Embed Size (px)
Citation preview

Molecular factors influencing
epithelial-mesenchymal
transition in breast cancer
Gisela Nilsson
Department of Medical Biochemistry and Cell Biology
Institute of Biomedicine
Sahlgrenska Academy at University of Gothenburg
Gothenburg 2014

Molecular factors influencing epithelial-mesenchymal transition in breast
cancer
© Gisela Nilsson 2014
ISBN 978-91-628-9116-9
Printed in Gothenburg, Sweden 2014
Kompendiet, Göteborg

To my family


Molecular factors influencing epithelial-mesenchymal
transition in breast cancer
Gisela Nilsson
Department of Medical Biochemistry and Cell Biology, Institute of
Biomedicine
Sahlgrenska Academy at University of Gothenburg, Göteborg, Sweden
ABSTRACT
Epithelial-mesenchymal transition (EMT) is a developmental process defined by loss
of epithelial characteristics and acquisition of mesenchymal phenotype. EMT or
similar processes are also implicated in carcinoma cell invasion and the progression
of breast carcinoma to metastasis. In a cell model system for mammary
carcinogenesis it has previously been shown that signaling from the oncogenic
receptor tyrosine kinase c-erbB2 (HER2), frequently overexpressed in mammary
cancers, induces EMT. In this system, c-erbB2-induced EMT was significantly
delayed by high cell-density and cell-cell-dissociation occurred before
downregulation of the epithelial adhesion molecule E-cadherin. Loss of E-cadherin
expression is generally viewed as a fundamental event in EMT. This thesis shows
that ectopic expression of E-cadherin concomitant with c-erbB2 signaling did not
hinder the progression of EMT. E-cadherin expressed in mesenchymal cells had a
weaker attachment to the cytoskeleton, implicating that rearrangement of the
cytoskeleton is an important mechanism in EMT-associated cell-cell-dissociation.
Expression of dominant negative E-cadherin weakened cell-cell adhesion but did not
enable EMT at high cell-density. These finding indicate that loss of E-cadherin is a
consequence rather than a cause of EMT and that density-dependent inhibition of
EMT is not mediated by E-cadherin. The expression of the transcription factor
nuclear factor I-C2 (NFI-C2) is lost during mammary tumor progression and NFI-C2
has been shown to counteract EMT by repressing the transcription factor Forkhead
box F1 (FoxF1). FoxF1 induces EMT and invasiveness in breast cancer cells. In this
thesis, Affymetrix microarray was used to find oppositely regulated targets of NFI-
C2 and FoxF1. The extracellular matrix enzyme lysyl oxidase (LOX) was found to
be negatively regulated by NFI-C2 and positively regulated by FoxF1 and
responsible for the increased invasiveness caused by FoxF1 overexpression. A
signaling pathway was identified where FoxF1-induced upregulation of LOX
activated focal adhesion kinase, subsequently suppressing Smad2 activity. In parallel,
overexpression of FoxF1 activated the p38 MAPK signaling pathway. These findings
give new insights into the regulation of signaling pathways known to be important
during breast tumor progression. Based on the findings that NFI-C2 is lost during
breast tumor progression and suppresses EMT, the prognostic value of NFI-C2 in a
mixed cohort of breast cancer patients was investigated. NFI-C2 was found to be a
powerful prognostic marker associated with good prognosis in breast cancer.
Keywords: breast cancer, epithelial-mesenchymal transition, c-erbB2, E-cadherin,
NFI-C2, FoxF1, LOX
ISBN: 978-91-628-9116-9


i
LIST OF PAPERS
This thesis is based on the following studies, referred to in the text by their
Roman numerals.
I. Nilsson GM., Akhtar N., Kannius-Janson M., Baeckström
D. Loss of E-cadherin expression is not a prerequisite for c-
erbB2-induced epithelial-mesenchymal transition.
International Journal of Oncology. 2014 Jul;45(1):82-94.
II. Nilsson G., Kannius-Janson M. Forkhead box F1 promotes
breast cancer cell migration by upregulating lysyl oxidase
and suppressing Smad2/3 signaling.
Manuscript.
III. Nilsson J., Nilsson G., Nemes S., Kovács A., Helou K.,
Jirström K., Kannius-Janson M. Nuclear factor I-C2 is a
powerful prognostic marker in breast cancer.
Manuscript.

ii
CONTENT
ABBREVIATIONS ............................................................................................. IV
INTRODUCTION ................................................................................................ 1
Mammary gland ............................................................................................ 1
Breast cancer ................................................................................................. 2
The invasion-metastasis cascade .................................................................. 3
Epithelial-mesenchymal transition ............................................................... 4
Molecular players involved in EMT ............................................................. 5
EMT in cancer .............................................................................................. 6
Important factors controlling EMT ............................................................... 7
E-cadherin ................................................................................................ 7
c-erbB2 .................................................................................................... 9
Nuclear factor I-C2 ................................................................................ 10
Forkhead box F1 .................................................................................... 12
AIMS .............................................................................................................. 14
SPECIFIC BACKGROUND ................................................................................. 15
Paper I ......................................................................................................... 15
Paper II ....................................................................................................... 16
Paper III ...................................................................................................... 17
RESULTS AND DISCUSSION ............................................................................ 18
Loss of E-cadherin expression is not a prerequisite for c-erbB2-induced
epithelial-mesenchymal transition (paper I). .............................................. 18
Forkhead Box F1 promotes breast cancer cell migration by upregulating
lysyl oxidase and suppressing Smad2/3 signaling (paper II). ..................... 20
Nuclear factor I-C2 is a powerful prognostic marker in breast cancer (paper
III). ............................................................................................................. 22
FUTURE ASPECTS ........................................................................................... 24
ACKNOWLEDGEMENTS .................................................................................. 26
REFERENCES .................................................................................................. 28

iii

iv
ABBREVIATIONS
BCSS breast cancer-specific survival
CDH-1 cadherin 1
CEL carboxyl ester lipase
CSC cancer stem cell
CTC circulating tumor cell
ECM extracellular matrix
EGF epidermal growth factor
EGFR epidermal growth factor receptor
EMT epithelial-mesenchymal transition
ER estrogen receptor
FAK focal adhesion kinase
Fox forkhead box
Jak2 janus tyrosine kinase 2
kDa kilodalton
LOX lysyl oxidase
MAPK mitogen activated protein kinase
MET mesenchymal-epithelial transition
miRNA micro-RNA
MMPs matrix metalloproteinases
NFI nuclear factor 1
NGF nerve growth factor
PDGF platelet-derived growth factor
PR progesterone receptor
RFS recurrence-free survival
RTK receptor tyrosine kinase
TGF-β transforming growth factor β
WAP whey acidic protein
Gisela Nilsson
1
INTRODUCTION
Mammary gland
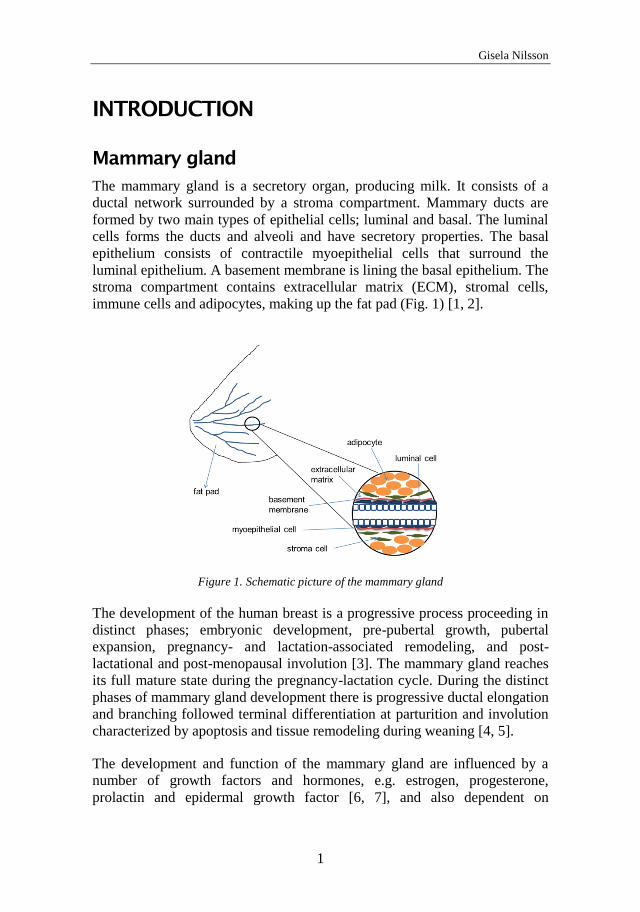
The mammary gland is a secretory organ, producing milk. It consists of a
ductal network surrounded by a stroma compartment. Mammary ducts are
formed by two main types of epithelial cells; luminal and basal. The luminal
cells forms the ducts and alveoli and have secretory properties. The basal
epithelium consists of contractile myoepithelial cells that surround the
luminal epithelium. A basement membrane is lining the basal epithelium. The
stroma compartment contains extracellular matrix (ECM), stromal cells,
immune cells and adipocytes, making up the fat pad (Fig. 1) [1, 2].
Figure 1. Schematic picture of the mammary gland
The development of the human breast is a progressive process proceeding in
distinct phases; embryonic development, pre-pubertal growth, pubertal
expansion, pregnancy- and lactation-associated remodeling, and post-
lactational and post-menopausal involution [3]. The mammary gland reaches
its full mature state during the pregnancy-lactation cycle. During the distinct
phases of mammary gland development there is progressive ductal elongation
and branching followed terminal differentiation at parturition and involution
characterized by apoptosis and tissue remodeling during weaning [4, 5].
The development and function of the mammary gland are influenced by a
number of growth factors and hormones, e.g. estrogen, progesterone,
prolactin and epidermal growth factor [6, 7], and also dependent on

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
2
interactions between stromal and epithelial cells. Signals from stromal cells,
as well as the composition of the ECM (laminin, fibronectin, collagen,
proteoglycans etc.) help epithelial cells to generate and maintain apico-basal
polarity [8]. In response to extracellular signals, integrins (transmembrane
receptors for ECM) activate growth and survival signals [9]. Moreover, many
of the factors important for mammary gland development are also implicated
in breast cancer. During the development of the mammary gland the
epithelium shows high levels of proliferation, reduced apico-basal polarity
and epithelial-mesenchymal plasticity, characteristics also associated with
tumorigenesis [10].
Breast cancer
Breast cancer development is a multi-step process categorized by different
stages [11]. These stages include cellular immortality, hyperplasia,
tumorigenicity and invasiveness. During these stages there is an initial
epithelial hyperplasia, which can develop into atypical hyperplasia, in situ
carcinoma, invasive carcinoma and metastasis. Breast cancer can begin in
different areas of the mammary epithelium; the ducts or the lobules and most
commonly derived from the luminal cells [12]. It is a heterogeneous disease
with different subtypes. These subtypes show differences in the molecular
profile, response to therapy and prognosis. The molecular subtypes are;
luminal A, luminal B, molecular apocrine, basal-like, HER2, normal breast-
like and claudin-low [13-15].
The mechanisms important for normal breast function are also important in
suppressing the development of tumors by regulating proliferation and
survival. Genetic and epigenetic changes leading to dysregulation of these
normal processes can cause uncontrolled proliferation and imbalance
between proliferation and apoptosis, promoting carcinogenesis. Inherited or
acquired mutations in oncogenes (e.g. HER2, MYC, CCND1, PIK3CA [16-
19]) and tumor suppressors (e.g. P53, BRCA1/2, RB1, PTEN [20-24]) are
associated with breast cancer. Hormones and growth factors controlling
normal mammary gland development together with their receptors and co-
regulators are also involved in the development of breast cancer. If these
factors are dysregulated it can lead to amplification of growth and survival
signals [25].
In addition, microRNAs (miRNAs) have been shown to play key roles in
carcinogenesis. miRNAs are small non-coding RNA molecules that control
post-transcriptional gene expression by binding to target messenger RNAs,
causing their degradation and downregulation of the target protein. Some
function as tumor suppressors and others act as oncogenes [26-28].

Gisela Nilsson
3
As a response to the genetic alterations in the epithelial cells, there are
alterations in the microenvironment, such as increased levels of growth
factors and cytokines, and increased matrix synthesis, cross-linking and ECM
stiffening. Matrix stiffening increases integrin signaling, promoting cell
growth, survival and tumor progression [29-32].
It has been suggested that different cell types or transformation of mammary
stem cells at different developmental stages are the cause of the different
breast cancer subtypes. The more aggressive breast cancer subtypes are
suggested to originate from mammary stem cells and the less aggressive
subtypes from more differentiated mammary cells [2, 33-35].
Cancer stem cells (CSCs) were first identified in hematopoietic cancers and
later in solid tumors such as breast tumors [36-38]. The existence of CSCs in
the breast was suggested in a study demonstrating that only a minority of
cancer cells have the ability to form new tumors. Tumorigenic (cancer-
initiating) and non-tumorigenic cancer cells were identified based on
differences in molecular markers [38]. Tumorigenic cancer cells have the
ability to self-renew and produce progeny that are able to differentiate, which
are properties of normal stem cells [39]. CSCs also have metastatic properties
such as increased motility [40].
The invasion-metastasis cascade
Metastases are formed by a multi-step process. Cancer cells that have
acquired the ability to disseminate from the primary tumor can travel through
the blood and lymphatic system to distant sites in the body where they can
form new colonies (metastases). The process when carcinoma cells acquire
the ability to break through the basement membrane and invade the nearby
stroma is termed localized invasion. The step when cancer cells enter the
blood and lymphatic vessels is called intravasation. The migrating cells may
die from anoikis, a form of apoptosis triggered by detachment from the ECM.
They can also survive for long periods and in some instances extravasate into
the surrounding tissue, leading to formation of micrometastases. If the
foreign tissue microenvironment is favorable, the cancer cells may begin to
proliferate and form a secondary tumor, a process termed colonization (Fig.
2) [41]. Carcinomas are benign as long as the tumor cells do not break
through the basement membrane and malign when they have acquired the
ability to metastasize which is the leading cause of death in cancer patients.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
4
Figure 2. Schematic picture of cancer cell invasion and metastasis. 1. Normal
epithelium. 2. Carcinoma in situ, 3. Invasive carcinoma. 4. Intravasation. 5.
Extravasation. 6. Micrometastasis. 7. Colonization.
Epithelial-mesenchymal transition
Epithelial and mesenchymal cell phenotypes are not always permanent.
During development it is critical that cells can change in morphology and
behavior for proper morphogenesis. They do this by converting between the
epithelial and mesenchymal phenotypes. These processes are termed
epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial
transition (MET) [42]. EMT and MET are critical during developmental
stages such as gastrulation [43], neural crest formation [44], and heart valve
formation [45], as well as other morphogenetic events. EMT has also been
observed during branching morphogenesis in the mammary gland [46].
EMT involves activation of genes that are critical for creating the
mechanisms needed for cell migration through the ECM. Epithelial cells are
attached to each other by different junctions (tight junctions, adherens
junctions and desmosomes). During EMT, these junctions dissolve, allowing
epithelial cells to separate, lose apical- basal polarity and gain motility. The
epithelial cell-cell adhesion molecule E-cadherin, and epithelial-specific
integrins, are replaced by mesenchymal specific N-cadherin and integrins
specific for a more transient adhesion. The epithelial intermediate filaments,
cytokeratins, are replaced by vimentin [47]. The underlying basement
membrane is degraded by matrix metalloproteinases (MMPs) and the cell
migrates into the surrounding stroma (Fig. 3). After migration to a distant
site, the mesenchymal cells may return to a epithelial phenotype by
undergoing MET [48].

Gisela Nilsson
5
Figure 3. Epithelial-mesenchymal transition. To migrate and to invade a cell needs
to detach from the neighboring cells in the epithelium. E-cadherin and epithelial
adhesion complexes are replaced by mesenchymal adhesion complexes. Epithelial
cells lose their polarity and gain motility and a mesenchymal morphology. The
cytoskeleton is remodeled and vimentin is expressed. There is an increase in
expression and secretion of extracellular components, e.g. fibronectin. The figure is
adapted from [49].
Besides being critical during development, EMT is involved during wound
healing. Keratinocytes at the edge of a wound undergo EMT and MET during
the wound healing process [50]. EMT or EMT-like events have also been
implicated in fibrosis [51] and cancer metastasis [52]. There are similarities,
but also differences, between EMT events during development and other
processes, leading to the classification of three different EMT types. Type 1
refers to developmental EMTs; type 2 includes EMT events in wound healing
and fibrosis; and type 3 refers to the EMT associated with cancer metastasis.
One difference between type 2 and 3 to type 1 is the existence of an
inflammatory response, which does not exist in embryos [53].
Molecular players involved in EMT
EMT is driven by a network of transcription factors including Snail1, Snail2
(also known as Slug), and Snail3, Zeb1 and Zeb2, Twist1 and Twist2 and
others. Their expression is triggered in response to extracellular signals
derived from the cellular microenvironment. Potent inducers of EMT are
members of the transforming growth factor–β (TGF-β) family [54, 55],
epidermal growth factor (EGF) [56, 57], fibroblast growth factor (FGF) [58,
59] and platelet-derived growth factor (PDGF), but also Wnt [60], Notch
[61], Hedgehog [62] and nuclear factor-κB (NF-κB) [63]. Receptor tyrosin
kinases (RTKs), as well as other types of receptors, and a wide range of
signaling pathways transduce EMT initiating signals and several pathways
work in cooperation in order to full transition to a mesenchymal phenotype.
Activation of Ras and Src pathways are involved in dissociation of adherens

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
6
junctions and desmosomes and remodeling of cytoskeleton by regulating the
Rho family of GTPases [64]. Hypoxia (oxygen deprivation in the
microenvironment) can also drive EMT during development or tumor
progression. The hypoxic response is mainly mediated by hypoxia inducible
factor-1 (HIF-1) which can promote EMT by regulation of Twist [65].
miRNAs have also emerged as key players in EMT and MET. The miRNA-
200 family are proposed to be involved in reversible switching between
epithelial and mesenchymal state [66].
EMT in cancer
In cancer, genetic and epigenetic events lead to alterations in cancer cell
phenotype. These changes become most evident when cancer cells leave the
epithelium and initiate metastasis (Fig. 2 and 3). Carcinoma cells at the
invasive front of a tumor may lose epithelial traits and acquire a
mesenchymal phenotype. Signals supplied by stromal cells, that secrete
growth factors and cytokines, also contributes in the transition to
mesenchymal phenotype [30, 67].
Signaling pathways and downstream effectors involved in the phenotypic
changes needed for a carcinoma cell to detach from the epithelium seems to
be highly similar to the events that orchestrate developmental EMT.
Therefore, EMT or EMT-like events have been implicated in tumor invasion
and metastasis, especially in the early steps of metastasis by facilitating cell
dissemination and thereby generation of circulating tumor cells (CTCs). The
revers process, MET, is believed to be responsible for the formation of
metastases at distant organs [68]. Constitutive expression of transcription
factors associated with EMT, suppresses metastasis formation, suggesting
that EMT transcription factors must be downregulated for metastases to form
[69-71]. In addition, high expression of E-cadherin is seen in breast cancer
metastases [72, 73], suggesting that re-expression of epithelial markers is
necessary for formation of metastases.
It is difficult to study EMT in human tumors, since EMT is considered a
transient event and a tumor specimen represents only one instant. However,
EMT in cultured cells and animal models has been intensely studied.
Multiple signaling pathways and often cooperation of different pathways has
been shown to be involved in the initiation and progression of EMT in cancer
cell models. Overexpression of RTKs, e.g. EGF receptors, or constitutive
signaling from Ras, can drive EMT by activating Raf/MAPK- and PI3K
pathways leading to expression of transcription factors like Snail, Slug Twist
and Zeb1 [74-76]. There are many studies implicating TGF-β as a master

Gisela Nilsson
7
regulator of EMT, and cooperation of TGF-β with Ras or RTKs has been
shown to cause EMT [77].
Expression of mesenchymal markers have been observed in breast
carcinomas of the basal and triple-negative subtypes [78]. However, such
tumors also contain epithelial markers, suggesting that they have undergone a
partial EMT [79, 80].
It has been proposed that EMT can generate CSCs [81], however the origin
of CSCs is a subject of debate. It has been shown that in mammary epithelial
cells, expression of Twist1 or Snail, or treatment with TGF-β increases the
number of cells with CSCs properties (CD44high
/CD24low
) [82, 83]. EMT
markers and stem cell markers are coexpressed in CTCs from breast cancer
patients with metastasis [84, 85] .
Important factors controlling EMT
As described above there are many factors involved in EMT. The studies
included in this thesis have focused specifically on four different factors that
have been shown to be involved in breast tumor development and EMT. The
following sections in the introduction will be about these factors.
E-cadherin
E-cadherin (E for epithelia), also known as cadherin-1 (CDH-1), uvomorulin
and L-CAM, belongs to the type-1 classical cadherin family and is the major
cell adhesion molecule (CAM) involved in binding between neighboring
cells in adherens junctions.
Cadherins play a central role during the development and maintenance of
multicellular organisms. E-cadherin helps to provide mechanically strong
adhesive links between cells in the tissue and is important in defining apico–
basal polarity and maintaining epithelial morphology. E-cadherin is
expressed early in embryonic development and initiates polarization of cells
to form the blastocyst [86]. E-cadherin knockout studies in mice show
lethality at the blastocyst stage [87]. Conditional gene inactivation of E-
cadherin in the mouse mammary gland leads to altered epithelial
differentiation and cell death [88].
The extracellular domain of E-cadherin consists of five repetitive domains.
The outermost part of E-cadherin contains an adhesion recognition motif
important for binding specificity to identical cadherin molecules [89]. E-
cadherin molecules form Ca2+
dependent complexes, also involving catenins;
β-catenin/α-catenin or γ-catenin (plakoglobin)/α-catenin and p120 catenin,
mediating linkage to the actin cytoskeleton (Fig. 4) [90-92]. β-catenin and γ-
catenin both interact with E-cadherin but since their binding sites overlap

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
8
they cannot bind simultaneously. α-catenin is an actin binding protein and is
believed to link the catenin-cadherin complex to the cytoskeleton [93].
However, it has been demonstrated that α-catenin cannot bind actin and β-
catenin at the same time [94, 95], suggesting additional complexity.
Figure 4. Schematic drawing of the organization of E-cadherin in adherens
junctions. E-cadherin is a 120 kDa protein expressed on the surface of epithelial
cells. E-cadherin contains five extracellular domains, a single transmembrane
domain and a cytoplasmic domain which interacts with catenins connecting E-
cadherin to the cytoskeleton; β-catenin (β), γ-catenin (γ) and α-catenin (α). p120
catenin binds to juxtamembrane domain of E-cadherin. E-cadherins exerts
homophilic interactions between neighboring cells, forming a “zipper” structure.
Regulation of adherens junction formation and stability involves both
transcriptional and posttranscriptional regulation of E-cadherin. The levels of
E-cadherin expression [96] and the levels of cytosolic β-catenin regulate the
strength of adhesion [97]. The amount of E-cadherin available for adhesion
can also be regulated by recycling of endocytosed E-cadherin [98] and
proteolytic cleavage [99].
E-cadherin expression is lost during the development of most epithelial
cancers. Loss of E-cadherin expression is suggested to be one of the key steps
in EMT, invasion and metastasis [100]. Consequently, E-cadherin is referred
to as a tumor suppressor and forced re-expression of E-cadherin in carcinoma
cells can reverse malignant properties [101]. Loss of E-cadherin expression is
a typical feature of invasive lobular carcinoma [102]. However, other types of
breast cancer show variable downregulation of E-cadherin expression [103,
104].

Gisela Nilsson
9
c-erbB2
The c-erbB family of RTKs, includes epidermal growth factor receptor
(EGFR; c-erbB1, HER1), c-erbB2 (Neu, HER2), c-erbB3 (HER3) and c-
erbB4 (HER4), which are activated by a number of ligands, e.g. EGF, TGF-α
and heregulin. c-erbB receptors consist of an extracellular ligand binding
domain, a single transmembrane domain and an intracellular kinase and
substrate domain. Upon ligand binding they form a variety of homo- or
heterodimers leading to activation and phosphorylation [105]. Activation of
these receptors induces cell growth, proliferation, differentiation and survival
signals, including Rho GTPases, the Ras-MAP kinase cascade, PI3K and
phospholipase Cγ [106]. c-erbB2 and c-erbB3 activity require
heterodimerization with other c-erbBs since c-erbB2 does not bind growth
factor ligands, and c-erbB3 lacks intrinsic kinase activity [107]. The c-erbB
family plays crucial roles in heart and nervous system development [108-
111] and epithelial morphogenesis, for example in the development of
embryonic and adult mammary gland [112-114].
c-erbB2 is a glycosylated protein with a molecular weight of 185 kDa.
Amplification of c-erbB2 is a common feature in breast cancer.
Overexpression is seen in 15-30 % of breast tumors and is associated with
aggressive and invasive tumors and poor prognosis [16, 115]. Treatment with
the monoclonal antibody trastuzumab (Herceptin) against c-erbB2 has a
positive effect on overall survival and recurrence risk [116, 117]. Lapatinib, a
tyrosine kinase inhibitor, is also used in treatment of c-erbB2 positive breast
cancer [118]. Overexpression of the normal c-erbB2 protein, or point
mutations in its transmembrane domain, has been shown to have a
transforming effect on mammary epithelial cells. When overexpressed, c-
erbB2 can form active homodimers in the absence of ligand [119-121].
Association between c-erbB2 homodimerization and gene amplification has
been shown in vivo, which also was associated with poor prognosis [122]. c-
erbB2 affects cell adhesion and migration, implicating a role for c-erbB2 in
metastasis [123, 124]. Although the receptor commonly associated with EMT
is the TGF-β receptor, there are studies also implicating c-erB2 in EMT. In
an in vitro three-dimensional model system it was demonstrated that
overexpressed c-erbB2 induces proliferation and disrupts tight junctions and
epithelial polarity [125]. Overexpression of c-erbB2 in the mammary
epithelial cell line MTSV1-7 inhibited transcription of E-cadherin [126]. In
vivo selection of MCF-7 cells overexpressing c-erbB2 in nude mice, resulted
in a sub-line that had underwent EMT [127].
CTCs expressing EMT- and stem cell-associated markers have been detected
in the blood of patients with c-erbB2 positive metastatic breast cancer [128].
As mentioned, EMT has been proposed to generate CSCs [81]. CSCs often

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
10
exhibit resistance to therapy [129]. It has been suggested that the mechanisms
leading to resistance to the c-erbB2-targeting drugs trastuzumab and lapatinib
in c-erbB2 positive breast cancer may be linked to induction of EMT [130,
131].
In paper I, we have investigated the role of E-cadherin in c-erbB2-induced
EMT.
Nuclear factor I-C2
Nuclear factor I-C2 (NFI-C2) is a member of the Nuclear factor I (NFI)
transcription factor family. The NFI gene family consists of four members,
NFI-A, NFI-B, NFI-C and NFI-X. NFI gene transcripts are differentially
spliced [132] and expressed in unique but overlapping patterns [133-135].
The NFI proteins bind DNA as dimers at the consensus sequence
TTGGC(N5)GCCAA and function as transcriptional activators as well as
repressors of target genes [136]. NFI proteins are involved in regulation of
genes controlled by e.g. insulin [137], TGF-β [138, 139], cAMP [137],
steroid hormones [140], TNFα [141] and others. The unique expression
patterns of NFI proteins suggests that they may play distinct roles in
regulating tissue-specific gene expression during mammalian embryogenesis
[134] and they have been demonstrated to be involved in cell differentiation
[142, 143]. The oncogene Ras causes transformation of cells. It has been
demonstrated that NFI can suppress Ras-induced transformation.
Overexpression of NFI genes in chicken embryo inhibited transformation by
subsequent overexpression of Ras in these cells [144]. Conversely,
overexpression of Ras in mouse cells represses expression of NFI by
affecting mRNA stability [145]. These studies suggest that loss of NFI
function may promote tumor development. Furthermore, NFI proteins have
been implicated in the function of the mammary gland after the identification
of NFI binding sits in the genes for the milk proteins whey acidic protein
(WAP) and carboxyl ester lipase (CEL) [146, 147].
Identification of specific NFI family members and isoforms involved in
different processes has been hindered by the lack of specific antibodies. The
responsible specific NFI protein is therefore unidentified in most studies.
Our laboratory has demonstrated that the specific isoform NFI-C2 is the
protein responsible for activation of the WAP and CEL promoters in the
mouse mammary gland at mid-pregnancy [148]. Further studies suggest a
broader role for NFI-C2 in the mouse mammary gland. Besides activation of
milk genes, it was shown that NFI-C2 activates the tumor suppressor gene
p53 at mid-pregnancy [149]. From these studies it was suggested that NFI-C2
might participate both in the establishment of a functional gland and in the
protection of the gland against tumorigenesis during proliferation.
Gisela Nilsson
11
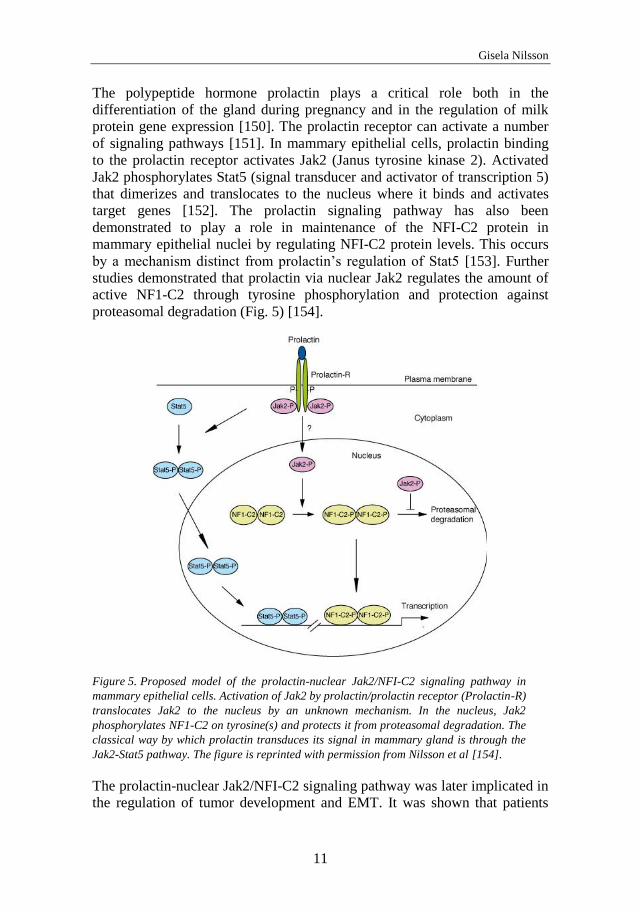
The polypeptide hormone prolactin plays a critical role both in the
differentiation of the gland during pregnancy and in the regulation of milk
protein gene expression [150]. The prolactin receptor can activate a number
of signaling pathways [151]. In mammary epithelial cells, prolactin binding
to the prolactin receptor activates Jak2 (Janus tyrosine kinase 2). Activated
Jak2 phosphorylates Stat5 (signal transducer and activator of transcription 5)
that dimerizes and translocates to the nucleus where it binds and activates
target genes [152]. The prolactin signaling pathway has also been
demonstrated to play a role in maintenance of the NFI-C2 protein in
mammary epithelial nuclei by regulating NFI-C2 protein levels. This occurs
by a mechanism distinct from prolactin’s regulation of Stat5 [153]. Further
studies demonstrated that prolactin via nuclear Jak2 regulates the amount of
active NF1-C2 through tyrosine phosphorylation and protection against
proteasomal degradation (Fig. 5) [154].
Figure 5. Proposed model of the prolactin-nuclear Jak2/NFI-C2 signaling pathway in
mammary epithelial cells. Activation of Jak2 by prolactin/prolactin receptor (Prolactin-R)
translocates Jak2 to the nucleus by an unknown mechanism. In the nucleus, Jak2
phosphorylates NF1-C2 on tyrosine(s) and protects it from proteasomal degradation. The
classical way by which prolactin transduces its signal in mammary gland is through the
Jak2-Stat5 pathway. The figure is reprinted with permission from Nilsson et al [154].
The prolactin-nuclear Jak2/NFI-C2 signaling pathway was later implicated in
the regulation of tumor development and EMT. It was shown that patients

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
12
with nuclear NFI-C2 in their breast cancer cells have better prognosis
compared to those without detectable NFI-C2, and that NFI-C2 suppresses
invasiveness and EMT by repressing the transcription factor Forkhead box F1
(FoxF1) [155]. Paper II and paper III are continuations of that study, which
will be discussed in more detail below.
Forkhead box F1
Forkhead box (Fox) proteins constitute a family of transcription factors that
are related through the presence of a conserved forkhead or winged-helix
DNA-binding domain. Fifty Fox genes have been identified in the human
genome (FoxA to FoxS). Fox proteins bind DNA at the recognition core
motif 5’ ((G/A)(T/C)(C/A)AA(C/T)A) 3’. Nucleotides outside this core motif
are important for the specificity between the different Fox proteins [156].
Temporal and spatial restriction of expression patterns provides an additional
level of specificity, as well as context dependence. For example, in the
developing lung, FoxF1 and FoxF2 have overlapping but not identical
expression patterns, suggesting that these transcription factors may have both
similar and independent functions [157, 158].
FoxF1 acts as a transcriptional activator or repressor of target genes [157,
159]. Early in development FoxF1 is required for differentiation of extra-
embryonic and lateral plate mesoderm and for mesodermal proliferation in
the primitive streak. Inactivation of FoxF1 in mouse embryos is lethal and the
embryos have abnormalities in the coelom, the amnion fails to expand and
there is a lack of vasculogenesis [160]. Later in development, FoxF1 is
expressed in the mesenchyme adjacent to the epithelium in the alimentary,
respiratory and urinary tracts, where it is believed to regulate mesenchymal-
epithelial interactions [157]. In the developing lung, FoxF1 transcription is
activated by Sonic hedgehog signaling [161, 162]. The effects of FoxF1 have
been shown to be dependent on gene dosage [163]. FoxF1 heterozygous
mouse pups show developmental defects in the gut, lungs, trachea, esophagus
and gallbladder [161, 163-165]. In the adult, expression of FoxF1 is found in
a range of tissues, e.g. in the gut, brain, eye and lung [166, 167]. In humans,
the neonatal lethal disorder Alveolar capillary dysplasia with misalignment of
pulmonary veins (ACDMPV) is linked to inactivating mutation of FoxF1
[168].
Fox proteins are important in a variety of biological processes, including
metabolism, development, differentiation, proliferation, apoptosis and
migration. As a consequence, loss or gain of Fox function can lead to
tumorigenesis. Fox proteins have been suggested to act as either tumor
suppressors or oncogenes [169].

Gisela Nilsson
13
The first study demonstrating a role of FoxF1 in tumorigenesis came from
our laboratory. There, it was shown that in a mouse mammary epithelial cell
line, FoxF1 overexpression induced EMT, migration and invasiveness. In
addition, it was demonstrated that forced expression of FoxF1 enhanced
xenograft tumorigenesis in nude mice [155]. FoxF1 has been shown to be
important for mesenchymal cell migration by directly repressing integrin β3
expression [159]. FoxF1 has also been shown to play a role in lung cancer
associated fibroblasts, where FoxF1 expression stimulated cell migration and
xenografted tumor growth [170]. In paper II, we have investigated the
mechanisms responsible for FoxF1-enhanced invasion.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
14
AIMS
The objective of this thesis was to study the influence of different molecular
factors on the EMT process associated with breast cancer cell invasion and
metastasis and their possible use as biomarkers.
The aim of paper I was to study the influence of oncogenic tyrosine kinase
receptor signaling on EMT; specifically, to investigate the role of E-cadherin
during c-erbB2-induced EMT, and in high cell-density dependent inhibition
of EMT.
In paper II, the objective was to study the role of NFI-C2 and FoxF1 in
mammary carcinogenesis by identifying factors regulated by these
transcription factors and involved in EMT and invasion and elucidate what
mechanisms are responsible for FoxF1-enhanced invasion.
Paper III aimed to investigate the prognostic value of NFI-C2 in breast
cancer.

Gisela Nilsson
15
SPECIFIC BACKGROUND
Paper I
The human mammary epithelial cell line HB2 [171] is a subclone from the
non-tumorigenic MTSV1-7 cell line. MTSV1-7 cells were obtained from
luminal epithelial cells from milk and immortalized with the SV40 T antigen
[172]. HB2 cells form spherical colonies in collagen and has been used for in
vitro studies of branching morphogenesis [171]. Constitutive overexpression
of c-erbB2 in MTSV1-7 cells causes irreversible changes in morphology and
repression of E-cadherin [126, 173]. To study the order of events in c-erbB2-
signlaing effects, an inducible system in MTSV1-7 cells and HB2 cells was
developed by Baeckström et al [174], using the hybrid receptor construct trk-
neu [175]. Trk-neu consists of the extracellular domain of the trkA nerve
growth factor (NGF) receptor and the transmembrane and cytoplasmic
domain of c-erbB2 (neu). c-erbB2 homodimerization and activation can be
induced by NGF treatment of the cells (Fig. 6).
Figure 6. The trk-neu hybrid receptor. The extracellular domain of trkA nerve
growth factor (NGF) is fused to the transmembrane and cytoplasmic domains of c-
erbB2 (neu). NGF treatment induces homodimerization and tyrosine kinase (TK)
activation and mimics c-erbB2 homodimerization.
Induced c-erbB2 signaling inactivated integrin α2β1 leading to scattered
growth in collagen. In addition, prolonged activation of c-erbB2 led to EMT
[174]. Further studies using the high expressing clone HB2/tnz34 showed
that c-erbB2-induced integrin inactivation was mediated by MEK/ERK
signaling and PI3K dependent ILK and PKB signaling pathways [176].
Subsequent studies demonstrated that the EMT caused by prolonged
induction of c-erbB2 signaling in HB2/tnz34 cells was irreversible and

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
16
associated with anchorage independent growth [177, 178]. Furthermore, c-
erbB2-induced EMT was suppressed when cells were grown at high density.
It was hypothesized that E-cadherin homophilic interaction can suppress
EMT. It was also observed that the initial cell-scattering associated with
EMT occurred before downregulation of E-cadherin [177].
In paper I, we aimed to analyze a possible role for E-cadherin in cell-cell
contact-inhibition of c-erbB2-induced EMT.
Paper II
In paper II we used three different cell lines; HC11, MDA-MB-436 and HB2.
HC11 cells are a mouse epithelial cell line cloned from the COMMA-1D cell
line, which is derived from mammary tissue of BALB/c mice at mid-
pregnancy [179, 180]. HC11 cells have been used for in vitro studies of
epithelial cell proliferation, signal transduction and differentiation. MDA-
MB-436 is a mesenchymal human breast carcinoma cell line derived from
metastatic site (pleural effusion) and have been used for studying triple-
negative breast cancer [181]. HB2 cells are described in the previous section.
As mentioned, in a previous paper it was demonstrated that the
prolactin/nuclear Jak2/NFI-C2-pathway plays a role in breast cancer by
regulating EMT, motility and invasion [154]. In order to study the effects of
NFI-C2 without the involvement of other effectors stimulated by prolactin, a
stable form of NFI-C2 (NFI-C2S) was stably transfected in HC11 cells. A
point mutation protects NFI-C2S from proteasomal degradation in the
absence of nuclear Jak2. NFI-C2S expression in HC11 cells increased
epithelial characteristics and impeded migration. NFI-C2S expression in
mesenchymal MDA-MB-436 cells lead to loss of some mesenchymal
characteristics, decreased in vitro invasion and abolished xenografted tumor
growth in nude mice, suggesting that NFI-C2 is a suppressor of tumor
progression and EMT. This was underscored by the observation that NFI-C2
status of primary breast cancer correlated with survival and its expression
was lost in lymph node metastases (discussed in the next section, paper III).
Affymetrix microarray analysis of the transcriptome of MDA-MB-436 cells
expressing NFI-C2S or control vector identified the FoxF1 gene to be one of
the most downregulated by NFI-C2, and it was also shown that FoxF1 is a
direct target of NFI-C2. FoxF1 overexpression in HC11 cells induced EMT,
increased in vitro invasion capacity and enhanced xenografted tumorigenesis
in nude mice.
In paper II, we wanted to further study the roles NFI-C2 and FoxF1 in EMT
and invasion, primarily by identifying targets that are oppositely regulated by
these factors and involved in EMT and invasion.

Gisela Nilsson
17
Paper III
Prognostic markers are used in the evaluation of a cancer patient’s risk of
relapse and disease progression. Prognostic markers include tumor size,
axillary lymph node status, histologic grade, patient age and menopausal
status. In addition, molecular markers are used to further help to predict
prognosis or treatment response, e.g. Ki67, estrogen receptor (ER),
progesterone receptor (PR) and HER2. However, these factors are unable to
accurate predict which tumor will recur and to indicate responses to different
therapies and therefore there is a need to find new prognostic markers.
It has previously been demonstrated that the presence of NFI-C2 in breast
tumors has a clinical relevance [155]. The status of NFI-C2 was analyzed by
immunohistochemistry in a tissue microarray containing 292 samples from
patients diagnosed with stage II invasive breast cancer. Tumor cells had a
weaker staining of NFI-C2 compared to normal glandular cells and only 1 of
159 lymph node metastases stained positive for NFI-C2. Patients with NFI-
C2 in their tumor cells (74 of 292) had better prognosis compared to those
without detectable NFI-C2, suggesting that NFI-C2 is a tumor suppressor.
Furthermore, there was a correlation of nuclear Jak2 and NFI-C2 in tumor
biopsies, indicating that the regulation of the nuclear levels of NFI-C2 by the
prolactin/Jak2/NFI-C2 pathway also occurs in cancer.
These findings, together with the finding that NFI-C2 suppresses EMT, make
NFI-C2 a candidate as a prognostic marker. In paper III, we aimed to
investigate the prognostic value of NFI-C2 in a mixed cohort of breast cancer
patients. We also analyzed the levels of NFI-C2 protein after tamoxifen
treatment in cellular extracts from T47D and 4T1 cells. T47D is a human
breast cancer cell line derived from metastatic site (pleural effusion). This
cell line has epithelial morphology and is non-invasive. 4T1 is a mouse
mammary tumor cell line with epithelial morphology. 4T1 cells form highly
metastatic tumors when injected to BALB/c mice and are frequently used as a
model for metastatic breast cancer. This is also a luciferase based model and
the metastatic process can be followed by using bioluminescence in live
animals.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
18
RESULTS AND DISCUSSION
Loss of E-cadherin expression is not a prerequisite
for c-erbB2-induced epithelial-mesenchymal
transition (paper I).
In paper I, we have further investigated earlier observations that cells
undergoing the first scattering phase in c-erbB2 induced EMT still expressed
surface-bound E-cadherin, and that the progression of EMT was delayed by
cell-cell contact [177]. These observations raised the questions whether loss
of E-cadherin expression is a cause or a consequence of EMT and if cell
density-dependent suppression of EMT is mediated by E-cadherin binding
between cells.
In order to analyze a possible modulation of c-erbB2-induced EMT by E-
cadherin, an inducible system for ectopic expression of E-cadherin in cells
stably expressing the trk/neu hybrid receptor (HB2/tnz34) was created. This
generated the clones TrE-ep1 and TrE-ep5 (ep for epithelial morphology).
TrE-ep5 cells were grown at low density with a combination of c-erbB2
signaling and ectopic E-cadherin expression. By subsequently analyzing
changes in expression and localization of epithelial- and mesenchymal
markers, using flow cytometry and immunofluorescence, we could show that
the forced expression of E-cadherin did not prevent the progression of EMT,
as compared to cells with c-erbB2 signaling only. EMT requires loss of cell-
cell adhesion and since E-cadherin is a major component in cell-cell contacts,
loss of its expression is considered to be a fundamental event. However, it
has been observed also in other model systems that EMT can occur without
simultaneous downregulation of E-cadherin. For example, EGF-induced
EMT in MDA-MB-468 cells occurred without complete downregulation of
E-cadherin [182, 183]. In the present study, we showed that cell scattering
following overnight EGF treatment of MDA-MB-468 cells occurred without
major changes in surface-bound E-cadherin, implicating that downregulation
of E-cadherin is not essential in at least the early phase of EMT.
In a fibroblastic clone (TrE-fib), isolated after prolonged c-erbB2 signaling in
the presence of E-cadherin, the endogenous CDH1 gene had been silenced.
Induced expression of ectopic E-cadherin did not have any effect on
morphology in these cells, suggesting that E-cadherin had lost its function.
Performing dissociation assays on confluent TrE-ep5 and TrE-fib cells
showed that induced expression of E-cadherin increased cell-cell adhesion in
TrE-ep5 cells. In contrast, in fibroblastic TrE-fib cells, E-cadherin did not

Gisela Nilsson
19
have a restoring effect on cell-cell adhesion. These results further supported
our assumption that the function of E-cadherin is lost in fibroblastic cells. In
addition, the attachment of E-cadherin to the cytoskeleton was weaker in
fibroblastic cells and β-catenin and α-catenin were localized to the cytoplasm
and nucleus, although the localization of E-cadherin was grossly the same in
epithelial and fibroblastic cells. It has previously been shown that c-erbB2
signaling in HB2 cells results in destabilization of the cortical cytoskeleton
[184]. Rearrangements of the cytoskeleton may lead to difficulties in
supporting E-cadherin mediated cell-cell adhesion, allowing cells to separate
even in the presence of surface-bound E-cadherin.
The observation that the progression of c-erbB2-induced EMT was delayed
by cell-cell contact raised the question whether cell-cell contact results in
intracellular signaling events that hinder the EMT process. If such cell-cell
contact-dependent signaling events are mediated by E-cadherin, then
interfering with E-cadherin function would allow cells grown at high density
to undergo c-erbB2-induced EMT. To test this hypothesis, we expressed a
mutant E-cadherin, where the adhesive function is abolished [90]. Cells
expressing this mutant construct, TrEwv
-ep, showed a decrease in cell-cell
adhesion. However, the combination of c-erbB2 signaling and mutant E-
cadherin expression in high density cultures did not result in progression of
EMT. This indicates that E-cadherin is not involved in mediation of density-
dependent inhibition of c-erbB2-induced EMT.
It has been demonstrated that MMP3 and Rac1b, factors that stimulate
alterations in the cytoskeleton, induce EMT in mammary epithelial cells. This
MMP3/Rac1b-induced EMT was dependent on the cells ability to spread
over a larger surface. When the available surface area was limited
MMP3/Rac1b-induced EMT was blocked [185], suggesting cell shape as an
important factor in the initiation of EMT.
Our findings suggest that loss of E-cadherin expression may be a
consequence rather than a cause of c-erbB2-induced EMT, and further that
density-dependent inhibition of c-erbB2-induced EMT is not mediated by E-
cadherin. We propose that cytoskeletal rearrangements in the initial phase of
EMT may be the mechanism leading to cell-cell separation by impairment of
E-cadherin function and cell-cell adhesion. These rearrangements might also
be important in density-dependent inhibition of EMT by eliciting signals, in
crowded cells, that control EMT progression.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
20
Forkhead Box F1 promotes breast cancer cell
migration by upregulating lysyl oxidase and
suppressing Smad2/3 signaling (paper II).
The aim of this study was to further investigate the roles of NFI-C2 and
FoxF1 in EMT and invasion. Using Affymetrix microarray analysis, we
could compare the transcriptome of HC11 wild type cells to that of cells
overexpressing NFI-C2S or FoxF1 and find targets that are oppositely
regulated by these factors, involved in EMT and invasion. In order to identify
factors that might be generally important in EMT and regulated by NFI-C2,
we used the data from a previous array where the transcriptome of MDA-
MB-436 cells was compared to that of MDA-MB-436 cells overexpressing
NFI-C2S [155]. 45 genes were downregulated 1.5 fold or more by NFI-C2 in
both these cell lines. Of these 45 genes, 17 were upregulated 1.5 fold or more
by FoxF1 overexpression in HC11 cells. The gene that was by far most
upregulated by FoxF1 was lysyl oxidase (LOX). LOX is an ECM enzyme that
catalyzes the cross-linking of collagens or elastin, thereby controlling the
structure and tensile strength of the ECM, but is also implicated in EMT and
tumor progression to metastasis [186-189]. LOX has been demonstrated to be
upregulated in invasive breast cancer cell lines and breast carcinomas [190,
191]. LOX was therefore a potential factor involved in the enhanced in vitro
invasion capacity observed following FoxF1 overexpression in HC11 cells.
We could detect high levels of secreted LOX in culture media from
HC11FoxF1 cells. In addition, we could show that the enhanced in vitro
invasion capacity of HC11FoxF1 cells was mediated by LOX, as treatment
with a LOX inhibitor or reduction of LOX expression using RNAi,
significantly decreased the capacity of HC11FoxF1 cells to migrate through
matrigel. Taken together, we found LOX to be downregulated by NFI-C2 and
upregulated by FoxF1 and responsible for the increased invasion capacity of
HC11FoxF1 cells. These findings together with the previous observations
that FoxF1 is repressed by NFI-C2 which expression is lost during tumor
progression, suggests that loss of NFI-C2 may lead to expression of factors
like FoxF1 and LOX leading to EMT and carcinoma cell dissemination.
Several of the 17 genes that were shown to be downregulated by NFI-C2 and
upregulated by FoxF1 are known to be involved in TGF-β signaling and
TGF-β associated EMT. In addition, LOX has been shown to be involved in
TGF-β induced EMT [192]. This raised the question whether FoxF1 is
involved in the regulation of TGF-β signaling pathways.
TGF-β is important for mammary gland development and function and in the
suppression of tumorigenesis. Breast cancer progression can convert the
function of TGF-β to become a tumor promoting factor [193, 194]. The

Gisela Nilsson
21
oncogenic activity of TGF-β is suggested to be a result from imbalance
between canonical (Smad2/3) and non-canonical (non-Smad) TGF-β
signaling systems, where the non-canonical pathways are believed to promote
EMT and invasive and metastatic properties of cancer cells. The non-Smad
pathways include various branches of MAP kinase pathways, e.g. p38 MAPK
[195].
We investigated whether TGF-β signaling pathways are affected by FoxF1
and found that the Smad2/3 pathway is suppressed while the p38 MAPK
signaling pathway is activated in HC11FoxF1 cells.
LOX has been demonstrated to control the activity of focal adhesion kinase
(FAK) [32, 186, 189, 196]. We found that FoxF1 activates FAK in a LOX-
dependent manner, as depletion of LOX by RNAi decreased the activity of
FAK. In addition, LOX depletion activated the Smad2/3 signaling pathway
by increasing the levels of Smad2/3 and the phosphorylation of Smad2.
However, FAK inhibitor treatment showed that FAK is involved in the
activation of Smad2 but not in the regulation of total Smad2/3 levels,
suggesting that downstream of LOX there are additional factors involved in
the regulation of Smad2/3 levels. The main findings are summarized in figure
7.
Figure 7. Schematic picture of signaling pathways regulated by FoxF1.
In this study we found LOX, which is upregulated in breast carcinomas and
shown to promote metastasis, to be highly expressed in response to FoxF1
overexpression. In addition, FoxF1-induced invasiveness was shown to be
dependent on LOX activity, suggesting that induction of EMT and
invasiveness by FoxF1 may have an important role during breast tumor
progression to metastasis. Furthermore, we found FoxF1 to be involved in
TGF-β signaling pathways known to be important during breast cancer
progression. However, the possible connection between FoxF1 and TGF-β

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
22
and whether these factors cooperate to influence metastatic activity needs to
be further investigated.
Nuclear factor I-C2 is a powerful prognostic marker
in breast cancer (paper III).
In this study, a tissue microarray was constructed from the tissue samples of
498 patients with primary invasive breast cancer diagnosed at the Malmö
University Hospital, Malmö, Sweden, between 1988 and 1992 [197]. Of
these 498 patients the expression level of NFI-C2 could be analyzed in 462
cases, using immunohistochemistry. 217 patients stained positive and 245
stained negative for NFI-C2. The expression of NFI-C2 was positively
associated with advanced age, ERα expression and PR expression, and
negatively associated with HER2 overexpression, lymph node status, tumor
size, Ki67 expression and histological grade. This shows that NFI-C2 is
positively associated with markers for good prognosis, suggesting that tumors
with NFI-C2 expression may be less aggressive than tumors without NFI-C2
expression.
Kaplan-Meier analysis of all patients showed a significantly lower risk for
recurrence and mortality from breast cancer for NFI-C2 positive compared to
NFI-C2 negative patients, demonstrating an association between NFI-C2 and
good prognosis. However, this association was only significant for patients
with ERα-positive tumors.
Kaplan-Meier analysis of the patients that had not received any adjuvant
treatment showed that NFI-C2 was associated with increased recurrence-free
survival (RFS) and breast cancer-specific survival (BCSS), demonstrating a
prognostic value of NFI-C2. Multivariate Cox analysis including age, tumor
size, ERα, PR, Ki67, HER2, and lymph node status demonstrated that NFI-
C2 was an independent predictor of RFS. NFI-C2 remained an independent
predictor of RFS in the ERα-positive group, indicating that NFI-C2 has an
additional value in predicting breast cancer recurrence besides that of the ER
status.
NFI-C2 suppresses EMT and its expression is lost during breast cancer
progression [155], suggesting that loss of NFI-C2 may facilitate the
dissemination of carcinoma cells from the primary tumor and thereby the
generation of CTCs. It has been demonstrated that the presence of CTCs
correlates with lymph node metastases [198]. We therefore investigated the
association between NFI-C2, lymph node status and RFS in the group of
patients that had received tamoxifen treatment. The group of untreated
patients could not be analyzed due to the low number with multiple lymph
node metastases. We found that NFI-C2 was positively associated to RFS in

Gisela Nilsson
23
patients with no lymph node metastases whereas in patients with multiple
lymph node metastases, NFI-C2 was negatively associated to RFS.
Furthermore, lymph node status was only prognostic in the group of patients
with NFI-C2 positive tumors. These data suggest that the role of NFI-C2
changes during breast cancer progression.
We investigated whether tamoxifen had an effect on NFI-C2 protein levels in
two different breast cancer cell lines and found that tamoxifen reduced the
levels of NFI-C2 in 4T1 cells, known to form metastases, while it had no
effect on NFI-C2 levels in the non-invasive T47D cell line. Removing
tamoxifen from 4T1 cells restored the levels of NFI-C2.
This study supports our hypothesis that NFI-C2 is a tumor suppressor and
shows that NFI-C2 is a prognostic factor associated with good prognosis in
breast cancer. However, it also provides data suggesting that the presence of
NFI-C2 may in some instances instead be negative. NFI-C2 protein levels
were differently affected by tamoxifen which might be dependent on the
cellular phenotype. NFI-C2 has been shown to promote MET [155], which is
believed to be a critical process in metastasis formation (described in the
introduction). In patients with no lymph node metastases the presence of
NFI-C2 would have a positive effect inhibiting EMT to occur. However, in
patients with many lymph node metastases and presumably also harboring
CTCs in their blood and lymphatic system, the presence of NFI-C2 in these
circulating cancer cells might lead to MET and an increased risk for
colonization and metastases formation. Our results indicate that during
tamoxifen treatment the NFI-C2 levels are low whereas they increase when
treatment is ended. If patients who have ended a tamoxifen treatment still has
CTCs and the levels of NFI-C2 would increase, this might lead to a higher
risk for metastases formation.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
24
FUTURE ASPECTS
The subject of this thesis was to investigate how different molecular factors
influence EMT. This thesis shows that c-erb2-induced EMT can proceed in
the presence of the adhesion molecule E-cadherin and that cell density-
dependent inhibition of EMT is not mediated by E-cadherin. Furthermore, it
demonstrates an involvement of NFI-C2 and FoxF1 in the regulation of
molecular factors and signaling pathways that are associated to EMT and
invasion. We identified a signaling pathway where FoxF1 upregulated LOX
in a FAK-dependent manner leading to suppression of Smad2/3 and
increased invasion. In addition, NFI-C2 was confirmed to be a prognostic
marker associated with good prognosis in breast cancer. In future work there
are several aspects that can be further investigated.
The occurrence of a mechanism that can hinder EMT is intriguing. If EMT
events are the cause of carcinoma cell dissemination and invasion,
understanding such a mechanism would give critical insights into the process
of metastases and possibly identify potential therapy targets. The effects of
cytoskeletal rearrangements and cell shape seem to be important for the EMT
process to occur and would be interesting to investigate further. Identifying
factors that induce cytoskeletal rearrangements and cell spreading (including
cell-surface targets other than E-cadherin) during EMT initiation would give
information about a possible association between cell morphology and
carcinoma progression.
To get further insight into the role of FoxF1 during carcinogenesis it would
be of importance to evaluate the effects of FoxF1 in vivo. In an ongoing
study, a transgenic mouse model for mammary carcinogenesis is used where
c-myc is specifically overexpressed in the mammary epithelium under control
of the WAP-promoter, causing tumor development after pregnancy [199]. In
this model, FoxF1 is specifically knocked-out in the epithelium by utilizing
the cre/lox system where cre is expressed under the control of the promoter of
keratin 14. This model system will give insights into the effects of FoxF1-
removal on tumor development and progression.
In addition, the 4T1 cell line can be used to study FoxF1 effect on metastatic
capability. We hypothesize that implantation of 4T1 cells overexpressing
FoxF1 into the fat pads of BALB/c mice may result in less formation of
metastases compared to control. FoxF1 overexpression would possibly result
in cancer cells with mesenchymal properties which would impede the ability
to colonize and form metastases.

Gisela Nilsson
25
The role of NFI-C2 as a prognostic marker was evaluated in a
nonrandomized trial. In order to evaluate the treatment predictive value of
NFI-C2, a randomized trial needs to be used containing a large number of
patients that has or has not received treatment.
To approach the question whether the level of NFI-C2 protein affects the
tumor cells' capacity to metastasize, the 4T1 cell line can be utilized. 4T1
cells where NFI-C2 has been knocked-down and control cells can be
implanted into the fat pads of BALB/c mice and their metastatic capability
compared. If NFI-C2 promotes MET then the NFI-C2 knock-down cells will
be more mesenchymal, and as a consequence of that, less capable to colonize
and form metastases.
To elucidate the dual effects of NFI-C2 positivity depending on tumor
progression stage, one can overexpress NF1-C2 in 4T1 cells. Implantation of
these cells in the mammary fat pads of BALB/c mice might give rise to
tumors with less metastatic capability compared to control. On the other
hand, if NFI-C2 overexpressing cells instead were injected into the tail,
resulting in cells entering the bloodstream, this might instead increase the
metastatic capability. We hypothesize that NFI-C2 overexpression would
generate cancer cells with epithelial properties enabling attachment at distant
sites and metastasis formation.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
26
ACKNOWLEDGEMENTS
I would like to thank all the people who in different ways have contributed to
this thesis. In particular I would like to thank:
Dan Baeckström, my supervisor, for taking me on as a PhD student in the
organized DB-lab, for all your support, for always taking time for questions
and for your positive thinking.
The former members of the DB-lab: Shahram for being a good friend and
for all the help in the beginning, Karin for nice company and for always
being positive and Noreen for nice collaboration.
Marie Kannius Janson, my co-supervisor, and Jeanette Nilsson, for letting
me continue my PhD studies in your lab and for your support and
encouragement. It has been a very educating and fun period for me.
Elin for teaching me immunohistochemistry and mouse models and for being
a good friend.
Levent Akyürek and Staffan Johansson for your help during the period
when you were my co-supervisors.
I would like to thank all my former co-workers at medkem, especially:
Gunnar Hanson for your support and for letting me use your equipment, and
all former and present members of the MUCIN-lab.
Isabella, Monica and Ka-Wei for technical advice and nice company during
my time at medkem.
Erika, Daniel, Nicola, Anna and Muna for nice fika-times.
I would also like to thank my co-workers at CMB, especially:
Peter Carlsson for valuable comments and for sharing your Fox-expertise.
Ali and Azadeh for sharing your knowledge and for contributing to the
friendly environment at CMB.
All the people in Per Sunnerhagens lab. Johanna for stimulating
discussions and for making me such beautiful pottery.
Marc Pilon, Julie Grantham and everyone in your labs for contributing to
the stimulating and enjoyable environment at CMB.
Catarina for being such a kind and fun person.
Valida for keeping the lab tidy.
Cecilia, it was nice to have you around all these years.

Gisela Nilsson
27
My mother and father for always being there for me and my family, without
you this would not have been possible. Veronica and Bobby for being the
best siblings. Also, thanks Veronica for all the help during my last period of
writing.
Maria and Monica, my beautiful friends, for your interest in my work and
for sharing fun times outside work.
Daniel for your love and support. Polly and Elliot, I love every day I get to
spend with you!

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
28
REFERENCES
1. Hassiotou, F. and D. Geddes, Anatomy of the human mammary gland: Current status of knowledge. Clin Anat, 2013. 26(1): p. 29-48.
2. Visvader, J.E., Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev, 2009. 23(22): p. 2563-77.
3. Russo, J. and I.H. Russo, Development of the human breast. Maturitas, 2004. 49(1): p. 2-15.
4. Sternlicht, M.D., Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res, 2006. 8(1): p. 201.
5. Williams, J.M. and C.W. Daniel, Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol, 1983. 97(2): p. 274-90.
6. Hennighausen, L. and G.W. Robinson, Signaling pathways in mammary gland development. Dev Cell, 2001. 1(4): p. 467-75.
7. Hynes, N.E. and C.J. Watson, Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb Perspect Biol, 2010. 2(8): p. a003186.
8. Fata, J.E., Z. Werb, and M.J. Bissell, Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res, 2004. 6(1): p. 1-11.
9. Naylor, M.J., et al., Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol, 2005. 171(4): p. 717-28.
10. Ewald, A.J., et al., Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell, 2008. 14(4): p. 570-81.
11. Beckmann, M.W., et al., Multistep carcinogenesis of breast cancer and tumour heterogeneity. J Mol Med (Berl), 1997. 75(6): p. 429-39.
12. Li, C.I., R.E. Moe, and J.R. Daling, Risk of mortality by histologic type of breast cancer among women aged 50 to 79 years. Arch Intern Med, 2003. 163(18): p. 2149-53.
13. Perou, C.M., et al., Molecular portraits of human breast tumours. Nature, 2000. 406(6797): p. 747-52.
14. Lam, S.W., C.R. Jimenez, and E. Boven, Breast cancer classification by proteomic technologies: current state of knowledge. Cancer Treat Rev, 2014. 40(1): p. 129-38.
15. Prat, A., et al., Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res, 2010. 12(5): p. R68.
16. Slamon, D.J., et al., Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science, 1987. 235(4785): p. 177-82.

Gisela Nilsson
29
17. Nass, S.J. and R.B. Dickson, Defining a role for c-Myc in breast tumorigenesis. Breast Cancer Res Treat, 1997. 44(1): p. 1-22.
18. Steeg, P.S. and Q. Zhou, Cyclins and breast cancer. Breast Cancer Res Treat, 1998. 52(1-3): p. 17-28.
19. Bachman, K.E., et al., The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther, 2004. 3(8): p. 772-5.
20. Levine, A.J., J. Momand, and C.A. Finlay, The p53 tumour suppressor gene. Nature, 1991. 351(6326): p. 453-6.
21. Smith, S.A., et al., Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat Genet, 1992. 2(2): p. 128-31.
22. Wooster, R., et al., Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science, 1994. 265(5181): p. 2088-90.
23. Varley, J.M., et al., The retinoblastoma gene is frequently altered leading to loss of expression in primary breast tumours. Oncogene, 1989. 4(6): p. 725-9.
24. Li, J., et al., PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 1997. 275(5308): p. 1943-7.
25. Brisken, C. and B. O'Malley, Hormone action in the mammary gland. Cold Spring Harb Perspect Biol, 2010. 2(12): p. a003178.
26. Mulrane, L., et al., miRNA dysregulation in breast cancer. Cancer Res, 2013. 73(22): p. 6554-62.
27. Melo, S.A. and M. Esteller, Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett, 2011. 585(13): p. 2087-99.
28. Zhang, L., et al., microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A, 2006. 103(24): p. 9136-41.
29. Allinen, M., et al., Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell, 2004. 6(1): p. 17-32.
30. Arendt, L.M., et al., Stroma in breast development and disease. Semin Cell Dev Biol, 2010. 21(1): p. 11-8.
31. Kuperwasser, C., et al., Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A, 2004. 101(14): p. 4966-71.
32. Levental, K.R., et al., Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell, 2009. 139(5): p. 891-906.
33. Prat, A. and C.M. Perou, Mammary development meets cancer genomics. Nat Med, 2009. 15(8): p. 842-4.
34. Stingl, J. and C. Caldas, Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer, 2007. 7(10): p. 791-9.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
30
35. Lim, E., et al., Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med, 2009. 15(8): p. 907-13.
36. Bonnet, D. and J.E. Dick, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med, 1997. 3(7): p. 730-7.
37. Ignatova, T.N., et al., Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia, 2002. 39(3): p. 193-206.
38. Al-Hajj, M., et al., Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A, 2003. 100(7): p. 3983-8.
39. Reya, T., et al., Stem cells, cancer, and cancer stem cells. Nature, 2001. 414(6859): p. 105-11.
40. Oskarsson, T., E. Batlle, and J. Massague, Metastatic Stem Cells: Sources, Niches, and Vital Pathways. Cell Stem Cell, 2014. 14(3): p. 306-321.
41. Vanharanta, S. and J. Massague, Origins of metastatic traits. Cancer Cell, 2013. 24(4): p. 410-21.
42. Nieto, M.A., Epithelial plasticity: a common theme in embryonic and cancer cells. Science, 2013. 342(6159): p. 1234850.
43. Solnica-Krezel, L., Conserved patterns of cell movements during vertebrate gastrulation. Curr Biol, 2005. 15(6): p. R213-28.
44. Duband, J.L., et al., Epithelium-mesenchyme transition during neural crest development. Acta Anat (Basel), 1995. 154(1): p. 63-78.
45. Runyan, R.B., J.D. Potts, and D.L. Weeks, TGF-beta 3-mediated tissue interaction during embryonic heart development. Mol Reprod Dev, 1992. 32(2): p. 152-9.
46. Lee, K., et al., Snail1, Snail2, and E47 promote mammary epithelial branching morphogenesis. EMBO J, 2011. 30(13): p. 2662-74.
47. Hay, E.D., The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn, 2005. 233(3): p. 706-20.
48. Chaffer, C.L., E.W. Thompson, and E.D. Williams, Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs, 2007. 185(1-3): p. 7-19.
49. Micalizzi, D.S., S.M. Farabaugh, and H.L. Ford, Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia, 2010. 15(2): p. 117-34.
50. Yan, C., et al., Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol, 2010. 176(5): p. 2247-58.
51. Iwano, M., et al., Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest, 2002. 110(3): p. 341-50.

Gisela Nilsson
31
52. Vernon, A.E. and C. LaBonne, Tumor metastasis: a new twist on epithelial-mesenchymal transitions. Curr Biol, 2004. 14(17): p. R719-21.
53. Lopez-Novoa, J.M. and M.A. Nieto, Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med, 2009. 1(6-7): p. 303-14.
54. Bhowmick, N.A., et al., Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell, 2001. 12(1): p. 27-36.
55. Boyer, A.S., et al., TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol, 1999. 208(2): p. 530-45.
56. Lu, Z., et al., Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell, 2003. 4(6): p. 499-515.
57. Camenisch, T.D., et al., Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med, 2002. 8(8): p. 850-5.
58. Ciruna, B. and J. Rossant, FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell, 2001. 1(1): p. 37-49.
59. Acevedo, V.D., et al., Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell, 2007. 12(6): p. 559-71.
60. Heller, R.S., et al., Expression patterns of Wnts, Frizzleds, sFRPs, and misexpression in transgenic mice suggesting a role for Wnts in pancreas and foregut pattern formation. Dev Dyn, 2002. 225(3): p. 260-70.
61. Timmerman, L.A., et al., Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev, 2004. 18(1): p. 99-115.
62. Katoh, Y. and M. Katoh, Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med, 2008. 22(3): p. 271-5.
63. Huber, M.A., et al., NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest, 2004. 114(4): p. 569-81.
64. Ellenbroek, S.I. and J.G. Collard, Rho GTPases: functions and association with cancer. Clin Exp Metastasis, 2007. 24(8): p. 657-72.
65. Yang, M.H., et al., Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol, 2008. 10(3): p. 295-305.
66. Gregory, P.A., et al., MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle, 2008. 7(20): p. 3112-8.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
32
67. Christofori, G., New signals from the invasive front. Nature, 2006. 441(7092): p. 444-50.
68. Liu, H., et al., The biological and clinical importance of epithelial-mesenchymal transition in circulating tumor cells. J Cancer Res Clin Oncol, 2014.
69. Ocana, O.H., et al., Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell, 2012. 22(6): p. 709-24.
70. Tsai, J.H., et al., Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell, 2012. 22(6): p. 725-36.
71. Celia-Terrassa, T., et al., Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest, 2012. 122(5): p. 1849-68.
72. Jeschke, U., et al., Expression of E-cadherin in human ductal breast cancer carcinoma in situ, invasive carcinomas, their lymph node metastases, their distant metastases, carcinomas with recurrence and in recurrence. Anticancer Res, 2007. 27(4A): p. 1969-74.
73. Saha, B., et al., Overexpression of E-cadherin protein in metastatic breast cancer cells in bone. Anticancer Res, 2007. 27(6B): p. 3903-8.
74. Edme, N., et al., Ras induces NBT-II epithelial cell scattering through the coordinate activities of Rac and MAPK pathways. J Cell Sci, 2002. 115(Pt 12): p. 2591-601.
75. Huber, M.A., N. Kraut, and H. Beug, Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol, 2005. 17(5): p. 548-58.
76. Peinado, H., D. Olmeda, and A. Cano, Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer, 2007. 7(6): p. 415-28.
77. Taylor, M.A., J.G. Parvani, and W.P. Schiemann, The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia, 2010. 15(2): p. 169-90.
78. Carey, L., et al., Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol, 2010. 7(12): p. 683-92.
79. Sarrio, D., et al., Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res, 2008. 68(4): p. 989-97.
80. Blick, T., et al., Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis, 2008. 25(6): p. 629-42.
81. Ansieau, S., EMT in breast cancer stem cell generation. Cancer Lett, 2013. 338(1): p. 63-8.
82. Mani, S.A., et al., The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 2008. 133(4): p. 704-15.

Gisela Nilsson
33
83. Morel, A.P., et al., Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One, 2008. 3(8): p. e2888.
84. Aktas, B., et al., Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res, 2009. 11(4): p. R46.
85. Baccelli, I., et al., Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol, 2013. 31(6): p. 539-44.
86. Hyafil, F., et al., A cell surface glycoprotein involved in the compaction of embryonal carcinoma cells and cleavage stage embryos. Cell, 1980. 21(3): p. 927-34.
87. Larue, L., et al., E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A, 1994. 91(17): p. 8263-7.
88. Boussadia, O., et al., E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev, 2002. 115(1-2): p. 53-62.
89. Blaschuk, O.W., et al., Identification of a cadherin cell adhesion recognition sequence. Dev Biol, 1990. 139(1): p. 227-9.
90. Chitaev, N.A. and S.M. Troyanovsky, Adhesive but not lateral E-cadherin complexes require calcium and catenins for their formation. J Cell Biol, 1998. 142(3): p. 837-46.
91. Ozawa, M., H. Baribault, and R. Kemler, The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J, 1989. 8(6): p. 1711-7.
92. Shibamoto, S., et al., Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J Cell Biol, 1995. 128(5): p. 949-57.
93. Rimm, D.L., et al., Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A, 1995. 92(19): p. 8813-7.
94. Yamada, S., et al., Deconstructing the cadherin-catenin-actin complex. Cell, 2005. 123(5): p. 889-901.
95. Drees, F., et al., Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell, 2005. 123(5): p. 903-15.
96. Steinberg, M.S. and M. Takeichi, Experimental specification of cell sorting, tissue spreading, and specific spatial patterning by quantitative differences in cadherin expression. Proc Natl Acad Sci U S A, 1994. 91(1): p. 206-9.
97. Hinck, L., W.J. Nelson, and J. Papkoff, Wnt-1 modulates cell-cell adhesion in mammalian cells by stabilizing beta-catenin binding to

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
34
the cell adhesion protein cadherin. J Cell Biol, 1994. 124(5): p. 729-41.
98. Le, T.L., A.S. Yap, and J.L. Stow, Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol, 1999. 146(1): p. 219-32.
99. Potter, S.W., G. Gaza, and J.E. Morris, Estradiol induces E-cadherin degradation in mouse uterine epithelium during the estrous cycle and early pregnancy. J Cell Physiol, 1996. 169(1): p. 1-14.
100. Birchmeier, W. and J. Behrens, Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta, 1994. 1198(1): p. 11-26.
101. Semb, H. and G. Christofori, The tumor-suppressor function of E-cadherin. Am J Hum Genet, 1998. 63(6): p. 1588-93.
102. Berx, G. and F. Van Roy, The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res, 2001. 3(5): p. 289-93.
103. Prasad, C.P., et al., Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer, 2009. 9: p. 325.
104. Caldeira, J.R., et al., CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer, 2006. 6: p. 48.
105. Lemmon, M.A. and J. Schlessinger, Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem Sci, 1994. 19(11): p. 459-63.
106. Alroy, I. and Y. Yarden, The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand-receptor interactions. FEBS Lett, 1997. 410(1): p. 83-6.
107. Pinkas-Kramarski, R., et al., Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J, 1996. 15(10): p. 2452-67.
108. Gassmann, M., et al., Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature, 1995. 378(6555): p. 390-4.
109. Lee, K.F., et al., Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature, 1995. 378(6555): p. 394-8.
110. Kramer, R., et al., Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc Natl Acad Sci U S A, 1996. 93(10): p. 4833-8.
111. Grinspan, J.B., et al., Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci, 1996. 16(19): p. 6107-18.

Gisela Nilsson
35
112. Jones, F.E., et al., Heregulin induces in vivo proliferation and differentiation of mammary epithelium into secretory lobuloalveoli. Cell Growth Differ, 1996. 7(8): p. 1031-8.
113. Schroeder, J.A. and D.C. Lee, Dynamic expression and activation of ERBB receptors in the developing mouse mammary gland. Cell Growth Differ, 1998. 9(6): p. 451-64.
114. Sebastian, J., et al., Activation and function of the epidermal growth factor receptor and erbB-2 during mammary gland morphogenesis. Cell Growth Differ, 1998. 9(9): p. 777-85.
115. van de Vijver, M., et al., Amplification of the neu (c-erbB-2) oncogene in human mammmary tumors is relatively frequent and is often accompanied by amplification of the linked c-erbA oncogene. Mol Cell Biol, 1987. 7(5): p. 2019-23.
116. Goldenberg, M.M., Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin Ther, 1999. 21(2): p. 309-18.
117. Moja, L., et al., Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst Rev, 2012. 4: p. CD006243.
118. Nolting, M., T. Schneider-Merck, and M. Trepel, Lapatinib. Recent Results Cancer Res, 2014. 201: p. 125-43.
119. Di Fiore, P.P., et al., erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science, 1987. 237(4811): p. 178-82.
120. Stern, D.F., M.P. Kamps, and H. Cao, Oncogenic activation of p185neu stimulates tyrosine phosphorylation in vivo. Mol Cell Biol, 1988. 8(9): p. 3969-73.
121. Qian, X., et al., Intermolecular association and trans-phosphorylation of different neu-kinase forms permit SH2-dependent signaling and oncogenic transformation. Oncogene, 1995. 10(1): p. 211-9.
122. Spears, M., et al., In situ detection of HER2:HER2 and HER2:HER3 protein-protein interactions demonstrates prognostic significance in early breast cancer. Breast Cancer Res Treat, 2012. 132(2): p. 463-70.
123. Feldner, J.C. and B.H. Brandt, Cancer cell motility--on the road from c-erbB-2 receptor steered signaling to actin reorganization. Exp Cell Res, 2002. 272(2): p. 93-108.
124. Boivin, B., et al., Receptor protein-tyrosine phosphatase alpha regulates focal adhesion kinase phosphorylation and ErbB2 oncoprotein-mediated mammary epithelial cell motility. J Biol Chem, 2013. 288(52): p. 36926-35.
125. Muthuswamy, S.K., et al., ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol, 2001. 3(9): p. 785-92.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
36
126. D'Souza, B. and J. Taylor-Papadimitriou, Overexpression of ERBB2 in human mammary epithelial cells signals inhibition of transcription of the E-cadherin gene. Proc Natl Acad Sci U S A, 1994. 91(15): p. 7202-6.
127. Ai, M., et al., Brk/PTK6 cooperates with HER2 and Src in regulating breast cancer cell survival and epithelial-to-mesenchymal transition. Cancer Biol Ther, 2013. 14(3): p. 237-45.
128. Giordano, A., et al., Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol Cancer Ther, 2012. 11(11): p. 2526-34.
129. Valent, P., et al., Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer, 2012. 12(11): p. 767-75.
130. Bedard, P.L., F. Cardoso, and M.J. Piccart-Gebhart, Stemming resistance to HER-2 targeted therapy. J Mammary Gland Biol Neoplasia, 2009. 14(1): p. 55-66.
131. Wu, Y., et al., Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res, 2012. 10(12): p. 1597-606.
132. Kruse, U. and A.E. Sippel, The genes for transcription factor nuclear factor I give rise to corresponding splice variants between vertebrate species. J Mol Biol, 1994. 238(5): p. 860-5.
133. Apt, D., Y. Liu, and H.U. Bernard, Cloning and functional analysis of spliced isoforms of human nuclear factor I-X: interference with transcriptional activation by NFI/CTF in a cell-type specific manner. Nucleic Acids Res, 1994. 22(19): p. 3825-33.
134. Chaudhry, A.Z., G.E. Lyons, and R.M. Gronostajski, Expression patterns of the four nuclear factor I genes during mouse embryogenesis indicate a potential role in development. Dev Dyn, 1997. 208(3): p. 313-25.
135. Liu, Y., H.U. Bernard, and D. Apt, NFI-B3, a novel transcriptional repressor of the nuclear factor I family, is generated by alternative RNA processing. J Biol Chem, 1997. 272(16): p. 10739-45.
136. Gronostajski, R.M., Roles of the NFI/CTF gene family in transcription and development. Gene, 2000. 249(1-2): p. 31-45.
137. Cooke, D.W. and M.D. Lane, The transcription factor nuclear factor I mediates repression of the GLUT4 promoter by insulin. J Biol Chem, 1999. 274(18): p. 12917-24.
138. Alevizopoulos, A., et al., Regulation of the transforming growth factor beta-responsive transcription factor CTF-1 by calcineurin and calcium/calmodulin-dependent protein kinase IV. J Biol Chem, 1997. 272(38): p. 23597-605.
139. Rossi, P., et al., A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-beta. Cell, 1988. 52(3): p. 405-14.

Gisela Nilsson
37
140. Chaudhry, A.Z., A.D. Vitullo, and R.M. Gronostajski, Nuclear factor I-mediated repression of the mouse mammary tumor virus promoter is abrogated by the coactivators p300/CBP and SRC-1. J Biol Chem, 1999. 274(11): p. 7072-81.
141. Alevizopoulos, A. and N. Mermod, Antagonistic regulation of a proline-rich transcription factor by transforming growth factor beta and tumor necrosis factor alpha. J Biol Chem, 1996. 271(47): p. 29672-81.
142. Delgado-Olguin, P., et al., NFI-C2 negatively regulates alpha-sarcoglycan promoter activity in C2C12 myoblasts. Biochem Biophys Res Commun, 2004. 319(3): p. 1032-9.
143. Behrens, M., et al., NFI in the development of the olfactory neuroepithelium and the regulation of olfactory marker protein gene expression. Eur J Neurosci, 2000. 12(4): p. 1372-84.
144. Schuur, E.R., et al., Nuclear factor I interferes with transformation induced by nuclear oncogenes. Cell Growth Differ, 1995. 6(3): p. 219-27.
145. Nebl, G., N. Mermod, and A.C. Cato, Post-transcriptional down-regulation of expression of transcription factor NF1 by Ha-ras oncogene. J Biol Chem, 1994. 269(10): p. 7371-8.
146. Li, S. and J.M. Rosen, Nuclear factor I and mammary gland factor (STAT5) play a critical role in regulating rat whey acidic protein gene expression in transgenic mice. Mol Cell Biol, 1995. 15(4): p. 2063-70.
147. Kannius-Janson, M., et al., Studies of the regulation of the mouse carboxyl ester lipase gene in mammary gland. Biochem J, 1998. 336 ( Pt 3): p. 577-85.
148. Kannius-Janson, M., et al., Nuclear factor 1-C2 contributes to the tissue-specific activation of a milk protein gene in the differentiating mammary gland. J Biol Chem, 2002. 277(20): p. 17589-96.
149. Johansson, E.M., et al., The p53 tumor suppressor gene is regulated in vivo by nuclear factor 1-C2 in the mouse mammary gland during pregnancy. Oncogene, 2003. 22(38): p. 6061-70.
150. Houdebine, L.M., et al., Hormonal action controlling mammary activity. J Dairy Sci, 1985. 68(2): p. 489-500.
151. Clevenger, C.V. and J.B. Kline, Prolactin receptor signal transduction. Lupus, 2001. 10(10): p. 706-18.
152. Watson, C.J. and T.G. Burdon, Prolactin signal transduction mechanisms in the mammary gland: the role of the Jak/Stat pathway. Rev Reprod, 1996. 1(1): p. 1-5.
153. Johansson, E.M., et al., Nuclear factor 1-C2 is regulated by prolactin and shows a distinct expression pattern in the mouse mammary epithelial cells during development. Mol Endocrinol, 2005. 19(4): p. 992-1003.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
38
154. Nilsson, J., G. Bjursell, and M. Kannius-Janson, Nuclear Jak2 and transcription factor NF1-C2: a novel mechanism of prolactin signaling in mammary epithelial cells. Mol Cell Biol, 2006. 26(15): p. 5663-74.
155. Nilsson, J., et al., Nuclear Janus-activated kinase 2/nuclear factor 1-C2 suppresses tumorigenesis and epithelial-to-mesenchymal transition by repressing Forkhead box F1. Cancer Res, 2010. 70(5): p. 2020-9.
156. Kaufmann, E. and W. Knochel, Five years on the wings of fork head. Mech Dev, 1996. 57(1): p. 3-20.
157. Mahlapuu, M., et al., FREAC-1 contains a cell-type-specific transcriptional activation domain and is expressed in epithelial-mesenchymal interfaces. Dev Biol, 1998. 202(2): p. 183-95.
158. Aitola, M., et al., Forkhead transcription factor FoxF2 is expressed in mesodermal tissues involved in epithelio-mesenchymal interactions. Dev Dyn, 2000. 218(1): p. 136-49.
159. Malin, D., et al., Forkhead box F1 is essential for migration of mesenchymal cells and directly induces integrin-beta3 expression. Mol Cell Biol, 2007. 27(7): p. 2486-98.
160. Mahlapuu, M., et al., The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development, 2001. 128(2): p. 155-66.
161. Mahlapuu, M., S. Enerback, and P. Carlsson, Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development, 2001. 128(12): p. 2397-406.
162. Astorga, J. and P. Carlsson, Hedgehog induction of murine vasculogenesis is mediated by Foxf1 and Bmp4. Development, 2007. 134(20): p. 3753-61.
163. Kalinichenko, V.V., et al., Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am J Physiol Lung Cell Mol Physiol, 2004. 286(3): p. L521-30.
164. Kalinichenko, V.V., et al., Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol, 2001. 235(2): p. 489-506.
165. Kalinichenko, V.V., et al., Haploinsufficiency of the mouse Forkhead Box f1 gene causes defects in gall bladder development. J Biol Chem, 2002. 277(14): p. 12369-74.
166. Kalinichenko, V.V., et al., The forkhead box F1 transcription factor is expressed in brain and head mesenchyme during mouse embryonic development. Gene Expr Patterns, 2003. 3(2): p. 153-8.
167. Pierrou, S., et al., Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J, 1994. 13(20): p. 5002-12.

Gisela Nilsson
39
168. Stankiewicz, P., et al., Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet, 2009. 84(6): p. 780-91.
169. Myatt, S.S. and E.W. Lam, The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer, 2007. 7(11): p. 847-59.
170. Saito, R.A., et al., Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res, 2010. 70(7): p. 2644-54.
171. Berdichevsky, F., et al., Branching morphogenesis of human mammary epithelial cells in collagen gels. J Cell Sci, 1994. 107 ( Pt 12): p. 3557-68.
172. Bartek, J., et al., Efficient immortalization of luminal epithelial cells from human mammary gland by introduction of simian virus 40 large tumor antigen with a recombinant retrovirus. Proc Natl Acad Sci U S A, 1991. 88(9): p. 3520-4.
173. D'Souza, B., et al., Collagen-induced morphogenesis and expression of the alpha 2-integrin subunit is inhibited in c-erbB2-transfected human mammary epithelial cells. Oncogene, 1993. 8(7): p. 1797-806.
174. Baeckstrom, D., P.J. Lu, and J. Taylor-Papadimitriou, Activation of the alpha2beta1 integrin prevents c-erbB2-induced scattering and apoptosis of human mammary epithelial cells in collagen. Oncogene, 2000. 19(40): p. 4592-603.
175. Sachs, M., et al., Motogenic and morphogenic activity of epithelial receptor tyrosine kinases. J Cell Biol, 1996. 133(5): p. 1095-1107.
176. Lindberg, L.E., S. Hedjazifar, and D. Baeckstrom, c-erbB2-induced disruption of matrix adhesion and morphogenesis reveals a novel role for protein kinase B as a negative regulator of alpha(2)beta(1) integrin function. Mol Biol Cell, 2002. 13(8): p. 2894-908.
177. Jenndahl, L.E., P. Isakson, and D. Baeckstrom, c-erbB2-induced epithelial-mesenchymal transition in mammary epithelial cells is suppressed by cell-cell contact and initiated prior to E-cadherin downregulation. Int J Oncol, 2005. 27(2): p. 439-48.
178. Jenndahl, L.E., J. Taylor-Papadimitriou, and D. Baeckstrom, Characterization of integrin and anchorage dependence in mammary epithelial cells following c-erbB2-induced epithelial-mesenchymal transition. Tumour Biol, 2006. 27(1): p. 50-8.
179. Perotti, C., et al., Characterization of mammary epithelial cell line HC11 using the NIA 15k gene array reveals potential regulators of the undifferentiated and differentiated phenotypes. Differentiation, 2009. 78(5): p. 269-82.
180. Danielson, K.G., et al., Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. Proc Natl Acad Sci U S A, 1984. 81(12): p. 3756-60.

Molecular factors influencing epithelial-mesenchymal transition in breast cancer
40
181. Cailleau, R., M. Olive, and Q.V. Cruciger, Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro, 1978. 14(11): p. 911-5.
182. Lo, H.W., et al., Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res, 2007. 67(19): p. 9066-76.
183. Davis, F.M., et al., Remodeling of purinergic receptor-mediated Ca2+ signaling as a consequence of EGF-induced epithelial-mesenchymal transition in breast cancer cells. PLoS One, 2011. 6(8): p. e23464.
184. Hedjazifar, S., et al., PKB mediates c-erbB2-induced epithelial beta1 integrin conformational inactivation through Rho-independent F-actin rearrangements. Exp Cell Res, 2005. 307(1): p. 259-75.
185. Nelson, C.M., et al., Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. J Cell Biochem, 2008. 105(1): p. 25-33.
186. Erler, J.T., et al., Lysyl oxidase is essential for hypoxia-induced metastasis. Nature, 2006. 440(7088): p. 1222-6.
187. El-Haibi, C.P., et al., Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A, 2012. 109(43): p. 17460-5.
188. Kirschmann, D.A., et al., A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res, 2002. 62(15): p. 4478-83.
189. Payne, S.L., et al., Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res, 2005. 65(24): p. 11429-36.
190. Perou, C.M., et al., Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A, 1999. 96(16): p. 9212-7.
191. Kirschmann, D.A., et al., Differentially expressed genes associated with the metastatic phenotype in breast cancer. Breast Cancer Res Treat, 1999. 55(2): p. 127-36.
192. Taylor, M.A., et al., Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-beta signaling in breast cancer cells. Neoplasia, 2011. 13(5): p. 406-18.
193. Moses, H. and M.H. Barcellos-Hoff, TGF-beta biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol, 2011. 3(1): p. a003277.
194. Pardali, K. and A. Moustakas, Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta, 2007. 1775(1): p. 21-62.

Gisela Nilsson
41
195. Parvani, J.G., M.A. Taylor, and W.P. Schiemann, Noncanonical TGF-beta signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia, 2011. 16(2): p. 127-46.
196. Postovit, L.M., et al., Hypoxia/reoxygenation: a dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J Cell Biochem, 2008. 103(5): p. 1369-78.
197. Borgquist, S., et al., Oestrogen receptors alpha and beta show different associations to clinicopathological parameters and their co-expression might predict a better response to endocrine treatment in breast cancer. J Clin Pathol, 2008. 61(2): p. 197-203.
198. Nakagawa, T., et al., Detection of circulating tumor cells in early-stage breast cancer metastasis to axillary lymph nodes. Clin Cancer Res, 2007. 13(14): p. 4105-10.
199. Sandgren, E.P., et al., Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res, 1995. 55(17): p. 3915-27.





























