Embed Size (px)
Citation preview

Mutation of FAS, XIAP, and UNC13D Genes in a PatientWith a Complex Lymphoproliferative Phenotype
abstractThis article presents a case report for a child presenting with mixedclinical features of autoimmune lymphoproliferative syndrome (ALPS),familial hemophagocytic lymphohistiocytosis (FHL), and X-linked lym-phoproliferative (XLP) disease. From 6 months, he exhibited spleno-megaly and lymphoadenopathy and from 4 years, he showedrecurrent severe autoimmune hemocytopenia and sepsislike boutsof fever, from which he eventually died at the age of 12. Intriguingly,the patient carried mutations in FAS, XIAP, and UNC13D genes, whichare involved in ALPS, XLP disease, and FHL, respectively. These muta-tions were inherited from the mother, who had rheumatoid arthritisbut no signs of ALPS. A role for other modifying genes was suggestedby the finding that the healthy father exhibited defective Fas function,without mutation of the FAS gene, and had transmitted to the patientan osteopontin (OPN) gene variant previously associated with ALPS.Therefore, several genes might influence the disease outcome in thisfamily. In vitro analyses revealed that the FAS and the XIAP mutationsdecreased expression of the corresponding proteins, and the UNC13Dmutation decreased granule secretion and Munc interaction with Rab-27a. These findings suggest that overlap may exist between ALPS, FHL,and XLP disease, in accordance with the notion that FHL and XLPdisease are due to defective natural killer (NK)/NK T–cell function,which involves Fas. Therefore, we propose that NK cell defects shouldbe evaluated in patients with ALPS-like characteristics, and hemato-poietic stem cell transplantation should be considered in individualswith severe refractory cytopenia and FHL-like manifestations. Pediat-rics 2013;132:e1052–e1058
AUTHORS: Elena Boggio, PhD,a Maurizio Aricò, MD,b MatteoMelensi, PhD,a Irma Dianzani, MD, PhD,a Ugo Ramenghi,MD,c Umberto Dianzani, MD, PhD,a and AnnalisaChiocchetti, MD, PhDa
aInterdisciplinary Research Center of Autoimmune Diseases andDepartment of Health Sciences, “A. Avogadro” University ofEastern Piedmont, Novara, Italy; bDepartment of PediatricHematology Oncology, Azienda-Ospedaliero Universitaria MeyerChildren’s Hospital, Florence, Italy; and cDepartment of Pediatrics,University of Torino, Turin, Italy
KEY WORDSALPS, FHL, XLP, FAS, XIAP, MUNC13-4
ABBREVIATIONSALPS—autoimmune lymphoproliferative syndromeFHL—familial hemophagocytic lymphohistiocytosisHMC-1—human mast cell 1NK—natural killerPBMC—peripheral blood mononuclear cellSNP—single-nucleotide polymorphismWB—Western blottingwt—wild-typeXIAP—X-linked inhibitor of apoptosisXLP—X-linked lymphoproliferative
All of the authors named in this article have made substantiveintellectual contributions to this study and meet the criteria forinclusion as authors. In particular, Drs Melensi, Aricò, andI. Dianzani performed the genetic analyses; Drs Boggio andMelensi performed the functional experiments; Dr Ramenghicharacterized the patient; Dr Chiocchetti analyzed the results;and Drs Chiocchetti and U. Dianzani designed the research andwrote the manuscript.
www.pediatrics.org/cgi/doi/10.1542/peds.2012-1838
doi:10.1542/peds.2012-1838
Accepted for publication May 29, 2013
Address correspondence to Umberto Dianzani, MD, PhD, IRCAD–Department of Health Sciences, Via Solaroli, 17, I-28100, Novara,Italy. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2013 by the American Academy of Pediatrics
(Continued on last page)
e1052 BOGGIO et al by guest on September 5, 2018www.aappublications.org/newsDownloaded from

Lymphoproliferation is a hallmark ofautoimmune lymphoproliferative syn-drome (ALPS), familial hemophagocyticlymphohistiocytosis (FHL), and X-linkedlymphoproliferative (XLP) disease. ALPSis due to defective function of the Fasdeath receptor, causing defective apo-ptosis of activated lymphocytes, andoften involves autoimmune manifes-tations.1,2 FHL is a result of defectiveperforin-mediated cytotoxicity, leadingto ineffective immune hyperactivationupon viral infection, with tissue dam-age and a fatal outcome.3 XLP diseaseis attributed to defective function ofSlam-associated protein (SAP), a naturalkiller (NK)–cell costimulator, causing in-creased susceptibility to Epstein-Barrvirus infection.4 However, 1 known XLPvariant is due to defective function of theX-linked inhibitor of apoptosis protein(xiap).5
METHODS
The expression of Fas in peripheralblood mononuclear cells (PBMCs) andCD63 inhumanmast cell 1 (HMC-1)cells,transfected with UNC13D constructsand stimulated with fMLP (formyl-methionyl-leucyl-phenylalanine), was eval-uated via flow cytometry6; the levels ofFas, xiap, and caspase-9 in transfectedcells were evaluated through Westernblotting (WB), as previously described.7
Caspase-8 and -9 activity was assessedin cell lysates on the basis of fluoro-metric assays.8,9 Transfection of HeLaand 293T cells was accomplished viaLipofectamine (Life Technologies, GrandIsland, NY), whereas transfection ofHMC-1 cells by electroporation (AmaxaCell Line Nucleofactor; Lonza GroupLtd, Basel, Switzerland).10 NK activitywas assessed by using a 51Cr releaseassay, and Fas-induced cell deathwas evaluated as previously de-scribed.11,12 (Mammalian Uncoordinated)Munc13-4 was coimmunoprecipitatedwith Rab-27a and evaluated throughWB.13
Patient Presentation
This article describes a patient pre-senting with clinical and genetic fea-tures of ALPS, FHL, and XLP disease(Table 1). From the age of 6 monthsonward, he exhibited unexplainedmassive splenomegaly and lymphoa-denopathy, and beginning at 4 years ofage he presented recurrent severethrombocytopenia, autoimmune neu-tropenia, and anemia. The patient un-derwent multiple bouts of sepsislikefever, which was responsive only tosteroids. His immunoglobulin levelswere in the normal range but includ-ed antinucleus and antiphospholipidantibodies. Both anti–Epstein-Barr vi-rus (EBNA) and antiadenovirus im-munoglobulin G were also evident,whereas immunoglobulin M and re-verse transcriptase–polymerase chainreaction analyses were consistentlynegative for these infections during theentire observation period. Moreover,analyses of markers of cytomegalovi-rus, morbillivirus, parvovirus B19,Toxoplasma gondii, and Bartonellainfections were negative. In peripheralblood samples, TCRab+ CD4/CD8double-negative T cells (a hallmark ofALPS) were found to be slightly in-creased (Table 1). Moreover, the pa-tient displayed decreased proportionsof NK T (CD3+TCRa24b11
+) and NK(CD16+CD56+) cells (Table 1), whereashis B- (CD19+), T helper– (CD3+CD4+),and T cytotoxic– (CD3+CD8+) cell levelswere in the normal range (data notshown). Histologic analysis of thepatient’s bone marrow and spleen didnot reveal signs of hemophagocytosis(Table 1).
At the age of 9 years, the patient’sspleen was palpable in the right iliacfossa and was removed, but throm-bocytopenia and episodes of hyper-pyrexia persisted. At the age of 12, hewas hospitalized for respiratory dis-tress but a chest radiograph didnot show any significant alterations,
whereas an electrocardiogram revealedtachycardia.
Because the patient was not hyper-thermic and no serologic signs ofbacterial infection were present, mac-rophage activation syndrome wassuspected. Macrophage activation syn-drome is a complication of childhoodsystemic inflammatory disorderscaused by excessive activation of T lym-phocytes and macrophages, leading towidespread hemophagocytosis and cy-tokine overproduction. Treatment withantibiotics, dexamethasone, and bi-carbonate was promptly initiated, butthe patient’s general condition de-teriorated, with disseminated intra-vascular coagulation being observed,and the patient died 12 hours afterhospital admission.
On the basis of characteristics de-scribedabove, upon receiving informedconsent from the parents we se-quenced the following genes: FAS andCASP10; PRF1, UNC13D, and STX11; andSH2D1A and XIAP, which are involved inALPS,1,2 FHL,3 and XLP disease,5 re-spectively. Additionally, we typed thevariations in the OPN gene, which havebeen found to be predisposing factorsfor ALPS development.14
The patient carried monoallelic muta-tions in FAS (926G.A), XIAP (1189delA),and UNC13D (2768C.G), which werenot detected in 100 normal donors, andthe OPN 1239A.C single-nucleotidepolymorphism (SNP), previously asso-ciated with ALPS.
The FAS variant has been observed inALPS patients (www.niaid.nih.gov/top-ics/ALPS/Pages/default.aspx) but rarelyin control populations (allele fre-quency of 0.002). This mutation causedan E194K substitution in the in-tracellular portion of the protein adja-cent to the death domain (Fig 1A). In thepatient’s PBMCs, Fas expression wasdecreased and Fas function was foundto be either defective or borderline inrepeated analyses, as determined by
CASE REPORT
PEDIATRICS Volume 132, Number 4, October 2013 e1053 by guest on September 5, 2018www.aappublications.org/newsDownloaded from
T cell apoptotic response (Fig 1A). Tofurther analyze the effect of the 926G.A variant, FLAG tag-fused wild-type (wt)
Fas (Faswt) and mutated Fas (FasE194K)cDNAs were transfected into 293T cells.WB analysis revealed similar expres-
sion of both cDNAs (Fig 1A). However,caspase-8 activity, which is triggeredby Fas signaling, was lower in the cells
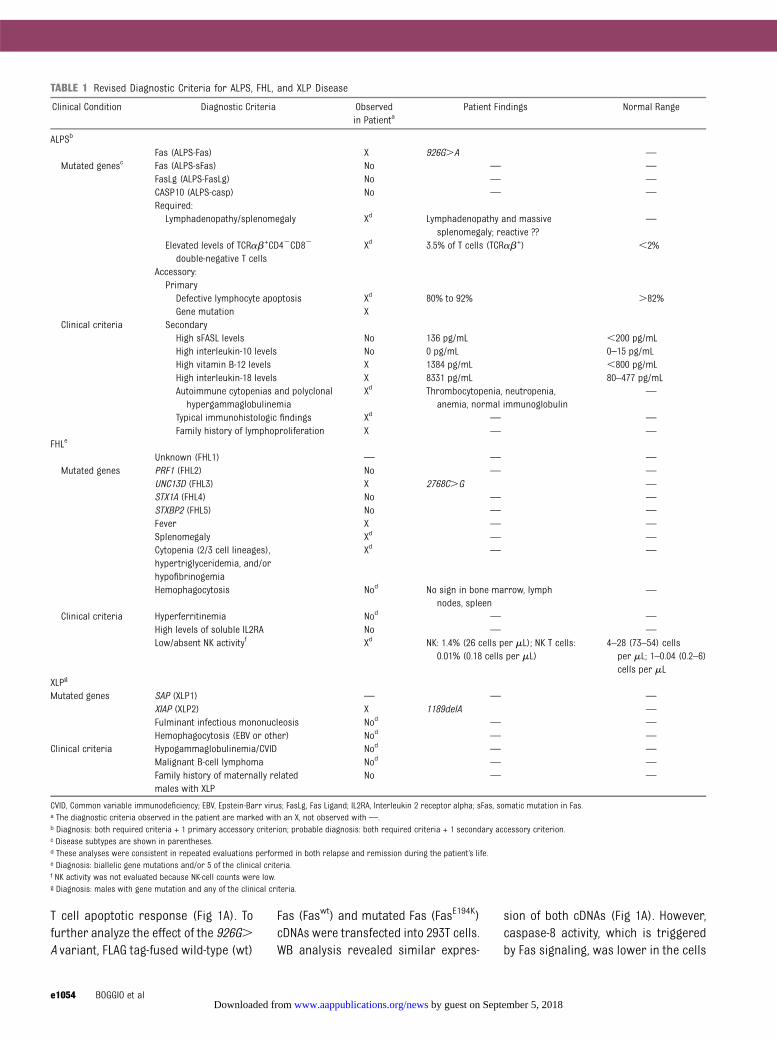
TABLE 1 Revised Diagnostic Criteria for ALPS, FHL, and XLP Disease
Clinical Condition Diagnostic Criteria Observedin Patienta
Patient Findings Normal Range
ALPSb
Fas (ALPS-Fas) X 926G.A —
Mutated genesc Fas (ALPS-sFas) No — —
FasLg (ALPS-FasLg) No — —
CASP10 (ALPS-casp) No — —
Required:Lymphadenopathy/splenomegaly Xd Lymphadenopathy and massive
splenomegaly; reactive ??—
Elevated levels of TCRab+CD42CD82
double-negative T cellsXd 3.5% of T cells (TCRab+) ,2%
Accessory:PrimaryDefective lymphocyte apoptosis Xd 80% to 92% .82%Gene mutation X
Clinical criteria SecondaryHigh sFASL levels No 136 pg/mL ,200 pg/mLHigh interleukin-10 levels No 0 pg/mL 0–15 pg/mLHigh vitamin B-12 levels X 1384 pg/mL ,800 pg/mLHigh interleukin-18 levels X 8331 pg/mL 80–477 pg/mLAutoimmune cytopenias and polyclonal
hypergammaglobulinemiaXd Thrombocytopenia, neutropenia,
anemia, normal immunoglobulin—
Typical immunohistologic findings Xd — —
Family history of lymphoproliferation X — —
FHLe
Unknown (FHL1) — — —
Mutated genes PRF1 (FHL2) No — —
UNC13D (FHL3) X 2768C.G —
STX1A (FHL4) No — —
STXBP2 (FHL5) No — —
Fever X — —
Splenomegaly Xd — —
Cytopenia (2/3 cell lineages),hypertriglyceridemia, and/orhypofibrinogemia
Xd — —
Hemophagocytosis Nod No sign in bone marrow, lymphnodes, spleen
—
Clinical criteria Hyperferritinemia Nod — —
High levels of soluble IL2RA No — —
Low/absent NK activityf Xd NK: 1.4% (26 cells per mL); NK T cells:0.01% (0.18 cells per mL)
4–28 (73–54) cellsper mL; 1–0.04 (0.2–6)cells per mL
XLPg
Mutated genes SAP (XLP1) — — —
XIAP (XLP2) X 1189delA —
Fulminant infectious mononucleosis Nod — —
Hemophagocytosis (EBV or other) Nod — —
Clinical criteria Hypogammaglobulinemia/CVID Nod — —
Malignant B-cell lymphoma Nod — —
Family history of maternally relatedmales with XLP
No — —
CVID, Common variable immunodeficiency; EBV, Epstein-Barr virus; FasLg, Fas Ligand; IL2RA, Interleukin 2 receptor alpha; sFas, somatic mutation in Fas.a The diagnostic criteria observed in the patient are marked with an X, not observed with —.b Diagnosis: both required criteria + 1 primary accessory criterion; probable diagnosis: both required criteria + 1 secondary accessory criterion.c Disease subtypes are shown in parentheses.d These analyses were consistent in repeated evaluations performed in both relapse and remission during the patient’s life.e Diagnosis: biallelic gene mutations and/or 5 of the clinical criteria.f NK activity was not evaluated because NK-cell counts were low.g Diagnosis: males with gene mutation and any of the clinical criteria.
e1054 BOGGIO et al by guest on September 5, 2018www.aappublications.org/newsDownloaded from
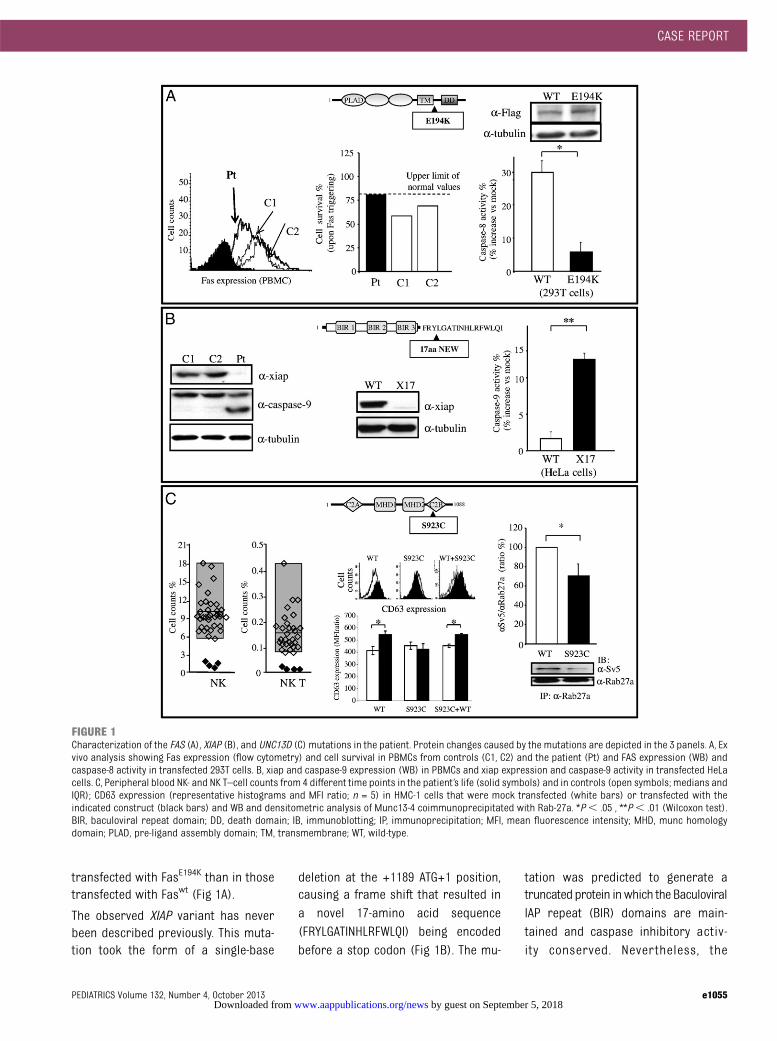
transfected with FasE194K than in thosetransfected with Faswt (Fig 1A).
The observed XIAP variant has neverbeen described previously. This muta-tion took the form of a single-base
deletion at the +1189 ATG+1 position,causing a frame shift that resulted ina novel 17-amino acid sequence(FRYLGATINHLRFWLQI) being encodedbefore a stop codon (Fig 1B). The mu-
tation was predicted to generate atruncated protein inwhich the BaculoviralIAP repeat (BIR) domains are main-tained and caspase inhibitory activ-ity conserved. Nevertheless, the
FIGURE 1Characterization of the FAS (A), XIAP (B), and UNC13D (C) mutations in the patient. Protein changes caused by the mutations are depicted in the 3 panels. A, Exvivo analysis showing Fas expression (flow cytometry) and cell survival in PBMCs from controls (C1, C2) and the patient (Pt) and FAS expression (WB) andcaspase-8 activity in transfected 293T cells. B, xiap and caspase-9 expression (WB) in PBMCs and xiap expression and caspase-9 activity in transfected HeLacells. C, Peripheral blood NK- and NK T–cell counts from 4 different time points in the patient’s life (solid symbols) and in controls (open symbols; medians andIQR); CD63 expression (representative histograms and MFI ratio; n = 5) in HMC-1 cells that were mock transfected (white bars) or transfected with theindicated construct (black bars) and WB and densitometric analysis of Munc13-4 coimmunoprecipitated with Rab-27a. *P, .05 , **P, .01 (Wilcoxon test).BIR, baculoviral repeat domain; DD, death domain; IB, immunoblotting; IP, immunoprecipitation; MFI, mean fluorescence intensity; MHD, munc homologydomain; PLAD, pre-ligand assembly domain; TM, transmembrane; WT, wild-type.
CASE REPORT
PEDIATRICS Volume 132, Number 4, October 2013 e1055 by guest on September 5, 2018www.aappublications.org/newsDownloaded from
premature termination codon couldalso predispose cells to nonsense-mediated mRNA decay.15 WB analysisrevealed that xiap expression wasundetectable in the patient ’s PBMCsand spontaneous caspase-9 activa-tion in these cells was increased(Fig 1B). To further analyze the func-tional effect of this variant, we cloned
wild-type (xiapwt) and mutated(xiapX17) cDNAs fused to Green Fluores-cent Protein (GFP) and transfected theminto HeLa cells, which express minimallevels of endogenous xiap. WB analysisshowed that compared with xiapwt,xiapX17 was nearly undetectable in thetransfected cells, and caspase-9 activitywas increased (Fig 1B).
The UNC13D variant identified in thepatient is reported in the 1000genomesdatabase as a rare variant with an al-lele frequency of ,0.01 and unknownpathologic significance but has notbeen validated by the National Heart,Lung, and Blood Institute’s Grand Op-portunity Exome Sequencing Project.UNC13D encodes Munc13-4, which is
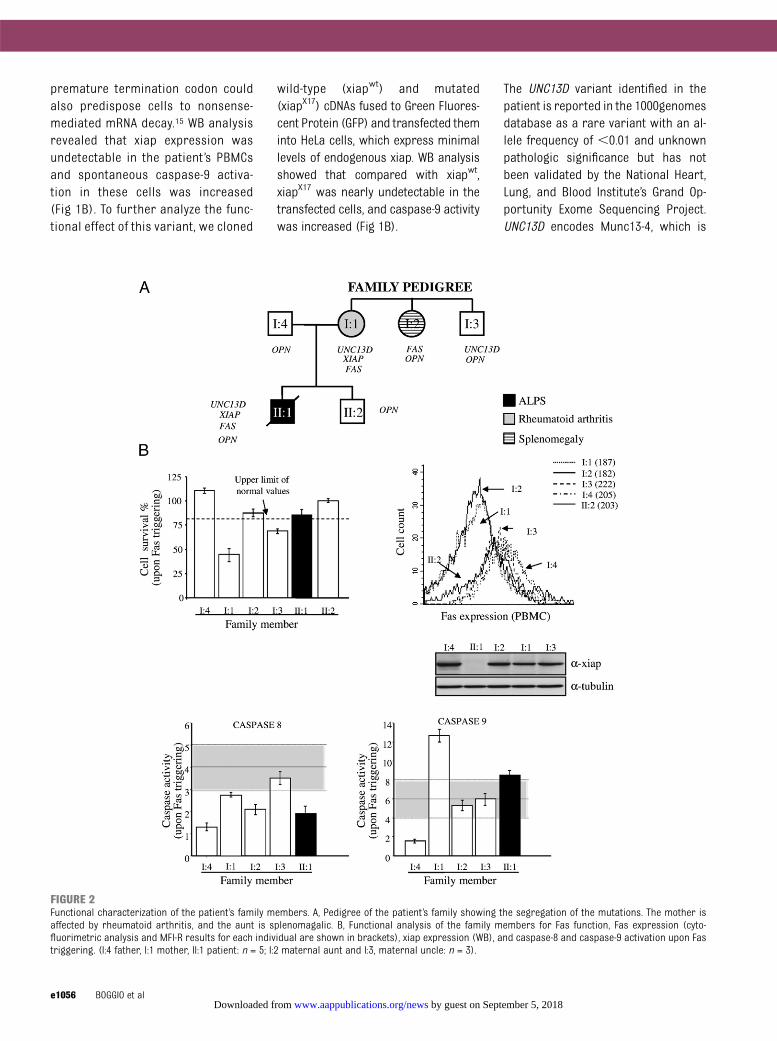
FIGURE 2Functional characterization of the patient’s family members. A, Pedigree of the patient’s family showing the segregation of the mutations. The mother isaffected by rheumatoid arthritis, and the aunt is splenomagalic. B, Functional analysis of the family members for Fas function, Fas expression (cyto-fluorimetric analysis and MFI-R results for each individual are shown in brackets), xiap expression (WB), and caspase-8 and caspase-9 activation upon Fastriggering. (I:4 father, I:1 mother, II:1 patient: n = 5; I:2 maternal aunt and I:3, maternal uncle: n = 3).
e1056 BOGGIO et al by guest on September 5, 2018www.aappublications.org/newsDownloaded from

involved in perforin secretion by CD8+
and NK cells, and the observed muta-tion caused an S923C amino acid sub-stitution (Fig 1C). The death of the patientmade it impossible to perform any exvivo functional analysis of Munc13-4.However, PBMC typing, which was re-peatedly performed during the patient’slife span, consistently revealed defectivefractions of NK T (CD3+TCRa24b11
+) andNK (CD16+CD56+) cells (Table 1, Fig 1C).
To directly assess the functional effectof the UNC13D variant, wild-type(Munc13-4wt) and mutant (Munc13-4S923C)cDNAs were cloned, fused to an SV5 tag,and transfected into the HMC-1 mas-tocytoma cell line. WB analysis showedthat the 2 constructs were expressedat similar levels (data not shown).Therefore, we evaluated the capacity offMLP to induce granule secretion, asdetected by increased surface expres-sion of CD63 (Fig 1C). CD63 expressionwas weaker in cells transfected withMunc13-4S923C than in those transfectedwith Munc13-4wt, and cotransfection ofboth forms did not reveal any dominantnegative effect of Munc13-4S923C onMunc13-4wt. A key step in Munc13-4 func-tion is its interaction with Rab27-a.16,17 Wetherefore assessed this interaction in thetransfected cells via coimmunoprecipi-tation, and we found that Rab27-a copre-cipitated greater amounts of Munc13-4wt
than Munc13-4S923C (Fig 1C).
DISCUSSION
Themutationsobserved in FAS,UNC13D,and XIAP may account for the patient’sphenotype, which met the diagnosticcriteria of ALPS but also included
features of XLP disease and FHL, such asbouts of macrophage activation and lowNK-/NK T–cell counts, which are not as-sociated with ALPS. All of the identifiedvariants were inherited from themother(I:1), who displayed rheumatoid arthritisbut no signs of these other diseases(Fig 2A). The normal Fas-induced celldeath phenotype observed in themothermight be explained by the opposingeffects of the FAS and XIAPmutations onthe caspase pathway, because the for-mer decreases caspase-8 activation,whereas the latter increases caspase-9activation, resulting in normal levels ofapoptosis. Indeed, the mother’s PBMCsshowed amoderate decrease in Fas andXiap expression, decreased caspase-8activation, increased capsase-9 activa-tion, and normal apoptosis (Fig 2B).
In contrast, the abnormal Fas functionobserved in the patient could be due toan additional defect in the Fas pathwayinherited from the father (I:4), who washealthy but showed a deficiency in Fasfunctionand incaspase-8and -9activity,together with a moderate decrease inFas expression (Fig 2B). Moreover, thefather also transmitted to the patientthe OPN SNP, which may contribute toALPS by increasing serum levels of thisantiapoptotic cytokine.
The phenotype of the patient’smaternalaunt (I:2), who displayed lymphadeno-megaly and defective function of Fasand carried the FAS mutation and theOPN SNP, showed that the FASmutationper se has a mild, but evident clinicaleffect. Finally, the maternal uncle (I:3)was healthy and carried the UNC13Dmutation (Fig 2A).
The patient’s phenotype suggests thatmutations in FAS, UNC13D, and XIAPmight cooperate in disrupting theability of the immune system to shutoff and interfere with the antiviral re-sponse. These processes involve thefunction of both Fas and NK/NK T cells,whose cytotoxicity is crucial for theclearance of virus-infected cells andthe fratricide of activated immunecells.18 The persistence of viral in-fection and the inability to switch offthe immune response may have con-tributed to the lymphocyte accumula-tion, autoimmune reactions, andbouts of fever displayed by the patient.This possibility is in line with ourprevious finding that monoallelicmutations in PRF1, although insuf-ficient to cause FHL, may act as dis-ease modifiers for ALPS in subjectscarrying genetic defects affecting Fasfunction.10
CONCLUSIONS
AlthoughALPS is anosologically definedsyndrome, ALPS-like syndromes resultin a heterogeneous clinical phenotype,possibly depending on the genes in-volved.19 Pediatricians should considera diagnosis of an ALPS-like syndromewhen cytopenia is associated withsplenomegaly or lymphadenomegaly.Ourdata suggest that NK-cell defectsmaybe detected in these patients, and wepropose that they should be routinelyevaluated in such cases. Finally, patientsshowing ALPS-like characteristics withNK-cell defects or FHL-like manifestationsshould be considered for hematopoieticstem cell transplantation.
REFERENCES
1. Oliveira JB, Bleesing JJ, Dianzani U, et al.Revised diagnostic criteria and classificationfor the autoimmune lymphoproliferativesyndrome (ALPS): report from the 2009 NIHInternational Workshop. Blood. 2010;116(14):e35–e40
2. Dianzani U, Chiocchetti A, Ramenghi U. Role ofinherited defects decreasing Fas function inautoimmunity. Life Sci. 2003;72(25):2803–2824
3. Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB.Familial haemophagocytic lymphohistiocytosis:advances in the genetic basis, diagnosis
and management. Clin Exp Immunol. 2011;163(3):271–283
4. Snow AL, Marsh RA, Krummey SM, et al.Restimulation-induced apoptosis of T cellsis impaired in patients with X-linked lym-phoproliferative disease caused by SAP
CASE REPORT
PEDIATRICS Volume 132, Number 4, October 2013 e1057 by guest on September 5, 2018www.aappublications.org/newsDownloaded from

deficiency. J Clin Invest. 2009;119(10):2976–2989
5. Rigaud S, Fondanèche MC, Lambert N, et al.XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Na-ture. 2006;444(7115):110–114
6. Dianzani U, Bragardo M, DiFranco D, et al.Deficiency of the Fas apoptosis pathway with-out Fas gene mutations in pediatric patientswith autoimmunity/lymphoproliferation.Blood. 1997;89(8):2871–2879
7. Ferretti M, Gattorno M, Chiocchetti A, et al.The 423Q polymorphism of the X-linked in-hibitor of apoptosis gene influencesmonocyte function and is associated withperiodic fever. Arthritis Rheum. 2009;60(11):3476–3484
8. Cerutti E, Campagnoli MF, Ferretti M, et al.Co-inherited mutations of Fas and caspase-10in development of the autoimmune lympho-proliferative syndrome. BMC Immunol. 2007;8(28):1–9
9. Clementi R, Chiocchetti A, Cappellano G, et al.Variations of the perforin gene in patientswith autoimmunity/lymphoproliferation and
defective Fas function. Blood. 2006;108(9):3079–3084
10. Dutta P, Koch A, Breyer B, et al. Identifica-tion of novel target genes of nerve growthfactor (NGF) in human mastocytoma cellline (HMC-1 (V560G c-Kit)) by transcriptomeanalysis. BMC Genomics. 2011;12(196):1–14
11. Clementi R, Dagna L, Dianzani U, et al.Inherited perforin and Fas mutations in a pa-tient with autoimmune lymphoproliferativesyndrome and lymphoma. N Engl J Med.2004;351(14):1419–1424
12. De Franco S, Chiocchetti A, Ferretti M, et al.Defective function of the Fas apoptoticpathway in type 1 diabetes mellitus corre-lates with age at onset. Int J ImmunopatholPharmacol. 2007;20(3):567–576
13. Elstak ED, Neeft M, Nehme NT, et al. TheMunc13-4-rab27 complex is specifically re-quired for tethering secretory lysosomesat the plasma membrane. Blood. 2011;118(6):1570–1578
14. Chiocchetti A, Indelicato M, Bensi T, et al.High levels of osteopontin associated withpolymorphisms in its gene are a risk factor
for development of autoimmunity/lympho-proliferation. Blood. 2004;103(4):1376–1382
15. Bhuvanagiri M, Schlitter AM, Hentze MW,Kulozik AE. NMD: RNA biology meets humangenetic medicine. Biochem J. 2010;430(3):365–377
16. Ménager MM, Ménasché G, Romao M, et al.Secretory cytotoxic granule maturation andexocytosis require the effector proteinhMunc13-4. Nat Immunol. 2007;8(3):257–267
17. Pivot-Pajot C, Varoqueaux F, de Saint BasileG, Bourgoin SG. Munc13-4 regulates gran-ule secretion in human neutrophils. JImmunol. 2008;180(10):6786–6797
18. Lünemann A, Lünemann JD, Münz C. Regu-latory NK-cell functions in inflammation andautoimmunity. Mol Med. 2009;15(9-10):352–358
19. Campagnoli MF, Garbarini L, Quarello P,et al. The broad spectrum of autoimmunelymphoproliferative disease: molecular bases,clinical features and long-term follow-upin 31 patients. Haematologica. 2006;91(4):538–541
(Continued from first page)
FINANCIAL DISCLOSURE: The authors have indicated they have no financial relationships relevant to this article to disclose.
FUNDING: This work was supported by the Associazione Italiana Ricerca sul Cancro (IG 10237; Milan), the Regione Piemonte (IMMONC Piattaforme Innovative), theFondazione Italiana Sclerosi Multipla (Genoa; 2010/R/12-2011/R/11), the Fondazione Cariplo (Milan), the Fondazione Amici di Jean (Turin), the Banca del Piemonte(Turin), the Associazione “Antonio Pinzino”, Associazione per la Ricerca sulle Sindromi Emofagocitiche (Palermo), and the Italian Ministry of Health, Bando MalattieRare 2008, A.O.U. Meyer, PRIN project 2009 (MIUR; Rome).
POTENTIAL CONFLICT OF INTEREST: The authors have indicated they have no potential conflicts of interest to disclose.
e1058 BOGGIO et al by guest on September 5, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2012-1838 originally published online September 16, 2013; 2013;132;e1052Pediatrics
Umberto Dianzani and Annalisa ChiocchettiElena Boggio, Maurizio Aricò, Matteo Melensi, Irma Dianzani, Ugo Ramenghi,
Lymphoproliferative Phenotype Genes in a Patient With a ComplexUNC13D, and XIAP, FASMutation of
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/132/4/e1052including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/132/4/e1052#BIBLThis article cites 19 articles, 8 of which you can access for free at:
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on September 5, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2012-1838 originally published online September 16, 2013; 2013;132;e1052Pediatrics
Umberto Dianzani and Annalisa ChiocchettiElena Boggio, Maurizio Aricò, Matteo Melensi, Irma Dianzani, Ugo Ramenghi,
Lymphoproliferative Phenotype Genes in a Patient With a ComplexUNC13D, and XIAP, FASMutation of
http://pediatrics.aappublications.org/content/132/4/e1052located on the World Wide Web at:
The online version of this article, along with updated information and services, is
ISSN: 1073-0397. 60007. Copyright © 2013 by the American Academy of Pediatrics. All rights reserved. Print the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on September 5, 2018www.aappublications.org/newsDownloaded from


![1 Pensions (FAS 87); Post Retirement Benefits (FAS 106); Post Employment Benefits (FAS 112); Disclosure about Pensions, etc. (FAS 132 [R]) – amendment](https://img.pdfslide.us/doc/110x75/56649d1f5503460f949f3b1c/1-pensions-fas-87-post-retirement-benefits-fas-106-post-employment-benefits.jpg)


























