Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMlSTRY Q 1993 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol . 268. , No. 2, Issue of 'January , 15, , pp. 1101-1108,1393 Printed in U. S. A .
Muta~ions of the FLP ~ e ~ o r n ~ i ~ a ~ ~ Gene That Cause a ~ ~ f i c i e n c y in DNA Bending and Strand Cleavage*
(Received for publication, June 10,1992)
Janusz KulpaS, Julie E. Dixong, Guohua Panll, and Paul D. Sadowskill From the Department of Molecular and Medical Genetics, University of Toronto, Toronto, Ontario M5S lA8, Canada
We have used site-directed mutagenesis to change several amino acids of the C-terminal portion of the FLP recombinase of Saccharomyces cereuisiae. These residues are a b ~ l u ~ l y conserved among the six FLP- like proteins from various yeast strains. We have ex- amined the ability of the altered proteins to catalyze recombination in vivo and in vitro and to perform various partial steps of the reaction in vitro. Two of the mutations produced a partial defect in DNA binding but the remainder resulted in normal binding. All of these mutations caused impairment of the ability of the protein to induce the type I1 bend of the FRT site, and some of these proteins were also defective in DNA strand cleavage. None of the mutations affected the ability of the proteins to perform synapsis between two FRT sites, but some were defective in strand ligation. Interestingly, some mutant proteins showed impair- ment of the initial stages of the r~ombination reaction on a linear substrate and yet they maintained the abil- ity to resolve a Holliday intermediate in the reaction. We conclude that this conserved region of the FLP protein is important for the early stage(s) of the recom- bination reaction.
The FLP gene of the 2-pm plasmid of Succhctromyces cere- visiue encodes a conservative site-specific recombinase that is involved in amplification of the plasmid (Futcher, 1986; Volk- ert and Broach, 1986; Reynolds et ul., 1987). The FLP protein is a member of the integrase family of recombinases whose members share 4 residues that are absolutely conserved (Argos et ai., 1986; Abremski and Hoess, 1992). These 4 residues (arginine 191, histidine 305, arginine 308, and tyrosine 343 of the 2 pm-FLP protein) are thought to be involved in the catalysis of DNA cleavage and religation in the recombination reaction (Prasad et ut., 1987; Parsons et ut., 1988,1990; Friesen and Sadowski, 1992; Pan and Sadowski 1992).
The integrase family includes five other FLP-like recom- binases encoded by 2 pm-like plasmids that exist in yeasts related to 5'. cereuisiae (Utatsu et at., 1987). Comparison of the amino acid sequences of these six recombinases reveals two highly conserved regions (region I which extends from amino acids 185-203 of FLP and region I1 which spans amino
* This work was supported by the Medical Research Council of Canada. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Recipient of a studentship from the Medical Research Council of Canada.
5 Recipient of a University of Toronto Open Fellowship. tl Recipient of an Ontario Graduate Scholarship. II To whom correspondence should be addressed. Tel.: 416-978-
6061: Fax: 416-978-6885.
acids 295-313 of FLP) as well as several absolutely conserved residues that lie C-terminal to region 11. We have used site- directed mutagenesis to change several of these conserved residues in the C terminus of the protein (the %onserved C- terminal region") in order to gain further insight into their function in the recombination reaction. All of the mutant proteins studied were defective in their ability to induce the type I1 bend in the target sequence, and several were also defective in their ability to cleave the target DNA. However, some mutant proteins that show defective bending and cutting are nevertheless able t o resolve a synthetic Holliday inter- mediate. We conclude that this region of the FLP protein is i m p o ~ a n t for bending and cutting of FLP recombination target (FRT)' and that DNA bending is required at an early stage in the reaction.
~ A T E ~ I A ~ ~ AND ~ E T H O D S
Enzymes-DNA restriction and modifying enzymes were pur- chased from Bethesda Research Laboratories, Boehringer Mannheim, New England Biolabs, and Pharmacia LKB Biotechnology Inc.
In Vitro Ol igon~leot~e-d i rec~d Mutagenesis-The mutagenesis was done on the plasmid pFLPflori (Friesen and Sadowski, 1992), a derivative of pCS3 that contains the FLP gene under control of the tac promoter (Schwartz and Sadowski, 1989). The oligonucleotides used for the site-directed mutagenesis were synthesized by the Bio- technology Service Center, Banting Institute at the University of Toronto. Nine different mutants of the FLP gene were created (Fig. 1) using the In Vitro Oligonucleotide-~rected Mutagenesis System, Version 2 from Amersham Corp. (Taylor et at., 1985; Sayers et al., 1988). Transformation of TG1 Escherichia coli cells (K12, supE, hsdD5/F'traD36, proA+B+, Zudq, lacZ) was done by electroporation using a Bio-Rad Gene Pulser apparatus set a t 25 microfarads and 2.5 kV; the Pulse Controller was set at 200 ohms.
DNA Sequencing-The double-stranded sequencing was done using the dideoxy sequencing method of Sanger et al. (1980).
FLP Protein Purification-The wild-type and mutant FLP pro- teins were partially purified using Bio-Rex 70 chromatography and ammonium sulfate precipitation, as described by Pan et al. (1991). The concentration of the FLP protein preparations was assessed by SDS-PAGE and Coomassie Blue staining. A wild-type FLP prepa- ration of greater than 90% purity was used as a size marker and a concentration standard. The concentration of the mutant FLP pro- tein was estimated by comparison with that of the purified FLP protein using a Bio-Rad 620 Video Densitometer and a Bio-Rad 3302A Integrator. When a group of mutant FLP proteins was pre- pared, the wild-type FLP protein was also purified in parallel by the same procedure. Within such a group of protein preparations, the recovery of mutant proteins was not appreciably different from the wild-type protein and the purity of the (FLP) proteins was compa- rahIe (-2-fold variation). Although the purity of the FLP proteins made at any one time was similar, the purity of proteins made at different times varied between 5 and 50%. We attribute this to differences in the success of induction of FLP synthesis and/or recoveries during the puri~cation.
FLP R e c o ~ b ~ ~ ~ ~ o n Assays-The in vivo recombination assay was
'The abbreviations used are: FRT, FLP recombination target; ___.
PAGE, polyacrylamide gel electrophoresis; kbp, kilohase pairs.

1102 Bending and Cleavage-defective FLP Mutants described by Schwartz and Sadowski (1989). This assay measures the abiIity of the FLP protein expressed in E. coli to excise the tetracycline resistance gene from a plasmid and to thereby render the cells tetracycline-sensitive.
The in vitro assay was described by Gronostajski and Sadowski (1985). FLP catalyzes recombination between two directly oriented FRT sites and shortens the 8.9-kbp linear plasmid molecule to 7.0 kbp (Fig. 2). The substrate was labeled at the EcoRI end using [a- s2P]dATP and reverse transcriptase. This substrate was incubated with FLP at 30 “C for 30 min, and the products of the reaction were analyzed by agarose gel electrophoresis followed by autoradiography. The reactions were quantitated by excision of the bands from the gel and Cerenkov counting.
DNA Binding Assay-Binding of FLP to the FRT site was detected by a gel mobility shift assay using a 100-bp EcoRI-Hind111 fragment of pBA112 plasmid containing the FRT site a t the EcoRI end (An- drews et al., 1987). The substrate was 3”labeled using [cY-~’P]~ATP and reverse transcriptase. The binding reactions were incubated a t 30 “C for 15 min, and the FLP-FRT complexes were resolved on a 5% polyacrylamide gel.
Measurement of DNA Strand Cleavage-In order to test for cleav- age of the FRT site by the FLP protein, the binding reactions were analyzed on an 8% denaturing polyacrylamide gel.
B e ~ i ~ A~say-FLP-induced bending of the FRT site was assayed as described by Schwartz and Sadowski (1990). The plasmid pCS38 was cut with BamHI generating a 300-bp fragment containing the FRT site at the end, or with PvuII producing a 300-bp substrate with the FRT site in the middle. The reaction and the gel electrophoresis were done as described for the FLP-DNA binding reaction. Bending angles were estimated as described by Schwartz and Sadowski (1990) using the standards of Thompson and Landy (1988).
Half-FRT Site Assay-The half-FRT site assay for detection of protein-protein interactions was used as described by Amin et al.
pGP25 (Proteau et al., 1986) with EcoRI and XbaI (which cuts within (1991). A half-FRT site a was generated by cleavage of the plasmid
the FRT core region). The EcoRI end was 32P-labeled at the 5’-end, and the resulting fragment contained symmetry element a and 3 bp of the core at one end of the fragment. The substrate was incubated with the FLP protein at room temperature for 15 min and then subjected to electrophoresis on a native polyacrylamide gel.
(1990). Synapsis Assay-Synapsis was assayed as described by Amin et ul.
Ligation Assay-The assay was done essentially as described by Pan and Sadowski (1992) except that the substrate consisted of a synthetic oligonucleotide bearing a tyrosine residue at the 3”PO.t terminus.2 The 5’-end of this oligonucleotide (18 nucleotides in length) was labeled with 32P using polynucleotide kinase. The 18-mer was annealed to a complementary 30-nucleotide substrate to form a synthetic half-FRT site. Ligation measures the formation of a 48- nucleotide hairpin structure that is detected on a denaturing poly- acrylamide gel (see diagram in Fig. 6). Reaction conditions were as described previously (Pan and Sadowski, 1992).
Resolution of Synthetic Holliday Intermediates-The substrate was a Holliday-like FRT site intermediate consisting of four arms each bearing one symmetry element radiating out from the core region. The substrate was assembled by annealing together one 5’ labeled and three unlabeled oIigonucleotidesi each one was about 80 nucleo- tides in length and the unlabeled oligonucleotides were in 5-fold excess over the 32P-labeled oligonucleotide. The synthetic x-structure was then purified by PAGE. Resolution gives either of two possible radiolabeled linear products (see Fig. 7). The details of the assay will be published elsewhere.
RESULTS
Site-directed Mutagenesis of the FLP Gene Oligonucleotide-directed mutagenesis was used to change
amino acid residues in the C-terminal conserved region of the FLP recombinase (Fig. 1). This region of FLP contains a number of amino acids that are conserved within the FLP family of recombinases from different species of yeast (Utatsu et al., 1987). The mutagenesis of some conserved amino acids within this region has already yielded insights into the mech-
~~
‘Pan, G., Leutke, K., Jaby, C. D., Brousseau, R., and Sadowski, P. D. (1993) J. Biol. Chem. 268, in press.
N S 3 2 3
3
9 6 D I I f ( I
I K H L 1; 3
S A H 3 3 3 3 3 3
2 3 9 5
3 3 4 9 6 9 5
a 0 1
7 : : H I Y D I I I , L 1 I I I I
2pm 303-kSEI&SF~SWXGLTELTNVV&SDXR---ASAV~TTXTii-345
pSB2 2 9 9 - 1 ( A H L ~ ~ S Y L G ~ S ~ S ~ T L Y G ~ S V ~ ~ E G V S ~ S R ~ - 3 ~ 4
pSRl ~~~-~(AHLGREVTASYLSNNEMDKEATLYGBWSMLREEGV~RVAXSR~-~~~
pSB3 2 9 7 - X S E L G ~ S F ~ E L D S W A N S L G ~ S S S Q N Q R E S G - ~ ~ ~ ~ - 3 4 1
pSMl 2 4 9 * K S ~ ~ S S Y L S H T N H G Q ~ S P ~ G ~ S A G K ~ S ~ ~ ~ - 2 9 4
PXDl 293-XSXFGRELXATFLSRSEXGXYVSSLGBWAGDREIQ-6AVARSliYS?i-337
I I
FIG. 1. The amino acid sequence of the conserved 303-346 region of the FLP recombinase of the 2-pm plasmid. The highlighted residues are conserved among the FLP recombinases of different species of yeast (Utatsu et al, 1987). The changes in the amino acid sequence made by o~gonucleotide-directed mutagenesis are indicated above the sequences.
anism of the FLP-mediated recombination reaction (Prasad et al., 1987; Parsons et ai., 1988,1990; Schwartz and Sadowski, 1989; Evans et al., 1990.
The objective of this study was to improve our understand- ing of the role(s) of the C-terminal conserved region of FLP and especially the role of the conserved amino acids within this region, in the recombination reaction. The conserved amino acid residues in this region that had not been changed or characterized previously were targeted using in vitro mu- tagenesis. The following changes were made (as indicated in Fig. 1): lysine 303 to glutamic acid (K303E): histidine 309 to leucine (H309L), leucine 315 to proline (L315P), asparagine 329 to histidine (N329H) and to aspartic acid (N329D), serine 336 to tyrosine (S336Y) and to phenylalanine (S336F), ala- nine 339 to aspartic acid (A339D) and histidine 345 to leucine (H345L). The sequences of the mutated FLP genes were confirmed by double-stranded DNA sequencing. The mutant FLP proteins were partially purified as described under “Ma- terials and Methods.” The mutant proteins were present in the purified fractions at a concentration similar to that of the wild-type FLP purified by the same procedure at the same time (data not shown). This observation suggests that none of the mutant proteins was unstable in E. coli.
Recombination Activity of the FLP Mutants In Viuo R e c o ~ b i ~ t i o n - T h e mutant FLP proteins were
first assayed for recombination proficiency in vivo. We used the FLP excision system developed by Schwartz and Sadowski (1989) which consists of a FLP expression plasmid (pFLPflori or derivatives) and a plasmid (pCS5.l) containing two FRT sites in direct orientation flanking the gene for tetracycline resistance (tetr). The wild-type FLP protein will excise the tet’ gene making the host cells sensitive to tetracycline whereas a mutant FLP protein that is defective in recombi- nation causes the cells to remain resistant to tetracycline.
The results of the in vivo recombination assays are pre- sented in Table I. The mutants K303E, H309L, L315P, and H345L gave colonies on agar plates containing tetracycline
The mutational changes are designated as follows: the amino acid of the wild-type FLP protein, followed by the amino acid number of the FLP protein, followed by the amino acid of the mutant protein. Thus, FLP K303E is a FLP protein in which lysine 303 has been changed to glutamic acid.

Bending and Cleavage-defective FLP Mutants 1103
TABLE I Summary of properties of FLP proteins
Recombination DNA binding
DNA DNA cleav- site Half-
Protein Liga- bSOlU- t iog tionh in vivo" in vitrob aged bending dimer$
FLP-WT K303E H309L L315P N329H N329D S336Y S336F A339D H345L
100% 0% 0% 0%
76% <5% 47% 85% <5%
0%
+ f + f + + + + + +
>144 N D
116 ND 115 94
122 121 110 125
+++ ND
ND k
+ ND +++ +++ +++
-
+++ ND + ND +++
++ (+) ND +++ +++
+ a The results of incubation at 37 "C for 24 h are shown. +, no colonies on tetracycline containing plates and therefore a recombination-
positive phenotype. -, all colonies were tet' and hence had a recombination defective-phenotype. f, 20% of colonies were tet' at 37 "C; all colonies were partially tet' at 30 "C. None of the mutations conferred a temperature-sensitive phenotype (not shown).
Percentage of wild-type protein (from Fig. 2).
+, cleavage product equal to wild-type protein. -, no detectable cleavage. f, cleavage is 4 0 % of wild-type level.
+. half-site dimers: trimers and tetramers detected. f. half-site dimers, but no trimers or tetramers detected. -, no dimers, trimers, or
e +, forms complexes 1-111 at same levels as wild-type FLP. f, impaired formation of complexes (see Fig. 3).
e Magnitude of type I1 bend (degrees).
tetramers formed. Based on data in Fig. 6.
h Based on data in Fig. 7. ' ND, not done.
and therefore are classified as defective in recombination. The FLP A339D showed a &fold decrease in the number of colo- nies under tetracycline selection and is categorized as partially defective in recombination. The remaining mutants of FLP (N329H, N329D, S336Y, and S336F) were as sensitive to the tetracycline selection as wild-type FLP and therefore are classified in Table I as proficient in recombination.
In Vitro Recombination-In order to quantitate the level of recombination for each of the mutant FLP proteins, in vitro assays were done as described by Gronostajski and Sadowski (1985) (see Fig. 2, bottom). As presented in Fig. 2, none of the mutant proteins mediated the recombination reaction as ef- ficiently as the wild-type FLP (for which 93% of the total recovered label was in the recombination product; see Fig. 2 A , lane 6). When the yield of recombination product was quan- titated by Cerenkov counting, the FLP mutant N329H showed 76% of the wild-type level of recombination (Fig. 2 A , lane 13), S336Y attained 47% (Fig. 2C, lane 6), and S336F showed 85% of the wild-type FLP recombination level (Fig. 2C, lane 11 ). When the concentration of FLP used in the reaction was higher than that required for maximum excision a decrease in the accumulation of product was observed. This effect might be due to contaminants in the protein preparations used in the experiments (Meyer-Leon et al., 1987). The two FLP mutants N329D (Fig. 2 A , lanes 15-20) and A339D (Fig. 2C, lanes 15-20) attained less than 5% of the wild-type FLP recombination level. (The recombination products were clearly visible when the autoradiographs were exposed four times longer than usual; not shown.) This low level of excision was apparently sufficient to give wild-type levels of recombi- nation in vivo (see Table I). The four remaining FLP mutants K303E, H309L, L315P, and H345L did not mediate any detectable recombination with a linear substrate.
Since the assays in Fig. 2 measured the yield of recombinant product after 30 min, the mutant proteins which were only partially defective in recombination (N329H, S336Y, and S336F) were compared with the wild-type FLP in a time course experiment. The mutant FLP proteins N329H, S336Y, and S336F had slower rates of accumulation of the excision product than the wild-type FLP (not shown). These rates
were probably due to impairment at some steps of the recom- bination reaction rather than a reduced half-life since all mutant proteins were still active beyond 40 min. In the first 35 min of the recombination reaction, each mutant accumu- lated more than 90% of the total product present after 2 h.
The in vitro recombination data confirmed the in vivo results in that the mutant FLP proteins that were deficient in in vivo recombination (K303E, H309L, L315P, and H345L) were also deficient in vitro. However, the mutants that were partially defective in vitro were as proficient as wild-type FLP in in vivo recombination. This suggests that the in vitro assay is more sensitive than the in vivo assays.
FRT Site Binding by the Mutant FLP Proteins As described above, the FLP mutants used in this study are
partially or completely defective in recombination. In order to understand the nature of these defects, we used a number of in vitro assays for particular steps in the FLP-mediated recombination reaction.
The formation of the FLP-FRT complexes can be regarded as the first step in the FLP-promoted recombination reaction. The mutant FLP proteins were assayed in vitro for formation of FLP-DNA complexes using the mobility shift assay de- scribed by Andrews et al. (1987). A 100-bp EcoRI-Hind111 fragment containing the FRT sequence was incubated with the same concentration of the wild-type and mutant FLP proteins. As shown in Fig. 3A wild-type FLP (lane 3 ) formed three complexes, CI, CII, and CIII, corresponding to the binding of FLP to one, two and three symmetry elements (Andrews et al., 1987; Beatty and Sadowski, 1988).
The mutant FLP proteins H309L (lane 5), N329H (lane 7), N329D (lane 8), S336Y (lane 9), S336F (lane IO), A339D (lane 11 ), and H345L (lane 12) formed the three complexes as efficiently as the wild-type FLP. The two remaining mutant FLP proteins, K303E and L315P, showed lower affinity for the FRT site (lanes 4 and 6). Quantitation of the distribution of the radioactivity showed that at high protein concentra- tions of FLP L315P only 2% of the labeled substrate was present in complex 111. When FLP K303E was used, 7% of the total labeled substrate was found in complex 111 uersus

1104 Bending and Cleavage-defective FLP Mutants
l- : z -1 n -1 m YI n - t
0 0 D 0 S E Y L X
1 2 3 4 5 8 7 8 0 10 11 12 13 14 15 18 17 = _ 1
P D
.x 0
0
0 LD c s %
0
0 LD
s *-~aObbv
5 I,.
p t -b ZOkbp
6 FIG. 2. In vitro recombination activity of the FLP mutants.
Reactions containing varying concentrations of the mutant proteins were incubated with 340 PM of the 8.9-kbp substrate containing two FRT sites in direct orientation. Autoradiogram of dried agarose gel. S, substrate; P, 7.0-kbp excision product; mock, substrate incubated with protein made from cells containing the parent expression plas- mid lacking the FLP gene. A , the following concentrations of each protein (nM) were used: 0.0024, 0.012, 0.06, 0.12, 0.6, and 2.4; lunes 3-8, wild-type FLP; lanes 9-14, N329H; lanes 15-20, N329D. B, lanes 3 (0.03 nM) and 4 (0.12 nM), wild-type FLP; lanes 5 (0.24 nM) and 6 (1.2 nM), K303E lanes 7 (0.06 nM), 8 (0.12 nM), 9 (0.24 nM), 10 (0.6 nM) H309L; lanes 11 (0.12 nM) and 12 (0.6 nM), L315P; lanes 13 (0.016 nM), I4 (0.06 nM), 25 (0.12 nM), 16 (0.6 nM), and 17 (2.7 nM) H345L. C, the same increasing concentrations (0.03-2.4 nM) as indi- cated in Fig. 2A were used for: S336Y (lanes 3-8), S336F (lanes 9- I4 ) , and A339D (lanes Ifj-20). Schemutic diagram of the assay is shown at the bottom.
77% for the wild-type FLP. The results presented in Fig. 3A show that K303E and L315P had significantly decreased ability to bind the target DNA whereas all the other mutant FLP proteins bound to the FRT site with an affinity similar to wild-type FLP.
DNA Cleavage Activity of the FLP Mutants In order to undergo strand exchange and recombination,
the two FRT sites must be cleaved. The mutant FLP proteins were assayed for their cleavage activity using a 3'-labeled 100- bp substrate. After incubating the fragment with FLP, half of the reaction was run on a non-denaturing gel to ensure that approximately equal levels of binding complexes were formed in each reaction (Fig. 3A). The other half of the binding reaction was analyzed for the presence of cleavage product on a denaturing gel (Fig. 3B). Cleavage of the 3"labeled strand by wild-type FLP yields a 64-bp fragment (lane 3 ) . The mutants N329H (lane 7), S336Y (lane 9) , and S336F (lane 10) showed levels of cleavage that were comparable to that of the wild-type FLP. Two mutant proteins, N329D (lane 8 ) and A339D (lane 11 ), yielded very low levels of cleavage that were estimated to be less than 10% of the wild-type. The cleavage products for these proteins were only seen after exposure of the autoradiograph four times longer than usual (not shown).
1 2 3 4 s 6 7 8 D l 0 1 1 1 2
b 4
Y l 2 3 4 6 0 7 8 0 1011 12
Cll l
c I1
C I
s
0 4 t v 4
X b . 1 4
FIG. 3. DNA binding and FRT cleavage activity of the FLP mutants. A , half of the FLP-FRT site binding reaction was run on a non-denaturing polyacrylamide gel to assess formation of protein- DNA complexes. 1 nM concentration of the 100-bp linear substrate was reacted with 180 nM concentration of the FLP protein at 30" for 30 min. S, substrate; CI-CHI, FLP-substrate complexes 1-111; lane 3, wild-type FLP; lanes 4-12, FLP mutants. B, the other half of the binding reaction was analyzed for the cleavage products on an 8% denaturing polyacrylamide gel. M , marker lane with XbuI cut sub- strate generating a 61-bp band 100 bp, substrate; 64 bp, product of FLP cleavage at the boundary of the core and symmetry element u.
As expected, mutant proteins K303E (lane 4 ) and L315P (lane 6) did not show any cleavage products presumably because they have diminished DNA binding. The remaining two proteins H309L and H345L formed complexes I1 and I11 (Fig. 3A, lanes 5 and 12, respectively) at wild-type levels but did not show any cleavage (Fig. 3B, lanes 5 and 12).
Target DNA Bending by the FLP Mutants
The FLP protein induces two types of bends in the FRT site (Schwartz and Sadowski, 1990). The type I bend (-65") is caused by the binding of FLP to a single symmetry element, and the type I1 bend (>144") is thought to be due to protein- protein interactions between two FLP molecules bound across the 8-bp core. The ability of the mutant proteins to bend the target DNA was assayed using a 300-bp fragment in which the FRT site was either at the end ( E lanes in Fig. 4) or in the middle of the substrate ( M lanes in Fig. 4). The FLP- induced bend in the middle of the substrate will cause greater retardation of the complex in a native polyacrylamide gel than the FLP-induced bend near the end of the substrate (Schwartz and Sadowski, 1990). The ratio of the mobility of the com- plexes formed by the FLP protein with those two substrates ( p ~ / p E ) can be determined from the position in the gel and compared to a standard curve derived from several DNA molecules containing a sequence-directed bend of known mag- nitude (Schwartz and Sadowski, 1990; Thompson and Landy, 1988). This assay allows one to estimate the angle of the FLP- induced bend.
The DNA in complex I in a reaction with wild-type FLP had a bend of 65" and the DNA in complex I1 had a bend of more than 140" (Fig. 4A, lanes 3 and 4 ) . These results are in good agreement with the data obtained by Schwartz and

Bending and Cleavage-defective FLP Mutants 1105
.. E Y E Y C Y E I "-
E 4c w
FIG. 4. Bending of the FRT site by the mutant FLP proteins. 300 p~ of the "P-labeled 300-bp substrate was incubated with 4.8 nM of the FLP protein at 30" for 15 min and analyzed by electropho- resis on a polyacrylamide gel. E, the 300-bp substrate with the FRT site at the end; M, the 300-bp substrate with the FRT site in the middle; S , mixture of both substrates without FLP; mock, mixed substrate incubated with mock preparation of extract; dots, position of complex I1 in the M lanes; arrowheads, position of complex I1 in the E lanes; the measurements were taken from these points to calculate the pM/pE ratio. A , lanes 3 and 4, wild-type FLP; lanes 5 and 6, H309L; lanes 7 and 8, N329H; lanes 9 and 10, N329D. B, lanes 3 and 4, S336Y; lanes 5 and 6, S336F; lanes 7 and 8, A339D; lanes 9 and 10, H345L; lanes I 1 and 12, wild-type FLP.
Sadowski (1990). As can be seen by inspection of the mobili- ties of the complex I bands in Fig. 4, the magnitude of the bend in the target DNA of complex I was found, for all of the mutant proteins, to be close to that of the wild-type FLP (calculated bend angles ranged from 60 to 72" in two separate experiments, data not shown). However, all of the mutant proteins were defective in the formation of the type I1 bend. The magnitude of the type I1 bend induced by N329D was only 94" which was the smallest in this series of mutants (Fig. 4A, lanes 9, 10, and Table I). The FLP proteins S336Y and S336F gave the largest bends of 122 and 121", respectively (Fig. 4B, lanes 3-6; Table I). Not only were the angles of the type I1 bends induced by the mutant proteins reduced, but the type I1 complexes were much more diffuse than those produced by wild-type FLP. The differences between the top and the bottom parts of the diffuse complexes were not investigated further in this study although they could be due to heterogeneity of the angles of the type I1 bend (Schwartz, 1990).
Since all the mutant proteins tested were deficient in for- mation of type I1 bend, the conserved 303-345 region of FLP is probably directly involved in this kind of DNA bending. The defect in bending may be due to impaired protein-protein interactions across the core.
Formation of Complexes with the Half-FRT Site Qian et al. (1990) showed that the FLP protein can bind to
half-FRT sites containing only one symmetry element and part of the core and promote formation of dimeric, trimeric, and tetrameric complexes. The formation of these complexes requires cleavage of the DNA by FLP (Qian et al., 1990; Amin et al., 1991) as well as protein-protein interactions (Friesen and Sadowski, 1992). In this study we used the half-site a (symmetry element a and part of the core; see Fig. 5) posi- tioned at the end of a 94-bp fragment (Amin et al., 1991). The formation of four complexes, complex I, dimer, trimer, and tetramer, with the half-site a by the wild-type FLP is shown
CI
S
1
r 1
T
n
FIG. 5. Formation of complexes with the half-FRT site. The 92P-end-labeled 94-bp linear substrate (0.9 nM) with the half-site a at the end was incubated with increasing concentrations of FLP protein (4.8,24, and 60 nM). S, substrate; CI, FLP-substrate binding complex; D, dimeric complex; TR, trimeric complex; TT, tetrameric complex. A , lanes 3-5, wild-type FLP; lanes 6-8, H309L; lanes 9-11, N329H; and lanes 12-14, N329D. B, lanes 3-5, S336Y; lanes 6-8, S336F; lanes 9-11, A339D; lanes 12-14, H345L; lane 15, wild-type FLP (60 nM).
in Fig. 5A, lanes 3-5. As expected, all of the mutant proteins formed complex I, which corresponds to FLP binding to one symmetry element. Three mutant proteins, N329H (Fig. 5A, lanes 9-11), S336Y (Fig. 5B, lanes 3-5), and S336F (Fig. 5B lanes 6-8) were able to form a small amount of dimeric complex as compared to wild-type FLP. The N329H protein also showed formation of a small amount of trimeric complex. None of the mutant proteins formed the tetrameric complex. Since the N329H, S336Y, and S336F proteins produced nor- mal levels of cleavage product, the paucity of dimeric com- plexes is likely the result of defective protein-protein inter- actions. The inability of the remaining mutant proteins (N329D, A339D, and H345L) to form half-site multimers could be due to their inability to cleave the DNA and/or to defective protein-protein interactions.
Formation of the Synaptic Complexes
In the FLP-mediated recombination reaction, protein-pro- tein interactions are required for synapsis, the process by which two FRT sites are aligned in order to undergo strand exchange. The transient synaptic complexes can be trapped by using glutaraldehyde as a cross-linking agent (Amin et al., 1990). When assayed by this method, all mutant proteins formed normal amounts of synaptic complexes (data not shown). Since all of the FLP mutant proteins formed synaptic
1106 Bending and Cleavage-defective FLP Mutants
complexes, the C-terminal conserved region is probably not involved in the synaptic type of protein-protein interactions.
Strand Ligation Activity After DNA cleavage and exchange of strands, FLP pro-
motes the rejoining of the DNA in the process of ligation. We have recently developed an assay for strand ligation which occurs independently of the ability of FLP to cleave and covalently attach to the DNA (Pan and Sadowski, 1992). It was therefore of interest to explore the capacity of the mutant proteins to promote ligation. Accordingly, each protein was incubated with an activated ligation substrate that consisted of a half-site containing an eight-nucleotide single-stranded 5"hydroxylated core sequence and a recessed 3'-phosphoty- rosine. Ligation of such a substrate yields a hairpin structure (Fig. 6, top). One protein (N329D) was defective in ligation whereas two other proteins (H309L and N329H) showed partial defects (Fig. 6). Three other proteins yielded the same efficiency of ligation as the wild-type protein (S336F, A339D, and H345L).
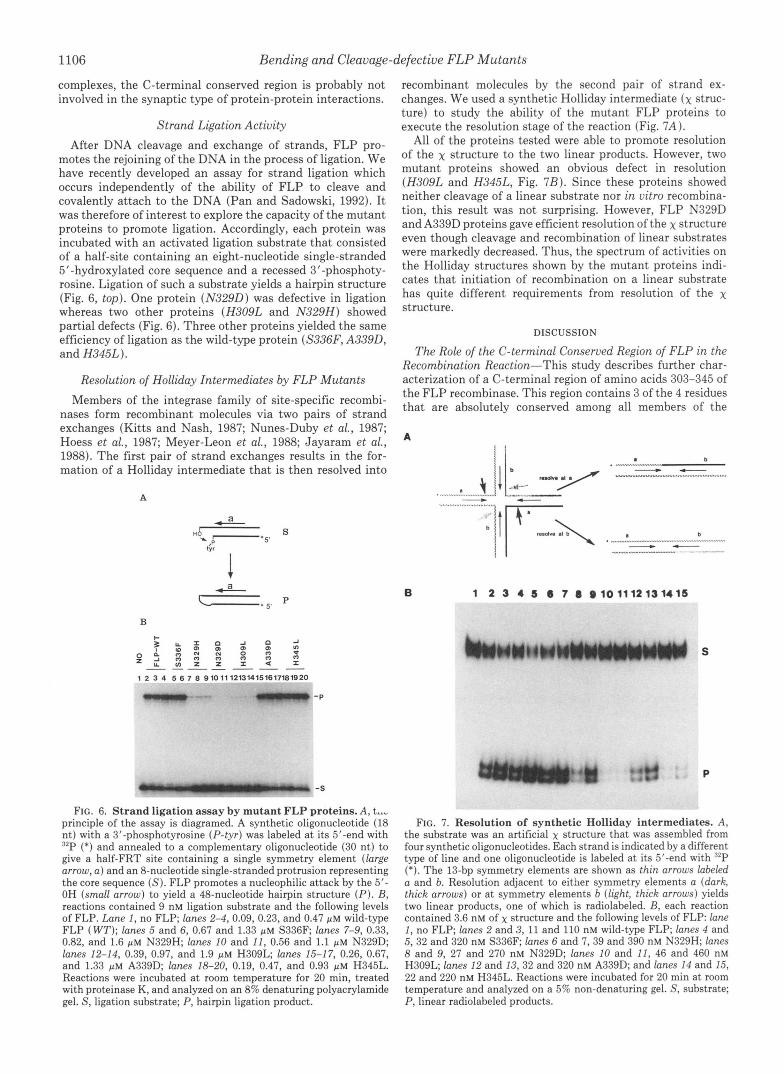
Resolution of Holliday Intermediates by FLP Mutants Members of the integrase family of site-specific recombi-
nases form recombinant molecules via two pairs of strand exchanges (Kitts and Nash, 1987; Nunes-Duby et al., 1987; Hoess et al., 1987; Meyer-Leon et al., 1988; Jayaram et al., 1988). The first pair of strand exchanges results in the for- mation of a Holliday intermediate that is then resolved into
A
B
recombinant molecules by the second pair of strand ex- changes. We used a synthetic Holliday intermediate (x struc- ture) to study the ability of the mutant FLP proteins to execute the resolution stage of the reaction (Fig. 7 A ).
All of the proteins tested were able to promote resolution of the x structure to the two linear products. However, two mutant proteins showed an obvious defect in resolution (H309L and H345L, Fig. 7B). Since these proteins showed neither cleavage of a linear substrate nor in vitro recombina- tion, this result was not surprising. However, FLP N329D and A339D proteins gave efficient resolution of the x structure even though cleavage and recombination of linear substrates were markedly decreased. Thus, the spectrum of activities on the Holliday structures shown by the mutant proteins indi- cates that initiation of recombination on a linear substrate has quite different requirements from resolution of the x structure.
DISCUSSION
The Role of the C-terminal Conserved Region of FLP in the Recombination Reaction-This study describes further char- acterization of a C-terminal region of amino acids 303-345 of the FLP recombinase. This region contains 3 of the 4 residues that are absolutely conserved among all members of the
A
b . ""
b
6 1 2 3 4 5 6 7 8 @101112131415
1 2 3 4 5 6 7 6 910 11 121314151617181920
'P
-S
FIG. 6. Strand ligation assay by mutant FLP proteins. A , t..- principle of the assay is diagramed. A synthetic oligonucleotide (18 nt) with a 3'-phosphotyrosine (P- tyr ) was labeled at its 5'-end with "P (*) and annealed to a complementary oligonucleotide (30 nt) to give a half-FRT site containing a single symmetry element (large arrow, a ) and an 8-nucleotide single-stranded protrusion representing the core sequence ( S ) . FLP promotes a nucleophilic attack by the 5'- OH (small arrow) to yield a 48-nucleotide hairpin structure ( P ) . B, reactions contained 9 nM ligation substrate and the following levels of FLP. Lane I, no FLP; lanes 2-4, 0.09, 0.23, and 0.47 p M wild-type FLP ( WT); lanes 5 and 6, 0.67 and 1.33 p~ S336F; lanes 7-9, 0.33, 0.82, and 1.6 p M N329H; lanes 10 and 11, 0.56 and 1.1 p M N329D; lanes 12-14, 0.39, 0.97, and 1.9 p M H309L; lanes 15-17, 0.26, 0.67, and 1.33 p~ A339D; lanes 18-20, 0.19, 0.47, and 0.93 p M H345L. Reactions were incubated at room temperature for 20 min, treated with proteinase K, and analyzed on an 8% denaturing polyacrylamide gel. S, ligation substrate; P, hairpin ligation product.
D
FIG. 7. Resolution of synthetic Holliday intermediates. A , the substrate was an artificial x structure that was assembled from four synthetic oligonucleotides. Each strand is indicated by a different type of line and one oligonucleotide is labeled at its 5'-end with 32P (*). The 13-bp symmetry elements are shown as thin arrows labeled a and b. Resolution adjacent to eit!ler symmetry elements a (dark, thick arrows) or at symmetry elements b (light, thick arrows) yields two linear products, one of which is radiolabeled. B, each reaction contained 3.6 nM of x structure and the following levels of FLP: lane
5,32 and 320 nM S336F; lanes 6 and 7,39 and 390 nM N329H; lanes I , no FLP; lanes 2 and 3, 11 and 110 nM wild-type FLP; lanes 4 and
8 and 9, 27 and 270 nM N329D; lanes 10 and 11, 46 and 460 nM H309L; lanes 12 and 13,32 and 320 nM A339D; and lanes 14 and 15, 22 and 220 nM H345L. Reactions were incubated for 20 min at room temperature and analyzed on a 5% non-denaturing gel. S, substrate; P, linear radiolabeled products.

Bending and Cleavage-defective FLP Mutants 1107
integrase family (His-305, Arg-308, Tyr-343 of FLP, Argas et al., 1986; Abremski and Hoess, 1992) as well as a number of amino acid residues that are conserved among the FLP-like proteins from different species of yeast (Utatsu et al., 1987). Previous experiments have suggested that His-305 plays a role in strand transfer after cleavage and in religation (Par- sons et al., 1988). Arg-308 is probably essential for tightness of the FLP-FRT-binding complex, for cleavage of the DNA target, and possibly for strand ligation (Parsons et al., 1990). The Tyr-343 residue is required for nicking of the DNA strands and covalent attachment of FLP to the 3'-end via a phosphotyrosine linkage (Evans et al., 1990). Previous studies have also suggested that this region might be involved in both DNA binding and DNA bending because mutations at posi- tion 305, 307, and 308 affected the ability of FLP to bind to its target site (Parsons et al., 1988, 1990; Schwartz and Sa- dowski, 1989), and a change of the glycine at position 328 abolished the type I1 bend (Schwartz and Sadowski, 1990). The results of the present study are summarized in Table I.
The diminished binding displayed by the K303E and L315P proteins correlates with the impaired DNA binding previously shown for the following FLP mutants: H305L, H305P (Par- sons et d . , 1988), G307R (Schwartz and Sadowski, 1989), and R308G, R308Q, R308P (Parsons et al., 1990). Those muta- tions are clustered in the N-terminal part of this region that is predicted to be in an a-helical configuration using the algorithms of Chou and Fassman (1978) and Garnier et al. (1978) (not shown). This a-helix could contribute to the binding domain of FLP. This domain was recently localized to a region between amino acids 148 and 350 (Pan et al., 1991, Chen et al., 1991).
The C-terminal conserved region is also involved in the bending of the target DNA by FLP. This region was previously implicated in bending because two mutant FLP proteins (G328R and G328E) were defective in forming the type I1 bend of the FRT site (Schwartz and Sadowski 1989,1990). In the present study, all of the mutants described are impaired in their capacity to promote the type I1 bend. The most deficient mutant in this respect, N329D, showed a type I1 bend of only 94" (Table I), close to the type I1 bend of 93" displayed by the G328E mutant of FLP (Schwartz and Sa- dowski, 1990). The N329D protein also resembled the proteins changed at position 328 in that all proteins were unable to initiate recombination by cutting a linear substrate. The N329D FLP was able to resolve a Holliday intermediate quite effectively (see below).
Other amino acid changes in this region also caused a defect in the DNA strand cleavage of linear substrates (H309L, A339D, and H345L). While all the mutant proteins studied showed defective ability to form the type I1 bend, clearly a type I1 bend of 115-122" (N329H, S336Y, and S336F) is sufficient to promote near-normal levels of recombination. Thus, the role for the type I1 bend in strand cleavage and recombination is unclear since there is not a perfect correla- tion between bending, cleavage, and recombination. It is pos- sible that the need for the type I1 bend is masked in certain mutant proteins by the nature of the substrates used in the in vivo and in uitro assays. These assays measure the excision of the DNA between two directly oriented FRT sites, whereas the natural substrate for FLP (2-pm plasmid) contains its two FRT sites in inverted orientation.
All of the mutations in the region of amino acids 305-345 also affect the ability of the FLP proteins to promote inter- actions between half-FRT sites. Qian et al. (1990) observed that two half-FRT sites occupied by FLP molecules can form a dimeric nucleoprotein complex that is held together by
protein-protein interactions. None of the mutant proteins described in this study was able to form dimeric complexes at the same level as wild-type FLP. The half-FRT site experi- ment strongly suggests direct involvement of the C-terminal conserved region in the FLP-FLP interactions needed to form dimers. These interactions may be the same as those required for the type I1 bend.
Three of the mutant proteins (S336F, A339D, and H345L) had no defect in strand ligation whereas two others (H309L and N329H) had partial defects. Even the N329D protein performs ligation efficiently when presented with a Holliday junction substrate.
Two mutant proteins (N329D and A339D) showed a partic- ularly interesting phenotype, i.e. they were severely deficient in their ability to initiate recombination on a linear substrate, but they were quite proficient in resolving x structures. Since both proteins showed defects in forming the type I1 bend, it is possible that bending plays a role in the initial strand exchange. Once the first pair of exchanges has occurred, the need for type I1 bending is diminished because the x structure has a conformation that permits resolution by these mutant proteins. This observation suggests that the initiation and resolution steps of the reaction are not equivalent.
In this study we have changed 3 residues that are conserved not only in all the six FLP-like proteins but in several other members of the integrase family. Position 309 is a histidine in 23 of 28 member proteins, position 315 is a leucine in 17 members (a methionine in four and phenylalanine in three), and position 345 is a histidine in 19 members (Abremski and Hoess, 1992). Mutation of each of these residues in FLP abolished recombination and strand cleavage and impaired the formation of the type I1 bend. The proximity of the 2 histidines to residues already implicated in the catalytic mech- anism of the enzyme (R308, Y343) makes it possible that the histidines are also involved in catalysis of strand breakage. The leucine at position 315 may have a structural role in correctly positioning the active site on the FRT site for proper DNA binding. We conclude that the C-terminal conserved region is involved in the early stages of the FLP recombina- tion reaction.
Acknowledgments-We thank Helena Friesen for her critical read- ing of the manuscript and Frieda Chan for her typing of the paper.
REFERENCES Abremski, K. E., and Hoess, R. H. (1992) Prot. Eng. 6,87-91 Amin, A,, Roca, H., Luetke, K., and Sadowski, P. D. (1991) Mol. Cell. Biol. 11 ,
Amin, A. A., Beatty, L. G., and Sadowski, P. D. (1990) J. Mol. Biol. 2 1 4 , 55-
Andrews, B. J., Beatty, L. G., and Sadowski, P. D. (1987) J. Mol. Biol. 193 ,
Argos, P., Landy, A., Abremski, K., Egan, J. B., Haggard-Ljungquist, E., Hoess, R. H., Kahn, M. L., Kalionis, B., Narayana, S. V. L., Pierson, L. S., 111, Sternberg, N., and Leong, J. M. (1986) EMBO J. 6,433-440
44974508
72
345-358
Beatty, L. G., and Sadowski, P. D. (1988) J. Mol. Biol. 2 0 4 , 283-294 Chen, J.-W., Evans, B. R., Yang, S.-H., Teplow, D. B., and Jayaram, M. (1991)
Chou, P. Y., and Fassman, G . D. (1978) Adu. Enzymol. 47,45-148 Evans, B. E., Chen, J.-W., Parsons, R. L., Bauer, T. K., Teplow, D. B., and
Friesen, H., and Sadowski, P. D. (1992) J. Mol. Bzol. 226,313-326 Futcher, A. B. (1986) J. Theor. Bid. 119 , 197-204 Garnier, 0. J., Osguthorpe, D. J., and Rohson, B. (1978) J. Mol. Biol. 120,97-
Gronostajski, R. M., and Sadowski, P. D. (1985) J. Biol. Chem. 2 6 0 , 12328-
Proc. Natl. Acad. Sci. U. S. A . 8 8 , 5944-5948
Jayaram, M. (1990) J. Biol. Chem. 266,18504-18510
109
12335 Hie-siyk. H., Wierzbicki, A,, and Abremski, K. (1987) Proc. Natl. Acad. Sci.
Jayaram, M., Crain, K. L., Parsons, R. L., and Harshey, R. M. (1988) Proc.
Kitts, P. A., and Nash, H. A. (1987) Nature 329,346-348 Meyer-Leon, L., Gates, C. A., Attwood, J. M., Wood, E. A., and Cox, M. M.
Merer-Leon, L., Huang L.-C., Umlauf, S. W., Cox, M. M., and Inman, R. B.
Nunes-Duby, S. E., Matsumoto, L., and Landy, A. (1987) Cell 60,779-788 Pan, G., and Sadowski, P. D. (1992) J. Biol. Chem. 2 6 7 , 12397-12399
U. S. A. 84,6840-6844
Natl. Acad. Sci. U. S. A. 8 6 , 7902-7906
(1987) Nucleic Acids Res. 16,6469-6488
(1988) Mol. Cell. Btol. 8 , 3784-3796

1108 Bending and Cleavage-defective FLP Mutants Pan,-H:, Clary, D., and Sadowski, P. D. (1991) J. Biol. Chem. 2 6 6 , 11347-
Parsons R. L Prasad P. V., Harshey, R. M., and Jayaram, M. (1988) Mol.
Parsons R. L. Evans, B. R., Zheng, L., and Jayaram, M. (1990) J. Biol. Chem.
Prasad, P. V., Youn , L.-J., and Jayaram, M. (1987) Proc. Nut/. A c d . Sci.
11534
Cell. diol. 8,"3303-3$10
2 6 5 , k527-4533
U. S. A. 84 2189-8193 Proteau, G., didenberg, D., and Sadowski, P. D. (1986) Nucleic Acids Res. 14 ,
Qi_a?,Z.-H., Inman, R. B., and Cox, M. M. (1990) J. Biol. Chem. 265,21779- 4787-4802
Re nolds, A. E., Murray, A. W., and Szostak, J. W. (1987) Mol. Cell. Biol. 7 , I1 IL(0
$566-3573
Sanger, F., Coulson, A. R., Barrel), B. G., Smith, A. J. H., and Roe, B. A. (1980)
Sayers, J. R., Schmidt, W., and Eckstein, F. (1988) Nucleic Acids Res. 16 , 791-
Schwartz, C. J. E. (1990) Ph.D. thesis, University of Toronto Schwartz, C. J. E., and Sadowski, P. D. (1989) J. Mol. Biol. 205,647-658 Schwartz, C. J. E., and Sadowski, P. D. (1990) J. Mol. Biol. 216 , 289-298 Taylor, J. W., Ott, J., and Eckstein, F. (1985) Nucleic Acids Res. 13,8764-8785 Thompson, J. F., and Landy, A. (1988) Nucleic Acids Res. 16,9687-9705 Utatsu, I., Sakamoto, S., Imura, T., and Toh-e, A. (1987) J. Bacteriol. 169 ,
Volkert, F. C., and Broach, d . R. (1986) Cell 46,541-550
J. Mol. Biol. 143, 161-178
802
5537-5545





























