derive
I state [6].
www.sciencedirect.com
tion correlated with the activation of PLD, as occurred withthe native A3 receptor transfected in the same cell system.Thus, a neoceptor–neoligand pair has been demonstratedto induce stages of a response to stress in a tissue that isknown to respond similarly when the native parent recep-tor is present.
The anti-inflammatory A2A receptor was also convertedto a neoceptor [3]. The approach ofmutating the conservedHis278 in TM7 (which was successfully applied to the A3
receptor) could not be used because mutation of the corre-sponding His residue to Asp or Glu did not lower thepotency of native adenosine at the A2A receptor. However,a hydrophilic residue (T88 in TM3) on the other side of theputative subdomain for ribose binding to the adenosinereceptors was selected for mutation. This residue inthe A2A receptor is exclusively associated with agonistbinding [3]. The ability of the T88D (3.36) mutant A2A
receptor to bind to the nonselective adenosine receptorantagonist CGS15943was unaffected; however, it failed tobind to the nonselective adenosine receptor agonistNECA, even at a concentration of 10 mM. This mutantreceptor recognized a strategically 50-modified aminoderivative (MRS3366; Figure 2a). Moreover, the preciseposition and spacer length of the amino groupwere crucialfor achieving a selective affinity enhancement at the neo-ceptor. Other mutant A2A receptors displayed evengreater degrees of enhancement for neoligands. Forexample, MRS3417 was functionally enhanced 110-foldat the N181D (5.42) mutant receptor, although this com-bination was not truly orthogonal because this modifiedreceptor was still capable of being activated by adenosinereceptor agonists.
In addition to mutating the ligand-binding pocket, itis also possible to mutate sites that are involved inphosphorylation and desensitization so that theseprocesses are retarded in neoceptors. This is particularlyimportant with regard to A3 receptors because they
ands. Docking of the agonist Cl�IB-MECA in the native human A3 receptor (a), and
d from a human A3 receptor model based on rhodopsin and resembling the meta

Opinion TRENDS in Pharmacological Sciences Vol.28 No.3 115
undergo exceptionally rapid desensitization. It should alsobe possible to mutate promoter regions of the neoceptortranscript. This could enable induction of the neoceptormRNA – thus adding an additional layer of control to theresponse to the neoligand.
Potential applications of neoceptorsLigand docking in rhodopsin-based molecular models ofGPCRs has been controversial. Neoceptors provide ameans of verifying the accuracy of predictions based onthe molecular modeling of GPCRs, which have been sub-ject to discrepancies, especially with regard to agonistdocking [6]. A gain-of-function mutation and a comp-lementary ligand provide powerful evidence that the pre-dicted binding pocket is correct. Moreover, neoceptors canbe used for mechanistic probing of the role of a specificGPCR in cells or tissues.
Therapeutic applications are also envisioned for genetherapy, which would be dependent on site-specific genedelivery (e.g. in the cardiovascular system) [30] (Table 1).Novel proposed applications include donor stem cells,which have great potential for repairing or regeneratingdiseased tissues such as the heart or other organs.Methods to enhance the duration of survival of exogenousstem cells when implanted in recipient subjects couldgreatly increase the ability of these cells to achieve repair.Although the native A3 receptor has a potent cytoprotec-tive effect, its ubiquitous presence causes major side-effects. The use of a tailor-made neoligand is proposedfor selectively activating, as needed, the expression of acytoprotective neoceptor in the donor stem cells. Anotherpotential therapeutic application of the orthogonalneoceptor–neoligand pair is in skeletal muscle, basedon cytoprotection by adenosine functioning at the A1
receptor [31].Additionally, there is increasing evidence that
mutations in genes encoding GPCRs are an importantcause of human disease. The neoceptor approach andbinding-site modeling could be used to design small mol-ecules to rescue disease-related mutations specifically.
Table 1. Future therapeutic applications ofadenosine-receptor-derived neoceptors
Receptor Target tissue Effect Refsa
A1 Skeletal myocytes Anti-ischemic protection [31]
A1 Atrioventricular node Antiarrhythmic [32]
A1 Kidneys Reduced hyperfiltration [33]
A1, A3 Cardiac myocytes Anti-ischemic protection [4]
A1, A3 Hematopoietic stem
cells
Myeloprotection,
enhanced duration of
survival
[34]
A2A Neutrophils, T cells Anti-inflammatory [35]
A2A Platelets Antithrombotic [36]
A2A Liver, bowel Cytoprotection [37,38]
A2A, A2B Endothelial cells,
vascular smooth
muscle cells
Vasodilatation,
angiogenesis
[39,40]
A3 Cancer cells, tumors Cytostatic, anticancer
effect
[22]
A3 Synoviocytes Anti-arthritic [23]
A3 Lungs Anti-ischemic protection [24]aThe basis of these projected applications in the physiological effects of the native
adenosine receptors is described in Ref. [25] and as indicated.
www.sciencedirect.com
Chemical names
CGS15943: N-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-
amine
I-AB-MECA: N6-(4-amino-3-iodobenzyl)-5’-N-methylcarboxamidoa-
denosine
MRS1960: 3’-amino-3’-deoxyadenosine
MRS3176: N6-(5-chloro-2-methoxybenzyl)-2’-aminomethyl-2’-deox-
yadenosine
MRS3366: 5’-N-(2-aminoethyl)carboxamidoadenosine
MRS3417: N6-(2-methylbenzyl)-5’-N-(2-aminoethyl)carboxamidoa-
denosine
MRS3481: N6-(3-iodobenzyl)-2’-deoxy-2’-ureidoadenosine
NECA: 5’-N-ethylcarboxamidoadenosine
Concluding remarksNeoceptors represent a rational design approach for newpharmacological tools and possible therapies. Usingiterative steps of modeling and ligand design, one couldidentify and refine a neoceptor–neoligand pair. The suc-cess of the neoceptor strategy for the adenosine receptorsvalidates the use of GPCR homology modeling and pro-vides a means of investigating signaling, designing smallmolecules to rescue disease-related mutations, andsmall-molecule-directed gene therapy. The neoceptor–neoligand pairing could provide spatial specificity bydelivering the neoceptor to a target site and temporalspecificity by administering the neoligand when needed.The success of this approach depends on orthogonality ofthe interaction, which is not required by the RASSLapproach, to avoid undesirable, nonselective activationof the native receptor. This process can now be applied toother GPCRs, even in the absence of X-ray crystallo-graphic structures.
AcknowledgementsWe thank the NIDDK Intramural Research Program, NIH, for support,and Soo-Kyung Kim for helpful discussions.
References1 Anderson, S.D. and Brannan, J.D. (2004) Long-acting b2-adrenoceptor
agonists and exercise-induced asthma: lessons to guide us in the future.Paediatr. Drugs 6, 161–175
2 Jacobson, K.A. et al. (2001) Neoceptor concept based on molecularcomplementarity in GPCRs: A mutant adenosine A3 receptor withselectively enhanced affinity for amine-modified nucleosides. J. Med.Chem. 44, 4125–4136
3 Jacobson, K.A. et al. (2005) A neoceptor approach to unravelingmicroscopic interactions between the human A2A adenosine receptorand its agonists. Chem. Biol. 12, 237–247
4 Gao, Z.G. et al. (2006) Orthogonal activation of the reengineeredA3 adenosine receptor (neoceptor) using tailored nucleoside agonists.J. Med. Chem. 2006, 2689–2702
5 Visiers, I. et al. (2002) Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 343,329–371
6 Kim, S.K. et al. (2006) Docking studies of agonists and antagonistssuggest an activation pathway of the A3 adenosine receptor. J. Mol.Graph. Model. 25, 562–577
7 Kristiansen, K. (2004) Molecular mechanisms of ligand binding,signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesisapproaches to receptor structure and function. Pharmacol. Ther.103, 21–80
8 Kim, J. et al. (1995) Site-directed mutagenesis identifies residuesinvolved in ligand recognition in the human A2A adenosine receptor.J. Biol. Chem. 270, 13987–13997

116 Opinion TRENDS in Pharmacological Sciences Vol.28 No.3
9 Kuhlmann, B. et al. (2005) Design of a novel globular protein fold withatomic-level accuracy. Science 302, 1364–1368
10 Kenski, D.M. et al. (2005) Chemical genetic engineering of G protein-coupled receptor kinase 2. J. Biol. Chem. 280, 35051–35061
11 Tedesco, R. et al. (2001) The estrogen receptor: a structure-basedapproach to the design of new specific hormone–receptorcombinations. Chem. Biol. 8, 277–287
12 Strader, C.D. et al. (1991) Allele-specific activation of geneticallyengineered receptors. J. Biol. Chem. 266, 5–8
13 Redfern, C.H. et al. (1999) Conditional expression and signaling of aspecifically designed Gi-coupled receptor in transgenic mice. Nat.Biotechnol. 17, 165–169
14 Baker, D. (2006) Proteins by design. Scientist 20, 26–3215 Koh, J.T. and Biggins, J.B. (2005) Ligand–receptor engineering and its
application towards the complementation of genetic disease and targetidentification. Curr. Top. Med. Chem. 5, 413–420
16 Doyle, D.F. et al. (2001) Engineering orthogonal ligand–receptor pairsfrom ‘near drugs’. J. Am. Chem. Soc. 123, 11367–11371
17 Elling, C.E. et al. (2006) Metal ion site engineering indicates a globaltoggle switch model for seven-transmembrane receptor activation.J. Biol. Chem. 281, 17337–17346
18 Bruysters, M. et al. (2005) A Gq/11-coupled mutant histamine H1
receptor F435A activated solely by synthetic ligands (RASSL).J. Biol. Chem. 280, 34741–34746
19 Beukers, M.W. et al. (2004) Random mutagenesis of the humanadenosine A2B receptor followed by growth selection in yeast.Identification of constitutively active and gain of function mutations.Mol. Pharmacol. 65, 702–710
20 Ohta, A. and Sitkovsky, M. (2001) Role of G-protein-coupled adenosinereceptors in downregulation of inflammation and protection fromtissue damage. Nature 414, 916–920
21 Linden, J. (2005) Adenosine in tissue protection and tissueregeneration. Mol. Pharmacol. 67, 1385–1387
22 Madi, L. et al. (2004) The A3 adenosine receptor is highly expressed intumor versus normal cells: potential target for tumor growthinhibition. Clin. Cancer Res. 10, 4472–4479
23 Fishman, P. et al. (2006) The PI3K–NF-kB signal transductionpathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res. Ther. 8, R33
24 Rivo, J. et al. (2004) Activation of A3 adenosine receptors attenuateslung injury after in vivo reperfusion. Anesthesiology 101, 1153–1159
25 Jacobson, K.A. and Gao, Z.G. (2006) Adenosine receptors astherapeutic targets. Nat. Rev. Drug Disc. 5, 247–264
Reproduction of materia
Interested in reproducing part or all of an article pub
If so, please contact our Global Rights Departmen
material will be used. To submit a perm
Elsevi
Global Rights D
PO Box
Oxford OX5
Phone: +44 (0)1
Fax: +44 (0)18
permissions@e
Alternatively, p
www.elsevier.com/lo
www.sciencedirect.com
26 Van Rompaey, P. et al. (2005) Exploring human adenosine A3 receptorcomplementarity and activity for adenosine analogues modified in theribose and purine moiety. Bioorg. Med. Chem. 13, 973–983
27 Jeong, L.S. et al. (2004) Design and synthesis of 30-ureidoadenosine-50-uronamides: effects of the 30-ureido group on binding to the A3
adenosine receptor. Bioorg. Med. Chem. Lett. 14, 4851–485428 Swaminath, G. et al. (2005) Probing the b2 adrenoceptor binding site
with catechol reveals differences in binding and activation by agonistsand partial agonists. J. Biol. Chem. 280, 22165–22171
29 Liang, B.T. (1996) Direct preconditioning of cardiac ventricularmyocytes via adenosine A1 receptor and KATP channel.Am. J. Physiol. 271, H1769–H1777
30 Fishbein, I. et al. (2005) Site specific gene delivery in the cardiovascularsystem. J. Control. Release 109, 37–48
31 Zheng, J. et al. (2005) Adenosine A1 receptors mediate potent anti-ischemic skeletal muscle protection in amouse hindlimbmodel. J.Mol.Cell. Cardiol. 38, 64
32 Belardinelli, L. et al. (1995) Ionic basis of the electrophysiologicalactions of adenosine on cardiomyocytes. FASEB J. 9, 359–365
33 Lee, H.T. et al. (2004) A1 adenosine receptor knockout mice exhibitincreased renal injury following ischemia and reperfusion. Am. J.Physiol. Renal Physiol. 286, F298–F306
34 Budak-Alpdogan, T. et al. (2005) Hematopoietic stem cell gene therapywith drug resistance genes: an update. Cancer Gene Ther. 12, 849–863
35 Lappas, C.M. et al. (2005) Adenosine A2A agonists in development forthe treatment of inflammation. Expert Opin. Investig. Drugs 14, 797–806
36 Camaioni, E. et al. (1997) Adenosine receptor agonists: synthesis andbiological evaluation of the diastereoisomers of 2-(3-hydroxy-3-phenyl-1-propyn-1-yl)NECA. Bioorg. Med. Chem. 5, 2267–2275
37 Day, Y.J. et al. (2004) Protection from ischemic liver injury byactivation of A2A adenosine receptors during reperfusion: inhibitionof chemokine induction. Am. J. Physiol. Gastrointest. Liver Physiol.286, G285–G293
38 Odashima, M. et al. (2005) Activation of A2A adenosine receptorattenuates intestinal inflammation in animal models ofinflammatory bowel disease. Gastroenterology 129, 26–33
39 Feoktistov, I. et al. (2004) Hypoxia modulates adenosine receptors inhuman endothelial and smooth muscle cells toward an A2B angiogenicphenotype. Hypertension 44, 649–654
40 Yang, D. et al. (2006) The A2B adenosine receptor protects againstinflammation and excessive vascular adhesion. J. Clin. Invest. 116,1913–1923
l from Elsevier articles
lished by Elsevier, or one of our article figures?
t with details of how and where the requested
ission request online, please contact:
er
epartment
800
1DX, UK
865 843 830
65 853 333
lsevier.com
lease visit:
cate/permissions





























