Embed Size (px)
Citation preview

Molecular evolution of key genes for type II secretionin Legionella pneumophilaemi_2646 1..17
Joana Costa,1,2 Ana Filipa d’Avó,1
Milton S. da Costa1,2 and António Veríssimo1,2*1Centro de Neurociências e Biologia Celular,Universidade de Coimbra, 3004-517 Coimbra, Portugal.2Department of Life Sciences, University of Coimbra,Apartado 3046, 3001-401 Coimbra, Portugal.
Summary
Given the role of type II protein secretion system(T2S) in the ecology and pathogenesis of Legionellapneumophila, it is possible that this system is a targetfor adaptive evolution. The population genetic struc-ture of L. pneumophila was inferred from the partialsequences of rpoB and from the complete sequenceof three T2S structural components (lspD, lspE andpilD) and from two T2S effectors critical for intracel-lular infection of protozoa (proA and srnA) of 37strains isolated from natural and man-made environ-ments and disease-related from worldwide sources. Aphylogenetic analysis was obtained for the concat-enated alignment and for each individual locus.Seven main groups were identified containing thesame L. pneumophila strains, suggesting an ancientdivergence for each cluster and indicating that theoperating selective pressures have equally affectedthe evolution of the five genes. Although linkage dis-equilibrium analysis indicate a clonal nature for popu-lation structure in this sample, our results indicatethat recombination is a common phenomenon amongT2S-related genes on this species, as 24 of the 37L. pneumophila isolates contained at least one locusin which recombination was identified. Furthermore,neutral selection acting on the analysed T2S-relatedgenes emerged as a clear result, namely on T2S effec-tors, ProA and SrnA, indicating that they are probablyimplicated in conserved virulence mechanismsthrough legionellae hosts.
Introduction
Legionella pneumophila is a ubiquitous bacterium innatural and water distribution systems known for its ability
to cause pneumonia in humans. The bacterium’s survivaland spread depends on the ability to replicate insidephagocytic cells. In humans, L. pneumophila reaches thelungs after inhalation of contaminated aerosol dropletswhere is phagocytosed by alveolar macrophages, whichare the major site for bacterial replication (Fields, 2008;Moliner et al., 2010; Newton et al., 2010).
Legionella pneumophila has a rather exceptionalnumber and wide variety of secretion systems for efficientand rapid deliverance of effector molecules into thephagocytotic host cells, underlining the importance ofprotein secretion for this pathogen (De Buck et al., 2007).Legionella pneumophila type II secretion system (T2S)was first identified based on the presence of PilD (Lileset al., 1998), a homologue of the Pseudomonas prepilinpeptidase, which has the ability to process pilin and theso-called pseudopilins that are implicated both in the bio-genesis of type IV pili and a functional type II secretionsystem (Lory and Strom, 1997; Ayers et al., 2010). T2S isa multistep process where proteins intent for secretion arefirst translocated across the inner membrane by the Secor Tat pathway, and upon delivery into the periplasm,where the unfolded proteins then assume their tertiaryconformation. Finally, the proteins are translocated acrossthe outer membrane by a multiprotein complex, the T2Sapparatus (Cianciotto, 2009). T2S is present in many butnot all Gram-negative bacteria, indicating its role as animportant yet specialized secretion system (Cianciotto,2005). It was showed that the system is operative in bothpathogens and non-pathogens playing important roles inpathogenesis and/or contributing to bacterial fitness indifferent ecological niches (Cianciotto, 2005; Evans et al.,2008). T2S is critical for L. pneumophila intracellular infec-tion of protozoa cells and promotes the intracellular infec-tion of lung epithelial cells, dampens the cytokine outputfrom infected macrophages and epithelia, and limits thelevels of cytokine transcripts in infected macrophages(Cianciotto, 2009; McCoy-Simandle et al., 2011). More-over, at least 25 proteins have been shown to be T2Ssubstrates (DebRoy et al., 2006; Cianciotto, 2009; Pearceand Cianciotto, 2009; Stewart et al., 2009). Although func-tional redundancy is prevalent among T2S substrates, ametalloprotease – ProA, and a ribonuclease – SrnA,were shown to be required for the optimal infection ofamoeba (DebRoy et al., 2006; Rossier et al., 2008; 2009).Double mutants lacking both ProA and SrnA exhibited an
Received 25 July, 2011; revised 9 September, 2011; accepted 23October, 2011. *For correspondence. E-mail [email protected]; Tel.(+351) 239824024; Fax (+351) 239826798.
Environmental Microbiology (2011) doi:10.1111/j.1462-2920.2011.02646.x
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd

infectivity defect that was even greater than the corre-sponding single mutants, indicating that the role of T2S inintracellular infection is due to the combined effect ofmultiple secreted effectors (Rossier et al., 2009). Interest-ingly, ProA exhibited differential importance among theamoebae tested, suggesting that legionellae might haveevolved some of its factors to especially target some oftheir protozoan hosts (Rossier et al., 2008). Given the roleof protozoa in L. pneumophila survival in water, thesedata further establish T2S as a major factor in L. pneu-mophila persistence in the environment. Moreover, T2Spreviously thought to be dispensable, is involved invirulence-related phenotypes under conditions mimickingthe spread of Legionnaires’ disease from environmentalniche and is the only system known to be necessary foroptimal survival in low temperature aquatic habitats(Söderberg et al., 2004; 2008).
Given the role of T2S on the persistence of L. pneumo-phila in the environment and our interest in understandingthe population structure and evolution of L. pneumophila,our goal was to determine if the existence of distinct hostsand ecological niches led to an increase in the allelicdiversity of T2S and to identify the molecular mechanismsoperating in the evolution of this secretion system. For thispurpose we included 37 L. pneumophila strains isolatedfrom natural and man-made environments and disease-related to determine the genetic structure of L. pneumo-phila inferred from the gene sequence of three structuralcomponents of T2S (lspD, lspE and pilD) and two effec-tors (proA and srnA) that are critical for intracellular infec-tion of protozoa. Legionella pneumophila strains wereselected based on the previously determined allelic diver-sity inferred from dotA analysis to capture the maximumgenetic variability, as L. pneumophila natural environmen-tal isolates were clustered in a discrete group understrong diversifying selection with the highest allelic diver-sity, probably reflecting fitness variation in the persistenceof those strains in distinct environmental niches and/ortropism to various protozoan hosts (Costa et al., 2010a).
Our results indicate that the operating selective pres-sures have equally affected the evolution of all five T2S-related genes in L. pneumophila and that recombination isan important evolutionary mechanisms influencing thepopulation genetic structure of L. pneumophila. Thedetection of neutral selection acting on both ProA andSrnA T2S effectors indicate that they are probably impli-cated in conserved virulence mechanisms throughlegionellae hosts.
Results and discussion
Sequence analysis of T2S-related genes
The almost-complete lspD (2301 bp), lspE (1485 bp), pilD(864 bp), proA (1581 bp) and srnA (900 bp) gene
sequences were determined from 37 L. pneumophilastrains (Table 1) to determine the mechanisms shapingT2S crucial gene evolution. Strains were selected fromseveral others in order to capture the maximum geneticvariability, as they represented the determined allelicdiversity from the complete dotA sequence, envisioningevolutionary constrains exerted by distinct niches and/orhosts (Costa et al., 2010a). The genome of L. pneumo-phila strain Alcoy (D’Auria et al., 2010) and L. pneumo-phila strain 130b (Schroeder et al., 2010) becameavailable only recently. Comparative gene sequenceanalysis confirmed that the T2S-related genes from Alcoyand 130b strains were closely related to the homologuesfrom Corby and 797-PA-H strains respectively.
Due to the positions of primers and sequence ambiguityterminal sequences at the 5′ and 3′ ends, respectively,could not be included in the analysis, namely 48 and 27nucleotides of T2S outer membrane secretin lspD, 42 inthe metalloprotease proA, and 9 and 21 nucleotides in theribonuclease srnA. All L. pneumophila studied strainsyielded the five analysed genes with the expected size.
After performing the alignment of the gene sequencesagainst the corresponding genes found in the fourL. pneumophila genome sequenced strains and perform-ing the corresponding translation, three stop codons wereidentified. Namely, in the prepilin peptidase pilD fromstrains Ma36 and Por3 (Table 2) and in the T2S ATPaselspE from strain NMex49 (Table 2). Several methodolo-gies were applied in order to ascertain the existenceof those stop codons, including further DNA extractionsfrom fresh L. pneumophila cultures, additional genere-amplifications with proof-reading DNA polymerases fol-lowed by further sequencing analysis. In spite of all pro-tocols those mutations were always identified. Thenonsense mutation in pilD from Ma36 and Por3 strainswas due to a deletion of nucleotide number 9, resulting ina missense protein. On the other hand, several substitu-tions and deletions between nucleotide 5 and 17 of lspEfrom strain NMex49 resulted in a truncated protein.Because those mutations occurred at the beginning of thegenes, it is likely to result in loss of protein function, ascritical parts of the amino acid chain were missing. Func-tional studies on L. pneumophila T2S implicating the dis-ruption of both lspDE, which encoded the T2S outermembrane secretin and the ATPase, resulted in a severegrowth defect both in protozoa and human cells (Rossierand Cianciotto, 2001). From these results we can notconclude that the effect on the ability of NMex49 strain toinfect and survive is caused by the lspE single mutation.Phylogenetic, biochemical and structural evidencesupport the hypothesis that the widely distributed type IVApilus (T4S) system, involved in twitching motility, and theT2S system, involved in exoprotein release, descendedfrom a common ancestor (Peabody et al., 2003; Ayers
2 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

et al., 2010). Given the homologies between T2S and typeIVA secretion system it is possible that the ATPasesexhibit uniform functions (Peabody et al., 2003). For thesereasons, strain NMex49 could have an operational T2S,but lacks a functional lspE, by functional superimposingactivities of type IVA ATPase. Furthermore, it has beendemonstrated that the L. pneumophila pilD mutant and itsparent grow in bacteriological media, although they differin virulence-related phenotypes (Liles et al., 1999).Indeed, L. pneumophila pilD mutant did not produce T4Sand had greatly impaired growth within Hartmannella ver-miformis and in human macrophage-like U937 cells,explained by loss of T2S secretion (Liles et al., 1999;Rossier and Cianciotto, 2001). It is possible that strainsMa36 and Por3, lacking a functional pilD, are able tosurvive, in particular niches, most likely as free-living bac-
teria or intracellularly, in co-infections, with other legionel-lae possessing a functional T2S.
Genetic structure of T2S-related genes
Maximum likelihood (ML) phylogenetic trees wereobtained for each locus separately with PhyML 3.0(Guindon and Gascuel, 2003) and the best evolutionarymodel according to TOPALi and jModeltest (Milne et al.,2004; Posada, 2008) was GTR + I + G for lspD, lspE, pilD,rpoB and srnA and TrN + I + G for proA genes (Fig. 1).
Sequences from an internal fragment of the rpoB gene,previously obtained from the same L. pneumophila strains(Costa et al., 2010a), were included in the analysis(Fig. 1A) because the inferred rpoB tree agrees with phy-logenetic and phenotypic analyses (Brenner et al., 1988;
Table 1. L. pneumophila unrelated strains, isolated from distinct environments, type and reference strains included in this study.
Strain designationEnvironmentaltype Subspecies Reference of the source
rpoB
Accession number
Agn2 Natural L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652358a
Felg244 Natural L. pneumophila subsp. pneumophila Costa et al. (2010b) FN652410a
Ice30 Natural L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652434a
Alf 18 Natural L. pneumophila subsp. pneumophila Marrão et al. (1993) FN652367a
NMex1 Natural L. pneumophila subsp. pneumophila Marrão et al. (1993) FN652468a
NMex49 Natural L. pneumophila subsp. pneumophila Marrão et al. (1993) FN652474a
Aço20 Natural L. pneumophila subsp. pneumophila Veríssimo et al. (1991) FN652349a
Aço13 Natural L. pneumophila subsp. pneumophila Veríssimo et al. (1991) FN652347a
Ice27 Natural L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652433a
FACO3 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652399a
Ma1 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652453a
Ma36 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652462a
Por3 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652476a
Ma29 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652458a
Por9 Man-made L. pneumophila subsp. pneumophila Costa et al. (2010a) FN652477a
IMC23 Man-made L. pneumophila subsp. pneumophila Veríssimo et al. (1990) FN652428a
Philadelphia 1 (ATCC 33152T) Disease-related L. pneumophila subsp. pneumophila Chien et al. (2004) AF367748b
Togus 1 (ATCC 33154) Disease-related L. pneumophila subsp. pneumophila McKinney et al. (1979) AY036039b
Bloomington 2 (ATCC 33155) Disease-related L. pneumophila subsp. pneumophila McKinney et al. (1979) AY036040b
Chicago 2 (ATCC 33215) Disease-related L. pneumophila subsp. pneumophila McKinney et al. (1980) AY036041b
Chicago 8 (ATCC 33823) Disease-related L. pneumophila subsp. pneumophila Bibb et al. (1983) AY036042b
Concord 3 (ATCC 35096) Disease-related L. pneumophila subsp. pneumophila Bissett et al. (1983) AY036043b
IN-23-G1-C2 (ATCC 35289) Disease-related L. pneumophila subsp. pneumophila Edelstein et al. (1984) AY036044b
Leiden 1 (ATCC 43283) Disease-related L. pneumophila subsp. pneumophila Brenner et al. (1988) AY036045b
797-PA-H (ATCC 43130) Disease-related L. pneumophila subsp. pneumophila Thacker et al. (1986) AY036046b
570-CO-H (ATCC 43290) Disease-related L. pneumophila subsp. pneumophila Thacker et al. (1987) AY036047b
82A3105 (ATCC 43736) Disease-related L. pneumophila subsp. pneumophila Lindquist et al. (1988) AY036048b
1169-MN-H (ATCC 43703) Disease-related L. pneumophila subsp. pneumophila Benson et al. (1988) AY036049b
Los Angeles 1 (ATCC 33156T) Disease-related L. pneumophila subsp. fraseri McKinney et al. (1979) AY036050b
Dallas 1E (ATCC 33216) Disease-related L. pneumophila subsp. fraseri England et al. (1980) AY036051b
Lansing 3 (ATCC 35251) Disease-related L. pneumophila subsp. fraseri Brenner et al. (1988) AY036052b
U8W (ATCC 33737T) Disease-related L. pneumophila subsp. pascullei Brenner et al. (1988) AJ746049c
U7W (ATCC 33736) Disease-related L. pneumophila subsp. pascullei Brenner et al. (1988) AJ746050c
MICU B (ATCC 33735) Disease-related L. pneumophila subsp. pascullei Brenner et al. (1988) AJ746051c
Lens Disease-related L. pneumophila subsp. pneumophila Cazalet et al. (2004) lpl0362d
Paris Disease-related L. pneumophila subsp. pneumophila Cazalet et al. (2004) lpp0387d
Corby Disease-related L. pneumophila subsp. pneumophila Glöckner et al. (2008) NC_009494e
a. Sequences determined in Costa and colleagues (2010a) were used in this study.b. Sequences determined in Ko and colleagues (2002) were used in this study.c. Sequences determined in Costa and colleagues (2005) were used in this study.d. Genome sequence determined in Glöckner and colleagues (2008) was used in this study.e. Genome sequences determined in Cazalet and colleagues (2004) were used in this study.
Molecular evolution of Legionella pneumophila T2S system 3
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

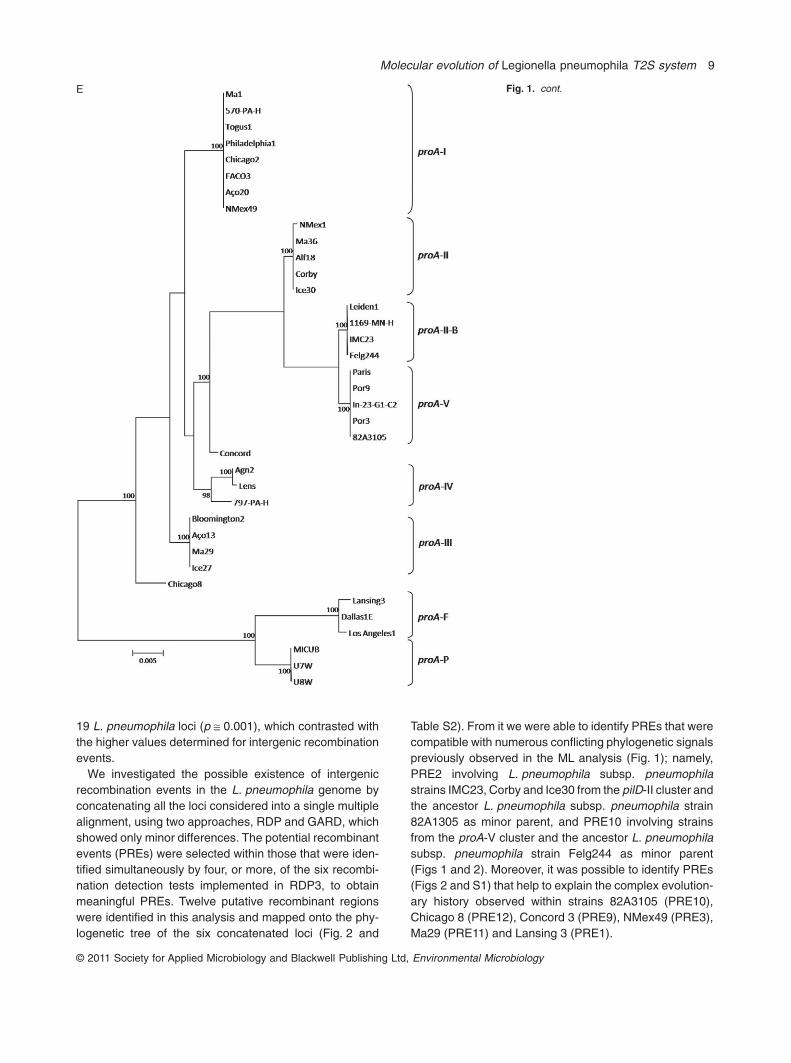
Hookey et al., 1996; Veríssimo et al., 1996; Ko et al.,2002), that allow the separation of the three L. pneumo-phila subspecies. The analysis of the rpoB gene from the37 strains matched the three different L. pneumophilasubspecies, namely, L. pneumophila subsp. pneumophila(cluster I to V), L. pneumophila subsp. fraseri (cluster F)and L. pneumophila subsp. pascullei (cluster P), compris-ing 83.8%, 8.1% and 8.1% of all strains respectively(Fig. 1A and Table 2). In fact, this topology was congruentin all five T2S-related genes, as it was possible to identifythe three corresponding lineages (Fig. 1).
From the gene sequence analysis of each of the fiveT2S-related genes it was also possible to identify sevenmajor clusters with very high bootstrap values (cluster I toV, F and P) consistent in all genes (Fig. 1), comprising83.8% of all strains. One important observation in thisstudy was that each of these seven clusters included thesame L. pneumophila strains, regardless the gene taken
into account (Table 2). These findings suggest that oper-ating selective pressures acting in the five T2S-relatedgenes have equally affected the strains within eachcluster. Although the observed similarity in strain compo-sition within the defined clusters, the topology of the phy-logenetic trees inferred from the five T2S-related geneswas not congruent because, and, depending on the con-sidered gene, most clusters had different relationshipswith each others (Fig. 1). These observations could besimply the result of neutral selection acting on the fiveT2S-related genes.
An exception was detected in the ML phylogenetic treeinferred from pilD gene sequences, as three strains(IMC23, Corby and Ice30) previously belonging to cluster IIwere separately grouped, forming a cluster closely relatedwith group pilD-III within L. pneumophila subsp. pneumo-phila strains, pilD-IIA (Fig. 1D and Table 2). A similar evo-lutionary drift was observed in the ML tree inferred from
Table 2. Distribution of L. pneumophila strains into clusters according with T2S-related gene sequences.
Strain designation Environmental type
Clusters
rpoB lspD lspE pilD proA srnA
Philadelphia 1 (ATCC 33152T) Disease-related I I I I I ITogus 1 (ATCC 33154) Disease-related I I I I I IChicago 2 (ATCC 33215) Disease-related I I I I I I570-CO-H (ATCC 43290) Disease-related I I I I I IFACO3 Man-made I I I I I IMa1 Man-made I I I I I IAço20 Natural I I I I I ICorby Disease-related II II II II-A II IILeiden 1 (ATCC 43283) Disease-related II II II II II-B II1169-MN-H (ATCC 43703) Disease-related II II II II II-B IIIMC23 Disease-related II II II II-A II-B IIMa36 Man-made II II II a II IIFelg244 Natural II II II II II-B IIIce30 Natural II II II II-A II IIAlf 18 Natural II II II II II IINMex1 Natural II II II II II IIBloomington 2 (ATCC 33155) Disease-related III III III III III IIIAço13 Natural III III III III III IIIIce27 Natural III III III III III III797-PA-H (ATCC 43130) Disease-related IV IV IV IV IV IVLens Disease-related IV IV IV IV IV IVAgn2 Natural IV IV IV IV IV IVParis Disease-related V V V V V VIN-23-G1-C2 (ATCC 35289) Disease-related V V V V V VPor3 Man-made V V V a V VPor9 Man-made V V V V V VLos Angeles 1(ATCC 33156T) Disease-related F F F F F FDallas 1E (ATCC 33216) Disease-related F F F F F FU8W (ATCC 33737T) Disease-related P P P P P PU7W (ATCC 33736) Disease-related P P P P P PMICU B (ATCC 33735) Disease-related P P P P P P82A3105 (ATCC 43736) Disease-related V II V II-A V IIIChicago 8 (ATCC 33823) Disease-related II – III III – –Concord 3 (ATCC 35096) Disease-related – V V – – –NMex49 Natural I II a I I IMa29 Man-made II III III III III –Lansing 3 (ATCC 35251) Disease-related F F I F F F
a. Truncated translated sequence.–, unclustered gene sequences.
4 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

proA gene because four strains belonging to cluster II in theother dendrograms, were clustered in a distinct group,proA-IIB, closely related to the proA-III cluster (Fig. 1E andTable 2). These incongruences are discussed below in thecontext of intergenic recombination.
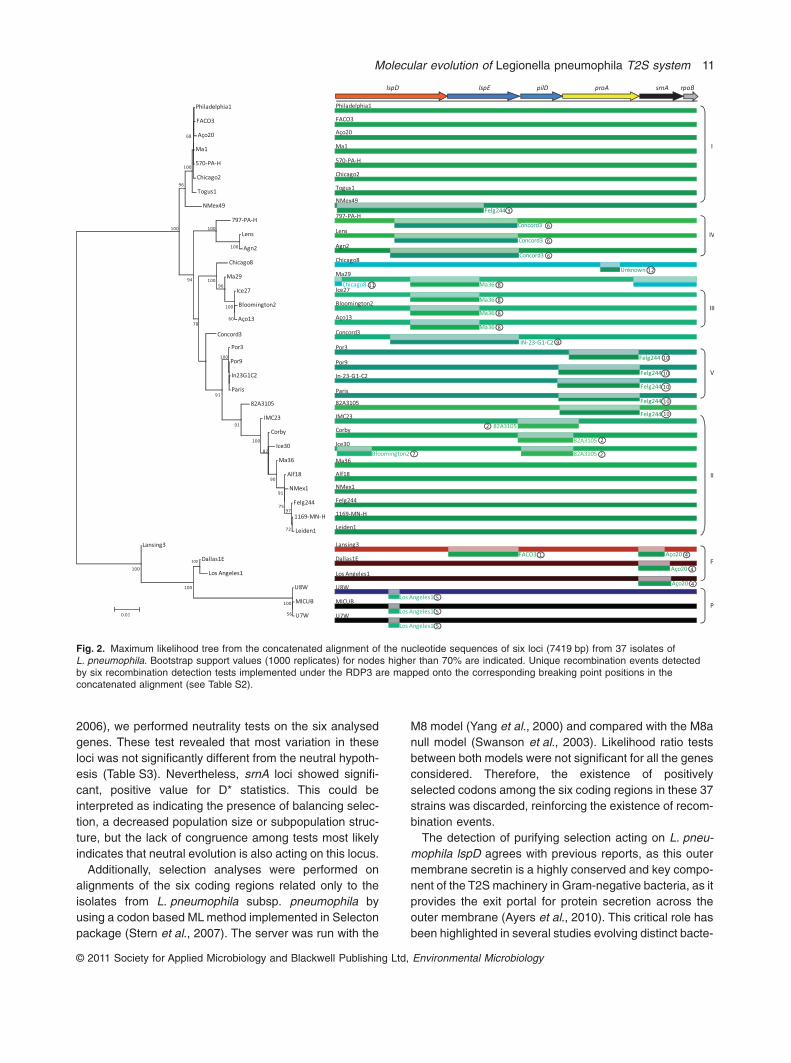
The concatenated alignment of the 6 loci included7419 bp and the best evolutionary model according toTOPALi and jModeltest was GTR + I + G. A ML phyloge-netic tree was obtained from the concatenated alignmentof the 6 loci (Fig. 2). Given the cluster congruenceobserved among the individual gene trees their main fea-tures were encased in the concatenated tree, so it was
possible to identify the previous seven clusters with veryhigh bootstrap values (Fig. 2).
From the 37 L. pneumophila strains, six exhibited amuch more complex evolutionary history because theywere included in distinct clusters, envisioning the exist-ence of several evolutionary reticulated events acting onthese T2S-related genes (Fig. 1 and Table 2).
Genetic variability of T2S-related genes
Within the analysed genes, the overall nucleotide diversityof pilD varied from 0 to 0.128 with an average of
A Fig. 1. Maximum likelihood phylogenetic treesof 37 L. pneumophila isolates, type andreferences strains (Table 1) from (A) rpoB (B)lspD (C) lspE (D) pilD (E) proA and (F) srnAlocus. Bootstrap support values (1000replicates) for nodes higher than 90% areindicated next to the corresponding node.
Molecular evolution of Legionella pneumophila T2S system 5
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

0.052 � 0.004, standing for the most diverse gene(Table S1). The diversity of lspD and lspE nucleotidesequences varied from 0.0 to 0.094 with an average of0.036 � 0.002, and from 0.001 to 0.110 with an averageof 0.043 � 0.003 respectively (Table S1). Interestingly,the less diverse genes encoded the T2S protein effectorsproA and srnA, crucial for protozoa infection, with agenetic pairwise differences varying from 0 to 0.052 withan average of 0.025 � 0.002 and from 0 to 0.082 with anaverage of 0.035 � 0.004 respectively (Table S1).
Genetic variability of 37 L. pneumophila unrelatedstrains was estimated based on the T2S-related loci using
genetic diversity parameters, not directly dependent onsample size (Table 3). The highest haplotype (h) and hap-lotype diversity (Hd) was found in lspE gene, presenting36 distinct alleles. The lowest haplotype diversity wasfound in srnA (0.905) with 15 haplotypes. The highestaverage number of pairwise differences (k = 79.827) andhighest total number of polymorphic sites (S = 305) werefound in LspD gene. On the other hand, pilD was the mostdiverse (p = 0.04916) and variable gene, with 18.4% poly-morphic sites. Moreover, the population mutation ratiowas also higher in pilD (q = 0.0447). Nucleotide substitu-tions accounting for differences among alleles were
B Fig. 1. cont.
6 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

mostly synonymous for locus lspD, in which we detected281 silent mutations (86.5%). Contrarily, almost 34% (52of 153) of all nucleotide changes detected in pilD werereplacement substitutions. In addition, proA and srnAgenes were the most conserved, with 8.2% (130/1581nucleotides) and 12.3% (111/900 nucleotides) variablesites, respectively, of which 16 and 14 predicted aminoacids were replacements respectively (Table 3). More-over, these genes also presented low levels of p, k and q(Table 3).
A comparison of the rate of non-synonymous substitu-tions (dN) to the rate of synonymous substitutions (dS)was performed, as it can be used as an indicator of theselective pressure acting on a protein-coding gene (Nei
and Gojobori, 1986; Yang and Bielawski, 2000). The lowdN/dS ratios obtained for all T2S-related genes indicatedthat these alleles were under purifying selection (Table 3).In this case, natural selection acts to selectively eliminatemutations with deleterious effects on protein structuresby causing change to functionally important amino acidresidues.
Because nucleotide substitutions may exert their influ-ence on the function of the final protein product at any ofseveral levels (e.g. DNA, mRNA or protein), dN/dS ratiosreflect general restrictions on gene and protein variability.On the other hand, dG values reflect variation purely inprotein structural and functional features, indicating somerestrictions on the amino acid substitutions at the level of
C Fig. 1. cont.
Molecular evolution of Legionella pneumophila T2S system 7
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

the final functioning product (Morozova et al., 2004). Fourphysico-chemical properties (volume, polarity, charge andhydrophobicity) were used to characterize the results ofamino acid substitutions in comparisons of translatedhomologous sequences (Bogardt et al., 1980;Kawashima and Kanehisa, 2000). Corresponding dGvalues were obtained using Miyata’s matrix (Miyata et al.,1979) and were calculated per one amino acid substitu-tion so that they would not depend on the rates of nucle-otide substitutions per se (Morozova et al., 2004). Low dGvalues indicate that only substitutions in amino acids withsimilar physico-chemical properties have been permitted.Contrarily, high dG values indicate that the amino acidsubstitution could introduce severe modifications on theprotein structure and function. Not all amino acid substi-
tutions in the genes with low dN/dS ratios are conserva-tive, as assessed by changes in amino acid physico-chemical properties. In fact, despite displaying relativelylow dN/dS values, the high calculated dG values for allT2S-related genes indicated that some of the amino acidsubstitutions resulted in drastic changes on protein prop-erties (Table 3).
Recombination events
We did not find any evidence of intragenic recombination inthe analyses performed separately for each locus withRDP3 (Martin et al., 2005a). These results were in agree-ment with the very low values of intragenic recombinationrates determined by Coscollá and colleagues (2011) from
D Fig. 1. cont.
8 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology
19 L. pneumophila loci (p @ 0.001), which contrasted withthe higher values determined for intergenic recombinationevents.
We investigated the possible existence of intergenicrecombination events in the L. pneumophila genome byconcatenating all the loci considered into a single multiplealignment, using two approaches, RDP and GARD, whichshowed only minor differences. The potential recombinantevents (PREs) were selected within those that were iden-tified simultaneously by four, or more, of the six recombi-nation detection tests implemented in RDP3, to obtainmeaningful PREs. Twelve putative recombinant regionswere identified in this analysis and mapped onto the phy-logenetic tree of the six concatenated loci (Fig. 2 and
Table S2). From it we were able to identify PREs that werecompatible with numerous conflicting phylogenetic signalspreviously observed in the ML analysis (Fig. 1); namely,PRE2 involving L. pneumophila subsp. pneumophilastrains IMC23, Corby and Ice30 from the pilD-II cluster andthe ancestor L. pneumophila subsp. pneumophila strain82A1305 as minor parent, and PRE10 involving strainsfrom the proA-V cluster and the ancestor L. pneumophilasubsp. pneumophila strain Felg244 as minor parent(Figs 1 and 2). Moreover, it was possible to identify PREs(Figs 2 and S1) that help to explain the complex evolution-ary history observed within strains 82A3105 (PRE10),Chicago 8 (PRE12), Concord 3 (PRE9), NMex49 (PRE3),Ma29 (PRE11) and Lansing 3 (PRE1).
E Fig. 1. cont.
Molecular evolution of Legionella pneumophila T2S system 9
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

Of the 37 isolates analysed, 24 contained at least onerecombinant region, and some of them presented two.Because recombination was present in more than half ofthe strains in this data set, it seems that horizontalexchange of genetic material is widespread in L. pneumo-phila. These several intergenic recombination eventsresulted in mosaic gene patterns in which different genesegments exhibit different evolutionary histories, whichmay contribute to novel biological properties. Therefore,these results may indicate that frequent recombinationdoes play a major role in diversification of this secretion
system, and that it may have had a role in the formation ofecologically distinct types. Nevertheless, Coscollá andcolleagues (2011) established in the syntenic genome ofL. pneumophila that recombination events are not distrib-uted evenly and several recombination hot spots weredetected, not including the T2S-related genes, raising thepossibility that the detected PREs could be merely theresult of simply shuffling genes in the genome without anyselective benefits.
In order to discard any influence of positive selection inthe detection of recombination events (Reed and Tishkoff,
F Fig. 1. cont.
10 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology
2006), we performed neutrality tests on the six analysedgenes. These test revealed that most variation in theseloci was not significantly different from the neutral hypoth-esis (Table S3). Nevertheless, srnA loci showed signifi-cant, positive value for D* statistics. This could beinterpreted as indicating the presence of balancing selec-tion, a decreased population size or subpopulation struc-ture, but the lack of congruence among tests most likelyindicates that neutral evolution is also acting on this locus.
Additionally, selection analyses were performed onalignments of the six coding regions related only to theisolates from L. pneumophila subsp. pneumophila byusing a codon based ML method implemented in Selectonpackage (Stern et al., 2007). The server was run with the
M8 model (Yang et al., 2000) and compared with the M8anull model (Swanson et al., 2003). Likelihood ratio testsbetween both models were not significant for all the genesconsidered. Therefore, the existence of positivelyselected codons among the six coding regions in these 37strains was discarded, reinforcing the existence of recom-bination events.
The detection of purifying selection acting on L. pneu-mophila lspD agrees with previous reports, as this outermembrane secretin is a highly conserved and key compo-nent of the T2S machinery in Gram-negative bacteria, as itprovides the exit portal for protein secretion across theouter membrane (Ayers et al., 2010). This critical role hasbeen highlighted in several studies evolving distinct bacte-
Fig. 2. Maximum likelihood tree from the concatenated alignment of the nucleotide sequences of six loci (7419 bp) from 37 isolates ofL. pneumophila. Bootstrap support values (1000 replicates) for nodes higher than 70% are indicated. Unique recombination events detectedby six recombination detection tests implemented under the RDP3 are mapped onto the corresponding breaking point positions in theconcatenated alignment (see Table S2).
Molecular evolution of Legionella pneumophila T2S system 11
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

ria, namely L. pneumophila (revised in Rossier and Cian-ciotto, 2001; Ayers et al., 2010). It has been proposed thatLspD is composed of two distinct regions: a variableN-terminal domain and a C-terminal domain involved inmultimerization (Nouwen et al., 2000). We have identifiedthe insertion of a putative SPOR domain at the beginning ofLspD N-terminal, between Asn16 and Gln80 (Fig. S1), withhigh homology with pfam05036 (Marchler-Bauer et al.,2009). The SPOR domain has been involved in peptidogly-can binding (Mishima et al., 2005), and is predicted to bepresent in at least 3488 (putative) proteins from over 1148bacterial species (PF05036; Pfam 23) (Finn et al., 2010),raising the question on how far SPOR properties havebeen conserved. Several reports predict that many otherbacterial SPOR domain proteins specifically recognize thesame or closely related target molecule(s) that accumu-lates transiently at sites of cell constriction (Gerding et al.,2009). We identified this SPOR domain in the LspD secre-tins in all genome sequences of L. pneumophila andLegionella longbeachae strains (Cazalet et al., 2004;Chien et al., 2004; Glöckner et al., 2008; Kozak et al.,2010). Interestingly, a very short LspD was identified inLegionella drancourtii LLAP12 (482 amino acids), lackingthe SPOR domain (results not shown). Given these resultsit is possible that the presence of this domain could berelated with legionellae lifestyle, as it is missing in thestrictly intra-amoebal L. drancourtii (Moliner et al., 2009).Moreover, it is tempting to speculate that the existence ofthis SPOR domain in L. pneumophila LspD could berelated with a specific targeting of these secretins but moredetailed analyses are necessary to verify it. Additionally, asin most described LspD secretins (Ayers et al., 2010), themajority of polymorphisms and non-synonymous substitu-tions were identified in the corresponding N-terminal, whilethe C-terminal domain was highly conserved among
L. pneumophila strains. A highly polymorphic region,between Tyr407 and Met421, was detected in strains belong-ing to L. pneumophila subsp. pascullei, located in thelinking region between two catalytic sites of the thirdsecretin-N domain. Given the location of the correspondingnon-synonymous substitutions, little impact in protein func-tion is expected. Furthermore, the detection of purifyingselection acting on L. pneumophila lspE also agrees withthe findings of Peabody and colleagues (2003), in whichprotein constituents of various secretion systems haveundergone sequence divergence at different rates, with theATPases and multispanning transmembrane proteinsbeing the most slowly diverging.
As previously mentioned, T2S protein secretion plays arole in a wide variety of functions that are important for theecology and pathogenesis of L. pneumophila. Perhaps,most dramatic is the critical role that this secretionpathway has in L. pneumophila intracellular infection ofprotozoa (Cianciotto, 2009). In this study, we provide phy-logenetic data and conduct sequence comparisons thatallowed determining the forces shaping L. pneumophilaT2S-related molecular gene evolution. The detection ofneutral selection acting on all L. pneumophila T2S-relatedgenes emerges as a clear result from various analysisperformed in the present study. In fact the two T2S effec-tors, ProA and SrnA, were identified as being required foramoeba infection (Rossier et al., 2008; 2009). The detec-tion of neutral selection acting on both ProA and SrnA T2Seffectors indicate that they were probably implicated invirulence mechanisms that are conserved in legionellaehosts. Given the role of T2S in L. pneumophila pathogen-esis, survival and persistence the recombination eventsfrequently detected could play an important role onincreasing the fitness of L. pneumophila strains in definedenvironmental niches.
Table 3. Summary of genetic diversity parameters for the five T2S-related loci of L. pneumophila strains.
lspD lspE pilD proA srnA
Sequence, n 37 36 35 37 37Sequence length, L 2301 1485 864 1581 900Haplotypes, h 26 36 27 15 15Haplotype diversity, Hd 0.971 1.000 0.983 0.911 0.905(standard deviation) (0.015) (0.007) (0.011) 0.024 (0.027)Nucleotide diversity, p 0.0347 0.0415 0.0492 0.0242 0.0336(standard deviation) (0.0052) (0.006) (0.007) (0.002) (0.004)Polymorphic sites, S 305 241 159 130 111(%) (13.3) (16.2) (18.4) (8.2) (12.3)q (from S) 0.0318 0.0391 0.0447 0.0197 0.0296(standard deviation) (0.0094) (0.0117) (0.0136) (0.0059) (0.0091)Pairwise differences, k 79.827 61.584 42.476 38.288 30.153Synonymous mutations 281 217 101 121 106(%) (86.5) (85.8) (66.0) (88.0) (88.3)Non-synonymous mutations 44 36 52 16 14(%) (13.5) (14.2) (34.0) (12.0) (11.6)dN/dS 0.05 0.048 0.159 0.034 0.075dG per one amino acid change 1.41 1.19 1.37 1.41 0.996
12 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

Experimental procedures
L. pneumophila strains
Thirty three unrelated strains of L. pneumophila wereselected for almost-complete sequencing of the lspD, lspE,pilD, proA and srnA genes to determine the genetic structureand molecular evolution (Table 1). These included 16 isolatesfrom nine sites comprising natural and man-made environ-ments, and 17 disease-related L. pneumophila type and ref-erence strains, 11 from L. pneumophila subsp. pneumophila,three L. pneumophila subsp. fraseri strains and threeL. pneumophila subsp. pascullei. The sequences from fourL. pneumophila subsp. pneumophila genome sequencedstrains (Cazalet et al., 2004; Chien et al., 2004; Glöckneret al., 2008) were also included in this work. Previously pub-lished sequences of partial rpoB gene from the studiedstrains were also used for comparison purposes (Table 1).
DNA extraction, polymerase chain reaction (PCR),cloning and DNA sequencing
The extraction of genomic DNA from the previously selectedL. pneumophila strains was carried out as previouslydescribed by Costa and colleagues (2005). PCRs wereperformed to amplify the five loci, lspD (2376 bp), lspE(1485 bp), pilD (845 bp), proA (1632 bp), and srnA (963 bp)using the primer sets described in Table S4. In general, PCRwas carried out using 150–200 ng DNA, 2.0 mM MgCl2, 1 ¥reaction buffer, 0.2 mM each dNTP, 5 pmol each primer, and1U Taq polymerase (Invitrogen) in 50 ml reaction volumeswith the following PCR profile: 5 min at 95°C; 30 cycles of95°C, 30 s; 52°C, 30 s; 72°C, 1 min and 30 s; 7 min at 72°C.For the lspD gene amplification an extension step of 2 minand 30 s was used. Moreover, in some cases it was neces-sary to adjust the annealing temperatures for individualstrains or genes. The amplified PCR products were detectedon 1.0% agarose gels stained with ethidium bromide andwere purified for sequencing by using a JetQuick gel extrac-tion kit (Genomed GmbH, Germany). To obtain nearly fullylength genes, the PCR products were cloned using pGEM-TEasy Vector Systems (Promega) according to the manufac-turer instructions. Positive clones were selected on Luria–Bertani agar plates containing 20 mg ml-1 Xgal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside), 0.5 mM IPTG(isopropyl-b-D-1-thiogalactopyranoside), and 100 mg ml-1
ampicillin. Plates were incubated overnight at 37°C in selec-tive media. Positive clones were confirmed by PCR with thesame primers used for amplification, and plasmidic DNA wasextracted using Zyppy Plasmid Miniprep Kit (Zymo Research,USA) according to the manufacturer instructions. Genesequences were determined by Macrogen Corporation(Netherlands).
Sequence analysis
The quality of the sequences was manually checkedusing the Sequence Scanner software (https://products.appliedbiosystems.com). Phylogenetic analyses were per-formed using MEGA5 package (Tamura et al., 2011). Align-
ment against the corresponding genes found in four genomesequenced L. pneumophila strains obtained from the publicdatabases (see accession numbers in Table S5), was per-formed using the multiple alignment CLUSTAL software(Higgins, 1994), included on MEGA5 package. For codingloci alignments were performed with the amino acidsequences and gaps were later introduced in the correspond-ing nucleotide alignments, thus keeping the correct frame fortranslation. Additionally, for further phylogenetic analyses andto test for recombination in this data set, the 6 loci wereconcatenated in a single multiple alignment with BioEdit7.0.9.0 (Hall, 1999).
Maximum likelihood phylogenetic trees were obtained forthe concatenated alignment and for each individual locus withPhyML 3.0 (Guindon and Gascuel, 2003), using the mostappropriate model of nucleotide substitution and likelihoodscores assessed by TOPALi V2.5 (Milne et al., 2004) and byjModeltest (Posada, 2008). The best model was determinedby using the Akaike Information Criterion (AIC) (Akaike, 1974;Posada and Buckley, 2004). Supports for the nodes wereevaluated by bootstrapping with 1000 pseudoreplicates.
Genetic variability analyses were performed with DnaSPsoftware (Librado and Rozas, 2009).
Recombination analysis
Intergenic recombination was analysed in the alignment ofthe six concatenated loci and intragenic recombination wasscreened for each individual locus alignment with theprogram RDP3 (Martin et al., 2005a). This program identi-fies recombinant sequences and recombination breakingpoints using several methods. We choose six of them: RDP(Martin and Rybicki, 2000), GENECONV (Padidam et al.,1999), BootScan (Martin et al., 2005b), Maximum Chi-squared Test (MaxChi; Maynard Smith, 1992), CHIMAERA(Posada and Crandall, 2001) and Sister Scan (SiScan;Gibbs et al., 2000). The analysis was performed with defaultsettings for the detection methods, a Bonferroni correctedP-value cut-off of 0.05, and a requirement that each poten-tial event had to be detected simultaneously by four or moremethods. The breakpoint positions and recombinantsequence(s) inferred for every detected potential recombi-nation event were manually checked and adjusted wherenecessary using the extensive phylogenetic and recombina-tion signal analysis features available in RDP3. The GARDmethod (Kosakovsky Pond et al., 2006) implemented indatamonkey server (Delport et al., 2010) was also used tosearch for evidences of phylogenetic incongruence, and toidentify the number and location of breakpoints correspond-ing to recombination events.
Neutrality tests and positive selection analysis
Tajima’s D (Tajima, 1989), Fu and Li’s D* and F* (Fu and Li,1993) and Fu’s Fs (Fu, 1997) statistics were calculated fortesting the mutation neutrality hypothesis (Kimura, 1983), aspreviously described by Coscollá and colleagues (2006).These statistics were calculated with the program DNASP4.0(Librado and Rozas, 2009) using a statistical significancelevel a = 0.025 and applying the false discovery rate (Ben-
Molecular evolution of Legionella pneumophila T2S system 13
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

jamini and Hochberg, 1995; Benjamini and Yekutieli, 2005) tocorrect for multiple comparisons and 1000 replicates in acoalescent simulation.
Estimates of the number of non-synonymous and synony-mous substitutions at each locus (dN/dS) were calculatedusing the modified Nei–Gojobori method (Nei and Gojobori,1986) with Jukes-Cantor correction (Jukes and Cantor, 1969)implemented in MEGA5 package (Tamura et al., 2011).
In order to investigate the presence of positively selectedcodons in the six coding loci, the estimates of both positiveand purifying selection at each amino acid site were calcu-lated from the ratio of non-synonymous to synonymous sub-stitutions, known as w, as previously described (Costa et al.,2010a). Nucleotide sequences alignment from L. pneumo-phila subsp. pneumophila strains were constructed using theMEGA5 package (Tamura et al., 2011) and analyses wereconducted using the Selecton version 2.1 software (Doron-Faigenboim et al., 2005; Stern et al., 2007). The significanceof the w scores was obtained by using a Likelihood Ratio Testthat compares two nested models: a null model that assumesno selection (M8a) (Swanson et al., 2003) and an alternativemodel that does (M8) (Yang et al., 2000).
Four physico-chemical properties (volume, polarity, chargeand hydrophobicity) were used to characterize the results ofamino acid substitutions in comparisons of translatedhomologous sequences (Bogardt et al., 1980; Kawashimaand Kanehisa, 2000). Corresponding dG values wereobtained using Miyata’s matrix (Miyata et al., 1979) and werecalculated per one amino acid substitution so that they wouldnot depend on the rates of nucleotide substitutions per se(Morozova et al., 2004).
Nucleotide sequence accession numbers
The near complete 165 sequences from L. pneumophilastrains determined in this study were deposited in theEMBL Nucleotide Sequence Database with Accession No.HE565705–HE565737 (lspD), HE565738–HE565769 (lspE),HE565770–HE565800 (pilD), HE565801–HE565833 (proA)and HE565834–HE565866 (srnA).
Acknowledgements
This work was supported by Project PTDC/BIA-MIC/105247/2008. J. Costa acknowledges scholarship from FCT(SFRH/BPD/34007/2006). We acknowledge the valuablecontributions of two anonymous reviewers.
References
Akaike, H. (1974) A new look at the statistical model identifi-cation. IEEE Trans Autom Control 19: 716–723.
Ayers, M., Howell, P.L., and Burrows, L.L. (2010) Architectureof the type II secretion and type IV pilus machineries.Future Microbiol 5: 1203–1218.
Benjamini, Y., and Hochberg, Y. (1995) Controlling the falsediscovery rate: a practical and powerful approach to mul-tiple testing. J R Stat Soc B 57: 289–300.
Benjamini, Y., and Yekutieli, D. (2005) False discovery rate –adjusted multiple confidence intervals for selected param-eters. J Am Stat Assoc 100: 71–93.
Benson, R.F., Thacker, W.L., Wilkinson, H.W., Fallon, R.J.,and Brenner, D.J. (1988) Legionella pneumophila sero-group 14 isolated from patients with fatal pneumonia. J ClinMicrobiol 26: 382.
Bibb, W.R., Arnow, P.M., Dellinger, D.L., and Perryman, S.R.(1983) Isolation and characterization of a seventh sero-group of Legionella pneumophila. J Clin Microbiol 17: 346–348.
Bissett, M.L., Lee, J.O., and Lindquist, D.S. (1983) New sero-group of Legionella pneumophila, serogroup 8. J ClinMicrobiol 17: 887–891.
Bogardt, R.A., Jones, B.N., Dwulet, F.E., Garner, W.H.,Lehman, L.D., and Gurd, F.R. (1980) Evolution of theamino acid substitution in the mammalian myoglobin gene.J Mol Evol 15: 197–218.
Brenner, D.J., Staigerwalt, A.G., Epple, P., Bibb, W.F., McKin-ney, R.M., Starnes, R.W., et al. (1988) Legionella pneumo-phila serogroup Lansing 3 isolated from a patient with fatalpneumonia, and descriptions of L. pneumophila subsp.pneumophila subsp. nov., L. pneumophila subsp. fraserisubsp. nov., and L. pneumophila subsp. pascullei subsp.nov. J Clin Microbiol 26: 1695–1703.
Cazalet, C., Rusniok, C., Brüggemann, H., Zidane, N.,Magnier, A., Ma, L., et al. (2004) Evidence in the Legionellapneumophila genome for exploitation of host cell functionsand high genome plasticity. Nat Genet 36: 1165–1173.
Chien, M., Morozova, I., Shi, S., Sheng, H., Chen, J., Gomez,S.M., et al. (2004) The genomic sequence of the accidentalpathogen Legionella pneumophila. Science 305: 1966–1968.
Cianciotto, N.P. (2005) Type II secretion: a protein secretionsystem for all seasons. Trends Microbiol 13: 581–588.
Cianciotto, N.P. (2009) Many substrates and functions of typeII secretion: lessons learned from Legionella pneumophila.Future Microbiol 4: 797–805.
Coscollá, M., Gosalbes, M.J., Catalán, V., and González-Candelas, F. (2006) Genetic variation in environmentalsamples of Legionella pneumophila from the ComunidadValenciana (Spain). Environ Microbiol 4: 1056–1063.
Coscollá, M., Comas, I., and González-Candelas, F.(2011) Quantifying nonvertical inheritance in the evolu-tion of Legionella pneumophila. Mol Biol Evol 28: 985–1001.
Costa, J., Tiago, I., da Costa, M.S., and Veríssimo, A. (2005)Presence and persistence of Legionella spp. in groundwa-ter. Appl Environ Microbiol 71: 663–671.
Costa, J., Tiago, I., da Costa, M.S., and Veríssimo, A. (2010a)Molecular evolution of Legionella pneumophila dotA gene,the contribution of natural environmental strains. EnvironMicrobiol 12: 2711–2729.
Costa, J., da Costa, M.S., and Veríssimo, A. (2010b) Coloni-zation of a therapeutic spa with Legionella spp.: a publichealth issue. Res Microbiol 161: 18–25.
D’Auria, G., Jiménez-Hernández, N., Peris-Bondia, F., Moya,A., and Latorre, A. (2010) Legionella pneumophila pange-nome reveals strain-specific virulence factors. BMCGenomics 17: 181–194.
De Buck, E., Anné, J., and Lammertyn, E. (2007) The role ofprotein secretion systems in the virulence of the intracellu-lar pathogen Legionella pneumophila. Microbiology 153:3948–3953.
14 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

Debroy, S., Aragon, V., Kurtz, S., and Cianciotto, N.P. (2006)Legionella pneumophila Mip, a surface-exposed peptidyl-proline cis-trans-isomerase, promotes the presence ofphospholipase C-like activity in culture supernatants. InfectImmun 74: 5152–5160.
Delport, W., Poon, A.F., Frost, S.D., and Kosakovsky Pond,S.L. (2010) Datamonkey 2010: a suite of phylogeneticanalysis tools for evolutionary biology. Bioinformatics 26:2455–2457.
Doron-Faigenboim, A., Stern, A., Mayrose, I., Bacharach, E.,and Pupko, T. (2005) Selecton: a server for detecting evo-lutionary forces at a single amino-acid site. Bioinformatics21: 2101–2113.
Edelstein, P.H., Bibb, W.F., Gorman, G.W., Thacker, W.L.,Brenner, D.J., Wilkinson, H.W., et al. (1984) Legionellapneumophila serogroup 9: a cause of human pneumonia.Ann Intern Med 101: 196–198.
England, A.C., 3rd, McKinney, R.M., Skaliy, P., and Gorman,G.W. (1980) A fifth serogroup of Legionella pneumophila.Ann Intern Med 93: 58–59.
Evans, F.F., Egan, S., and Kjelleberg, S. (2008) Ecology oftype II secretion in marine gammaproteobacteria. EnvironMicrobiol 10: 1101–1107.
Fields, B.S. (2008) Legionella in the environment. InLegionella pneumophila: Pathogenesis and Immunity.Hoffman, P., Friedman, H., and Bendinelli, M. (eds). NewYork, USA: Springer Science and Business Media, pp.85–94.
Finn, R.D., Mistry, J., Tate, J., Coggill, P., Heger, A., Polling-ton, J.E., et al. (2010) The Pfam protein families database.Nucleic Acids Res 38: D211–D222.
Fu, Y.X. (1997) Statistical tests of neutrality of mutationsagainst population growth, hitchhiking and backgroundselection. Genetics 147: 915–925.
Fu, Y.X., and Li, W.H. (1993) Maximum likelihood estimationof population parameters. Genetics 134: 1261–1270.
Gerding, M.A., Liu, B., Bendezú, F.O., Hale, C.A., Bernhardt,T.G., and de Boer, P.A. (2009) Self-enhanced accumulationof FtsN at division sites and roles for other proteins with aSPOR domain (DamX, DedD, and RlpA) in Escherichia colicell constriction. J Bacteriol 191: 7383–7401.
Gibbs, M.J., Armstrong, J.S., and Gibbs, A.J. (2000) Sister-scanning: a Monte Carlo procedure for assessing signals inrecombinant sequences. Bioinformatics 16: 573–582.
Glöckner, G., Albert-Weissenberger, C., Weinmann, E.,Jacobi, S., Schunder, E., Steinert, M., et al. (2008) Identi-fication and characterization of a new conjugation/type IVAsecretion system (trb/tra) of Legionella pneumophila Corbylocalized on two mobile genomic islands. Int J Med Micro-biol 298: 411–428.
Guindon, S., and Gascuel, O. (2003) A simple, fast, andaccurate algorithm to estimate large phylogenies bymaximum likelihood. Syst Biol 52: 696–704.
Hall, T.A. (1999) BioEdit: a user-friendly biological sequencealignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98.
Higgins, D.G. (1994) CLUSTAL V: multiple alignment of DNAand protein sequences. Methods Mol Biol 25: 307–318.
Hookey, J.V., Saunders, N.A., Fry, N.K., Birtles, R.J., andHarrison, T.G. (1996) Phylogeny of Legionellaceae basedon small-subunit ribosomal DNA sequences and proposal
of Legionella lytica comb. nov. for Legionella-like amoebalpathogens. Int J Syst Bacteriol 46: 526–531.
Jukes, T.H., and Cantor, C.R. (1969) Evolution of proteinmolecules. In Mammalian Protein Metabolism. Munro, H.N.(ed.). New York, USA: Academic Press, pp. 21–132.
Kawashima, S., and Kanehisa, M. (2000) AAIndex: aminoacid index database. Nucleic Acids Res 28: 374.
Kimura, M. (1983) The Neutral Theory of Molecular Evolu-tion. Cambridge, UK: Cambridge University Press.
Ko, K.S., Lee, H.K., Park, M.Y., Park, M.S., Lee, K.H., Woo,S.Y., et al. (2002) Population genetic structure ofLegionella pneumophila inferred from RNA polymerasegene (rpoB) and DotA gene (dotA) sequences. J Bacteriol184: 2123–2130.
Kosakovsky Pond, S.L., Posada, D., Gravenor, M.B., Woelk,C.H., and Frost, S.D. (2006) GARD: a genetic algorithm forrecombination detection. Bioinformatics 22: 3096–3108.
Kozak, N.A., Buss, M., Lucas, C.E., Frace, M., Govil, D.,Travis, T., et al. (2010) Virulence factors encoded byLegionella longbeachae identified on the basis of thegenome sequence analysis of clinical isolate D-4968.J Bacteriol 192: 1030–1044.
Librado, P., and Rozas, J. (2009) DnaSP v5: A software forcomprehensive analysis of DNA polymorphism data. Bio-informatics 25: 1451–1452.
Liles, M.R., Viswanathan, V.K., and Cianciotto, N.P. (1998)Identification and temperature regulation of Legionellapneumophila genes involved in type IV pilus biogenesisand type II protein secretion. Infect Immun 66: 1776–1782.
Liles, M.R., Edelstein, P.H., and Cianciotto, N.P. (1999) Theprepilin peptidase is required for protein secretion by andthe virulence of the intracellular pathogen Legionella pneu-mophila. Mol Microbiol 31: 959–970.
Lindquist, D.S., Nygaard, G., Thacker, W.L., Benson, R.F.,Brenner, D.J., and Wilkinson, H.W. (1988) Thirteenth sero-group of Legionella pneumophila isolated from patientswith pneumonia. J Clin Microbiol 26: 586–587.
Lory, S., and Strom, M.S. (1997) Structure-function relation-ship of type-IV prepilin peptidase of Pseudomonas aerugi-nosa – a review. Gene 192: 117–121.
McCoy-Simandle, K., Stewart, C.R., Dao, J., Debroy, S.,Rossier, O., Bryce, P.J., and Cianciotto, N.P. (2011)Legionella pneumophila type II secretion dampens thecytokine response of infected macrophages and epithelia.Infect Immun 79: 1984–1997.
McKinney, R.M., Thacker, L., Harris, P.P., Lewallen, K.R.,Herbert, G.A., Edelstein, P.H., and Thomason, B.M. (1979)Four serogroups of Legionnaires’ disease bacteria definedby direct immunofluorescence. Ann Intern Med 90: 621–624.
McKinney, R.M., Wilkinson, H.W., Sommers, H.M., Fikes,B.J., Sasseville, K.R., Yungbluth, M.M., and Wolf, J.S.(1980) Legionella pneumophila serogroup six: isolatedfrom cases of Legionellosis, identification by immunof-luirescence staining, and immunological response to infec-tion. J Clin Microbiol 12: 395–401.
Marchler-Bauer, A., Anderson, J.B., Chitsaz, F., Derbyshire,M.K., DeWeese-Scott, C., Fong, J.H., et al. (2009) CDD:specific functional annotation with the Conserved DomainDatabase. Nucleic Acids Res 37: D205–D210.
Molecular evolution of Legionella pneumophila T2S system 15
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

Marrão, G., Verissimo, A., Bowker, R.G., and Costa, M.S.(1993) Biofilms as major sources of Legionella spp. inhydrothermal areas and their dispersion into stream water.FEMS Microbiol Ecol 12: 25–33.
Martin, D., and Rybicki, E. (2000) RDP: detection of recom-bination amongst aligned sequences. Bioinformatics 16:562–563.
Martin, D.P., Williamson, C., and Posada, D. (2005a) RDP2:recombination detection and analysis from sequence align-ments. Bioinformatics 21: 260–262.
Martin, D.P., Posada, D., Crandall, K.A., and Williamson, C.(2005b) A modified bootscan algorithm for automatedidentification of recombinant sequences and recombina-tion breakpoints. AIDS Res Hum Retroviruses 21:98–102.
Maynard Smith, J. (1992) Analysing the mosaic structure ofgenes. J Mol Evol 34: 126–129.
Milne, I., Wright, F., Rowe, G., Marshal, D.F., Husmeier, D.,and McGuire, G. (2004) TOPALi: software for automaticidentification of recombinant sequences within DNA mul-tiple alignments. Bioinformatics 20: 1806–1807.
Mishima, M., Shida, T., Yabuki, K., Kato, K., Sekiguchi, J.,and Kojima, C. (2005) Solution structure of the pepti-doglycan binding domain of Bacillus subtilis cell wall lyticenzyme CwlC: characterization of the sporulation-relatedrepeats by NMR. Biochemistry 44: 10153–10163.
Miyata, T., Miyazawa, S., and Yasunaga, T. (1979) Two typesof amino acid substitutions in protein evolution. J Mol Evol12: 219–236.
Moliner, C., Raoult, D., and Fournier, P.E. (2009) Evidencethat the intra-amoebal Legionella drancourtii acquired asterol reductase gene from eukaryotes. BMC Res Notes27: 51–59.
Moliner, C., Fournier, P.E., and Raoult, D. (2010) Genomeanalysis of microorganisms living in amoebae reveals amelting pot of evolution. FEMS Microbiol Rev 34: 281–294.
Morozova, I., Qu, X., Shi, S., Asamani, G., Greenberg, J.E.,Shuman, H.A., and Russo, J.J. (2004) Comparativesequence analysis of the icm/dot genes in Legionella.Plasmid 51: 127–147.
Nei, M., and Gojobori, T. (1986) Simple methods for estimat-ing the numbers of synonymous and nonsynonymousnucleotide substitutions. Mol Biol Evol 3: 418–426.
Newton, H.J., Ang, D.K., van Driel, I.R., and Hartland, E.L.(2010) Molecular pathogenesis of infections caused byLegionella pneumophila. Clin Microbiol Rev 23: 274–298.
Nouwen, N., Stahlberg, H., Pugsley, A.P., and Engel, A.(2000) Domain structure of secretin PulD revealed bylimited proteolysis and electron microscopy. EMBO J 19:2229–2236.
Padidam, M., Sawyer, S., and Fauquet, C.M. (1999) Possibleemergence of new geminiviruses by frequent recombina-tion. Virology 265: 218–225.
Peabody, C.R., Chung, Y.J., Yen, M.R., Vidal-Ingigliardi, D.,Pugsley, A.P., and Saier, M.H., Jr (2003) Type II proteinsecretion and its relationship to bacterial type IV pili andarchaeal flagella. Microbiology 149: 3051–3072.
Pearce, M.M., and Cianciotto, N.P. (2009) Legionella pneu-mophila secretes an endoglucanase that belongs to thefamily-5 of glycosyl hydrolases and is dependent upon typeII secretion. FEMS Microbiol Lett 300: 256–264.
Posada, D. (2008) jModelTest: phylogenetic model averag-ing. Mol Biol Evol 25: 1253–1256.
Posada, D., and Buckley, T.R. (2004) Model selection andmodel averaging in phylogenetics: advantages of the AICand Bayesian approaches over likelihood ratio tests. SystBiol 53: 793–808.
Posada, D., and Crandall, K.A. (2001) Evaluation of methodsfor detecting recombination from DNA sequences: com-puter simulations. Proc Natl Acad Sci USA 98: 13757–13762.
Reed, F.A., and Tishkoff, S.A. (2006) Positive selection cancreate false hotspots of recombination. Genetics 172:2011–2014.
Rossier, O., and Cianciotto, N.P. (2001) Type II protein secre-tion is a subset of the PilD-dependent processes that facili-tate intracellular infection by Legionella pneumophila.Infect Immun 69: 2092–2098.
Rossier, O., Dao, J., and Cianciotto, N.P. (2008) The type IIsecretion system of Legionella pneumophila elaboratestwo aminopeptidases, as well as a metalloprotease thatcontributes to differential infection among protozoan hosts.Appl Environ Microbiol 74: 753–761.
Rossier, O., Dao, J., and Cianciotto, N.P. (2009) A type IIsecreted RNase of Legionella pneumophila facilitatesoptimal intracellular infection of Hartmannella vermiformis.Microbiology 155: 882–890.
Schroeder, G.N., Petty, N.K., Mousnier, A., Harding, C.R.,Vogrin, A.J., Wee, B., et al. (2010) Legionella pneumophilastrain 130b possesses a unique combination of type IVsecretion systems and novel Dot/Icm secretion systemeffector proteins. J Bacteriol 192: 6001–6016.
Söderberg, M.A., Rossier, O., and Cianciotto, N.P. (2004)The type II protein secretion system of Legionella pneumo-phila promotes growth at low temperatures. J Bacteriol186: 3712–3720.
Söderberg, M.A., Dao, J., Starkenburg, S.R., and Cianciotto,N.P. (2008) Importance of type II secretion for survival ofLegionella pneumophila in tap water and in amoebae atlow temperatures. Appl Environ Microbiol 74: 5583–5588.
Stern, A., Doron-Faigenboim, A., Erez, E., Martz, E.,Bacharach, E., and Pupko, T. (2007) Selecton 2007:advanced models for detecting positive and purifyingselection using a Bayesian inference approach. NucleicAcids Res 35: W506–W511.
Stewart, C.R., Rossier, O., and Cianciotto, N.P. (2009)Surface translocation by Legionella pneumophila: a form ofsliding motility that is dependent upon type II protein secre-tion. J Bacteriol 191: 1537–1546.
Swanson, W.J., Nielsen, R., and Yang, Q. (2003) Pervasiveadaptive evolution in mammalian fertilization proteins. MolBiol Evol 20: 18–20.
Tajima, F. (1989) Statistical method for testing the neutralmutation hypothesis by DNA polymorphism. Genetics 123:585–595.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M.,and Kumar, S. (2011) MEGA5: molecular evolutionarygenetics analysis using maximum likelihood, evolutionarydistance, and maximum parsimony methods. Mol Biol Evol28: 2731–2739. doi: 10.1093/molbev/msr121.
Thacker, W.L., Benson, R.F., Wilkinson, H.W., Ampel, N.M.,Wing, E.J., Staigerwalt, A.G., and Brenner, D.J. (1986)
16 J. Costa, A. F. d’Avó, M. S. da Costa and A. Veríssimo
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology

11th serogroup of Legionella pneumophila isolated from apatient with fatal pneumonia. J Clin Microbiol 23: 1146–1147.
Thacker, W.L., Wilkinson, H.W., Benson, R.F., and Brenner,D.J. (1987) Legionella pneumophila serogroup 12 isolatedfrom human and environmental sources. J Clin Microbiol25: 569–570.
Veríssimo, A., Vesey, G., Rocha, G.M., Marrão, G., Col-bourne, J., Dennis, P.J., and Costa, M.S. (1990) A hotwater supply as the source of Legionella pneumophila inincubators of a neonatology unit. J Hosp Infect 15: 255–263.
Veríssimo, A., Marrão, G., da Silva, F.G., and Costa,M.S. (1991) Distribution of Legionella spp. in hydro-thermal areas in continental Portugal and the island ofSão Miguel, Azores. Appl Environ Microbiol 57: 2921–2927.
Veríssimo, A., Morais, P.V., Diogo, A., Gomes, C., and daCosta, M.S. (1996) Characterization of Legionella speciesby numerical analysis of whole-cell protein electrophoresis.Int J Syst Bacteriol 46: 41–49.
Yang, Z., and Bielawski, J.P. (2000) Statistical methods fordetecting molecular adaptation. Trends Ecol Evol 15: 496–503.
Yang, Z., Nielsen, R., Goldman, N., and Pedersen, A.M.(2000) Codon-substitution models for heterogeneousselection pressure at amino acid sites. Genetics 155: 431–449.
Supporting information
Additional Supporting Information may be found in the onlineversion of this article:
Fig. S1. Domain search with LspD from L. pneumophilaPhiladelphia 1 using The Pfam database (Finn et al., 2010.Nucleic Acids Res 38: D211–D222).Table S1. Genetic pairwise differences, average and stan-dard deviation (SD) values for (A) and between (B–D) lspD,lspE, pilD, proA, srnA and rpoB clusters.Table S2. Potential recombinant events (PRE) identified withRDP3 from the alignment of 6 loci concatenated obtainedfrom 37 L. pneumophila strains. The minimum number ofindependent recombination events (IREs) within each identi-fied PRE was inferred by a minimum of four methods andwere mapped on the phylogenetic tree (Fig. 2).Table S3. D (Tajima), D* and F* (Fu and Li) and Fs (Fu)statistics obtained from the data set.Table S4. Primers and their sequences designed in thisstudy.Table S5. Locus tag from the gene sequences obtained fromthe four L. pneumophila subsp. pneumophila genomesequenced strains.
Please note: Wiley-Blackwell are not responsible for thecontent or functionality of any supporting materials suppliedby the authors. Any queries (other than missing material)should be directed to the corresponding author for the article.
Molecular evolution of Legionella pneumophila T2S system 17
© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology





























