Embed Size (px)
Citation preview

- 1 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
Monitoring Bacterial Community Shifts in the SBR Treating Agricultural Processing Wastewater
Sirinan Thacharoen1* Mujalin K. Pholchan2 and Piyanuch Niamsup3
1 Graduate student Program in Biotechnology, Faculty of Science, Maejo University, Chiang Mai 50290,
Thailand; 2 *Lecturer, Program in Environmental technology, Faculty of Science, Maejo University, Chiang Mai 50290, Thailand; 3Assistant Professor, Program in Biotechnology, Faculty of Science, Maejo
University, Chiang Mai 50290, Thailand *Phone : (+66) 856220285, Fax : (+66) 5387-8225, E-mail : [email protected]
ABSTRACT Agricultural industrial wastewater is typically treated by the biological processes, especially the activated sludge system. However, the performance of the systems relies not only on system configurations but also on microbial community and environmental conditions. The aim of this study is an application of PCR-DGGE technique to estimate and monitor the change of bacterial diversity in the Sequencing Batch Reactor (SBR) treating the corn processing wastewater. The DGGE patterns showed that the bacterial community structures in the SBR were dissimilar due to organic loading and wastewater characteristics. Bacterial communities in the SBR system were dominated by the phyla Proteobacteria and Bacteroidetes which were usually observed in the activated sludge processes and very diverse. The results obtained can be used as a guide for future treatment plant development and for the a better system performance. Keywords: Sequencing Batch Reactor (SBR), PCR-DGGE, activated sludge, bacterial diversity, bacterial community INTRODUCTION Agricultural industrial wastewater is typically treated by biological processes especially the activated sludge systems. Sequencing Batch Reactor (SBR) is one of the activated sludge processes that involve the aerobic microbial metabolism. This microorganism is a key factor for enhancing the performance of biological wastewater treatment. Thus, the performance of the systems relies not only on systems configurations but also on microbial community and environmental conditions. Previous studies on microbial diversity focused on the relationship between microbial diversity and bioreactor performance, in lab scale system treated the synthetic wastewater [1]. The combination of Polymerase Chain Reaction (PCR) amplification of 16S rRNA genes with denaturing gradient gel electrophoresis (DGGE) analysis has permits direct visualization and rapid comparison of the structure of bacterial community in the environmental samples [2]. However, only a few studies have reported on microbial communities and system stabilities in full scale biological treatment plants[3]. The aim of this study was an application of PCR-DGGE technique to investigate the monitor changes in the bacterial community in the SBR receiving the wastewater from the agricultural processing factory during a year. The understanding of the microbial communities in the SBR system and the correlation between system performances and bacterial diversity may be helpful in solving the system failures. This could enable us to rapidly monitor and access the process performances and also to optimize the biological degradation processes in the existing SBR system. METHODOLOGY 1. Sampling and chemical analysis The wastewater and sludge samples were obtained from the wastewater treatment plant of the sweet corn processing factory in Hangchat, Lampang, Thailand. The raw wastewater entering to the SBR was collected every month from February to December 2012. Samples of raw wastewater were analyzed for COD, NH3-N, NO3-N, TKN, PO4-P, SS and VSS according to the Standard Methods [4]. Sludge sample were taken from the SBR monthly, preserved immediately in 50% ethanol and stored at -20ºC prior to the molecular analysis.

- 2 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
A
B Fig.1 Layout of the wastewater treatment plant (A) Sequencing Batch Reactor; SBR (B)
2. Nucleic acid extraction and PCR amplification of 16S rDNA gene Total community DNA was extracted from the sludge using DNA extraction kit (Nucleospin® soil, Germanny) following the manufacturer’s instruction. The variable V3 region of 16S rDNA was amplified by using primers targeted to conserved regions of the 16S rDNA gene:341F (TACGGGAGGCAGCAG) and 518R (ATTACCGCGCTGCTGG) [5]. Changes in the microbial diversity were monitored by performing DGGE of polymerase chain reaction amplified 16S rRNA gene segments from the bacterial community according to Pholchan and co workers [1]. Primer 341F had at its 5’ end an additional 40-nucleotide GC-rich sequence to facilitate separation by DGGE. Each sample contained 1 μL of extracted DNA and 49 μL of master mix (1 μL of 0.01 mM stock forward primer (1st base, Malaysia), 1 μL of 0.01 mM stock reward primer (1st base, Malaysia), 25 μL of Quick tag (HS Dyemix), 22 μL of molecular grade water (nuclease-free water, AppliChem)). The 16S rDNA gene fragment were amplified in PCR reaction mixture using a PTC-200 Peltier Thermal Cycler as follows: initial denaturation at 95°C for 5 min , denature 94°C 0.5 min followed by a touchdown procedure. In particular, the annealing temperature was initially set at 65°C and then decreased by 0.5°C every cycle until it reached 54°C; then 35 additional cycles were elongation 72°C 1 min followed by primer extension for 5 min at 72°C. Once the PCR was completed, the templates were stored at –20ºC. 3. Denaturing gradient gel electrophoresis (DGGE) Changes in the microbial diversity were investigated through PCR-DGGE analysis. DGGE was performed using a Bio-Rad DCode system (Bio-Rad, USA), using a 10% polyacrylamide gel with 30% to 55% denaturing concentrations in 1X TAE buffer (Diluted from 50X TAE buffer; Bio-Rad, USA) as previously described (Pholchan et al., 2010). Electrophoresis was performed at the constant voltage of 200 V 60º C for 4 h. After electrophoresis, the gel was stained for 15 min in 250 ml 1X TAE containing ethidium bromide (Bio-Rad, USA). DGGE bands were visualized under an UV transilluminator Genesnap program (SynGene, UK). Individual band patterns were compared with each other using the pairwise similarity coefficient of Dice (S) as follows :S = 2j / (�+b) where, � is the number of DGGE bands in pattern 1, b is the number of bands in pattern 2 and j is the number of common bands. Then the cluster analysis of band patterns was performed using the unweighted-pair group method using average linkages (UPGMA) (Genetool version 3.02.00 SynGene, UK) 4. DNA sequence and phylogenetic analysis Individual selected bands were excised from DGGE gels, placed into sterilized 1.5 ml tube containing 30 µl of nuclease free water and stored overnight at 4ºC. 10 µl of eluted DNA was used as the template 10 µl for amplification with the primer 341F_GC and 518R [5], as described above. For sequencing analysis (First base, Malaysia), PCR products were purified with RBC BIOSCIENCE PCR fragments extraction kit (Taiwan) and used as the templates for sequencing reactions. To determine the phylogenetic affiliation, similarity search was performed using the BLAST program. The nucleotide sequences were aligned with the

- 3 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
MUSCLE program and the phylogenetic trees were completed using the Mega6 program with neighbor-joinin method [6]. RESULTS AND DISCUSSIONS Physical and chemical characterization The wastewater and sludge samples in this study were obtained from the full scaled SBR of corn-processing factory during February to December 2012.The flow rate of wastewater range from 450-800 m3/day. The physical and chemical characteristics of influents during the sampling period are shown in fig. 2. The COD has found in the range of 833-5333 mg.L-1 with the highest amount shown in December (5333 mg.L-1) . The total nitrogen ranged from 4.2-144.27 mgL-1.
A
B
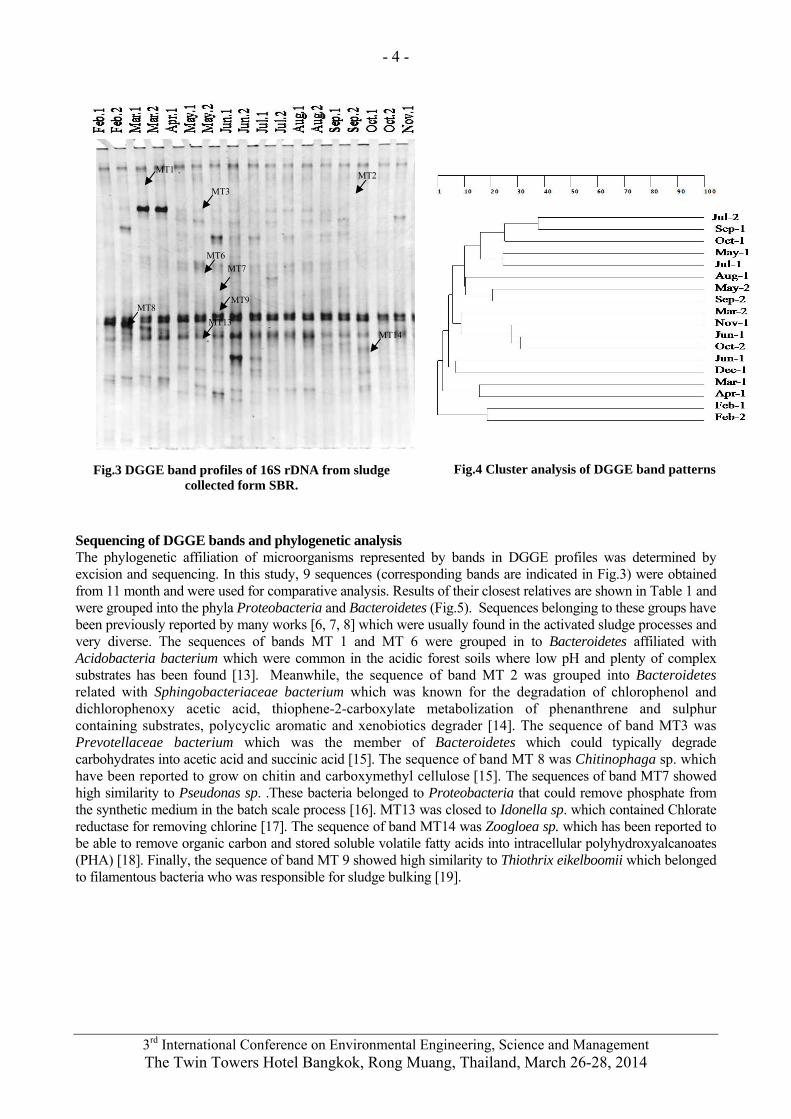
Fig.2 Changes of chemical parameters: COD and COD removal efficiency (A) and TKN, NH3-N and NO3-N (B) from February to December 2012 DGGE analysis of bacteria community The dynamics of the bacterial communities in the SBR and the cluster analysis are shown in Fig.3 and Fig.4. In general, DGGE analysis showed that there were changes in band profile over operational time as well as variation time in band intensity. Results also showed that the number of bands per lane varied from 9-15. Some bands were found to be common in almost all samples (e.g., bands MT6 and MT7), while others were only present in some months (e.g. bands MT1, MT2, MT3, and MT9). It is speculated that these common bands were the essential microorganisms who played the key role in the degradation process, while all unique bands were could link the change of wastewater property [7, 8]. These differences indicated that some variance of bacterial community compositions existed during the period. Cluster analysis of DGGE patterns were compared with one another by pair wise similarity coefficient of Dice (S) with ranged from 0.07 to 1.00. The cluster analysis revealed that bacterial community structures in the SBR of the sweet corn processing factory were dissimilar due to influent loading, wastewater characteristics and maybe operational problem (S values of 0.08-0.38). The samples could be grouped into five clusters; group 1, corresponding to samples collected from May, July, August, September, and October1; group 2 which included sample collected from March, Jun, October 2, and November; group 3 with sample collected from June and December; group 4 obtained from March and April; and group 5 which included sample collected in February. Interestingly, both wastewater loadings and influents were found to be strongly correlated with differences on community structures. High COD loading group (665-800 m3/day) were found on samples obtained in cluster group 1 and 4, while medium loading groups (500-700 m3/day) were found on sample in group 2, 3, and 5. These results indicated that the major shifts in community occurred on changes of influent loading basis, similar to the results of Yi and co workers [7, 8, and 9]. They found that the influent BOD or substrate concentrations played the important key role in changing microbial diversity. Also, there is some evidence demonstrating the bacterial community exhibited some changes due to influent load and COD of influent [10, 11, and 12].
- 4 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
Fig.3 DGGE band profiles of 16S rDNA from sludge collected form SBR.
Fig.4 Cluster analysis of DGGE band patterns
Sequencing of DGGE bands and phylogenetic analysis The phylogenetic affiliation of microorganisms represented by bands in DGGE profiles was determined by excision and sequencing. In this study, 9 sequences (corresponding bands are indicated in Fig.3) were obtained from 11 month and were used for comparative analysis. Results of their closest relatives are shown in Table 1 and were grouped into the phyla Proteobacteria and Bacteroidetes (Fig.5). Sequences belonging to these groups have been previously reported by many works [6, 7, 8] which were usually found in the activated sludge processes and very diverse. The sequences of bands MT 1 and MT 6 were grouped in to Bacteroidetes affiliated with Acidobacteria bacterium which were common in the acidic forest soils where low pH and plenty of complex substrates has been found [13]. Meanwhile, the sequence of band MT 2 was grouped into Bacteroidetes related with Sphingobacteriaceae bacterium which was known for the degradation of chlorophenol and dichlorophenoxy acetic acid, thiophene-2-carboxylate metabolization of phenanthrene and sulphur containing substrates, polycyclic aromatic and xenobiotics degrader [14]. The sequence of band MT3 was Prevotellaceae bacterium which was the member of Bacteroidetes which could typically degrade carbohydrates into acetic acid and succinic acid [15]. The sequence of band MT 8 was Chitinophaga sp. which have been reported to grow on chitin and carboxymethyl cellulose [15]. The sequences of band MT7 showed high similarity to Pseudonas sp. .These bacteria belonged to Proteobacteria that could remove phosphate from the synthetic medium in the batch scale process [16]. MT13 was closed to Idonella sp. which contained Chlorate reductase for removing chlorine [17]. The sequence of band MT14 was Zoogloea sp. which has been reported to be able to remove organic carbon and stored soluble volatile fatty acids into intracellular polyhydroxyalcanoates (PHA) [18]. Finally, the sequence of band MT 9 showed high similarity to Thiothrix eikelboomii which belonged to filamentous bacteria who was responsible for sludge bulking [19].
MT1 MT2
MT3
MT6 MT7
MT8 MT13
MT14
MT9

- 5 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
Table 1 Similarity to the closest relative of amplified 16S rRNA gene sequences excised from DGGE gels Band
number Similarity
(%) Organism Phylum Isolation source Accession
number
MT1
98 Acidobacteria bacterium IGE-017
Bacteroidetes
surface soil
GU187032.1
MT2 88
99
Sphingobacteriaceae bacterium A22.1 uncultured bacterium
Bacteroidetes
-
Glacier foreland sediments
JN662529.1
KC925223.1
MT3
98 Prevotellaceae bacterium WR041 Bacteroidetes rice straw residue in a methanogenic reactor of cattle farm waste
AB298732.2
MT6
92 Acidobacteriaceae bacterium PA-34
Bacteroidetes acidic mine drainage
KF663609.1
MT7
87 Pseudomonas sp. 37zhy
Proteobacteria Pseudomonas sp. 37zhy
AM411060.1
MT8
89 Chitinophaga sp. A5I53
Bacteroidetes soil of sugar cane culture
FJ890894.1
MT9
100 Thiothrix eikelboomii
Proteobacteria bulking activated sludge
AB042542.1
MT13
89 Ideonella sp. CUW2
Proteobacteria polycyclic aromatic hydrocarbon
JX983162.1
MT14
96 Zoogloea sp. AR30
Proteobacteria Zoogloea sp. AR30
JN585324.1

- 6 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
Fig.5 Neighbor-joining phylogenetic tree based on partial 16S rDNA sequences derived from DGGE bands.

- 7 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
CONCLUSION The study of bacterial community shifts from the SBR is essential to understand the full scaled biological treatment process as well as to solve the system operational problems for improving the planting design. Considering that little information is available regarding to the microbial community in the complex system such as the biological wastewater treatment plants, the PCR-DGGE approach applied has become an effective tool to get a better understanding of the community structure. The PCR-DGGE method applied here has provided insights into the shifts over 11 months of bacterial communities from the SBR treating the wastewater from the sweet corn processing factory. The following conclusions can be drawn from the present study: 1. This result strongly indicated that the bacterial community structures were dissimilar and changed obviously during the period. 2. The different bacterial compositions were influenced by wastewater loading and characteristics. The results obtained can be used as a guide for the future treatment plant development and for a better system performance. ACKNOWLEDGEMENT This work was financially supported by Thailand Research Fund, Bangkok, Thailand, (Project number MRG 5480103) and Research and Researchers for Industries-RRI, Bankok, Thailand, (Project number MSD56I0138). The authors gratefully acknowledge to the Majestic Food Industry Co., Ltd. for their cooperation. REFERENCE [1] Pholchan, M.K., Joana de C. Baptista, R.J. Davenport and T.P. Curtis. 2010. Systematic study of the
effect of operating variables on reactor performance and microbial diversity in laboratory-scale activated sludge reactors. Water Res. 44:134-352
[2] Sanz, J.S. and T. Kochling. 2007. Molecular biology techniques used in wastewater treatment: An overview. Process Biochem. 42: 119–133
[3] Wang, X., X. Wen., C. Criddle., H. Yan., Y. Zhang and K. Ding. 2010. Bacterial community dynamics in two full-scale wastewater treatment systems with functional stability. Appl. Microbiol. 109: 1218–1226
[4] APHA, AWWA, and WEF. 2005. Standard Methods for the Examination of Water and Wastewater, twentieth ed. American Public Health Association, Washington, DC.
[5] Muyzer, G., E.C. de Waal and A.G. Uitterlinden.1993. Profiling of complex microbial populations by Denaturing Gradient Gel Electrophoresis analysis of Polymerase Chain Reaction-Amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59: 695-700.
[6] Edgar, R.C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32(5):1792-1797.
Moura, A., M. Tacao, I, Henriques, J. Dias., P. Ferreira and A. Correia. 2009. Characterization of bacterial diversity in two aerated lagoons of a wastewater treatment plant using PCR-DGGE analysis. Microbiol. Res. 164(5):560-569.
[7] Von Wintzingerode, F., B. Selent.,W. Hegemann and U.B. Göbel. 1999. Phylogenetic analysis of an anaerobic, trichlorobenzene transforming microbial consortium. Appl. Environ. Microb. 65: 283-286.
[8] Zhang, R.G., K.M. Pappas, J.L. Brace, C.P. Miller, T. Oulmassov, J.M. Molyneaux, J.C.Anderson, J.K. Bashkin, C.S. Winans and A. Joachimiak. 2002. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature . 417:971–974.
[9] Yi, T., E.H. Lee., S. Kang., J. Shin and K.S. Cho. 2012. Structure and dynamics of microbial community in full-scale activated sludge reactors. J. Ind. Microbiol. Biot. 39: 19–25.
[10] Yu, Z., and William,W.M. 2001. Bacterial Diversity and Community Structure in an Aerated Lagoon Revealed by Ribosomal Intergenic Spacer Analyses and 16S Ribosomal DNA Sequencing. Appl Environ Microbiol. 67(4): 1565–1574.
[11] Hesham, A.E.L., Qi, R., and Yang, M. 2011. Comparison of bacterial community structures in two systems of a sewage treatment plant using PCR-DGGE analysis. Journal of Environmental Sciences. 23(12): 2049-2054.

- 8 -
3rd International Conference on Environmental Engineering, Science and Management The Twin Towers Hotel Bangkok, Rong Muang, Thailand, March 26-28, 2014
[12] Kwiatkoska, A.C., Magdalena, Z., and Irena, W.B. 2012. Impact of Operational Parameters on Bacterial Community in a Full-Scale Municipal Wastewater Treatment Plant. Polish Journal of Microbiology. 61(1): 41–49.
[13] Dalahmeh, S. 2013. Bark and Charcoal Filters for Greywater Treatment. Doctoral Thesis, Swedish
University of Agricultural Sciences. P. 31. [14] Hesham, A.E.L., Qi, R., and Yang, M. 2011. Comparison fo bacterial community structures in two
systems of a sewage treatment plant using PCR-DGGE analysis. Journal of Environmental Sciences. 23(1): 2049-2054.
[15] Peihong, S., Han, F., Su, S., Zhang, J., Chen, Z., Li, J., Gan, J., Feng, B., and Wu, B. 2014. Using pig manure to promote fermentation of sugarcane molasses alcohol wastewater and its effects on microbial community structure. Bioresource Technology. 155:323–329.
[16] Hayes, A.C., Steven, N.L.,2 and Allen, D.G. 2010. Growth Kinetics of Hyphomicrobium and Thiobacillus spp. in Mixed Cultures Degrading Dimethyl Sulfide and Methanol. Appl. Environ. Microbiol.76 (16): 5423–543.
[17] Krishnaswamy,U., Muthusamy, M., and Perumalsamy, L. 2009. Studies on the Efficiency of the Removal of Phosphate Using Bacterial Consortium for the Biotreatment of Phosphate Wastewater. European Journal of Applied Sciences. 1(1): 06-15.
[18] David, W., Sébastien, G., Samuel, L., Emmanuelle, R., Pierre, R., Sirous, E., and Christof, H. 2010. Predominance of Zoogloea sp. during aerobic granular sludge biofilms development for wastewater treatment. SSM, 69 th Annual Assembly, Zurich, Switzerland.
[19] Wagner, M., Loy, A., Nogueira, R., Purkhold, U., Lee, N., and Daims, H. 2002. Microbial community composition and function in wastewater treatment plants. Antonie van Leeuwenhoek. 81: 665-680.





























