Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/nmd
ScienceDirect
Neuromuscular Disorders 24 (2014) 195–200
Monoclonal antibodies for clinical trials of Duchennemuscular dystrophy therapy
Le Thanh Lam a, Nguyen Thi Man a, Glenn E. Morris a,b,⇑
a Wolfson Centre for Inherited Neuromuscular Disease, RJAH Orthopaedic Hospital, Oswestry SY10 7AG, UKb Institute for Science and Technology in Medicine, Keele University, Keele, UK
Received 15 November 2013; accepted 27 November 2013
Abstract
Most pathogenic mutations in Duchenne and Becker muscular dystrophies involve deletion of single or multiple exons from thedystrophin gene, so exon-specific monoclonal antibodies (mAbs) can be used to distinguish normal and mutant dystrophin proteins.In Duchenne therapy trials, mAbs can be used to identify or rule out dystrophin-positive “revertant” fibres, which have aninternally-deleted dystrophin protein and which occur naturally in some Duchenne patients. Using phage-displayed peptide libraries,we now describe the new mapping of the binding sites of five dystrophin mAbs to a few amino-acids within single exons. The phagedisplay method also confirmed previous mapping of MANEX1A (exon 1) and MANDRA1 (exon 77) by other methods. Of the 79dystrophin exons, mAbs are now available against single exons 1, 6, 8, 12, 13, 14, 17, 21, 26, 28, 38, 41, 43, 44, 45, 46, 47, 50, 51, 58,59, 62, 63, 75 and 77. Many have been used in clinical trials, as well as for diagnosis and studies of dystrophin isoforms.� 2013 Elsevier B.V. All rights reserved.
Keywords: Duchenne muscular dystrophy; Dystrophin; Monoclonal antibody; Epitope mapping; Clinical trial; Exon-skipping; Therapy; Phage display
1. Introduction
One of the long-term aims of the MDA MonoclonalAntibody Resource (www.glennmorris.org.uk/mabs.htm)has been to produce monoclonal antibodies (mAbs)against dystrophin that recognise specific amino-acidsequences spread throughout its 79 exons and 3684amino-acids.
We have paid particular attention to mAbs against theexons that are commonly deleted in Duchenne musculardystrophy; these can be used to characterise mutantBecker dystrophins at the protein level in muscle biopsies[1]. The antibodies have also been used to characterisesome of the short forms of dystrophin, including Dp71[2–4], Dp140 [5] and Dp260 [6], and to identify
0960-8966/$ - see front matter � 2013 Elsevier B.V. All rights reserved.
http://dx.doi.org/10.1016/j.nmd.2013.11.016
⇑ Corresponding author at: Wolfson Centre for InheritedNeuromuscular Disease, RJAH Orthopaedic Hospital, Oswestry SY107AG, UK. Tel.: +44 1691 404155; fax: +44 1691 404170.
E-mail address: [email protected] (G.E. Morris).
nNOS-binding regions of dystrophin [7]. Exon-specificmAbs can also be used to distinguish tissue-specificisoforms of full-length dystrophin. Thus, our MANEX1mAbs were mapped to the first three amino-acids of themuscle-specific isoform of dystrophin (first exon encodesLWWEEVEDCY) and so will not recognize either thebrain isoform (first exon encodes ED) or the isoform incardiac Purkinje cells (first exon encodes SEVSSD) [8].They have also been used to investigate the nature andorigin of revertant fibres in Duchenne patients [9] andmdx mice [10]. More recently, they have found a majorapplication in monitoring the success of clinical trials ofvarious experimental treatments for Duchenne MD. Thedystrophin in revertant fibres always has missing exons,whereas dystrophin supplied in gene or cell therapy trialsis usually full-length and will contain the exons deletedby the Duchenne mutation. Dystrophin-positive fibres inan early myoblast therapy trial were shown to be due torevertant fibres using exon-specific mAbs [11] and mostsubsequent trials have used this method to demonstrate

196 L.T. Lam et al. / Neuromuscular Disorders 24 (2014) 195–200
successful dystrophin replacement [12,13]. In an earlystudy, our exon-specific mAbs were used to distinguishrevertant fibres in a Duchenne patient fromdystrophin-positive fibres that might have arisen fromstem cells in donor bone marrow transplants givenseveral years earlier [14]. The exon-specific mAbs havealso been used in trials of gene therapy [15], antisenseoligo-based [16], morpholino-based [17,18] and stopcodon readthrough [19,20] therapies. MANDYS106 hasbeen particularly popular for “exon-skipping” approacheswhich aim to convert severe Duchenne patients intomilder, Becker-like patients [16–18].
These applications require mAbs that work well forboth immunolocalization and western blotting. Syntheticpeptide immunogens do not work well for either globularregions or the triple-helical rod regions of dystrophin,although we had some success with peptides from thefour non-helical linkers, or “hinges”, in the dystrophinrod [21]. For this reason, we have produced mostdystrophin mAbs by using large recombinant fragmentsas immunogens and mapping the epitopes subsequentlyto single exons or groups of exons.
Fig. 1. Use of MANDYS126 to isolate phage expressing surface peptideswhich mimic its epitope. The four steps shown are described in the resultssection. (A) There are few mAb-positive clones after the first round of bio-panning with a mixture of 13 mAbs. (B) After two rounds of biopanning,several E. coli. colonies reacted with the mAb mixture and ten of these(circled in B) were selected. (C) The ten colonies were streaked onto 13horizontal strips and each strip was incubated with a single mAb from themixture of 13. Only MANDYS126 gave a strong reaction. (D) Four of theten colonies were cloned again and 10 clones were tested for mAb reaction– this was repeated until all 10 clones were positive to ensure that thephage contains a single sequence only. One colony from each of theseclonings was amplified for isolation and sequencing of phage DNA.
2. Materials and methods
2.1. Epitope mapping
Epitope mapping using phage-displayed random peptidelibraries in filamentous phage was performed as previouslydescribed [22] using a modification of an earlier method[23]. Monoclonal antibody mixtures were diluted 1:50with Tris-buffered saline (TBS) and immobilised ontosterile 35 mm Petri dishes coated directly with 1 ml of1:200 dilution of rabbit-anti-[mouse Ig] in TBS(DAKOpatts, Denmark). Biopanning was performedusing a 15-mer peptide library in phage f88–4,maintained in the K91Kan strain of Escherichia coli andgenerously supplied by Smith (University of Missouri).Any remaining binding sites on the dishes were blockedusing 4% BSA in sterile TBS. A sample of the phagelibrary (1013 virions) was pre-incubated in dishes coatedwith the rabbit anti-mouse antibodies alone to ensure anybinding was specific for the target mAbs. Following thefirst round of biopanning, the bound phage were elutedand amplified by infection of K91Kan E. coli cells. Tworounds of biopanning were performed. Individualcolonies of the phage-infected cells after the secondround were grown on nitrocellulose membrane (BA85)and screened by western blotting to reveal positiveclones. Positive clones were subjected to western blottingwith individual mAbs from the mixture used forbiopanning. After blocking non-specific sites with 5%skimmed milk protein in TBS, membranes wereincubated with mAb supernatant (1/100 dilution in TBS).Antibody-reacting clones were visualized followingdevelopment with biotinylated horse anti-mouse Ig in a

Dystrophin mAbs:
MANEX1216D:DWLTKTEERTRKMEEEPLGPDLEDLKRQVQQHKVLQEDLEQ Dystrophin
DSSPYLMSPLGLDFD Peptide1 SPDDPPLPDLLYRSG Peptide2
MANEX1216B:QQHKVLQEDLEQEQVRVNSLTHMVVVVDESSGDHATAA Dystrophin
FAPDLTRFPSVVVST Peptide3
MANDYS19:ALKEKGQGPMFLDAD DystrophinSSADGGQGPHLLVRY Peptide4
MANDYS17:KDLSEMHEWMTQAEEEYLERDFEYKTPDELQKAVEEMKR Dystrophin
EHFAPSSPDFLERHF Peptide5 NVSPDALEWLVGSKC Peptide6
MANDYS126:KASIPLKELEQFNSDIQKLLEPLEAEIQQGVNL Dystrophin Exon 38
DRRFFQSDILALFSP Peptide7LPPPQQFHQDMMKLF Peptide8RLPLDTFHSDLSRLT Peptide9
MANEX1A:MLWWEEVEDCYEREDVQKKT Dystrophin Exon1
DTADLWWNSGTFLPA Peptide10
MANDRA1:EQLNNSFPSSRGRNTPGKPMREDTM Dystrophin Exon77THASYMSPSSAFTLQ Peptide11
Fig. 2. Alignment of peptide sequences with human dystrophin sequence. For each of the seven mAbs, the full 15-aa sequence of the peptide from thephage library is aligned with a partial dystrophin that matches. The matching amino-acids are shown bold and underlined in both peptide and dystrophinsequences.
Table 1Mapping of five monoclonal antibodies epitopes encoded by single dystrophin exons, using a phage-displayed peptide library.
Name of mAb Previous assignment New mapping assignment
MANDYS1216D Within exons 12-16 Exon 12: aa478-486, PLGPDLEDL
MANDYS1216B Within exons 12-16 Exon 13: aa514-516, VVV
MANDYS19 Within exons 20-21 Exon 21: aa901-904, GQGP
MANDYS17 Within exons 26-27 Exon 26: aa1181-1188, LERDFEYK
MANDYS126 Within exons 38-39 Exon 38: aa1787-1795, FNSDIQKL
L.T. Lam et al. / Neuromuscular Disorders 24 (2014) 195–200 197
Vectastain ABC kit (Vector Labs, Burlingame, CA) anddiaminobenzidine substrate (Sigma; 0.4 mg/ml). PhageDNA was purified from positive clones by the phenol/chloroform method and sequenced using primer:50-AGTAGCAGAAGCCTGAAGA-30.
2.2. Immunofluorescence microscopy
Culture supernatants containing monoclonal antibodieswere diluted 1:10 in PBS and incubated on 5–7 micronfrozen muscle sections without fixation for 1 h. Primaryantibody was then removed by washing four times with
PBS. Sections were then incubated with 5 lg/ml goatanti-mouse ALEXA 488 (Molecular Probes, Eugene,Oregon, USA) secondary antibody diluted in PBScontaining 1% horse serum, 1% fetal bovine serum and0.1% BSA, for 1 h. DAPI (diamidino phenylindole: 1 lg/ml) was added for the final 5 min of incubation tocounterstain nuclei before mounting in Hydromount(Merck). Images were obtained using a Leica SP5confocal microscope with a 20� objective under identicalcapture conditions and laser power. Human musclesections were obtained with the appropriate informedconsent and ethical approval.
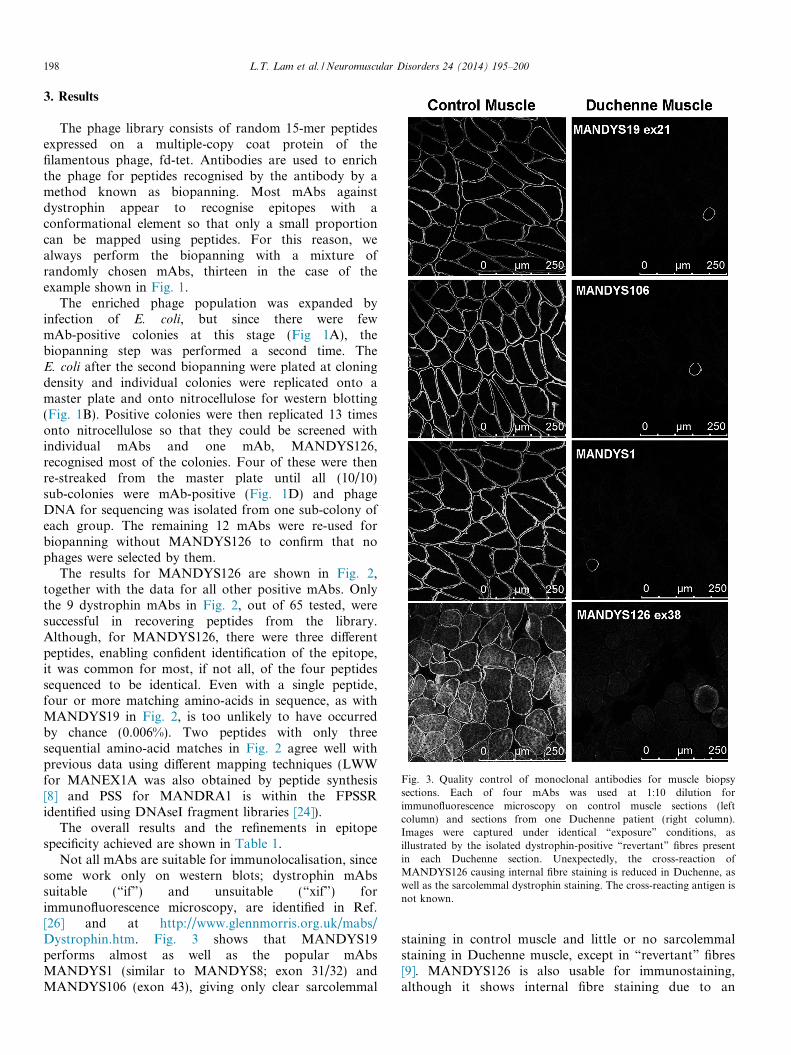
Fig. 3. Quality control of monoclonal antibodies for muscle biopsysections. Each of four mAbs was used at 1:10 dilution forimmunofluorescence microscopy on control muscle sections (leftcolumn) and sections from one Duchenne patient (right column).Images were captured under identical “exposure” conditions, asillustrated by the isolated dystrophin-positive “revertant” fibres presentin each Duchenne section. Unexpectedly, the cross-reaction ofMANDYS126 causing internal fibre staining is reduced in Duchenne, aswell as the sarcolemmal dystrophin staining. The cross-reacting antigen isnot known.
198 L.T. Lam et al. / Neuromuscular Disorders 24 (2014) 195–200
3. Results
The phage library consists of random 15-mer peptidesexpressed on a multiple-copy coat protein of thefilamentous phage, fd-tet. Antibodies are used to enrichthe phage for peptides recognised by the antibody by amethod known as biopanning. Most mAbs againstdystrophin appear to recognise epitopes with aconformational element so that only a small proportioncan be mapped using peptides. For this reason, wealways perform the biopanning with a mixture ofrandomly chosen mAbs, thirteen in the case of theexample shown in Fig. 1.
The enriched phage population was expanded byinfection of E. coli, but since there were fewmAb-positive colonies at this stage (Fig 1A), thebiopanning step was performed a second time. TheE. coli after the second biopanning were plated at cloningdensity and individual colonies were replicated onto amaster plate and onto nitrocellulose for western blotting(Fig. 1B). Positive colonies were then replicated 13 timesonto nitrocellulose so that they could be screened withindividual mAbs and one mAb, MANDYS126,recognised most of the colonies. Four of these were thenre-streaked from the master plate until all (10/10)sub-colonies were mAb-positive (Fig. 1D) and phageDNA for sequencing was isolated from one sub-colony ofeach group. The remaining 12 mAbs were re-used forbiopanning without MANDYS126 to confirm that nophages were selected by them.
The results for MANDYS126 are shown in Fig. 2,together with the data for all other positive mAbs. Onlythe 9 dystrophin mAbs in Fig. 2, out of 65 tested, weresuccessful in recovering peptides from the library.Although, for MANDYS126, there were three differentpeptides, enabling confident identification of the epitope,it was common for most, if not all, of the four peptidessequenced to be identical. Even with a single peptide,four or more matching amino-acids in sequence, as withMANDYS19 in Fig. 2, is too unlikely to have occurredby chance (0.006%). Two peptides with only threesequential amino-acid matches in Fig. 2 agree well withprevious data using different mapping techniques (LWWfor MANEX1A was also obtained by peptide synthesis[8] and PSS for MANDRA1 is within the FPSSRidentified using DNAseI fragment libraries [24]).
The overall results and the refinements in epitopespecificity achieved are shown in Table 1.
Not all mAbs are suitable for immunolocalisation, sincesome work only on western blots; dystrophin mAbssuitable (“if”) and unsuitable (“xif”) forimmunofluorescence microscopy, are identified in Ref.[26] and at http://www.glennmorris.org.uk/mabs/Dystrophin.htm. Fig. 3 shows that MANDYS19performs almost as well as the popular mAbsMANDYS1 (similar to MANDYS8; exon 31/32) andMANDYS106 (exon 43), giving only clear sarcolemmal
staining in control muscle and little or no sarcolemmalstaining in Duchenne muscle, except in “revertant” fibres[9]. MANDYS126 is also usable for immunostaining,although it shows internal fibre staining due to an

L.T. Lam et al. / Neuromuscular Disorders 24 (2014) 195–200 199
unidentified cross-reaction (Fig. 3). Of the remaining mAbsmapped in Fig. 2, MANEX1A and MANDRA1 also workvery well on frozen sections (data not shown), but the otherthree are recommended for western blotting only [26].
4. Discussion
Many different approaches have been used in theattempt to replace the missing dystrophin protein inDuchenne muscle. To assess whether any of thesetreatments has been effective in clinical trials, it isnecessary to use antibodies against dystrophin todetermine whether dystrophin has been produced duringthe therapy. Although improved muscle function is theultimate aim, it is important to confirm that suchimprovement is due to new dystrophin production, or, ifno improvement was observed, to determine whether newdystrophin is present, though ineffective. Wheretreatments are performed by local injection, it is usual totake a muscle biopsy near the site of injection and todetermine its dystrophin status by immunocytochemistry.This technique can detect even a small number ofdystrophin-positive fibres in a biopsy, whereas westernblotting may only work well when the proportion ofpositive fibres is quite high.
Because of the important requirement for goodimmunostaining of the sarcolemma, we have generallyused large dystrophin fragments as immunogens andscreened for mAbs that work well forimmunocytochemistry of human muscle biopsies. Thismay explain why only 9 out of 65 dystrophin mAbstested recognised 15-mer peptides in the phage library;the other 56 mAbs tested may require longer amino-acidsequences to mimic the dystrophin epitope that theyrecognise. These 56 mAbs include the popularMANDYS106 mAb, which has been mapped to exon 43by a dystrophin fragmentation method [24]. Less likely isthe possibility that the relevant peptide is missing fromthe library; sufficient phage particles (1013 which is>factorial 15) in the initial biopanning step should ensurethat all possible random 15-mers are available forselection, but this assumes a truly random library (thelibraries are generated by chemical synthesis of 45 basesusing a mixture of all 4 bases at each synthetic step). Wedeveloped the method of biopanning with a mixture of10–20 mAbs when it became clear that only a smallproportion of mAbs raised against large recombinantimmunogens can be mapped in this way (because manyhave a conformational element in their epitope whichpeptides cannot mimic).
It is interesting that MANDYS17 has been mapped toamino-acids 1181–1188 for the first time here (Table 1),because MANDYS18 was previously mapped to1181–1187 by a transposon mutagenesis method [25].Using the transposon method, MANDYS18 did not bindto aa815–1181, but did bind to aa815–1187, whereasMANDYS17 bound neither of those two fragments but
did bind aa815–1205. The two mAbs recognise asequence that lies on a turn between two helices of thetriple-helical coiled-coil structure of the dystrophin roddomain [25], so it is possible that MANDYS17 prefers alarger epitope than MANDYS18 for efficient binding. Asin all the data presented here, peptide mapping resultsshould preferably supported by at least one [24], andpreferably two [25], alternative mapping methods.
All the dystrophin mAbs described in this study arefreely available for non-profit research from the MDAMonoclonal Antibody Resource (www.glennmorris.org.uk/mabs.htm).
Acknowledgements
This study was supported by a TranslationalInfrastructure Research Grant from the MuscularDystrophy Association (USA). We thank George P.Smith (University of Missouri) for the generous gift ofphage libraries and Prof. Caroline Sewry (RJAHOrthopaedic Hospital, Oswestry, UK) for muscle biopsysections and comments on the manuscript.
References
[1] Le TT, Nguyen thi Man, Hori S, Sewry CA, Dubowitz V, Morris GE.Characterization of genetic deletions in Becker muscular dystrophyusing monoclonal antibodies against a deletion-prone region ofdystrophin. Am J Med Genet 1995;58:177–86.
[2] Lederfein D, Levy N, Augier N, et al. 71kD protein is a majorproduct of the Duchenne muscular dystrophy gene in brain and othernon-muscle tissues. Proc Natl Acad Sci USA 1992;89:5346–50.
[3] Hugnot JP, Gilgenkrantz H, Vincent N, et al. Novel products of thedystrophin gene: a distal transcript initiated from a unique alternativefirst exon encoding a 75 kDa protein widely distributed in non-muscletissues. Proc Natl Acad Sci USA 1992;89:7506–10.
[4] Blake DJ, Love DR, Tinsley J, et al. Characterization of a 4.8kbtranscript from the Duchenne muscular dystrophy locus expressed inSchwannoma cells. Hum Mol Genet 1992;1:103–9.
[5] Morris GE, Simmons C, Nguyen thi Man. Apo-dystrophins (Dp140and Dp71) and dystrophin splicing isoforms in developing brain.Biochem Biophys Res Commun 1995;215:361–7.
[6] d’Souza VN, Nguyen thi Man, Morris GE, Karges W, Pillers DM,Ray PN. A novel dystrophin isoform is required for normal retinalelectrophysiology. Hum Mol Genet 1995;4:837–42.
[7] Lai Y, Zhao J, Yue Y, Duan D. a2 and a3 helices of dystrophin R16and R17 frame a microdomain in the a1 helix of dystrophin R17 forneuronal NOS binding. Proc Natl Acad Sci USA 2013;110:525–30.
[8] Le TT, Nguyen thi Man, Love DR, Helliwell TR, Davies KE, MorrisGE. Monoclonal antibodies against the muscle-specific N-terminus ofdystrophin: characterization of dystrophin in a muscular dystrophypatient with a frameshift deletion of exons 3–7. Am J Hum Genet1993;53:131–9.
[9] Le TT, Nguyen thi Man, Helliwell TR, Morris GE. Characterizationof revertant muscle fibres in Duchenne muscular dystrophy usingexon-specific monoclonal antibodies against dystrophin. Am J HumGenet 1995;56:725–31.
[10] Lu QL, Morris GE, Wilton SD, et al. Massive idiosyncratic exonskipping corrects the nonsense mutation in dystrophic mouse muscleand produces functional revertant fibres by clonal expansion. J CellBiol 2000;148:985–96.
[11] Partridge TA, Lu QL, Morris GE. Hoffmann EP Is myoblasttransplantation effective? Nature Med 1998;4:1208–9.

200 L.T. Lam et al. / Neuromuscular Disorders 24 (2014) 195–200
[12] Mendell JR, Kissel JT, Amato AA, et al. Myoblast transfer in thetreatment of Duchenne’s muscular dystrophy. N Engl J Med1993;333:832–8.
[13] Skuk D, Roy B, Goulet M, et al. Dystrophin expression in myofibersof Duchenne muscular dystrophy patients following intramuscularinjections of normal myogenic cells. Mol Ther 2004;9:475–82.
[14] Gussoni E, Bennett RR, Gilgoff I, et al. Detection of donor nuclei inthe muscles of a Duchenne muscular dystrophy patient 13 yearsfollowing bone marrow transplantation. J Clin Invest2002;110:807–14.
[15] Romero NB, Braun S, Benveniste O, et al. Phase I study ofdystrophin plasmid-based gene therapy in Duchenne/Beckermuscular dystrophy. Hum Gene Therapy 2004;15:1065–76.
[16] Goemans NM, Tulinius M, van den Akker JT, et al. Systemicadministration of PRO051 in Duchenne’s muscular dystrophy. NEngl J Med 2011;364:1513–22.
[17] Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration ofdystrophin expression with the morpholino oligomer AVI-4658 inDuchenne muscular dystrophy: a single-blind, placebo-controlled,dose-escalation, proof-of-concept study. Lancet Neurol 2009;8:918–28.
[18] Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping anddystrophin restoration in patients with Duchenne muscular dystrophyafter systemic phosphorodiamidate morpholino oligomer treatment:an open-label, phase 2, dose-escalation study. Lancet2011;378:595–605.
[19] Welch EM, Barton ER, Zhuo J, et al. PTC124 targets geneticdisorders caused by nonsense mutations. Nature (Lond)2007;447:87–91.
[20] Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-inducedreadthrough of stop codons in Duchenne muscular dystrophy. AnnNeurol 2010;67:771–80.
[21] Ahmed N, Nguyen thi Man, Morris GE. Flexible hinges indystrophin. Biochem Soc Trans 1998;26:S310.
[22] Pereboev A, Morris GE. Reiterative screening of phage displaypeptide libraries for epitope mapping. In: Epitope Mapping ProtocolsHumana Press; (Methods in Mol. Biol. 66) 1996. pp. 195–206.
[23] Scott JK, Smith GP. Searching for peptide ligands with an epitopelibrary. Science 1990;249:386–90.
[24] Nguyen thi Man, Morris GE. Use of epitope libraries to identifyexon-specific monoclonal antibodies for characterization of altereddystrophins in muscular dystrophy. Am J Hum Genet1993;52:1057–66.
[25] Sedgwick SG, Nguyen thi Man, Ellis JM, Crowne H, Morris GE.Rapid mapping by transposon mutagenesis of epitopes on themuscular dystrophy protein, dystrophin. Nucleic Acids Res1991;19:5889–94.
[26] Morris GE, Nguyen M, Sewry CA. Monitoring Duchenne musculardystrophy gene therapy with epitope-specific monoclonal antibodies.In: Dongsheng D, editor. Muscle Gene Therapy; 2011: 709, pp. 39–62.





























