Embed Size (px)
Citation preview

Multiscale Modeling of Catalysis in Chemistry and Biology
Weitao Yang, Duke University
TheoryBiologicalNano
F di
NanoMaterial
FundingNSFNIHDOEONR
CTMS, DukeCTMS, DukeSept, 2009

Studies of Biological Systems
-DevelopmentFunding: NIH Steven Burger
Xiangqiang Hu
-Enzyme/Solution Reaction Mechanism-Design Takumi Hori
Yingkai Zhang (NYU) Haiyan Liu (USTC, China)Andres Cisneros (Wayne State) Mingliang Wang
X. “Fox” Zeng
Fitzgerald, Rudolph, McCarfferty (Duke), Carter (UNC)
Andres Cisneros (Wayne State) Mingliang Wang
(Duke), Carter (UNC)Ganglong Cui
Kazuhiro Fujimoto
Hao Hu (HKU)
Jerry Parks (ORNL) Zhenyu LuPan Wu Zhigang Sun

Chemical ReactionsChemical ReactionsA chemical reaction is a process that leads to the
transformation of one set of chemical substances totransformation of one set of chemical substances to another.
Splitting of water – important for hydrogen energy2 H2O → 2 H2 + O2
Where:chemistry, biology (life processes), materials, engineering

CatalysisCatalysisCatalysis is the process in which the rate of a chemical
reaction is increased by means of a catalystreaction is increased by means of a catalyst.

Enzymes and Life Processes
•The greatest majority of biochemical reactions doThe greatest majority of biochemical reactions do not take place spontaneously.
•The catalysts of biochemical reactions are enzymes and are responsible for bringing about y p g galmost all of the chemical reactions in living organisms.
•There are an estimated 75,000 human enzymes
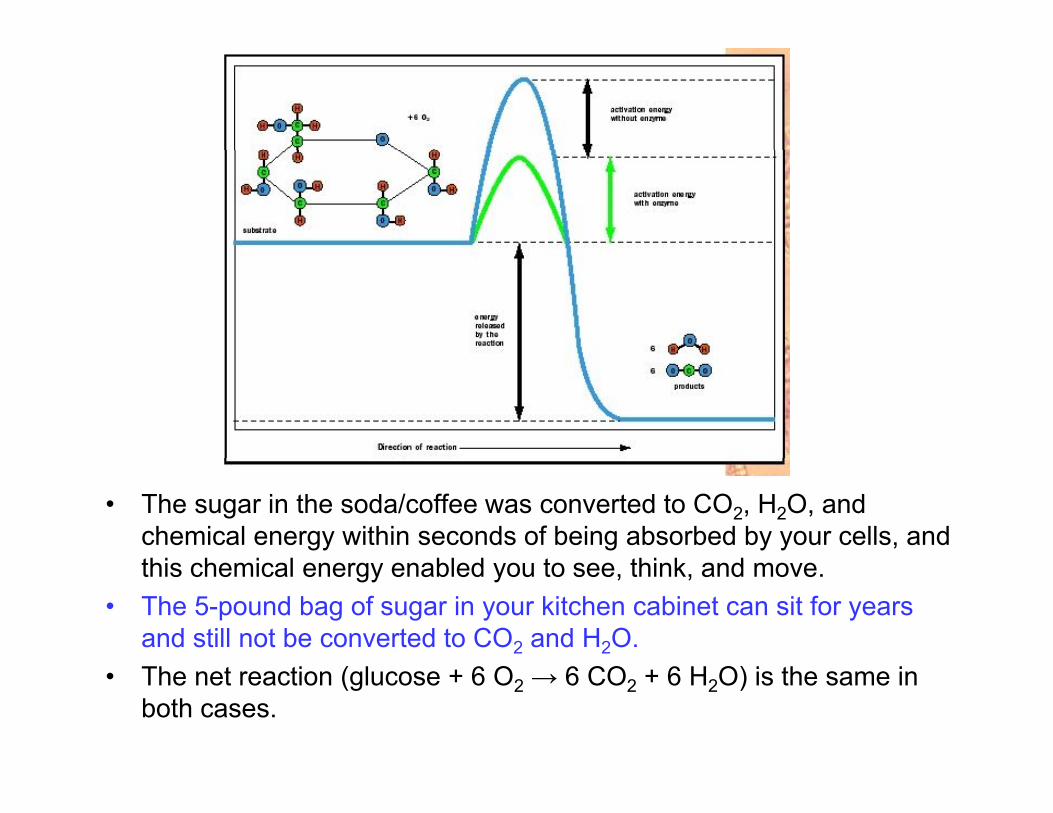
• The sugar in the soda/coffee was converted to CO2, H2O, and chemical energy within seconds of being absorbed by your cells, and thi h i l bl d t thi k dthis chemical energy enabled you to see, think, and move.
• The 5-pound bag of sugar in your kitchen cabinet can sit for years and still not be converted to CO2 and H2O.
• The net reaction (glucose + 6 O2 → 6 CO2 + 6 H2O) is the same in both cases.

Mechanistic study of enzyme catalysisExperiments
• Structural determinationBi h i l ki ti t• Biochemical kinetics measurement
• Mutagenesis study
• Cannot directly determine transition-state structureCannot directly determine transition state structure• Infer mechanisms from indirect evidences• Provide basis for other studies
Computer simulations• Statistic mechanics principles• Quantum mechanics or molecular mechanics
• Rely on experimental information (structures, …)• Can determine precise transition-state or intermediate structures• Provide molecular details for the mechanism, structure and dynamics

How to model molecules and h i l tichemical reactions
M l l El t N l i•Molecules = Electrons + Nuclei
•Electron mass = 9.10938188 × 10-31 kilograms -- Quantum
•Proton (H) mass = 1.67262158 × 10-27 kilograms –Quantum/Classical
•Electron mass/proton mass = 1/1836, Born-Oppenheimer Approximation:
(1) Electrons motions fast and quantum, remain in the ground state, even during chemical reactions
(2) Nuclei move in the force mediated by the electrons (the Born-Oppenheimer surface). (Quantum or Classical)

• Reaction is a rare eventReaction is a rare event
Di t d i t k l ti• Direct dynamics takes long time
• Reaction rate theory reduces/eliminates the need for dynamics. y

Chemical reactivity is dominated by the mechanistic topology of the local reaction chemistry space.

Theory of Reaction Rate exp( / )catkTk G kT eo y o eact o ate
G
--The free energy of activation
G
G
activation--Quasi-equilibrium
G
Th t i i--The transmission
coefficient--Dynamics
1 --Transition State Theory

Steps of Enzyme ReactionsSteps of Enzyme Reactions
1 E S ES 1. E S ES 2. ( )catES EP k3
cat
EP E P 3. EP E P

Methods for CalculationMethods for Calculation
kT exp( / )catkTk G kT
Ideally1. Accurate QM for ( )E R
ln exp( ( ) / )G kT dR E R kT
2. Statistical mechanics for free energy
ln exp( ( ) / )G kT dR E R kT 3. Quantum dynamics for transmission coeff. y

Possible Methods
QM/MM Multi-Scale instead of QMQM/MM, Multi-Scale, instead of QM• Empirical Valence Bond
S i i i l QM• Semiempirical QM • CPMD DFT• Gaussian AO based DFTStatistical Mechanics MethodsStatistical Mechanics Methods• MD Umbrella sampling
R ti th h MD• Reaction path approach + MD

Our Ab initio QM/MM Approach to Large Systems
Combined QM and Molecular Mechanics—O(1) in practice!
– A Pseudobond approach to QM/MM interface JCP (1999)( )
– Iterative OptimizationJCP (2000)JCP (2000)
– QM/MM-FE: Free-energy calculationJCP (2000)JCP (2000)
– QM/MM-MFEPJCTC (2007), JCP (2008)

Potential Energy Surface E(R): QM/MMPotential Energy Surface E(R): QM/MM
Reaction active part: small (QM)
The rest: large (MM)
Warshel and Levitt (1976)

What is the justification for QM/MM?What is the justification for QM/MM?
•Chemistry is local, separation of degrees of freedom based on length scale is reasonablefreedom based on length scale is reasonable.
•But, quantum mechanics is NON-LOCAL.
22
- +V(r)) (r)=E (r)2
(
•Mathematical justification?

Pseudobond ApproachYingkai Zhang, Taisung Lee and WY, JCP 1999g g g
Cps: an one-free-valence atom with an effective core potential
Cps – C: a pseudo-bond mimics the original C-C bond with the similar bond length and strength, and similar effects on the rest of the active parton the rest of the active part

QM/MM-FE: An iterative optimizing procedureYi k i Zh H i Li d WY JCP (2000)Yingkai Zhang, Haiyan Liu and WY, JCP. (2000)
Taking advantages and avoiding disadvantages
– Optimizing the small QM sub-system with quasi-Newton minimizer with ab initio QM/MM calculationsQ
– Optimizing the large MM subsystem with truncated Newton method with only MM calculationswith only MM calculations
– Converged very efficiently
min ,QM MMEr r
r r min minQM
r r,QM MMr r QM MMr r

−1502.65
−1502.66
−1502.65
−1502.67
−1502.66
ree)
−1502.68
−1502.67
E (
Har
tree
−1502.69
E
0 2 4 6 8−1502.70
0 2 4 6 8Iteration

What we can do with the iterative optimization procedure
– Determining the reactant and product structure
– Mapping out the reaction path with the coordinate driving method (performing a series of constrained
i i i ti l d ti di t )minimization along a proposed reaction coordinate)
If the minimum energy path is smooth and continuous theIf the minimum energy path is smooth and continuous, the maximum on the minimum energy path is a transition state

QM/MM-FE: free energy calculation methodYingkai Zhang, Haiyan Liu and WY, JCP. (2000)
Approximate the dynamics of the QM sub-system • independent of MMp• harmonic
the QM energy differencethe QM energy difference
calculated from the QM frequencies
the free energy change from QM/MM interaction

5
0
−5
cal/m
ol)
−10
Fqm
/mm
(kc
a
−15
−10
Δ F
q
−1.5 −1.0 −0.5 0.0 0.5 1.0−20
−15
−1.5 −1.0 −0.5 0.0 0.5 1.0−20
Reaction Coordinate



4-Oxalocrontonate tautomerase (4OT)
•Reveal a Machsnism without acid
•Water
•Arg-39
•Prediction on mechanism andmutation effects confirmed by exptlater.
Cisneros, Liu, Zhang, and Yang, JACS, 125, 10384 (2003)
4-Oxalocrontonate tautomerase (4OT)
Cisneros, Liu, Zhang, and Yang, JACS, 125, 10384 (2003)

Cl i A id B i C t l d R ti ?
CO
Classic Acid-Basis Catalyzed Reactions?
CO2 CO2 CO2
OH
OHH OH
1
2
1
2
1
2O
H
H
OHH
H
O3
4
3
4
3
4
CO2 CO CO
HH H
H H
H
H5
6
5
6
5
6CO2 CO2 CO26 6

N
NH
Arg-39''
H
H
O H
H
O H
O2C OO N
NH
Arg-39''
H
H
O H
H
O H
O2C OO
A (2) (2)
(1)(1)
N
N
HH
H
HN
O N
N
HH
HH
H2N
OHH
Pro-1 Pro-1
B(2) (2)
O2C O
O
O N
N
N
Arg-39''
HH
H
H
HH
H
O H
H
O H
HN
O2C O
O
OH N
N
N
Arg-39''
HH
H
HH
H
O H
H
O H
H2N
H
B
(1) (1)
(2) (2)
H
O H
H
O H
Arg-39''
H
O H
O H
Pro-1 Pro-1
C(2)
(1)(1)
(2)
N
N
NH
Arg-39''
HH
H
H
H
HN
O2C O
O
O N
N
NH
Arg-39''
HH
H
HH
H
O
H2N
O2C O
O
O
HH
Pro-1 Pro-1
N
NH
Arg-39''
H
H
O H
H
O H
O2C OO N
NH
Arg-39''
H
H
O H
H
O H
O2C OO
D
(1)
(2)
(1)
(2)
N
NH
HH
H
O2C O
O N
NH
HH
HH
H2N
O2C O
OHH
Pro-1
HN
Pro-1

O
O
NH
NH2
N
O
O O H2N NH2
N
H
Arg-11'
Arg-39''H
O
O
NH
NH2
N
O
O H2N NH2
N
H
Arg-11'
Arg-39''H
O
ONH2 O
H
H
N
Arg-39''
Pro-1
H
H
O
H
N
H
Pro-1
H
O
H
H
H
TSI
O
O
NH
NH2
N
O
O O H2N NH2
N
H
Arg-11'
Arg-39''H
O
O
NH
NH2
N
O
O O H2N NH2
N
H
Arg-11'
Arg-39''H2
N
H
Arg-39''
Pro-1N
Pro-1
H
HH H
O
H
PR
H H H
O
H

Our calculations show that there is no general id i th ti
Two important factors:
acid in the reaction.
Two important factors:
Arg-39”—hydrogen bonded to the carboxylate group of theArg 39 hydrogen bonded to the carboxylate group of the substrate
An ordered water—gets closer to the site of the charge formed in the transition state and intermediate play the main role in transition state/intermediate stabilizationrole in transition state/intermediate stabilization

Electrostatic Interactions Dominate the Catalytic Contribution of Arg39 in 4-Oxalocrotonate Tautomerase
JACS 04 M t i B ik D K i (T h i S i )JACS 04, Metanis, Brik, Dawson,Keinan (Technion,Scripps)
X NH2+ i iX=NH2+,arginine
X= O, citrulline

JACS 04, Metanis, Brik, Dawson, Keinan, , , ,
… the Arg39Cit
•reduces kcat by 1660-fold, consistent with Arg39 interacting with the developing negative charge ofinteracting with the developing negative charge of the ketoacid group in the transition state.
•gives further evidence against the role of Arg39 as a general acid, in agreement with recent
i l dicomputational studies.

Protein backbone makes important contributions to 4OT catalysis: Understanding from experiment and theory
Ci W Sili ki Fit ld d Y Bi h i t (2004)Cisneros, Wang, Silinski, Fitzgerald, and Yang, Biochemistry (2004).
C l l ti QM/MM l l tiCalculations: QM/MM energy calculations,
Experiments: backbone chemical modification of NH to OExperiments: backbone chemical modification of NH to O, by Wang, Silinski and Fitzgerald at Duke
Leu8 (OL8)4OT(R61A)E C l l ti 1 0 1 5E Calculation 1.0 1.5
F Expt 1.8

Protein backbone makes important contributions to 4OT catalysis: Understanding from experiment and theory
Ci W Sili ki Fit ld d Y Bi h i t (2004)Cisneros, Wang, Silinski, Fitzgerald, and Yang, Biochemistry (2004).
C l l ti QM/MM l l tiCalculations: QM/MM energy calculations,
Experiments: backbone chemical modification of NH to OExperiments: backbone chemical modification of NH to O, by Wang, Silinski and Fitzgerald at Duke
Leu8 (OL8)4OT(R61A)E C l l ti 1 0 1 5E Calculation 1.0 1.5
F Expt 1.8 1.2

Issues in Dynamics and Statistical Sampling
– Most Ab initio QM/MM calculations are based on the determination of a minimum energy path
– Statistical mechanics and classical or quantum dynamics?dynamics?
O QM/MM FE h t i ti– Our QM/MM-FE has two approximations:• The QM dynamics and MM dynamics are independent• The motion of the QM subsystem is harmonic• The motion of the QM subsystem is harmonic

Reaction Path Potential for Reactions in EnzymesZhengyu Lu and WY, JCP (2004).
RPP follows the ideas from the reaction path Hamiltonian (RPH) of Miller, Handy and Adams. It is an analytical energy expression of the QM/MM potential energyof the QM/MM potential energy.
– An expansion is made in both the nuclear and the electronic degrees of freedom for the QM subsystem internal energydegrees of freedom for the QM subsystem internal energy
– The energy of the MM subsystem and the interaction between th QM d MM b t i h dthe QM and MM subsystems remains unchanged
– The QM charges are polarizable in response to the changes in g p p gboth the MM and the QM degrees of freedom
– The input data are energies, gradients, vibrational frequencies,The input data are energies, gradients, vibrational frequencies, and electron density response properties of the QM subsystem.

QM charges polarizable w.r.t. to QM and MM

The reaction catalyzed by triosephosphate isomerase (TIM)
O2-
O
HIS 95 HIS 95
N
N
HN
N
H
C1
HO C2CH2
PO42-
HH
C1
HO C2
O
CH2
PO42-
H
H
-
H H
C
O O
C
O O
H
GLU 165
-
GLU 165 GLU 165

The average time-dependent transmission coefficient
1
0.75
0.5
κ(t)
0.25
10 20 30 40 50Time (fs)
0
Wang, Lu, and Yang JCP (2004)

Nuclear Quantum Effects with the Reaction Path Potential: Proton Transfer in Triosephosphate Isomerasep p
(Mingliang Wang, Zhenyu Ju and WY, JCP, 2006)
• based on potential energy surface from the reaction path• based on potential energy surface from the reaction path potential method
• the normal mode centroid path integral molecular dynamics (Voth,Chandler and Miller 1989, Tuckerman et al 1996)
• In the simulation, the primary and secondary hydrogens, and the C and O atoms involving bond forming and bondand the C, and O atoms involving bond forming and bond breaking are treated quantum mechanically, while the motions of all other atoms are dealt with classically.

25
Classical
20
ClassicalQuantum
--potential energy surface from the reaction path
15
al/m
ol)
from the reaction path potential based on QM/MM
--the normal mode centroidth i t l MD
10
PMF
(kca
l/ path integral MD (Voth,Chandler and Miller 1989)
5
PM
0
5
-0.8 -0.4 0 0.4 0.8Reaction Coordinate R
C-H-R
O-H (Å)
0

20PMF of T
20PMF of TPMF of DPMF of H
15
cal/m
ol)
10
PMF
(kca
5
P
-0.8 -0.4 0 0.4 0.80
-0.8 -0.4 0 0.4 0.8Reaction Coordinate R
C-H-R
O-H (Å)
0

Quantum Effects in TIM
•Quantum Proton Transfer: Reaction activation energy reduced by 2.3 Kcal/mol, rate enhanced by a factor of y , y47.5
•Primary kinetic isotope effects for proton transfer in TIM•Primary kinetic isotope effects for proton transfer in TIM at temperature of 300 K
•The Swain-Schadd exponent ln( / ) / ln( / )H T D Tk k k k pwas predicted to be 3.01, less than the semi-classical limit value of 3.34, indicating that the quantum effects mainly arise from quantum vibrational motion rather thanmainly arise from quantum vibrational motion rather than tunneling.

Development in Determining Reaction Path for Enzymatic ReactionsReactions
• Parallel Iterative Reaction Path Optimization in Ab Initio Quantum Mechanical/Molecular Mechanical Modeling of E R ti " Li L Ci d Y (JCP 04)Enzyme Reactions", Liu, Lu, Cisneros, and Yang (JCP 04).
• Adapting the Nudged Elastic Band Method for Determining p g g gMinimum Energy Paths for Chemical Reactions in Enzymes", Xie, Liu and Yang (JCP 04).
• Reaction path determination for QM/MM modeling of enzyme reactions by combining first-order and second-order chain-of-replica methods", Cisneros, Liu, Lu and Y (JCP 05)Yang (JCP 05).
• QSM: Multiobjective optimization of reaction path, Steven Burger and Weitao Yang (JCP, 06).

Quadratic String Method (QSM)based on Multiobjective Path Optimization
B d Y JCP 2006Burger and Yang, JCP, 2006
Based on an extension of the string method of E, Ren and Vanden-EijndenCommun. Math. Sci. 2002

Iterative optimization based on a quadratic model

1.5
1.75
M ll B P t ti l1
1.25
Muller-Brown Potential
0.25
0.5
0.75
0
QSMSM
-1 -0.5 0 0.5
0
-50
SMVV SM
-100-100
0 0.5 1 1.5 2 2.5Arc Length
-150

0.5
QSMSMVV SM
0.3
0.4
||x-x
* ||
Muller-Brown Potential
0.1
0.2
2 QSM
0 20 40 60 80 100 120Iteration
0
0
2 QSMSMVV SM
||g⊥||
-4
-2
Log
||g ⊥
0 50 100Iteration
-6

CH2CHOH CH3CHO 0.5
0.6NEBQSMVV SM
CH2CHOH CH3CHO
0.3
0.4
0.5
||ΔT
S||
0.1
0.2
-1
0
VV SMQSMNEB
0 50 100 150 200Iteration
0
-3
-2
-1
g ||g
⊥||
NEB
-5
-4
-3
Log
0 50 100 150 200Iteration
-6
-5

Issues with structures and reaction path on total energy surfacegy
Depend on initial protein/solvent conformations– Depend on initial protein/solvent conformations
– Can fail to determine reaction path (defined onCan fail to determine reaction path (defined on total energy) for enzymatic reactions
– Complete failure for reactions in solutions
Not yet a robust method (like gas phase– Not yet a robust method (like gas phase calculations)

QM/MM Minimum Free Energy Path (QM/MM-MFEP)H H Zh L d WY JCTC 2007Hao Hu, Zhenyu Lu and WY, JCTC 2007
• Combine free energy calculations with reaction path determination
• Define the reaction path on the potential of mean force (PMF) surface -- One path represents an ensemble of paths for the whole systems
• Use chain-of-state methods like the QSM on the PMFUse chain of state methods, like the QSM, on the PMF.
• Eliminate the dependence of the reaction path on the finitial protein conformations.

'exp( ( ))Z E d d r r r r
Partition function and Free energy
0 exp( ( , ))QM MM QM MMZ E d d r r r r
0 01 ln( )A Z
'0 ( ) exp( ( , ))QM QM MM MMZ E d r r r r
Potential of Mean Force
0
0 0
( ) p( ( , ))
1( ) ln( ( ))
QM QM MM MM
QM QMA Z
r r
00
1 ln( ( ))( ) ( , )QM
QM QM MMZ
A E
r
r r r
, , ,MM
QM i QM i QM i r
r r r
Okuyama-Yoshida, Nagaoka, YamabeOkuyama Yoshida, Nagaoka, YamabeSchenter, Garrett, and TruhlarZiegler

PMF

Sequential Sampling and Optimization for QM/MM-MFEPHao Hu, Zhenyu Lu and WY, JCP 2008,
Main computational cost of QM/MM MFEP method is• Main computational cost of QM/MM-MFEP method is the statistical sampling required for the calculation of the QM PMF and its gradients.
• In the sequential sampling and optimization approach:approach: – sampling MM phase space with fixed QM – optimizing the QM subsystem in the fixed ensemble of MM– iterating until convergence.
• The use of a fixed-size, finite MM conformationalThe use of a fixed size, finite MM conformational ensemble enables the precise evaluation of the QM PMF and its gradients

The idea of sequential optimizations
min ,QM MM
QM MMEr r
r rQM/MM-FE,QM MMr r
min min
1i ( ) i l ( ( ))A E
QM MMr r
01min ( ) min ln exp( ( , ))
QM QMMM
QM QM MMA E
r r r
r r rQM/MM-MFEP
minQM
rQMMMr

The idea of sequential optimizationse dea o seque t a opt at o s
QM/MM-FE QM/MM-MFEPQM/MM FE
,min ,
QM MMQM MME
r rr r 0
1min ( ) min ln exp( ( , ))QM QM
MM
QM QM MMA E
r rr
r r r
Iteration:a) MM optimization with
fixed QM geometry
Iteration:a) MM sampling with
fi d QM tfixed QM geometry.
b) QM optimization with
fixed QM geometry.
a) QM optimization withb) QM optimization with fixed MM conformation.
a) QM optimization with fixed MM ensemble.

sequential optimizations: QM/MM-FE, Zhang, Liu and WY, 1999 JCP
1) Set initial QM structure rQM(0), set counter n=0;Q
2) Increase counter n=n+1;a) Carry out MM optimization with QM geometry fixed at rQM
(n-1)
( ) ( 1)in nE
b) Carry out QM optimization with the MM conformation fixed at rMM(n)
( ) ( 1)arg min ,MM
n nMM QM MME
rr r r
c) Update QM geometry3) Go to step (2) until converged
( ) arg min ,QM
nQM QM MME (n)
rr r r
3) Go to step (2) until converged

Sequential QM/MM-MFEP
1) Set initial QM structure rQM(0), set counter n=0;
2) Increase counter n=n+1;a) Carry out MD simulation of the MM environment with QM geometrya) Carry out MD simulation of the MM environment with QM geometry
fixed at rQM(n-1)
( ) ( ), 1,..., MD sampling based on ( ) nMM ref MMN E r r
b) Carry out QM optimization with the MM ensemble fixed at ( ) ( )nMM r
( ) ( ) ( )1 1( ) ln exp , ( ) ( )N
n n nQM ref QM MM ref MMA A E E
r r r r
1
( )( ) ( )
( ),1
( ) p , ( ) ( )
, ( )exp , ( ) ( )( )
QM ref QM MM ref MM
nNQM MM n n
n QM MM ref MMQM iQM
N
EE EA
r rr r rrr
c) Update QM geometry
( ) ( ),
1exp , ( ) ( )
Nn nQM i
QM MM ref MME E
r r r r
) p g y3) Go to step (2) until converged
Finite, fixed-size MM ensemble gives the precise PMF and PMF gradient

Approximate total QM/MM energy ( , )QM MME r r
•Simple QM point chargesSimple QM point charges
QM i t h ith l i ti d t th MM t•QM point charges with polarization due to the MM atoms
•QM point charges with polarization due to the MM and QM atoms

Sequential QM/MM-MFEP Applications: SN2 reaction in water
• Classical SN2 reaction: CH3Cl + Cl-

Applications: SN2 reaction• Umbrella sampling (WHAM analysis)
d=d1-d2d d1 d2
MP2/6-31+G*MP2/6-311+G*B3LYP/6-31+G*B3LYP/6-311+G*
G‡(US)=20.7 kcal/mol

Sequential QM/MM-MFEP Applications: convergence behavior
• Convergence of relative free energy (SN2)

Free Energy ConvergenceFree Energy Convergence

Optimization of the reaction path on the QM PMF
•Can use any path optimization approach
•Quadratic String Method (QSM), Steven Burger and WY, JCP, 2006

Sequential QM/MM-MFEP Applications: SN2 reaction
• QSM path: before and after the determination of TSdetermination of TS
G‡(MFEP)=18.7 kcal/mol

Sequential QM/MM-MFEP Applications: SN2 reaction
• RS, TS structures
2.30 Å 2.32 Å
3.68 Å1.81 Å
3.68 Å~100
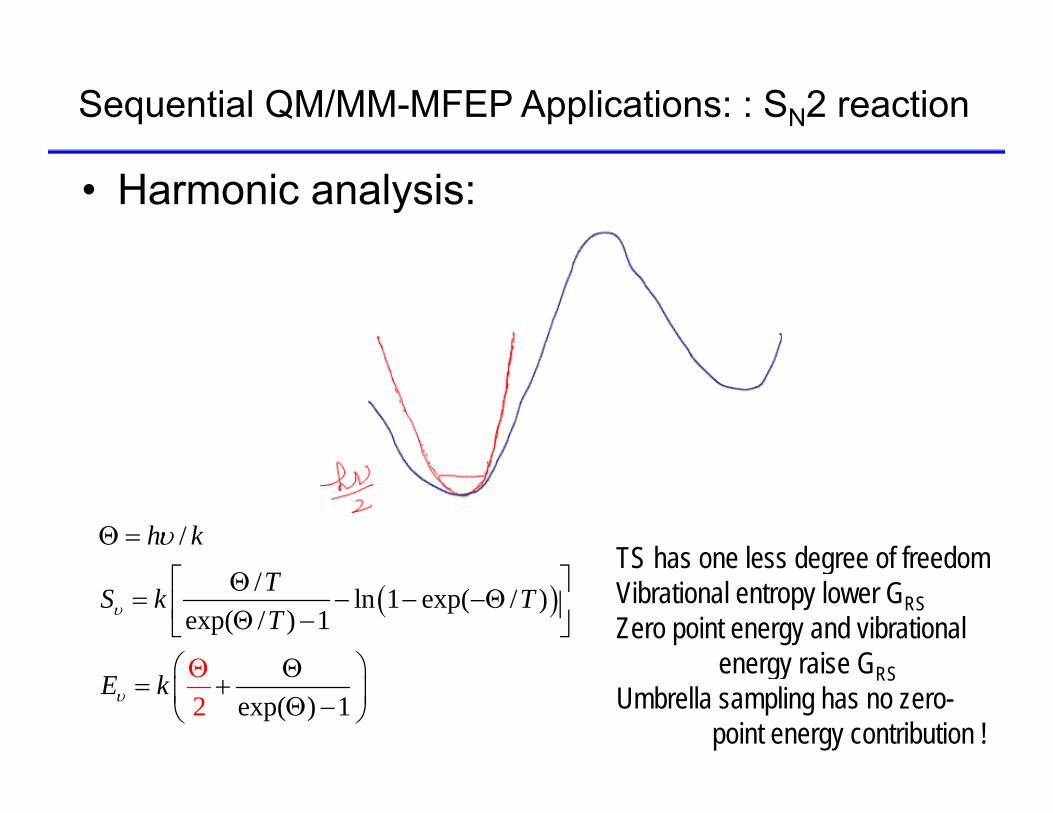
Sequential QM/MM-MFEP Applications: : SN2 reaction
• Harmonic analysis:
/h k TS has one less degree of freedom
/ ln 1 exp( / )exp( / ) 1
TS k TT
TS has one less degree of freedomVibrational entropy lower GRSZero point energy and vibrational
energy raise Gexp2 ( ) 1
E k
energy raise GRSUmbrella sampling has no zero-
point energy contribution !

Sequential QM/MM-MFEP Applications: SN2 reaction
• Activation free energyG‡(MFEP) = 18.7 kcal/mol
index TS RS1 -298.64 41.72( )
harmonic corrections:G‡(MFEP+Freq) = 20 4 kcal/mol
2 66.37 49.443 95.87 57.764 106.46 66.09
G‡(MFEP+Freq) 20.4 kcal/mol
Additional 3-fold rotational
5 113.75 77.406 125.46 109.977 129.38 168.40
multiplicity 8 214.60 175.909 217.02 182.3810 234.07 708.32
ln(1/3) 0.65 /kT kcal mol
G‡(MFEP) = 21.1 kcal/molv.s.
11 976.67 1039.9712 982.33 1052.3113 1125.74 1408.77
G‡(direct dynamics) = 20.7 kcal/mol 14 1425.90 1497.8615 1429.94 1514.0016 3220.21 3146.16

Ab initio QM/MM minimum free energy path
Convergence
Hu, et al. J. Chem. Phys., 2008; Hu & Yang, Annu. Rev. Phys. Chem., 2008

Ab initio QM/MM minimum free energy path
Advantages
C li d l i d i b• Complicated solution and enzyme reaction becomes gas-phase alike
• Remove the path-dependence of initial conformation
G d t ti ti• Good statistics
• Optimizing reaction path without explicit definition ofOptimizing reaction path without explicit definition of reaction coordinates
A j t t d b t li ti f b i iti QM/MM• A major step toward robust application of ab initio QM/MM to solution and enzymes.

Mechanism of OMP Decarboxylation in Orotidine5′-Monophosphate Decarboxylase (ODCase)
ODC t tODCase structure
O O O
N
O
H3
45 -CO2
NH
34
5 +H+ N
H3
45
N
O
O-
O12 6
NO
12 6-
NO
12 67
One of the most proficient enzymes known-- Radzicka, A.; Wolfenden, R. Science 1995
RP RP RP

ODCase mechanism
Mechanism without agreement
E i f di d b l i h i b• Experiments favor direct-decarboxylation mechanism, but failed to provide definite evidence
• Simulations have been inconclusive too– Pai & Gao (AM1/CHARMM27 simulation)– Warshel (EVB)– Lee & Kollman (QM/MM-FE)Lee & Kollman (QM/MM FE)– Lundberg & Siegbahn (QM)– Raugei (CPMD + Jazynski simulation)– Houk (CP2K + meta-dynamics)Houk (CP2K + meta dynamics)
– All DFT-based simulations yielded barrier (21 ~ 27 kcal/mol) significantly higher than experimental data (15.3 kcal/mol)s g ca t y g e t a e pe e ta data ( 5 3 ca / o )
Is DFT inappropriate for this reaction?

ODCase: gas-phase reaction
B3LYP/6-311+G* is in good agreement with MP2 method

ODCase: solution reaction
G‡ 40.2 kcal/mol (Expt. G‡ 38.5 kcal/mol)
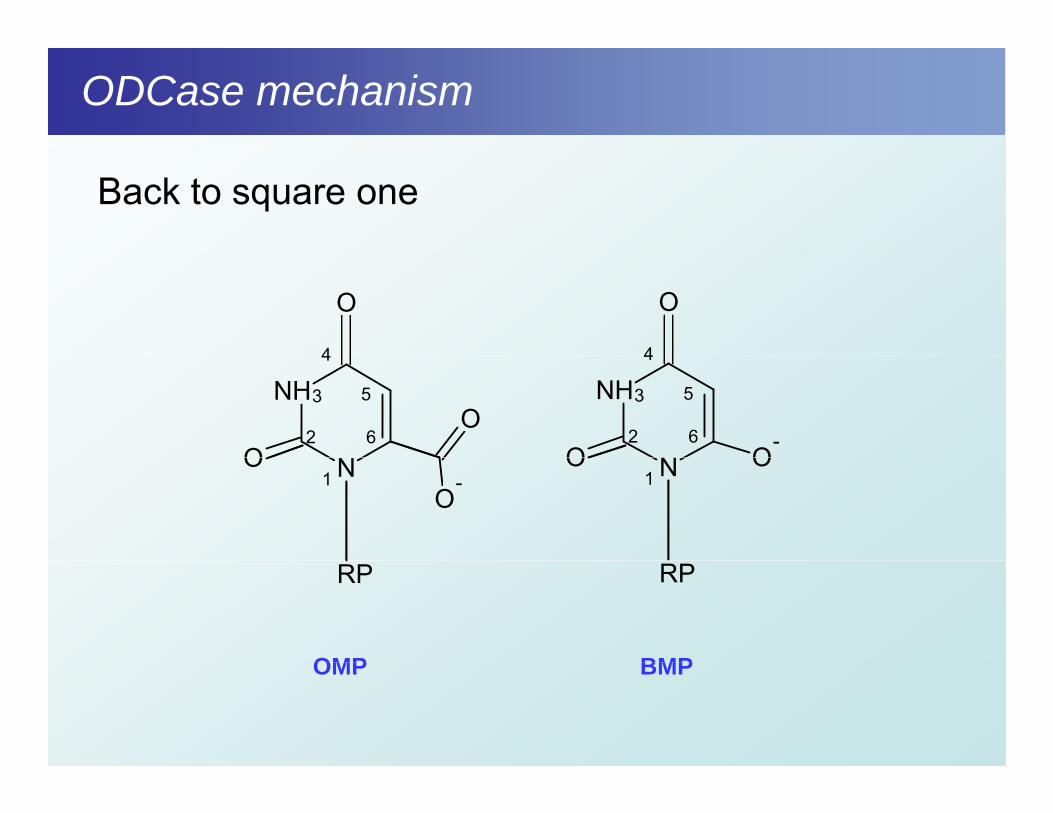
ODCase mechanism
Back to square one
O
4
O
4
NHO
O2
3
4
5
6
N
NH
O-
O2
3
4
5
6
NO
-O
1 N OO1
RP RP
OMP BMPOMP BMP

ODCase mechanism
BMP binds to a water molecule
ODCase mechanism
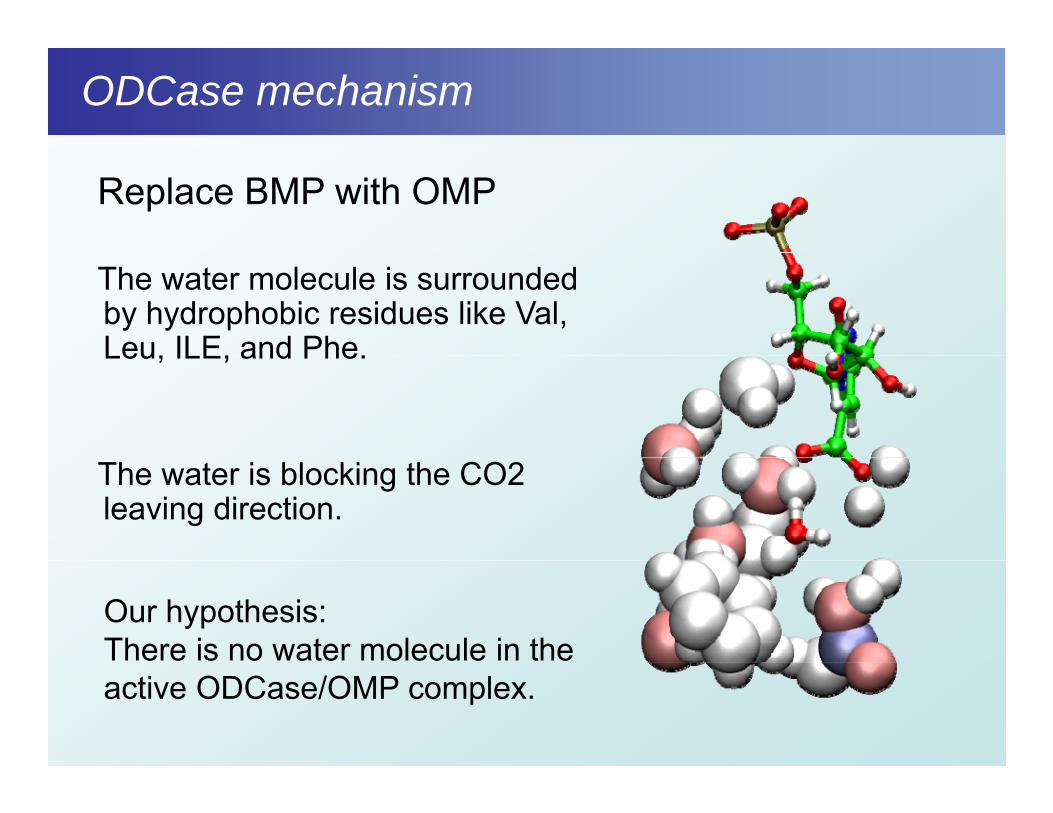
Replace BMP with OMP
The water molecule is surrounded by hydrophobic residues like Val, Leu, ILE, and Phe.Leu, ILE, and Phe.
The water is blocking the CO2 leaving direction.
Our hypothesis:There is no water molecule in the active ODCase/OMP complex.

ODCase: enzyme reaction
G‡ 16.5 kcal/mol (Expt. G‡ 15.3 kcal/mol)

ODCase: direct mechanism
Results
1. Reproduced experimental barrier
2. Supported direct decarboxylation mechanism
3. Highlighted the importance of proper recognition of protein-p p g pbound water molecules
Hao Hu, Amy Boone, and WY, JACS, 2008

Ann. Rev. Phys. Chem, 2008





























