Embed Size (px)
Citation preview

pubs.acs.org/Organometallics Published on Web 10/14/2009 r 2009 American Chemical Society
6206 Organometallics 2009, 28, 6206–6212
DOI: 10.1021/om9005627
MoO2Cl2 as a Novel Catalyst for C-P Bond Formation and for
Hydrophosphonylation of Aldehydes
Rita G. de Noronha,† Paulo Jorge Costa,‡ Carlos C. Rom~ao,† Maria Jos�e Calhorda,*,§ andAna C. Fernandes*,^
†Instituto de Tecnologia Quımica e Biol�ogica, Universidade Nova de Lisboa, Avenida da Rep�ublica, EAN,2781-157Oeiras, Portugal, ‡Departamento deQuımica, CICECO,Universidade de Aveiro, 3810-193Aveiro,Portugal, §Departamento de Quımica e Bioquımica, CQB, Faculdade de Ciencias da Universidade de Lisboa,1749-016 Lisboa, Portugal, and ^Centro de Quımica Estrutural, Complexo I, Instituto Superior T�ecnico,
Avenida Rovisco Pais, 1049-001 Lisboa, Portugal
Received July 1, 2009
This work reports the use ofMoO2Cl2 as a novel catalyst for C-P bond formation, exemplified here bythe hydrophosphonylation of aldehydes. A series of R-hydroxyphosphonates was prepared in excellentyields using the catalytic system HP(O)(OEt)2/MoO2Cl2 (5 mol %) under solvent-free conditions or atrefluxingTHF.DFTcalculations indicate that thePdObond coordinates tomolybdenumwhile theP-Hhydrogen is transferred to one ModO oxygen atom. The active species then reacts with the aldehyde toform theR-hydroxyphosphonates. The calculated activation barriers (free energies) are ca. 20 kcal mol-1.
Introduction
The syntheses ofR-hydroxyphosphonates and R-hydroxy-phosphonic acids have attracted much attention due totheir important biological activities as pesticides, antibiotics,anticancers, antivirals, and enzyme inhibitors, includingHIV protease.1 For example, the R-hydroxyphosphonatestype 1 are potent anti-HIV agents, the compounds 2 and 3 areenzyme inhibitors, and the R-hydroxyphosphonic acids 4-6
are anticancer agents (see Figure 1). R-Hydroxyphospho-nates are also useful precursors for the synthesis of otherbiologically important R-functionalized phosphonates suchas amino-, keto-, and acetoxyphosphonates.2
The addition of H-phosphonates to carbonyl compounds(Pudovik orAbramov reaction) is a crucial and highly valuablemethod for the synthesis of R-hydroxyphosphonates andfor C-P bond formation. The accepted mechanism for thisreaction involves basic deprotonation of the dialkyl phospho-nate, which exists in the tautomeric forms phosphonate and
phosphite, forming a dialkyl phosphate anion. This anion thenreacts as a nucleophile toward electrophilic carbonyl carbonatoms, leading to the synthesis of R-hydroxyphosphonates(Scheme 1).3 Various metal complexes have been usedas effective catalysts for this reaction, including aluminum,titanium, lanthanum, ytterbium, and niobium complexes.3
Due to the fast development of the chemistry and biologyof R-hydroxyphosphonates over the past decade, the intro-duction of novel catalysts that show wide applicability forthe hydrophosphonylation of aldehydes is strongly required.Recently, we and other authors have demonstrated that
high-valent oxo-molybdenum and -rhenium complexes acti-vate Si-H,4 B-H,5 and H-H6 bonds and are excellentcatalysts for hydrosilylation of aldehydes and ketones4,7 as
*Corresponding authors. E-mail: [email protected]. Tel: 351 217500196.Fax: 351 2175000 088. E-mail: [email protected]. Tel: 351218419388. Fax: 351 218464457.(1) (a) Szyma�nska, A.; Szymczak, M.; Boryski, J.; Stawi�nski, J.;
Kraszewski, A.; Collu, G.; Sanna, G.; Giliberti, G.; Loddo, R.; LaColla, P. Bioorg. Med. Chem. 2006, 14, 1924. (b) Pokalwar, R. U.;Hangarge, R. V.; Maske, P. V.; Shingare, M. S. Arkivoc 2006 (xi), 196. (c)Shi, D.-Q.; Sheng, Z.-L.; Liu, X.-P.; Wu, H.Heteroat. Chem. 2003, 14, 266.(d) Ganzhorn, A. J.; Hoflack, J.; Pelton, P. D.; Strasser, F.; Chanal, M.-C.;Piettre, S. R. Bioorg. Med. Chem. 1998, 6, 1865. (e) Frechette, R. F.;Ackerman, C.; Beers, S.; Look, R.; Moore, J. Bioorg. Med. Chem. Lett.1997, 7, 2169. (f) Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.;Rogers, W. L.; Smith, S. A.; DeForrest, J. M.; Oehl, R. S.; Petrillo, E. W. J.Med. Chem. 1995, 38, 4557. (g) Stowasser, B.; Budt, K.-H.; Jian-Qi, L.;Peyman, A.; Ruppert, D. Tetrahedron Lett. 1992, 33, 6625. (h) Patel, D. V.;Rielly-Gauvin, K.; Ryono, D. E. Tetrahedron Lett. 1990, 31, 5587.(2) (a)Kaboudin, B.TetrahedronLett. 2003, 44, 1051. (b) Firouzabadi,
H.; Iranpoor, N.; Sobhani, S. Synth. Commun. 2004, 34, 1463. (c) Miranda,L. S. M.; Vasconcellos, M. L. A. A. Synthesis 2004, 1767.
(3) (a) Merino, P.; Marqu�es-L�opez, E.; Herrera, R. P. Adv. Chem.Catal. 2008, 350, 1195. (b) Yang, F.; Zhao, D.; Lan, J.; Xi, P.; Yang, L.;Xiang, S.; You, J.Angew. Chem., Int. Ed. 2008, 47, 5646. (c) Zhou, X.; Liu,X.; Yang, X.; Shang, D.; Xin, J.; Feng, X.Angew. Chem., Int. Ed. 2008, 47,392. (d) Abell, J. P.; Yamamoto, H. J. Am. Chem. Soc. 2008, 130, 10521. (e)Thottempudi, V.; Chung, K. H. Bull. Korean Chem. Soc. 2008, 29, 1781. (f)Kolodiazhnyi, O. I.Tetrahedron: Asymmetry 2005, 16, 3295. (g) Kee, T. P.;Nixon, T. D. The Asymmetric Phospho-Aldol Reaction. Past, Present andFuture. New Aspects in Phosphorus Chemistry II; Topics in CurrentChemistry 223; Springer-Verlag: Berlin, 2003; pp 45-65.
(4) (a) Kennedy-Smith, J. J.; Nolin, K. A.; Gunterman, H. P.; Toste,F. D. J. Am. Chem. Soc. 2003, 125, 4056. (b) Nolin, K. A.; Krumper, J. R.;Puth, M. D.; Bergman, R. G.; Toste, F. D. J. Am. Chem. Soc. 2007, 129,14684.
(5) Fernandes, A. C.; Fernandes, J. A.; Almeida Paz, F. A.; Rom~ao,C. C. Dalton Trans. 2008, 6686.
(6) Reis, P. M.; Costa, P. J.; Rom~ao, C. C.; Fernandes, J. A.;Calhorda, M. J.; Royo, B. Dalton Trans. 2008, 1727.
(7) (a) Du, G. D.; Fanwick, P. E.; Abu-Omar, M. M. J. Am. Chem.Soc. 2007, 129, 5180. (b) Du, G. D.; Abu-Omar, M. M. Organometallics2006, 25, 4920. (c) Ison, E. A.; Trivedi, E. R.; Corbin, R. A.; Abu-Omar, M.M. J. Am.Chem. Soc. 2005, 127, 15374. (d) Royo, B.; Rom~ao, C. C. J.Mol.Catal A:Chem. 2005, 236, 107. (e) Fernandes, A. C.; Fernandes, R.; Rom~ao,C. C.; Royo, B. Chem. Commun. 2005, 213. (f) Costa, P. J.; Rom~ao, C. C.;Fernandes, A. C.; Royo, B.; Reis, P. M.; Calhorda, M. J. Chem.;Eur. J.2007, 13, 3934.

Article Organometallics, Vol. 28, No. 21, 2009 6207
well as for the reduction of imines,8 amides,9 esters,10 sulf-oxides,11 pyridine N-oxides,11a and alkynes6 to the corre-sponding amines, alcohols, sulfides, pyridines, and alkenes.We have also developed a novel method for the synthesis
of aromatic ketones and sulfones by Friedel-Crafts acyla-tion and sulfonylation using the high-valent oxo-complexMoO2Cl2 as catalyst.
12 This novel methodology showed thatMoO2Cl2 is a good catalyst for C-CandC-S bond-formingreactions and suggested its application toward other C-Xbond construction reactions, namely, the C-P bond. Inthis work, we studied the activation of the P-H bond ofH-phosphonates by MoO2Cl2 and the synthesis of R-hydro-xyphosphonates using the catalytic system HP(O)(OEt)2/MoO2Cl2 (5 mol %).The catalytic activity of MoO2Cl2 complexes in reduction
reactions initiated by the activation of H-Hor Si-Hbonds,for instance, has been explained by a heterolytic cleavagewith addition of the hydride to the metal and of the cation tothe oxide, formingOHorOR.The hydride intermediate is anactive species for the propagation of the reaction.6,7f Theseconclusions have been based on results of DFT13 calcula-tions and are particularly relevant in these Mo(VI) com-plexes, where an oxidative addition in the first step isimpossible, contrary to Re(VI) complexes, which catalyzesimilar reactions.14 TheDFT computational study described
in this work addresses the possibility of activating the P-Hbond in a similar way and with alternative pathways. Thismechanism was compared with that previously accepted anddescribed in Scheme 1.
Results and Discussion
In order to find the best reaction conditions for theactivation of the P-H bond of H-phosphonates byMoO2Cl2, we started by investigating the reaction of the testsubstrate 4-nitrobenzaldehyde with diethylphosphonatecatalyzed by varying loads of MoO2Cl2 in several solventsor under solvent-free conditions (Table 1). The progress ofthe reactions was monitored by thin-layer chromatographyand by 1H NMR. The reactions performed in THF showedthat the best result was obtained at reflux temperature in thepresence of 5 mol % of MoO2Cl2 (Table 1, entry 1). Similarreaction at room temperature afforded the product in lowconversion (Table 1, entry 2). In the presence of 1 mol % ofMoO2Cl2, the reaction required a long time at refluxingtemperature to give a high conversion (Table 1, entry 3).No reaction was observed in the absence of catalyst (Table 1,entry 4).The hydrophosphonylation carried out in acetonitrile
with 5 mol % of MoO2Cl2 at refluxing temperature gavethe product in 100% conversion after 22 h (Table 1, entry 5).In contrast, dichloromethane dramatically decreased theconversion of the R-hydroxyphosphonate to 18% (Table 1,entry 6).We also investigated the hydrophosphonylation of 4-
nitrobenzaldehyde under solvent-free conditions using dif-ferent amounts of MoO2Cl2 (Table 1, entries 7-9). All theseexperiments afforded the product in excellent conversions.However, the reaction performed with 5 mol% ofMoO2Cl2was very fast, requiring only 1.2 h to be completed (Table 1,entry 7).After gaining some initial insights about the reactions
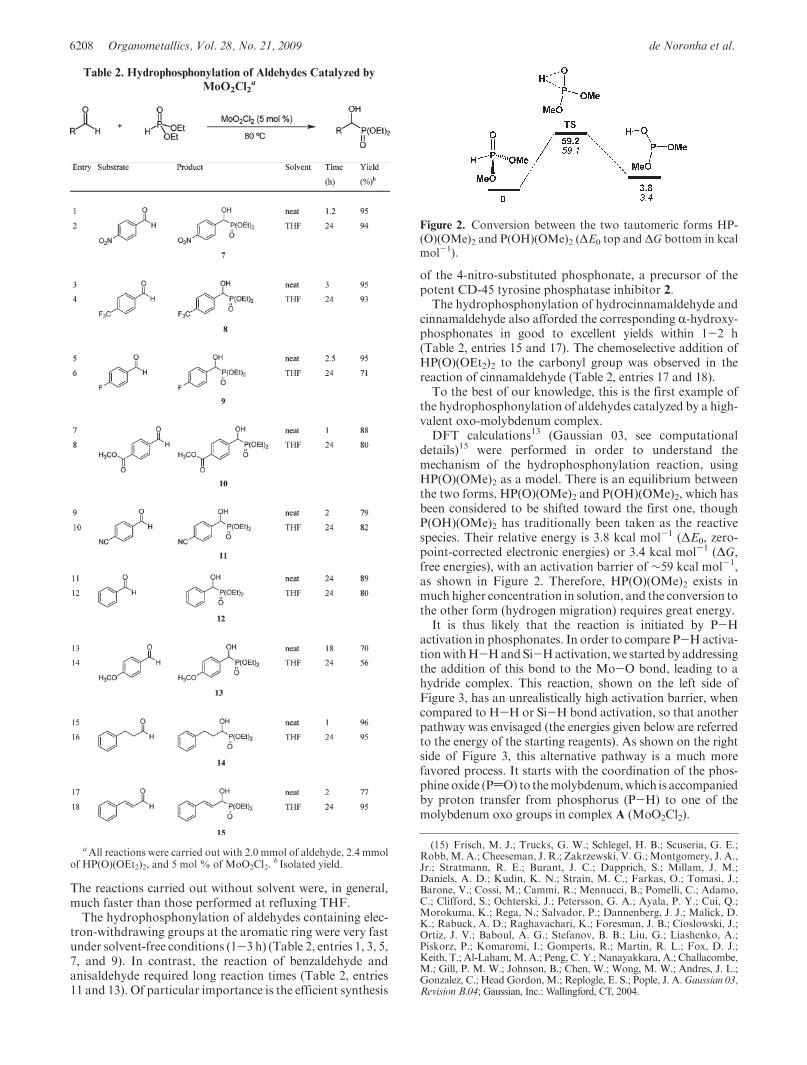
conditions, we explored the scope of this novel methodologywith a variety of aldehydes using the system HP(O)(OEt2)2/MoO2Cl2 (5 mol %). These reactions were carried out at80 �C under solvent-free conditions or at refluxing THF(Table 2). In both reaction conditions, the R-hydroxy-phosphonates were obtained in good to excellent yields.
Figure 1. Selected examples of biologically active R-hydroxy-phosphonates and R-hydroxyphosphonic acids.
Scheme 1. Catalytic Cycle for the Pudovik Reaction
Table 1. Hydrophosphonylation of 4-Nitrobenzaldehyde with
Diethylphosphonate Catalyzed by MoO2Cl2a
entryMoO2Cl2(mol %) solvent
temp(�C)
time(h)
conversion(%)b
1 5 THF reflux 24 942 5 THF rt 27 283 1 THF reflux 48 904 THF reflux 24 no reaction5 5 CH3CN reflux 22 1006 5 CH2Cl2 reflux 18 187 5 neat 80 1.2 1008 2.5 neat 80 3 989 1 neat 80 3.5 90
aAll reactions were carried out with 2.0 mmol of aldehyde, 2.4 mmolof HP(O)(OEt)2, and 5 mol % of MoO2Cl2 in 5 mL of the appropriatesolvent. bConversion was determined by 1H NMR.
(8) Fernandes, A. C.; Rom~ao, C. C.Tetrahedron Lett. 2005, 46, 8881.(9) Fernandes, A.C.; Rom~ao,C. C. J.Mol. CatalA:Chem. 2007, 272,
60.(10) Fernandes, A. C.; Rom~ao, C. C. J. Mol. Catal A: Chem. 2006,
253, 96.(11) (a) Fernandes, A. C.; Rom~ao, C. C. Tetrahedron 2006, 62, 9650.
(b) Fernandes, A. C.; Rom~ao, C. C. Tetrahedron Lett. 2007, 48, 9176.(12) Noronha, R. G.; Fernandes, A. C.; Rom~ao, C. C. Tetrahedron
Lett. 2009, 50, 1407.(13) Parr, R. G.; Yang, W. Density Functional Theory of Atoms and
Molecules; Oxford University Press: New York, 1989.(14) Chung,L.W.; Lee,H.G.; Lin, Z.;Wu,Y. J.Org. Chem. 2006, 71,
6000–6009.
6208 Organometallics, Vol. 28, No. 21, 2009 de Noronha et al.
The reactions carried out without solvent were, in general,much faster than those performed at refluxing THF.The hydrophosphonylation of aldehydes containing elec-
tron-withdrawing groups at the aromatic ring were very fastunder solvent-free conditions (1-3 h) (Table 2, entries 1, 3, 5,7, and 9). In contrast, the reaction of benzaldehyde andanisaldehyde required long reaction times (Table 2, entries11 and 13). Of particular importance is the efficient synthesis
of the 4-nitro-substituted phosphonate, a precursor of thepotent CD-45 tyrosine phosphatase inhibitor 2.The hydrophosphonylation of hydrocinnamaldehyde and
cinnamaldehyde also afforded the corresponding R-hydroxy-phosphonates in good to excellent yields within 1-2 h(Table 2, entries 15 and 17). The chemoselective addition ofHP(O)(OEt2)2 to the carbonyl group was observed in thereaction of cinnamaldehyde (Table 2, entries 17 and 18).To the best of our knowledge, this is the first example of
the hydrophosphonylation of aldehydes catalyzed by a high-valent oxo-molybdenum complex.DFT calculations13 (Gaussian 03, see computational
details)15 were performed in order to understand themechanism of the hydrophosphonylation reaction, usingHP(O)(OMe)2 as a model. There is an equilibrium betweenthe two forms, HP(O)(OMe)2 and P(OH)(OMe)2, which hasbeen considered to be shifted toward the first one, thoughP(OH)(OMe)2 has traditionally been taken as the reactivespecies. Their relative energy is 3.8 kcal mol-1 (ΔE0, zero-point-corrected electronic energies) or 3.4 kcal mol-1 (ΔG,free energies), with an activation barrier of ∼59 kcal mol-1,as shown in Figure 2. Therefore, HP(O)(OMe)2 exists inmuch higher concentration in solution, and the conversion tothe other form (hydrogen migration) requires great energy.It is thus likely that the reaction is initiated by P-H
activation in phosphonates. In order to compare P-Hactiva-tionwithH-HandSi-Hactivation,we started by addressingthe addition of this bond to the Mo-O bond, leading to ahydride complex. This reaction, shown on the left side ofFigure 3, has an unrealistically high activation barrier, whencompared to H-H or Si-H bond activation, so that anotherpathway was envisaged (the energies given below are referredto the energy of the starting reagents). As shown on the rightside of Figure 3, this alternative pathway is a much morefavored process. It starts with the coordination of the phos-phine oxide (PdO) to themolybdenum,which is accompaniedby proton transfer from phosphorus (P-H) to one of themolybdenum oxo groups in complex A (MoO2Cl2).
Table 2. Hydrophosphonylation of Aldehydes Catalyzed by
MoO2Cl2a
aAll reactions were carried out with 2.0 mmol of aldehyde, 2.4 mmolof HP(O)(OEt2)2, and 5 mol % of MoO2Cl2.
b Isolated yield.
Figure 2. Conversion between the two tautomeric forms HP-(O)(OMe)2 and P(OH)(OMe)2 (ΔE0 top and ΔG bottom in kcalmol-1).
(15) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb,M. A.; Cheeseman, J. R.; Zakrzewski, V. G.;Montgomery, J. A.,Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.;Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.;Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo,C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.;Morokuma, K.; Rega, N.; Salvador, P.; Dannenberg, J. J.; Malick, D.K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.;Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.;Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham,M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe,M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.;Gonzalez, C.; HeadGordon,M.; Replogle, E. S.; Pople, J. A.Gaussian 03,Revision B.04; Gaussian, Inc.: Wallingford, CT, 2004.

Article Organometallics, Vol. 28, No. 21, 2009 6209
Figure 4 gives some relevant distances in the intermediatesand transition states. In the low-energy path, the tetrahedralcoordination around molybdenum becomes trigonal bypir-amidal, with the new oxygen ligand axially trans to chlorine.The P-H bond has stretched from 1.407 A in the initialMoO2Cl2 (A) to 1.670 A in TSAC, while the MoO-Hdistance is already 1.205 A and the new Mo-O distance, at2.060 A, is close to the finalMo-Obond of 1.858 A inC.C isthe active intermediate in the second part of the reaction,namely, the activation of the aldehyde substrate. Themolybdenum has a square-pyramidal coordination, with ashort axialModObond (1.664 A), not too different from thecorresponding ModO bonds in the precursor A.In TSAC, the phosphorus atom retains a coordination
number four, with an approximately tetrahedral environ-ment, while in the alternative pathway phosphorus becomespentacoordinate, a change that may account for the highenergy of the transition state TSAB. The energy differencebetween the two possible intermediates B and C is not verylarge, but the kinetic barrier makes the formation of B
unlikely, so that C will be considered for the remainder ofthe mechanism.Benzaldehyde was taken as amodel to study the formation
of the P-Cbond in the following step. The activation energy
is small and it increases to 20 kcal mol-1 when one considersfree energies, as the first step (rate limiting) is associative. Inthe transition stateTSCD, the aldehyde approaches themetaland starts to bind through the oxygen atom (2.254 A), whilethe P-C bond starts to form (2.341 A). These two distanceshave reached normal bond lengths (1.996 and 1.861 A,respectively) in the distorted octahedral D. In order to formthe product, a proton migration has to take place, from theMo-OH group to the oxygen initially belonging to thealdehyde. This migration occurs in TSDE, with a smallbarrier (5-6 kcal mol-1). The hydrogen is weakly boundto Mo-O (1.084 A) and interacting with the other oxygen(1.409 A). The first bond has broken completely inE, leavingModO, and a normal OH bond has formed in the finalproduct (0.974 A).These and other relevant distances are given in Figure 6.
After the hydrogen migration, the final product is stillweakly bound to the catalyst, by one O-H 3 3 3O bondand a weak PdO 3 3 3Mo interaction, and approximately10 kcal mol-1 is needed to separate them and regeneratethe catalyst.This mechanism, based on the HP(O)(OMe)2 tautomer
present in higher concentrations in the reactionmedium, is ingeneral agreement with the experimental data, with lowreaction barriers. It is, however, interesting to examine thepossibility of activation of the O-H bond of the othertautomer, P(OH)(OMe)2. The reaction profile (Figure 7)shows the proton transfer from the OH bond of the phos-phite to one oxide of the catalyst, while the oxygen of the OHbond coordinates the metal, in a concerted addition of theO-H bond to the ModO bond of the MoO2Cl2 complex,leading to the formation of the same intermediate C. Thiskind of addition has been already observed in the first step ofolefin epoxidation by tert-butylhydroperoxide (TBHP) pro-moted by Mo(VI) catalysts bearing a cis-Mo(O)2 group.16
The electronic barrier of 17.2 kcal mol-1 is not very high, butalmost duplicates the value shown in Figure 3 when activat-ing the P-H bond. As expected for this type of associativemechanism, the free energies are higher, following the sametrend. The reaction mechanism should thus proceed by thefirst mechanism proposed (tautomer present in higheramounts and lower barriers). The second step of the reactionis the same in both cases. A few attempts were made in order
Figure 3. Alternative reaction pathways for the activation ofHP(O)(OMe)2 in the presence ofMoO2Cl2 (A) (ΔE0 top andΔGbottom in kcal mol-1).
Figure 4. Relevant distances in the complexes and transition states involved in the reactions between complex MoO2Cl2 (A) andHP(O)(OMe)2.

6210 Organometallics, Vol. 28, No. 21, 2009 de Noronha et al.
to test the alternative concerted reaction between the inter-mediate and the substrate (from C to E in Figure 5) withoutsuccess. Since the barriers shown in Figure 5 are very low andthe concerted pathway has always led to higher ones, thispath was not pursued.Experimental results showthat theyield increasesandreaction
timesdecreasewhenelectron-withdrawinggroupsarepresent. Inorder to check this acceleration effect, calculationswere repeatedfor other substrates containing fluorine or NO2 substituents inpara positions. The solvent effect was analyzed running single-point calculations in acetonitrile and dichloromethane (PCMmodel).17 The differences between the two aldehydes studied are
Figure 6. Relevant distances in the complexes and transition states involved in the formation of the P-Cbond, starting from the activeintermediate, C.
Figure 5. Reaction pathway for the reaction between the intermediate C and the aldehyde substrate (ΔE0 top and ΔG bottom inkcal mol-1).
Figure 7. Reaction pathway for the activation of P(OH)(OMe)2in the presence of MoO2Cl2 (A0) (ΔE0 top and ΔG bottom inkcal mol-1).
(16) Veiros, L. F.; Prazeres, A.; Costa, P. J.; Rom~ao, C. C.; K€uhn, F.E.; Calhorda, M. J. Dalton Trans. 2006, 1383–1389.
(17) Rapp�e, A.K.;Kasewit, C. J.; Colwell, K. S.; Goddard,W.A. III;Skiff, W. M. J. Am. Chem. Soc. 1992, 114, 10024.

Article Organometallics, Vol. 28, No. 21, 2009 6211
small, and only the behavior of the p-nitrobenzaldehyde will bediscussed. The relevant energies are collected in Table 3.Although the energies do not vary much, the trends reflect
the experimental data. Substitution of hydrogen by theelectron-withdrawing NO2 group leads to a stabilization ofthe products (from -9 to -12 kcal mol-1, last entry inTable 3), the intermediates, and the two transition states,when comparing the gas-phase results. The two solvents alsoshow a different influence. When going from dichloro-methane to acetonitrile, the energies of the products andintermediates decrease, while the activation barriers becomelower, in agreement with the faster reaction occurring in thislast solvent.
Conclusions
In conclusion, we have developed a simple, efficient, andsolvent-free procedure for the synthesis of R-hydroxypho-sphonates catalyzed byMoO2Cl2. This method proved to bechemoselective and effective for a variety of aldehydes.Otheradvantages of this novel methodology include good to highyields, fast reaction times, mild reaction conditions, andclean reactions. The computational studies indicate thatthe reaction proceeds with low activation barriers, startingwith coordination of the PdO bond of HP(O)(OMe)2 tomolybdenum and hydrogen transfer from P-H to the oxo inModO, formingMo-OH.The new intermediate reacts withthe aldehyde substrate in two steps. In the first, the aldehydebinds the metal through the carbonyl and the P-C bondforms; in the second, the hydrogen is transferred fromMo-OH to the oxygen of the final product. On the otherhand, it has been shown that the mechanism proposed in theliterature involving the P(OH)(OMe)2 tautomer, present insmaller amounts, leads to higher activation barriers, beingtherefore less likely to occur.The present work opens a new research area for high-
valent oxo-complexes as excellent catalysts for C-P bond-forming reactions. Application of oxo-complexes to otherC-P bond-forming reactions as well as to asymmetrichydrophosphonylation reactions is currently being investi-gated in our group.
Experimental Section
Solvents were purified by conventional methods and distilledunder nitrogen, prior to use. Diethylphosphonate andMoO2Cl2were obtained from Aldrich. Flash chromatography was per-formed onMNKieselgel 60 M 230-400 mesh. 1H, 13C, and 31PNMR spectra were measured on a Bruker Avance III 400 MHzspectrometer. Chemical shifts are reported in parts per million(ppm) downfield from an internal standard. Melting tempera-tures were measured with a Lecia Galen III hot stage apparatus
and are uncorrected. Microanalyses (C and H) were performedat Laborat�orio de An�alise, Instituto Superior T�ecnico.
General Procedure for theHydrophosphonylation of Aldehydes
with HP(O)(OEt)2 Catalyzed by MoO2Cl2. To a mixture ofaldehyde (2 mmol) and MoO2Cl2 (5 mol %) was added diethyl-phosphonate (2.4 mmol). The reaction mixture was stirred at80 �C under inert atmosphere, and the progress of the reactionwas monitored by TLC and 1H NMR. When the reaction wascomplete, water (3mL)was added and themixture was stirred at80 �C. After 1 h, the reaction mixture was cooled to ambienttemperature and extracted with ethyl acetate (2 � 10 mL). Thecombined organic layers were dried over Na2SO4 and filtered,and the solvent was removed under reduced pressure.Whenevernecessary, the residue was purified by flash chromatographywith appropriate mixtures of n-hexane and ethyl acetate.
Compound 7:3c 1HNMR (400MHz, CDCl3) δ 8.21 (d, J=8.3Hz, 2H), 7.66 (dd, J = 8.3, 2.1 Hz, 2H), 5.24 (s, 1H), 5.16 (d,J= 12.4 Hz, 1H), 4.22-4.04 (m, 4H), 1.28 (t, J= 8.0 Hz, 3H),1.25 (t, J = 8.0 Hz, 3H) ppm.; 13C NMR (101 MHz, CDCl3)δ 147.7 (d, J = 3.7 Hz), 144.5 (d, J = 2.3 Hz), 127.9 (d, J =5.2 Hz), 123.5 (d, J= 2.7 Hz), 70.3 (d, J= 158.4 Hz), 63.8 (dd,J = 67.0, 7.4 Hz), 16.6-16.5 (m) ppm; 31P NMR (162 MHz,CDCl3) δ 20.4 ppm.
Compound 8: mp 85-87 �C; 1H NMR (400 MHz, CDCl3) δ7.61 (s, 4H), 5.10 (d, J= 11.6 Hz, 1H), 4.57 (s, 1H), 4.25- 3.91(m, 4H), 1.27 (t, J=7.2 Hz, 3H), 1.24 (t, J=7.2 Hz, 3H) ppm;13C NMR (101 MHz, CDCl3) δ 141.0 (br s), 127.4 (d, J =5.4 Hz), 125.4-125.2 (m), 70.5 (d, J= 158.4 Hz), 63.6 (dd, J=43.2, 7.3 Hz), 16.6 (d, J = 5.7 Hz) ppm; 31P NMR (162 MHz,CDCl3) δ 21.1 ppm. Anal. Calcd for C12H16F3O4 P: C, 46.16; H,5.17. Found: C, 45.98; H, 5.01.
Compound 9:3c 1H NMR (400 MHz, CDCl3) δ 7.53-7.38 (m,2H), 7.04 (t, J=8.6 Hz, 2H), 4.99 (d, J=10.4 Hz, 1H), 4.23 (s,1 H), 4.18 - 3.87 (m, 4H), 1.26 (t, J = 7.2 Hz, 3H), 1.22 (t,J = 7.2 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3) δ 162.8(d, J = 248.0 Hz), 132.6 (s), 129.0 (dd, J = 8.1, 5.9 Hz), 115.4(d, J = 21.5 Hz), 70.3 (d, J = 160.3 Hz), 63.4 (dd, J = 33.3,7.2 Hz), 16.6 (d, J=4.3Hz) ppm; 31PNMR (162MHz, CDCl3)δ 21.7 ppm.
Compound 10:mp 109-111 �C; 1H NMR (400MHz, CDCl3)δ 8.01 (d, J=8.2Hz, 2H), 7.55 (dd, J=8.2, 1.8Hz, 2H), 5.09 (d,J=11.6 Hz, 1H), 4.85 (s, 1H), 4.14-3.97 (m, 4H), 3.90 (s, 3H),1.25 (t, J=7.1Hz, 6H), 1.21 (t, J=7.1Hz, 6H) ppm; 13CNMR(101 MHz, CDCl3) δ 167.1 (s), 142.1 (s), 129.8 (d, J= 3.0 Hz),129.6 (d, J = 3.0 Hz), 127.1 (d, J = 5.4 Hz), 70.9 (d, J =158.7 Hz), 63.5 (dd, J = 34.9, 7.2 Hz), 52.3 (s), 16.5 (d, J =5.6 Hz) ppm; 31P NMR (162 MHz, CDCl3) δ 21.2 ppm. Anal.Calcd for C13H19O6 P: C, 51.66; H, 6.34. Found: C, 51.23;H, 6.19.
Compound 11:3c 1HNMR(400MHz, CDCl3) δ 7.71-7.53 (m,4H), 5.10 (d, J=12.2Hz, 1H), 4.21-3.96 (m, 5H), 1.25 (dd, J=16.1, 7.1 Hz, 6H) ppm; 13CNMR (101MHz, CDCl3) δ 142.5 (d,J= 2.4 Hz), 132.1 (d, J= 2.8 Hz), 127.8 (d, J= 5.3 Hz), 118.9(d, J = 2.1 Hz), 111.8 (d, J = 3.5 Hz), 70.4 (d, J = 158.3 Hz),63.8 (dd, J=54.3, 7.3 Hz), 16.6 (m) ppm; 31P NMR (162MHz,CDCl3) δ 20.6 ppm.
Compound 12:3c 1HNMR(400MHz, CDCl3) δ 7.53-7.45 (m,2H), 7.42-7.28 (m, 3H), 5.03 (d, J = 10.7 Hz, 1H), 4.11-3.91(m, 4H), 1.27 (t, J=7.1 Hz, 3H), 1.22 (t, J=7.1 Hz, 3H) ppm;13C NMR (101 MHz, CDCl3) δ 136.8 (br s), 128.4 (d, J =5.4 Hz), 128.2 (br s), 127.3 (d, J = 5.8 Hz), 70.9 (d, J =158.7 Hz), 63.5 (dd, J = 34.9, 7.2 Hz), 16.5 (dd, J = 5.5,3.8 Hz) ppm; 31P NMR (162 MHz, CDCl3) δ 22.2 ppm.
Compound 13:3c 1H NMR (400MHz, CDCl3) δ 7.41 (dd, J=8.5, 2.1 Hz, 2H), 6.89 (d, J=8.5 Hz, 2H), 4.94 (d, J=10.0 Hz,1H), 4.15-3.90 (m, 4H), 3.80 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H),1.21 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3)δ 159.8 (s), 159.7 (s), 128.7 (d, J=6.1Hz), 114.0 (d, J=2.2Hz),70.7 (d, J = 160.6 Hz), 63.3 (dd, J = 17.7, 7.1 Hz), 55.5 (s),16.6 (m) ppm; 31P NMR (162 MHz, CDCl3) δ 22.3 ppm.
Table 3. Free Energies (kcal mol-1) Calculated for C6H5CHO
in the Gas Phase and 4-NO2-C6H4CHO in the Gas Phase,
Acetonitrile, and Dichloromethane
C6H5CHO 4-NO2-C6H4CHO
gas phase gas phase CH2Cl2 CH3CN
C þ aldehyde 0 0 0 0TSCD 20 19 20 18D 9 7 8 7TSDE 14 12 13 12E -10 -11 -10 -12MoO2Cl2 þ product (F) -9 -12 -11 -14

6212 Organometallics, Vol. 28, No. 21, 2009 de Noronha et al.
Compound 14:3c 1HNMR(400MHz,CDCl3) δ 7.32-7.14 (m,5H), 4.15 (m, 4H), 3.85 (dt, J=9.1, 4.5 Hz, 1H), 3.48 (br s, 1H),3.04-2.91 (m, 1H), 2.83-2.63 (m, 1H), 2.18-1.87 (m, 2H), 1.32(t, J = 7.2 Hz, 3H), 1.30 (t, J = 7.2 Hz, 3 H) ppm; 13C NMR(101MHz, CDCl3) δ 141.5 (s), 128.8 (s), 128.6 (s), 126.2 (s), 67.1(d, J= 160.7 Hz), 62.8 (dd, J= 13.8, 7.1 Hz), 33.1 (s), 31.9 (d,J = 14.1 Hz), 16.7 (dd, J = 5.5, 3.3 Hz) ppm; 31P NMR(162 MHz, CDCl3) δ 25.8 ppm.Compound 15:
3c 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J =7.5, 2H), 7.31 (t, J=7.7, 2H), 7.28-7.18 (m, 1H), 6.78 (dd, J=15.9, 4.5, 1H), 6.32 (dt, J = 15.9, 5.7, 1H), 4.68 (dd, J = 13.0,6.2, 1H), 4.23-4.15 (m, 4H), 1.32 (q, J = 6.9, 6H); 13C NMR(101 MHz, CDCl3) δ 136.6 (d, J = 2.9 Hz), 132.5 (d, J =13.1Hz), 128.7 (s), 128.0 (s), 126.8 (d, J=1.9Hz), 124.1 (d, J=4.3 Hz), 69.6 (d, J=161.3 Hz), 63.4 (dd, J=15.4, 7.2 Hz), 16.7(dd, J=5.5Hz) ppm; 31PNMR (162MHz, CDCl3) δ 22.3 ppm.
DFT Calculations. The DFT13 calculations were performedwith Gaussian03,15 using the B3LYP hybrid functional. Thisfunctional includes amixture of Hartree-Fock exchange18 with
DFT exchange-correlation, given by Becke’s three-parameterfuntional19 with the Lee, Yang, and Parr correlation functional,which accounts for both local and nonlocal terms.20 ForMo, thestandard LanL2DZ basis set with the associated ECP21 wasused, augmented with an f-polarization function (exponent1.043).22 The remaining elements were described by the6-31G(d,p)23 basis set. All intermediates and transitions stateswere optimized without any symmetry constraints, and fre-quency calculations were performed in all species at this levelof theory to confirm the nature of the stationary points. Thetransition-state structures, which yielded one imaginary fre-quency, were relaxed following the vibrational mode to confirmthe connecting reagents. The reported zero-point-correctedelectronic energies (E0) and gas-phase Gibbs free energies (G)were also obtained at this level of theory. The free energies indichloromethane and acetonitrile solution were obtained byperforming self-consistent reaction field (SCRF) calculationsusing the polarizable continuummodel (PCM) and the universalforce field (UFF)17 on the gas-phase-optimized structures. TheHP(O)(OEt)2 structure was modeled by HP(O)(OMe)2.
Acknowledgment. This research was supported byFCT, POCI, and FEDER through projects PTDC/QUI/71741/2006 and PTDCI/QUI/58925/2004. R.G.N.and P.J.C. thank FCT for postdoctoral grants (SFRH/BPD/27215/2006 and (SFRH/BPD/27082/2006, res-pectively). The NMR spectrometers are part of theNational NMR Network and were acquired with fundsfrom FCT and FEDER.
Supporting Information Available:1H, 13C, and 31P NMR
spectra of the products. This material is available free of chargevia the Internet at http://pubs.acs.org.
(18) Lee, C.; Yang, W.; Parr, W. R. G. Phys. Rev. B 1988, 37, 785.(19) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.(20) Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett.
1989, 157, 200.(21) (a) Dunning, T. H., Jr.; Hay, P. J. In Modern Theoretical
Chemistry, Vol. 3; Schaefer, H. F., III, Ed.; Plenum: New York, 1976; p 1.(b) Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270. (c) Wadt, W. R.;Hay, P. J. J. Chem. Phys. 1985, 82, 284. (d) Hay, P. J.; Wadt, W. R. J. Chem.Phys. 1985, 82, 2299.(22) Ehlers, A. W.; B€ohme, M.; Dapprich, S.; Gobbi, A.; H€ollwarth,
A.; Jonas, V.; K€ohler, K. F.; Stegmann, R.; Veldkamp,A.; Frenking, G.Chem. Phys. Lett. 1993, 208, 111.(23) (a) Ditchfield, R.; Hehre,W. J.; Pople, J. A. J. Chem. Phys. 1971,
54, 724. (b)Hehre,W. J.; Ditchfield, R.; Pople, J. A. J.Chem.Phys. 1972, 56,2257. (c) Hariharan, P. C.; Pople, J. A.Mol. Phys. 1974, 27, 209. (d) Gordon,M. S. Chem. Phys. Lett. 1980, 76, 163. (e) Hariharan, P. C.; Pople, J. A.Theor. Chim. Acta 1973, 28, 213.




![Ce [MoO ][MoO ]andCe[MoO : Two New Cerium Oxomolybdates, Each Exhibiting a Special ...znaturforsch.com/s66b/s66b0763.pdf · 2014. 12. 15. · Ce 2 [MoO 5][MoO 4]andCe 5 [MoO 4] 8:](https://img.pdfslide.us/doc/110x75/612bf67e5d9d87214d0e167f/ce-moo-moo-andcemoo-two-new-cerium-oxomolybdates-each-exhibiting-a-special.jpg)