Embed Size (px)
Citation preview

Multiple sequence alignment
Irit OrrShifra Ben-Dor

An example of Multiple AlignmentVTISCTGSSSNIGAG-NHVKWYQQLPGQLPGVTISCTGTSSNIGS--ITVNWYQQLPGQLPGLRLSCSSSGFIFSS--YAMYWVRQAPGQAPGLSLTCTVSGTSFDD--YYSTWVRQPPGQPPGPEVTCVVVDVSHEDPQVKFNWYVDG--ATLVCLISDFYPGA--VTVAWKADS--AALGCLVKDYFPEP--VTVSWNSG---VSLTCLVKGFYPSD--IAVEWWSNG--

An important contribution of MolecularBiology studies was the following discovery:
− Similar genes are conserved across widelydivergent species, often performingidentical or similar function.
− Sometimes these genes are mutated andtheir function altered according to naturalselection.

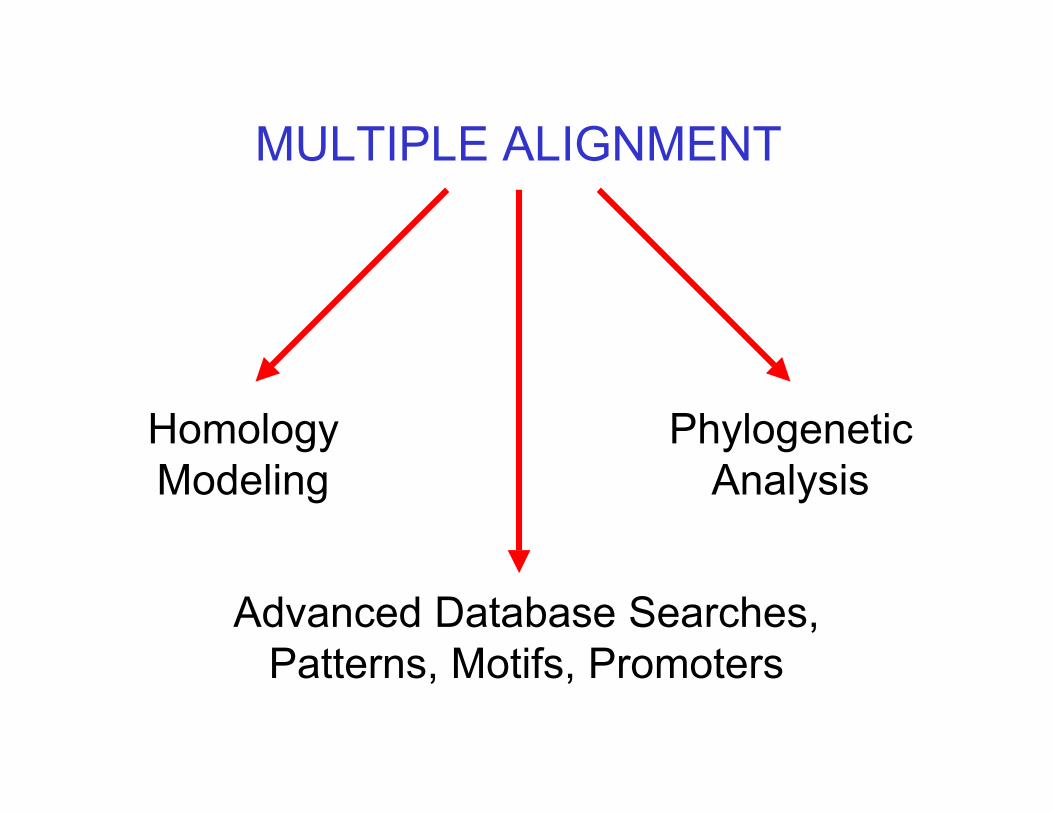
Why do we need multiplealignments?
Multiple alignment, whether made of DNA orprotein sequences, can yield much moreinformation than analysis of a singlesequence (or even 2).
When dealing with a new protein, withunknown function, the presence of severaldomains similar to domains in other “known”sequences, can imply a similar structure orfunction.

Why do we need multiplealignments?
It is known that selective pressure ofevolution results from the need toconserve a function.
In proteins, maintaining their functiongenerally requires a specific 3D structure.Thus, protein multiple alignments can givesome information about the 3D structure.

PAIRWISE ALIGNMENT
DATABASE SEARCHING
MULTIPLE ALIGNMENT
MULTIPLE ALIGNMENT
PhylogeneticAnalysis
HomologyModeling
Advanced Database Searches,Patterns, Motifs, Promoters

Why do we need multiplealignments?
– In order to reveal the relationship between agroup of sequences. (homology)
– In order to characterize protein families -to identify conserved regions of a specificfamily, and locate its variable regions.
– In order to retrieve information about domainsor active sites. Similar regions may indicatesimilar functions. (e.g promoter regions in DNA)

Why do we need multiplealignments?
– To plan point mutations based upon highlightedregions of multiple alignments, either verysimilar or very different.
– To build a family profile for use in a moresensitive database scan. Such a search canfind new (more distant) members of thefamily.
– Determination of the consensus sequence ofseveral aligned sequences, for further analysis.

Why do we need multiplealignments?
– Planning probes in order to fish outdistant members of a protein family.
– Multiple alignments are used for proteinmodeling programs.
– To help prediction of secondary andtertiary structures of new sequences.

Why do we need multiplealignments?
Multiple alignments are input forconstructing phylogenetic trees.

The Computational Challenge ofMSA
Finding optimal alignment between a groupof sequences that include: matches,mismatches and gaps is very difficult.
For Pairwise Alignments, DynamicProgramming methods are used, but theyare impractical with multiple alignments(too many calculations, too much CPU time).

The Computational Challenge ofMSA
The difficulties with aligning a group ofsequences varies with the degree ofsimilarity between the sequences.
High degree of variation of the comparedsequences – many alignments possible.
Many possibilities – very hard to find“optimal” alignment.

The Computational Challenge ofMSA
Approximate methods are used insteadof Dynamic programming methods.
Another computational challenge isplacement and scoring of gaps in thealigned sequences.

Approximate Methods
1) Progressive global alignment: Starts with the most similar sequences,
and builds the alignment by adding therest of the sequences.
2) Iterative methods: Starts by making initial alignments of
small groups of sequences, and thenrevises the alignment for better results.

Approximate Methods
3) Alignment based on small conserveddomains (or patterns), found in the sameorder within the aligned sequences.
4) Alignment based on statistical orprobabilistic models of the sequences.

Various Multiple Alignment algorithms:
ClustalwT-CoffeeMuscle
PRALINEMultAlignDiAlignMSA
BLOCKSHMMER
SAMPileup

Global multiple alignment methods
Clustalwhttp://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html MSA (limited to a small number of sequences)http://www.ibc.wustl.edu//ibc/msa.html PRALINEhttp://ibivu.cs.vu.nl/programs/pralinewww/

Iterative multiple alignment methods
DIALIGNhttp://www.gsf.de/biodv/dialign.html
MultAlignhttp://protein.toulouse.inra.fr/multalign.html

Local multiple alignment methods
BLOCKShttp://blocks.fhcrc.org/blocks/
HMMERhttp://hmmer.wustl.edu/
MEMEhttp://meme.sdsc.edu/meme/website/intro.html
SAMhttp://www.cse.ucsc.edu/research/compbio/sam.html

Multiple Alignment
The most practical and widely usedmethod for multiple alignment isprogressive global alignment.
How does it work?

Steps to create multiplealignment
Pairwise comparisons of all sequences Perform cluster analysis on the pairwise data to
generate a hierarchy for alignment. This may bein the form of a binary tree or simple orderingtree.
Start with the most related (similar) sequences,then the next most similar pair and so on.Once an alignment of two sequences has beenmade, then this is fixed.

Steps in Multiple Alignment

Steps to create multiplealignment
After the clustering is done, relationshipsbetween the sequences are modeled by atree.
If the program (e.g clustalw) builds anevolutionary tree, then the sequences representthe outer branches of the evolutionary tree.
Inner branches represent dissimilarities of thesequences at the outer branches.

Tips in choosing your sequencesGeneral considerations
Sequences taken directly from the database cancontain irrelevant data, (e.g: multiple genes,fragments of different lengths). Check yoursequences and use only the relevant parts of themfor the alignment.
If you align your own sequences, edit them andremove the unrelated data before alignment.
Try to use sequences with more or less the samelength for alignment.

Tips in choosing your sequencesGeneral considerations
For most uses of multiple alignments:
– The more sequences to align the better.– Don’t include similar (>80%) sequences.– Sub-groups should be pre-aligned
separately, and one member of eachsubgroup should be included in the finalmultiple alignment.

What you need to know aboutmultiple alignment programs
Almost all programs will align whatever sequencesthe user gives as input.
They will always return an alignment, even if thesequences are completely unrelated. The biologythinking should be done by you.
Most programs will insert gaps. However, ifinserted they are there to stay.
You need to check how the program you use treatsend gaps.

ClustalW- for multiple alignment
ClustalW is a global multiple alignment program forDNA or protein.
ClustalW was produced by Julie D. Thompson,Toby Gibson of EMBL, Germany and DesmondHiggins of EBI, Cambridge, UK.
ClustalW is cited: Improving the sensitivity ofprogressive multiple sequence alignment throughsequence weighting, positions-specific gappenalties and weight matrix choice. Nucleic AcidsResearch, 22:4673-4680.

ClustalW- for multiple alignment
ClustalW can create multiple alignments,manipulate existing alignments andcreate phylogenic trees.
The initial alignment can be done by 2methods:- slow/accurate- fast/approximate

ClustalW alignment Method
ClustalW alignment algorithm consists of 3steps:
1) Pairwise Alignments are performedbetween all sequences in the comparedgroup. Alignment scores are used to builda distance matrix. In calculating thedistance matrix, the program takes intoaccount the divergence of the sequences.

ClustalW alignment Method
2) A guide (phylogenetic) tree is created fromthe distance matrix using the Neighbour-Joining method.This guide tree has branches of differentlengths. Their length is proportional to theestimated divergence along each branch.

ClustalW alignment Method
3) Progressive alignment of the sequences isdone, following the branch order of theguide tree. The sequences are aligned fromthe tips to the root.
The alignment of the sequences is guided bythe phylogenetic relationships indicated bythe tree.

ClustalW alignment Method
At each stage of the progressive alignmentfull dynamic programming is applied, anduses a scoring matrix.
The program calculates sequence weightsfrom the guide tree, and chooses thescoring matrix accordingly (according tothe divergence of the comparedsequences).

Clustalw calculates the geneticdistances as follows:
# mismatches in the alignment
# matches in the alignment
Positions opposite a gap are not scored.

ClustalW alignment Method
Clustalw weights the sequences accordingto the distance of each sequence from theroot.
Clustalw calculates gaps in a novel way,designed to place them between conserveddomains.
Clustalw penalizes for gap opening andextension.

Running ClustalW
The input file for ClustalW is a single filecontaining all of the sequences for alignment.
It accepts the following formats:NBRF/PIR, EMBL/SwissProt, Pearson (Fasta),GDE, Clustal, GCG/MSF, RSF.

Using ClustalW****** MULTIPLE ALIGNMENT MENU ****** 1. Do complete multiple alignment now (Slow/Accurate) 2. Produce guide tree file only 3. Do alignment using old guide tree file
4. Toggle Slow/Fast pairwise alignments = SLOW
5. Pairwise alignment parameters 6. Multiple alignment parameters
7. Reset gaps between alignments? = OFF 8. Toggle screen display = ON 9. Output format options
S. Execute a system command H. HELP or press [RETURN] to go back to main menu
Your choice:

M_MELB VADYAEFQKNRHDQDATKRKLMEIANYVDKFYRSLNIR----IALVGLEVWTHGDKCEVSM_MELA VADNREFQRQGKDLEKVKQRLIEIANHVDKFYRPLNIR----IVLVGVEVWNDIDKCSISM_MELG VVDKERYDMMGRNQTAVREEMIRLANYLDSMYIMLNIR----IVLVGLEIWTDRNPINIIM_ADAM28 VLDNGEFKKYNKNLAEIRKIVLEMANYINMLYNKLDAH----VALVGVEIWTDGDKIKITM_ADAM10 QTDHLFFKYYG-TREAVIAQISSHVKAIDTIYQTTDFSGIRNISFMVKRIRINTTSDEKD * :. : .: :: :* : : :: .: . .
M_MELB ENPYSTLWSFLSWRR-KLLAQKSHDN---AQLITGRSFQGTTIGLAPLMAMCSVY-----M_MELA QDPFTRLHEFLDWRKIKLLPRKSHDN---AQLISGVYFQGTTIGMAPIMSMCTAE-----M_MELG GGAGDVLGNFVQWREKFLITRRRHDS---AQLVLKKGFGG-TAGMAFVGTVCSRS-----M_ADAM28 PDANTTLENFSKWRGNDLLKRKHHDI---AQLISSTDFSGSTVGLAFMSSMCSPY-----M_ADAM10 PTNPFRFPNIGVEKFLELNSEQNHDDYCLAYVFTDRDFDDGVLGLAWVGAPSGSSGGICE : .: : * .: ** * :. * . . *:* : : .
CLUSTAL W (1.82) multiple sequence alignment

ClustalW optionsYour choice: 5 ********* PAIRWISE ALIGNMENT PARAMETERS ********* Slow/Accurate alignments:
1. Gap Open Penalty :15.00 2. Gap Extension Penalty :6.66 3. Protein weight matrix :BLOSUM30 4. DNA weight matrix :IUB
Fast/Approximate alignments:
5. Gap penalty :5 6. K-tuple (word) size :2 7. No. of top diagonals :4 8. Window size :4
9. Toggle Slow/Fast pairwise alignments = SLOW
H. HELPEnter number (or [RETURN] to exit):

ClustalW optionsYour choice: 6
********* MULTIPLE ALIGNMENT PARAMETERS *********
1. Gap Opening Penalty :15.00 2. Gap Extension Penalty :6.66 3. Delay divergent sequences :40 %
4. DNA Transitions Weight :0.50
5. Protein weight matrix :BLOSUM series 6. DNA weight matrix :IUB 7. Use negative matrix :OFF
8. Protein Gap Parameters
H. HELP
Enter number (or [RETURN] to exit):

ClustalX - Multiple SequenceAlignment Program
ClustalX provides a window-based user interfaceto the ClustalW program.
It uses the Vibrant multi-platform userinterface development library, developed by theNational Center for Biotechnology Information(Bldg 38A, NIH 8600 Rockville Pike,Bethesda,MD 20894) as part of their NCBI SOFTWAREDEVELOPEMENT TOOLKIT.

ClustalX

ClustalX

ClustalX

ClustalX
ClustalX

ClustalX

Displaying a multiple alignmentin GCG
There are several programs to display the multiplealignment prettily.
The Pretty program prints sequences with theircolumns aligned and can display a consensus for thealignment, allowing you to look at relationshipsamong the sequences.
The PrettyBox program displays the alignmentgraphically with the conserved regions of thealignment as shaded boxes. The output is inPostscript format.

Example of PrettyBox Output

PrettyBox on Clustal output
You can also run Prettybox on the output ofClustalW. It requires a few steps:
1) Before you run the alignment:Choose output format options, and chooseGCG/MSF
2) Before you run prettybox:Change all of the weights to 1, or it will colorthe picture incorrectly.

Problems with Progressive alignments
In progressive alignment the ultimate multiplealignment is dependent on the initial pairwisealignments. The first sequences to be aligned are the most
similar (closely related on the tree). If the initial alignments are good, with very
few errors, the ultimate multiple alignment willbe good.
However, if the sequences aligned are distantlyrelated, much more errors can be madeaffecting the final alignment

Problems with progessive alignments
Figure from JMB Vol 302, pp205-217, 2000


Problems with Progressive alignments
Another problem with progressivealignment is that the ultimatemultiple alignment is dependent onchoosing the correct scoringmatrices, and the correct gappenalty.

T-Coffee References
T-Coffee: A novel method for fast and accuratemultiple sequence alignment. C.Notredame, D.Higgins, J. Heringa, Journal of Molecular Biology,Vol 302, pp205-217, 2000
COFFEE: A New Objective Function For Multiple SequenceAlignnent. C. Notredame, L. Holme and D.G. Higgins,Bioinformatics, Vol 14 (5) 407-422, 1998
T-Coffee in the WEBhttp://igs-server.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi

T-Coffee first step
Creating the primary librarySet of all pairwise alignments between all
sequences in the dataset Global alignments of all against all using
CLUSTALW Local alignments of all against all using LALIGN In the library – each alignment = a list of
pairwise residue matches

Slides taken from http://www.isrec.isb-sib.ch/DEA/module5/Course_Cedric/maln3.pdf

T-Coffee second step
After the primary library was created, theprogram assigns a WEIGHT to each pair ofaligned residues in the library
For each set of sequences – 2 primary librariesare computed along with their weight: Global +Local alignments
The library becomes a list of weighted pairwisealigned scores.

T-Coffee third step
Combination of the Global and Local weights toone Primary Library
Checking the weighted pairs: If the pair of seqs is duplicated (appears) in
the 2 libraries, it is merged into a single entrywith weight equal to the sum of the 2 librariesweights
Otherwise a new entry of this pair is created

T-Coffee fourth step
Library Extension Is the process where the program assigns a
weight for each pair of aligned residues inthe Primary Library.
This weight reflects the degree of a pairconsistency in all the seqs in the dataset
The Extension is done by the TripletApproach

The Triplet ApproachSlides taken fromhttp://www.isrec.isb-sib.ch/DEA/module5/Course_Cedric/maln3.pdf

Figure from JMB Vol 302, pp205-217, 2000

Figure from JMB Vol 302, pp205-217, 2000

Figure from JMB Vol 302, pp205-217, 2000

The complete extension of the Primary Library(check all triplets of the dataset) will assign aweight for each pair of residues that is a sum of allweights gathered for all the triplets that containthe pair.
The more sequences supporting a pair alignment – thehigher is its weight
By using pair weights specific to the dataset insteadof matrix scores the multiple alignment is muchmore powerful

T-Coffee fifth step Progressive Alignment of the extended
library set is done by dynamicprogramming algorithm to achieve thefinal multiple alignment of the dataset.

T-Coffee Summary
Good for a limited number of sequences Takes long time to run – not good for a
large dataset Does not deal well (misaligns) sequences
which vary a lot in their length

Muscle – Fast tool for MultipleAlignment
Muscle (Multiple Sequence Comparison by log-expectation) is cited:
Edgar, Robert C. (2004), MUSCLE: multiple sequencealignment with high accuracy and high throughput,Nucleic Acids Research 32(5), 1792-97.
Muscle on the WEBhttp://phylogenomics.berkeley.edu/muscle/muscle_form.htmlhttp://www.drive5.com/muscle/


Muscle first stageDraft Alignment
Building the Guide Tree (“k-mer clustering”): Calculates number of matching “words”, and
calculates distances without doing alignments,builds a distance matrix, and then a tree(UPGMA)
Progressive alignmentFollows the guide tree 1, from the tips to theroot, and at each node aligns either 2sequences, sequence/profile or profile/profile

Unaligned seqs
Calculate K-mers
K-mer distance matrix
UPGMA
Tree 1
Progressive Alignment
First Multiple Alignment

Muscle Second stageImproved alignment
Optimization (“tree refinement”):Using the multiple alignment as a base, compute
pairwise identities for each of the sequencepairs.
Build a distance matrix 2 (Kimura distance)Build a new tree (UPGMA).Progressive alignment is done following the guide
tree 2, resulting in Multiple Alignmnet 2.

First Multiple Alignment
Compute pairwise %
Kimura distance matrix
UPGMA
Tree 2
ProgressiveAlignment
Second MultipleAlignment

Muscle Third stageMultiple Alignment Refinement
This tree is divided into 2 subtrees. (takingan edge off the tree to create the twogroups)
The sequences in the subtree are used tobuild a multiple alignment and then aprofile.
By realigning the 2 profiles a new multiplealignment is built.

Muscle Third stageMultiple Alignment Refinement
If this new alignment improves the score, itis kept. Otherwise it is discarded.
This is done for all the edges in the tree(from the edges to the root.)
The whole step is iterated untilconvergence, or a user defined limit

Second MultipleAlignment Delete
an edge
subtree1
subtree 2
Realignprofiles
Compute subtree profile
Third Multiple Alignment
Better SP? Save
Not better? Delete

Muscle Summary Fast Works with a large group of
sequences Sequence length is not important

Bottom Line
Speed: Muscle > ClustalW >>T-Coffee
Accuracy (Generally): Muscle >= T-Coffee > ClustalW
Accuracy depends on the individualsequence family, and for some the order isdifferent…so use more than one algorithm!






























