Embed Size (px)
Citation preview

1
MRMPROBS tutorial
Edited in 20161116
Introduction
MRMPROBS is launched as a universal program for targeted metabolomics using not only multiple
reaction monitoring (MRM)- or selected reaction monitoring (SRM) but also SCAN and data
independent MSMS acquisition (DIA) data Originally the previous MRMPROBS program was
developed to deal with large scale MRM assayrsquos data sets monitoring 500-1000 small molecules in a
single run simultaneously The program provided 1) a user-friendly graphical user interface (GUI) for
data curation and 2) an objective evaluation system of small molecule identifications Here it was
expanded for DIA-MS data (like SWATH-MS) and for SCAN data (like GCMS and LCMS)
All data-processing workflow from data import to statistical analysis is supported This
tutorial will introduce the workflow for 1) MRM data 2) SWATH-MS (DIA) data and 3) GCMS
data for targeted metabolomics In this MRMPROBS project your feedback would be appreciated to
improve the identification and quantification systems as well as the user interface
Hiroshi Tsugawa
RIKEN Center for Sustainable Resource Science
hiroshitsugawarikenjp
MRMPROBS screenshot
2
Table of contents
Software environments 4
Required software programs and files 5
Project type and condition 6
ABF file conversion 7
Downloading the ABF converter 7
Check the conditions for file conversion 8
File conversion 10
Reference file format 11
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf) 11
Reference library for Project type 2 MRMPROBS key index = Function (mzML) 14
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf) 16
Reference format for DIA-MS data 16
Dictionary file for DIA-MS data processing 18
Reference format for GCMS and LCMS data 19
Starting MRMPROBS 20
Summary for MRM demonstration data sets 20
Starting up your project 21
Importing Abf files 22
Parameter 23
MRMPROBS viewer 25
Mouse operation in the chromatogram viewer 25
Library editor [optional] 27
Tool button 28
Tab 29
Button 30
List Box 31
Details on the MRMPROBS function 32
File menu 32
Data reprocessing 33
Statistical analysis 34
Missing value methods 35
Normalization 36
Window menu 38
View menu 39
Option menu 40
Export menu 41
3
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file 42
Appendix B Third option of MRMPROBS via mzML file 46
4
Software environments
Microsoft Windows 7 or later
NET Framework 40 or later
5
Required software programs and files
Reifycs Analysis Base File Converter (ABF file converter)
Download link httpwwwreifycscomAbfConverterindexhtml
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Demo files and the reference library (tab-delimited text file)
Example httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
MRMPROBS can import Analysis Base Framework (ABF) format data MRMPROBS extracts
chromatogram data together with the reference library including the name of the target metabolite its
retention-time and amplitude information and precursor mz and product mz The supported formats
for ABF conversion are Shimadzu Inc (LCD) Agilent Technologies (D) AB Sciex (WIFF) Waters
(RAW) and Thermo Fisher Scientific (RAW) MRMPROBS is also acceptable to a common data
format mzML converted by an open source file translator ProteoWizard The information is
described in Appendix B
6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

2
Table of contents
Software environments 4
Required software programs and files 5
Project type and condition 6
ABF file conversion 7
Downloading the ABF converter 7
Check the conditions for file conversion 8
File conversion 10
Reference file format 11
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf) 11
Reference library for Project type 2 MRMPROBS key index = Function (mzML) 14
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf) 16
Reference format for DIA-MS data 16
Dictionary file for DIA-MS data processing 18
Reference format for GCMS and LCMS data 19
Starting MRMPROBS 20
Summary for MRM demonstration data sets 20
Starting up your project 21
Importing Abf files 22
Parameter 23
MRMPROBS viewer 25
Mouse operation in the chromatogram viewer 25
Library editor [optional] 27
Tool button 28
Tab 29
Button 30
List Box 31
Details on the MRMPROBS function 32
File menu 32
Data reprocessing 33
Statistical analysis 34
Missing value methods 35
Normalization 36
Window menu 38
View menu 39
Option menu 40
Export menu 41
3
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file 42
Appendix B Third option of MRMPROBS via mzML file 46
4
Software environments
Microsoft Windows 7 or later
NET Framework 40 or later
5
Required software programs and files
Reifycs Analysis Base File Converter (ABF file converter)
Download link httpwwwreifycscomAbfConverterindexhtml
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Demo files and the reference library (tab-delimited text file)
Example httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
MRMPROBS can import Analysis Base Framework (ABF) format data MRMPROBS extracts
chromatogram data together with the reference library including the name of the target metabolite its
retention-time and amplitude information and precursor mz and product mz The supported formats
for ABF conversion are Shimadzu Inc (LCD) Agilent Technologies (D) AB Sciex (WIFF) Waters
(RAW) and Thermo Fisher Scientific (RAW) MRMPROBS is also acceptable to a common data
format mzML converted by an open source file translator ProteoWizard The information is
described in Appendix B
6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

3
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file 42
Appendix B Third option of MRMPROBS via mzML file 46
4
Software environments
Microsoft Windows 7 or later
NET Framework 40 or later
5
Required software programs and files
Reifycs Analysis Base File Converter (ABF file converter)
Download link httpwwwreifycscomAbfConverterindexhtml
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Demo files and the reference library (tab-delimited text file)
Example httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
MRMPROBS can import Analysis Base Framework (ABF) format data MRMPROBS extracts
chromatogram data together with the reference library including the name of the target metabolite its
retention-time and amplitude information and precursor mz and product mz The supported formats
for ABF conversion are Shimadzu Inc (LCD) Agilent Technologies (D) AB Sciex (WIFF) Waters
(RAW) and Thermo Fisher Scientific (RAW) MRMPROBS is also acceptable to a common data
format mzML converted by an open source file translator ProteoWizard The information is
described in Appendix B
6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

4
Software environments
Microsoft Windows 7 or later
NET Framework 40 or later
5
Required software programs and files
Reifycs Analysis Base File Converter (ABF file converter)
Download link httpwwwreifycscomAbfConverterindexhtml
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Demo files and the reference library (tab-delimited text file)
Example httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
MRMPROBS can import Analysis Base Framework (ABF) format data MRMPROBS extracts
chromatogram data together with the reference library including the name of the target metabolite its
retention-time and amplitude information and precursor mz and product mz The supported formats
for ABF conversion are Shimadzu Inc (LCD) Agilent Technologies (D) AB Sciex (WIFF) Waters
(RAW) and Thermo Fisher Scientific (RAW) MRMPROBS is also acceptable to a common data
format mzML converted by an open source file translator ProteoWizard The information is
described in Appendix B
6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

5
Required software programs and files
Reifycs Analysis Base File Converter (ABF file converter)
Download link httpwwwreifycscomAbfConverterindexhtml
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Demo files and the reference library (tab-delimited text file)
Example httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
MRMPROBS can import Analysis Base Framework (ABF) format data MRMPROBS extracts
chromatogram data together with the reference library including the name of the target metabolite its
retention-time and amplitude information and precursor mz and product mz The supported formats
for ABF conversion are Shimadzu Inc (LCD) Agilent Technologies (D) AB Sciex (WIFF) Waters
(RAW) and Thermo Fisher Scientific (RAW) MRMPROBS is also acceptable to a common data
format mzML converted by an open source file translator ProteoWizard The information is
described in Appendix B
6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

6
Project type and condition
1 MRMPROBS key index = metabolite name (abf)
Use half-width alphanumeric symbols for compound names in MRM method setting
Do not use the same compound name at different transition (precursor-product) sets
2 MRMPROBS key index = Function (mzML)
lsquoFunction IDrsquo is utilized to extract the chromatogram data Users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format (see Appendix B)
The above two projects are for MRM data sets
3 MRMPROBS key index = SCAN or DIA-MS (abf)
MRMPROBS can import GCMS LCMS and LC-DIA-MS (like SWATH) data sets
Prepare the reference library (see Appendix C) to extract the certain retention time range and mz
values
Use half-width alphanumeric symbols for compound names
4 MRM-DIFF (abf mzML)
See httpprimepscrikenjpMetabolomics_SoftwareMRM-DIFFindexhtml
7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

7
ABF file conversion
Downloading the ABF converter
1 Go to httpwwwreifycscomAbfConverterindexhtml
2 Check the requirements and license terms and download the converter
File converter is freely available
8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

8
Check the conditions for file conversion
To convert files of some MS vendors including Bruker LECO Shimadzu Thermo and Waters the
specific data access library needs to be installed on your PC
Also see FAQ for ABF converter
httpprimepscrikenjpMetabolomics_SoftwareMS-DIALindex3html
Summary of PC condition required for file conversion
Vendor Formats Required
Agilent D None but the files from Chemstation should be
converted to netCDF
Bruker D CompassXtract
LECO PEG All PEG files should be first converted to netCDF
(AIA)
Sciex WIFF None
Shimadzu for
GCMS QGD GCMS solution
Shimadzu for
LCMS LCD LCMS solutions
Thermo RAW MSFileReader
Waters RAW MassLynx Raw Data Reader Interface Library
netCDF CDF Microsoft Visual J 20
FAQ
Agilent Almost all of Agilentrsquos raw data (D) can readily be converted without any additional
steps However the only exception is GCMS data sets obtained from ChemStation which
cannot be directly converted to ABF Therefore you have to convert your files into netCDF
(AIA) in ChemStation Then convert your AIA files into ABF using our file converter
Bruker Go to
httpswwwbrukercomservicesupport-upgradessoftware-downloadsmass-spectrometryhtml
You have to register first and obtain the installer Finally download and install the version
compatible with your operating system environment (32-bit or 64-bit)
LECO All of your data has to be converted to netCDF (AIA) first Then convert them into ABF
using our file converter
Shimadzu The compound table of the lcd file must include the MRM conditions of the ldquolcmrdquo
file How to set this is described in Appendix A-1 In case that you had a direct conversion of
raw data please convert your data into the common data format (netCDF or mzML) For
GCMS convert your data into netCDF using GCMS solution For LC-ITTOFMS convert your
9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

9
data into mzML using ProteoWizard (httpproteowizardsourceforgenetindexshtml) Then
convert them into ABF
Thermo The following link explains how to install MSFileReader
httpfieldsscrippsedurawconv For GCMS data you may have to convert your data into
netCDF Then convert them into ABF using our converter We validated the direct conversion of
GC-QExactive raw data to ABF but some GCMS data (DSQ etc) had to be converted to
netCDF first
Waters 1 Download MassLyncs Raw Data Reader Interface Library
(httpwwwwaterscomwaterssupportListhtmcid=511442ampfilter=documenttype|DWNLamploc
ale=en_US) 2 Unzip the archive file `watersrawsdkredistzip` and copy `MassLynxRawdll`
(64-bit) to `ABFCvtSvrWtrRw` folder in the ABF converter Note that 32ndashbit environments are
not supported yet for file conversion
NetCDF When you get an error about the J dependency problem download and install the
Microsoft Visual J 20 library at
httpswwwmicrosoftcomen-usdownloaddetailsaspxid=15468
10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

10
File conversion
1 Start ldquoAnalysisBaseFileConverterexerdquo
2 Drag amp drop MS vendor files into this program
3 Click ldquoConvertrdquo
4 The ABF files are generated in the same directory as the raw data files
11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

11
Reference file format
Reference library for Project type 1 MRMPROBS key index = metabolite name (abf)
Five items are required as tab-delimited format The header names are flexible but the item order
should be kept
1 column Compound namea
2 column Precursor mz (accurate mz information is rounded into nominal mz information)
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []b
Notes aWhen you choose project 1 of MRMPROBS the name must be identical to the compound name in
the instrument setting window The compound name MUST be written by half-width alphanumeric
symbols
bAbout the amplitude ratio format
lsquo100rsquo value is recognized as the quantitative ion for targeted compounds and the other values are
recognized as the diagnostic ions for identification of targeted compounds
12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

12
The quantitative ion must be set to each compound record and the current program does not
accept multiple quantitative ions for one compound it means that lsquo100rsquo value can be used only
once The below is the examples for reference records
Example only one transition for one metabolite
Thymine 125 4205 558 100
Example multiple transitions for one metabolite
G6P 2589 9705 921 100
G6P 2589 7905 921 301
G6P 2589 19915 921 55
Again MRMPROBS does not quantify the metabolite by the total ion chromatogram Instead
one quantitative transition is used for peak quantification (we call it the ldquotargetrdquo transition)
Enter the ratio [] against 100 as the amplitude for the qualifier transition (or the confirmation
transition) This information is used for peak identification (we call it the ldquoqualifierrdquo transition)
Note 1 You can edit the reference library and update its information in MRMPROBS However an
empty value cannot be accepted when the library is imported If you do not know the suitable
retention time and amplitude information for the metabolites enter arbitrary values for the
metabolites
Note 2 Users do not have to include all metabolite information you entered in the MS instrument
13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

13
Note 3 Sometimes the tab-delimited file exported from Microsoft Excel includes unexpected hidden
trailing columns These unexpected columns after the lsquoRatiorsquo column cannot be handled by
MRMPROBS You can inspect the exported file by selecting a few rows (see below) If there are
selected characters after the last column (Ratio) edit the file in Excel to delete these columns and
re-export it again
Good example (no unexpected column)
Bad example (there are unexpected columns)
14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

14
Reference library for Project type 2 MRMPROBS key index = Function (mzML)
Six items are required as tab-delimited text format The header names are flexible but the item order
should be followed (Here in order easily to see the library the reference was described in the
Microsoft Excel)
1 column Compound namea
2 column Function IDb
3 column Precursor mz (accurate mz information is rounded into nominal mz information)
4 column Product mz
5 column Retention time [min]
6 column Amplitude ratios []c
Notes aWhen you use project 2 of MRMPROBS the name doesnrsquot have to be identical to the compound
name in the instrument setting The compound name MUST be written by half-width alphanumeric
symbols
bThe function ID is the most important ID to use this option In the mzML data there is a markup
indicating a lsquoFunction IDrsquo which is unambiguous key to contact to the specific MRM chromatogram
for the retention time range the precursor ion and product ion In order to easily to see the
relationship between the function ID and the MRM information use the SeeMS program which can
be downloaded at ProteoWizard webpage httpproteowizardsourceforgenet
1 Open SeeMS
15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

15
2 Select a mzML file
To find the identical function ID in your data use the Microsoft Excel sorting function and your
experiment condition file In the most of case the proteowizard is sorting the functions following the
order to 1 Precursor Ion 2 Product Ion 3 Retention time starting point
cAbout the amplitude ratio format
See the section of Reference library for Project type 1 MRMPROBS key index = metabolite name
(abf)
16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

16
Reference library for Project type 3 MRMPROBS key index = SCAN or DIA-MS (abf)
Reference format for DIA-MS data
Users can utilize MRMPROBS software for scan type data such as GCMS LCMS and LC-data
independent MSMS (DIA-MS) The below figure is the reference library for DIA-MS data Here
our objective is to utilized DIA-MS data as MRM (what we call DIA-MRM for example
SWATH-MRM for SCIEX machine) This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
1 column Compound name
2 column Precursor mz
3 column Product mz
4 column Retention time [min]
5 column Amplitude ratios []
6 column RT begin start time to draw the chromatogram
7 column RT end end time to draw the chromatogram
8 column MS1 tolerance mass accuracy for survey scan MS data
17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

17
9 column MS2 tolerance mass accuracy for MSMS spectra
10 column MS level put 1 for survey scan MS data (MS1) and put 2 for MSMS
11 column Class itrsquos used for the MRMPROBS viewer to filter out the chromatograms Set
lsquoNArsquo or something if not interest
Below is the description of the lsquobridgersquo from MS-DIAL to MRMPROBS
18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

18
Dictionary file for DIA-MS data processing
The dictionary file should contain MS1 scan range and precursor window in combination with its
experimental ID
In the case of SWATH data-independent analysis the experiment file can be made at PeakView
(Show-gtsample information) Do not change the column orders The word ldquoSCANrdquo should be kept
19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

19
Reference format for GCMS and LCMS data
MRMPROBS is improved to utilize single MS data such as GCMS and LCMS and the below
figure is the reference library for GCMS data The trick to import the single MS data sets is 1) to
assign the same values for product mz and MS2 tolerance as precursor mz and MS1 tolerance
respectively and 2) to assign lsquo1rsquo as MS level for all queries
This library can be easily exported by MS-DIAL software
httpprimepscrikenjpMetabolomics_SoftwareMS-DIAL
20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

20
Starting MRMPROBS
Summary for MRM demonstration data sets
1 Starting up your project
2 Importing Abf files
3 Setting parameters
4 Running the software (1-2 min sample)
The tutorial uses 40 demonstration files and the reference library which are downloadable from the
above link The common measurement conditions of the demonstration files were as follows
Liquid chromatography total 25 min run per sample with CELI L-column2 ODC (150 mmtimes21 mm
3 m)
Mass spectrometer MRM method with negative ion mode
Target metabolite number 60
Total transitions 166
The detail of experimental conditions is downloadable at the MRM Database section (Ion-pair
LC-QqQMS)
httpprimepscrikenjpMetabolomics_SoftwareMrmDatabaseindexhtml
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter
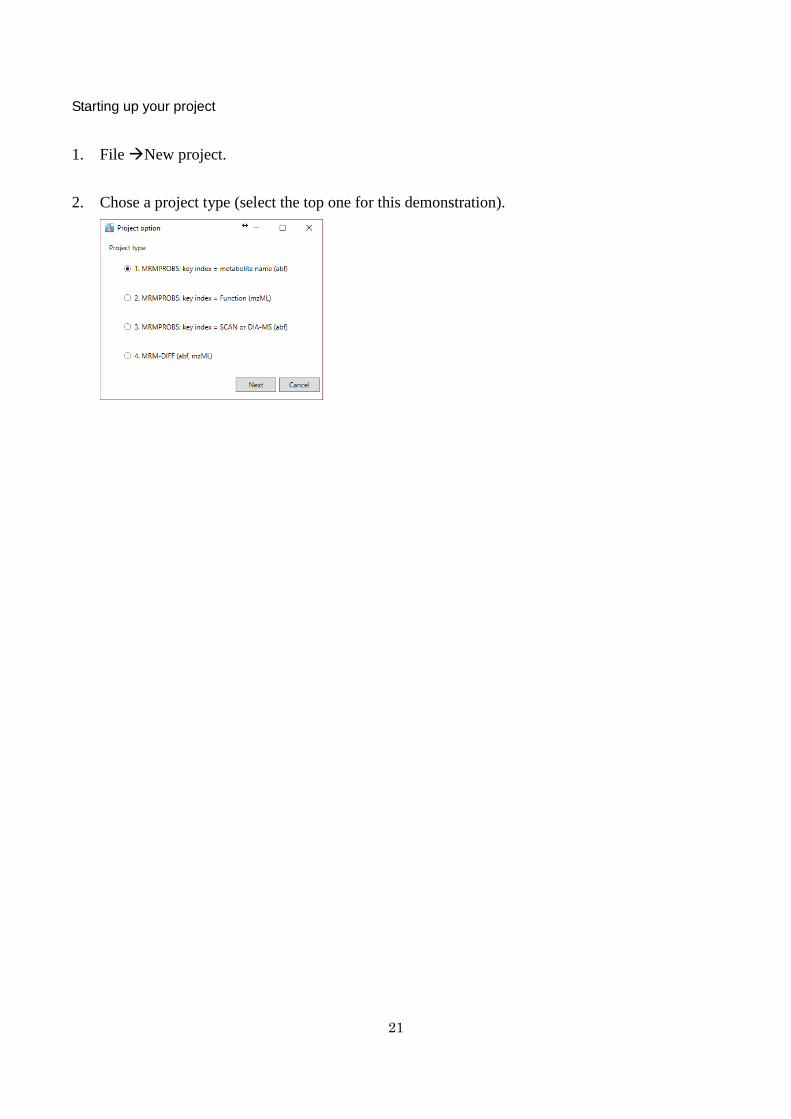
21
Starting up your project
1 File New project
2 Chose a project type (select the top one for this demonstration)
22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

22
Importing Abf files
Note
We recommend that the project folder be made for each batch experiment In the
MRMPROBS project two folders (raw processed) and one file (mth) are generated They
should be included in the same directory
The file name should be entered in half-width alphanumeric symbols
Select the file type of each file from ldquoSamplerdquo ldquoStandardrdquo ldquoBlankrdquo and QCrdquo QC is
required for the LOESS-based normalization method
Class ID is used for the color labels
Analytical order and class ID can be changed after data processing
23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

23
Parameter
Select lsquoExampleLibrarytxtrsquo and set the above parameters for this demonstration
Note
If you want to edit or update the retention time and amplitudes of metabolites in your
reference library from an example file such as a QC or standard mixture check ldquocreate new
library and choose the file name to which you want to refer In this demonstration
lsquo20_STD10uM_02rsquo is selected
[Recommended]
Peak detection
Smoothing method linear weighted moving average
Smoothing level 1-2
Minimum peak width 3-5
Minimum peak height 50-100
Peak identification
Retention time tolerance As long as the reverse phase or hydrophilic interaction
chromatography LC are used 01-02 min is recommended
Amplitude tolerance 15
24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

24
Minimum posterior Decide the minimum probability for peak identification MRMPROBS
calculates a probability for a peak ie ldquoprobability of true target metabolite given the calculated
scoresrdquo The detected peak less than this criterion is recognized as a false peak The
recommended value is 50-70
Note The first data processing including file import peak detection and peak identification requires
5-20 seconds (depending on machine specifications) per file
25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

25
MRMPROBS viewer
Mouse operation in the chromatogram viewer
Main window
View mode
① Chromatogram window drag holding left click chromatogram scroll drag holding right click
chromatogram zoom
1
3
4
2
26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

26
② Detected window left double-click the reverse triangle change the true peak right
double-click anywhere un-checked detected peaks
③ Retention time window drag holding right click warping on retention time range
④ Intensity window drag holding right click warping on intensity range
Edit mode
① Left click and drag on the peak edge [red square] change the location of the peak edge
② Right click and drag detect new peak
27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

27
Library editor [optional]
You can update the retention time and amplitude ratio of each metabolite by means of the QC
sample file or the standard mixture file You can also change the target transition for peak
quantification
The default identification is based on the highest peak in the EIC chromatogram If you need to
change the true peak double click the reverse triangle
If you change the target transition select the target MRM transition from the ldquoTarget MRMrdquo
combo box
The right-column information including the compound name target MRM RT and amplitude
ratios are generated by double-clicking the compound name in the list box on the left or by
re-selecting the true peak in the EIC chromatogram viewer
If the object metabolite is not detected click the ldquoViewrdquo button at the top left and switch to
ldquoEditrdquo mode In the ldquoEditrdquo mode hold the right mouse click and drag the peak area you want to
detect
If you want to change the retention time or amplitude information manually just type what you
want in the textbox and click the ldquoUpdaterdquo button on the top right
If you want to save the updated library click the ldquoSaverdquo button on the top left side
Lastly after editing the reference library click the ldquoFinishrdquo button on the top left side
Note The details and the operation method for chromatogram viewer are described later
28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

28
Tool button
File start new project open existing project save as a project and save the project
Data processing for data re-processing per file per metabolite or in all data sets
Statistical analysis data normalization and statistical analysis
Window tile setting of chromatogram viewer
View sorting of chromatogram viewer
Option re-define class ID and analytical order choose the internal standard decide ldquoincluderdquo or
ldquoexcluderdquo data for statistical analysis
Identification non-meaningful in MRMPROBS project
Export The result is exported in tab-delimited text format
Help show version information
29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

29
Tab
Chromatogram All data manipulation tasks are performed here
Raw data matrix The peak quantification value of each file and each metabolite is shown here
Processed data matrix The normalized value of each file and each metabolite is shown here
Statistical result The result of statistical analysis is shown here
Raw data matrix
30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

30
Button
Reset Reset the display range of chromatograms
View If you push this ldquoViewrdquo button the chromatogram viewer is changed to ldquoEditrdquo mode In
the ldquoEditrdquo mode you can modify the peak edge and detect new peaks manually
None The properties of detected peaks are shown in this ComboBox You can confirm the total
score (probability) rt similarity area value and reference RT in the chromatogram viewer
Height You can set the quantification mode The default is set by peak height Instead you can
change it to area mode By using the ldquoAllrdquo option the quantification mode is reflected =
implemented in all files and all metabolites
Processed You can see raw chromatograms as well as smoothed chromatograms
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter
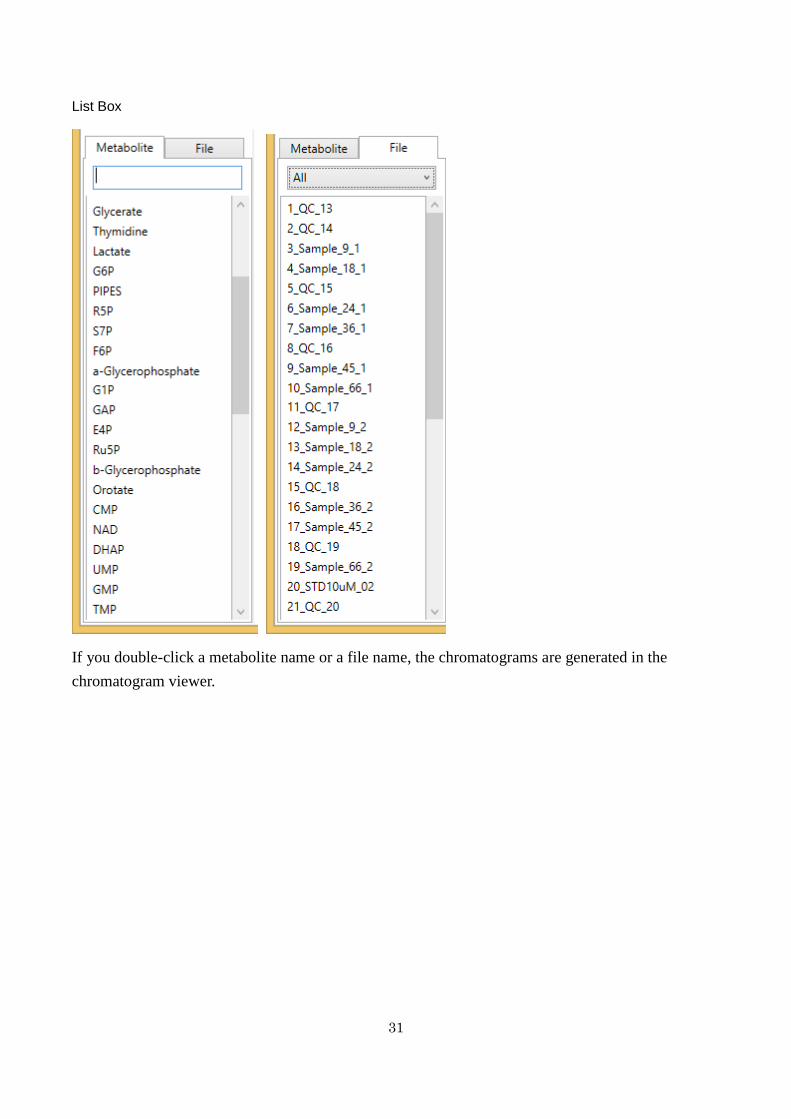
31
List Box
If you double-click a metabolite name or a file name the chromatograms are generated in the
chromatogram viewer
32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

32
Details on the MRMPROBS function
File menu
New project used for creating a new project
Open project used for opening an existing project Make sure that mth file raw folder and
processed folder are included in the same directory
Save as use to save as a new file
Save use to overwrite an existing project
33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

33
Data reprocessing
Data re-processing can be done by newly optimized parameters in this option Re-processing is also
performed per metabolite or per file The target MRM can also be changed The parameters are set
per metabolite and per file The required time for data re-processing is very short because file import
has been performed already
34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

34
Statistical analysis
The current program can apply two types of missing value approaches and can normalize a
quantification value by the internal standard and loesscubic spline with the analytical order
information If you want to use the internal standard you must set the optimal setting in the ldquoOption
menurdquo The current program can also do principal component analysis
35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

35
Missing value methods
Zero value This option gives zero (0) for ldquoNDrdquo in the raw data matrix
Data point value of retention time average This option gives the value described below for
ldquoNDrdquo in the raw data matrix
1 The process is performed per column ie per metabolite
2 If the value of a metabolite is ldquoNDrdquo in all files a zero (0) value is assigned
3 The retention time values except for ldquoNDrdquo files are stored and the average value is calculated
4 For each ldquoNDrdquo the intensity of ldquodata pointrdquo consistency with the average retention time of the
processed EIC chromatogram (after smoothing) is assigned as the quantification value
36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

36
Normalization
None after implementation of the missing value approach the values of the raw data matrix are
stored in the processed data matrix
Internal standard after implementation of the missing value approach the value divided by the
internal standard value set in the ldquoOption menurdquo is stored in the processed data matrix
LOESS after implementation of the missing value approach the signal intensities of each
metabolite are normalized with the QC samples information by means of loesscubic spline
Internal standard + LOESS After internal standard normalization loesscubic spline based
normalization is performed
After clicking the ldquoDonerdquo button the ldquoStatistical analysis settingrdquo button is activated
You can do principal component analysis Add the calculated number of the principal components
and choose the scale and transform method
37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

37
Zooming in and out can be done with the mouse wheel Each principal component is shown by
selecting the X axis or the Y axis combobox
38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

38
Window menu
The tile setting is possible depending on your computers resolution Please select your preference
39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

39
View menu
In this menu the chromatograms in the chromatogram viewer are sorted by file id analytical order
class id and file type
40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

40
Option menu
Here it is possible to set the properties of metabolites and files In particular this option menu is used
to create a data matrix for statistical analysis
In the file properties you can re-set the file type class ID and analytical order except for the
file name If you clear the check box of the included property it is no longer included in the
processed data matrix
In the metabolite properties you can set the internal standard It can be set independently for
each metabolite However please make sure that the metabolite name of the internal standard is
completely consistent with the metabolite name in the ldquointernal standardrdquo column Therefore we
recommend that you use copy and paste for the internal standard setting In this window although
copy and paste can be performed just by using the keyboard you can do ldquomultirdquo copy For example
copy a metabolite name by pushing Ctrl + C Select the rows you want to add in the internal standard
column by dragging and paste the clipboard contents by pushing Ctrl + V
41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

41
Export menu
A tab-delimited text file can be exported for a raw data matrix a processed data matrix the updated
library detected peak information detail and PCA results Moreover the PCA result can be exported
by some image formats
42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

42
Appendix A-1 how to obtain appropriate file conversion of the Shimadzu lcd file
Although you can do a content change of the lcd file after LC-QqQMS (MRM) analysis it is very
useful to construct a suitable method file (lcm format file) for the successful file convert of the
MRMPROBS software
1 Event name and channel (MRM transitions) rule
2 Update compound table
After the method construction of MRM transitions you should update the compound table mz by
the MRM event If you can analyze the samples by using the updated method file you do not have to
perform any other tasks for the stable file convert
For stable convert of Reifycs file convert
software the compound name should be
made just by ASCII format
MRM transitions should be
constructed for one metabolite
The completely same precursor and
product mz pair cannot be
acceptable in the file converter
43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

43
You can check the updated table by Method-gtData Processing Parameters-gtCompound tab
44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

44
3 If your data (lcd) were not collected by a suitable method described above you can improve
the lcd file by using the method file modified in the above way After the construction of the
modified method file please open ldquoPostrun Analysisrdquo of LabSolutions
After selecting the analysis files (lcd) push the ldquoApply to Methodrdquo button
Select the modified method file and improve your lcd file including the compound table mz If you
can do this the file (lcd) is successfully converted by Reifycs Inc software
45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

45
4File convert
Conditions You can convert from lcd files to abf files on your computer by installing LabSolutions
software ldquoTTFLDataExportVer5dllrdquo of LabSolutions ver 553 SP4 or later is required for the file
convert Check the ldquoTTFLDataExportVer5dllrdquo (Program Files (or 86)gtLabSolutions) file property
If the file size is less than 577536 bytes contact Shimadzu Inc for a file change
After ldquoAnalysisBaseFileConverterexerdquo is opened drag and drop the lcd files to this converter
Push the ldquoConvertrdquo button The ABF format files will be generated in the same folder as the lcd
files
Reifycs Inc software refers to this compound table for
the file convert from lcd file to abf file
46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

46
Appendix B Third option of MRMPROBS via mzML file
Required software and file
MSConvert
Download link httpproteowizardsourceforgenet
MRMPROBS
Download link httpprimepscrikenjpMetabolomics_SoftwareMRMPROBSindexhtml
Reference library for compound identification (tab-delimited text file)
MRMPROBS can import the mzML format file In the third option of MRMPROBS the
ldquofunction idrdquo is utilized to extract the chromatogram data The users should add the ldquofunction idrdquo
information to the reference library in addition to the normal library format
Download ProteoWizard
1 Select download type Windows installer (includes vendor reader support) is recommended
2 Read license agreements and download the proteowizard
(httpproteowizardsourceforgenetdownloadsshtml)
Setup ProteoWizard
1 Follow the wizard windows (Maybe you donrsquot miss it)
2 ldquoSeeMSrdquo should be also imported
47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter

47
Convert the vendorrsquos MS file to mzML via ProteoWizard
1 Open the MSConvertGUIexe
2 Select ldquoList of Filesrdquo
3 Select the vendorrsquos file via ldquoBrowserdquo button
4 In the ldquoOptionsrdquo never check any additional compression including ldquoUse numpress linear
compressionrdquo ldquoUse numpress short logged float compressionrdquo and ldquoUse numpress short
positive integer compressionrdquo Each of binary encoding precision is available
5 Click ldquoStartrdquo button
Note ProteoWizard doesrsquont support Shimadzu MS format If you want to use them please use the
abf converter



























![Vs2 review, 2008[1]](https://img.pdfslide.us/doc/110x75/5553f36fb4c90544428b491e/vs2-review-20081.jpg)

