Embed Size (px)
Citation preview

Molecular Dynamics Study of the Melting of Nitromethane (CH3NO2)
Investigators:
Paras M. Agrawal Betsy M. Rice Donald L. Thompson
Thanks:
(1) Dr. Dan Sorescu and the OSU group for the helpful discussions. (2) Mr. Jerry Clarke and Mr. John Vines (ARL) for making ‘movies’.

Melting of nitromethane
System selection
(1) Published work on the melting of nitromethane(a): Experimental(a) Yes Theoretical Not known to us.
(2) Good force field is available(b),(c)
(3) Moderate size (7-atom system)
(a) Piermarini, Block, and Miller, J. Phys. Chem. 93, 457 (1989); Jones and Giauque, J. Am. Chem. Soc. 69, 983 (1947). (b) Sorescu, Rice and Thompson, J. Phys. Chem. B 104, 8406 (2000). (c) Sorescu, Rice and Thompson, J. Phys. Chem. A 105, 9336 (2001).

Important methods for computing the melting point (Tm) from the interaction potential
(1) Thermodynamics
Based on the Gibbs free energy:
G(P, Tm) = Gs(P, Tm)
G = Gº + G
Calculating Gsº is difficult for polyatomic molecules.
Example: Melting of N2(a)
(2) Void nucleated melting
(3) Coexistence of solid and liquid
(a) Meijer, Frenkel, LeSar, and Ladd, J. Chem. Phys. 92, 7570 (1990).

N(2)
O(4)O(3)
C(1)
H(5)H(7)
Orthorhombic space group P212121
4 molecules per unit cell
H(6)

Interaction potential(a)
V = Vinter + Vintra
Vinter = [Aij exp(-Bij r) – Cij/r6 + qiqj/r]
Vintra = [Vstretch + Vbend + Vtorsion]
Vstretch= Di{[1-exp(-i(ri-ri0))]2-1}
6
1i
Vbend = [ ki(i-i0)2 ]
9
1i 21
Vtorsion = Vi[1+cos(mii- i)]
7
1i
(a) (i) Sorescu, Rice and Thompson, J. Phys. Chem. B 104, 8406 (2000). (ii) Agrawal, Rice, and Thompson, J. Chem. Phys, in press. (Preprint available at: http://theory.chem.okstate.edu/MURI/)

Basics of molecular dynamics (MD) simulations
Step1: Start with the appropriate positions and velocities of all atoms.
Step 2: Compute the force acting on each atom due to all other atoms from the known force field.
Step 3: Allow each atom to move according to the force computed in step 2. Determine new positions and velocities after time t = ~ 1 fs.
Step 4: Repeat a large number of cycles of Steps 2 and 3 and monitor the changes that take place in the system.
For next trajectory, start with step 1.

Special type of MD simulations
NPT simulations => N, P, and T are constant. N = Number of atoms in the system
P = Pressure. It is maintained constant by appropriately# varying the volume of the system. T = Temperature. It is kept constant by appropriately# scaling the velocities of all atoms. # For example, Nosé-Hoover method.

Void-nucleated melting:
Our procedure
Step 1:
N0 = 240 molecules of CH3NO2 are equilibrated at the desired
pressure (P = P0 ) and temperature (T = T0).
Equilibration time ~40000 time steps = 30 ps.
Step 2: n molecules are randomly removed.
Step 3:NPT simulations are run for ~15 ps.
N = N0-n P = P0 T = T0

Step 4:
The system is heated by redefining the desired temperature in the
NPT simulation at every time step during simulations:
Tdesired = To + 0.00015 time step
We increase the desired temperature at the rate of
0.00015 K per step
15 K per 105 steps
0.2 Kelvin per ps.

Does Pressure remain constant?? Yes
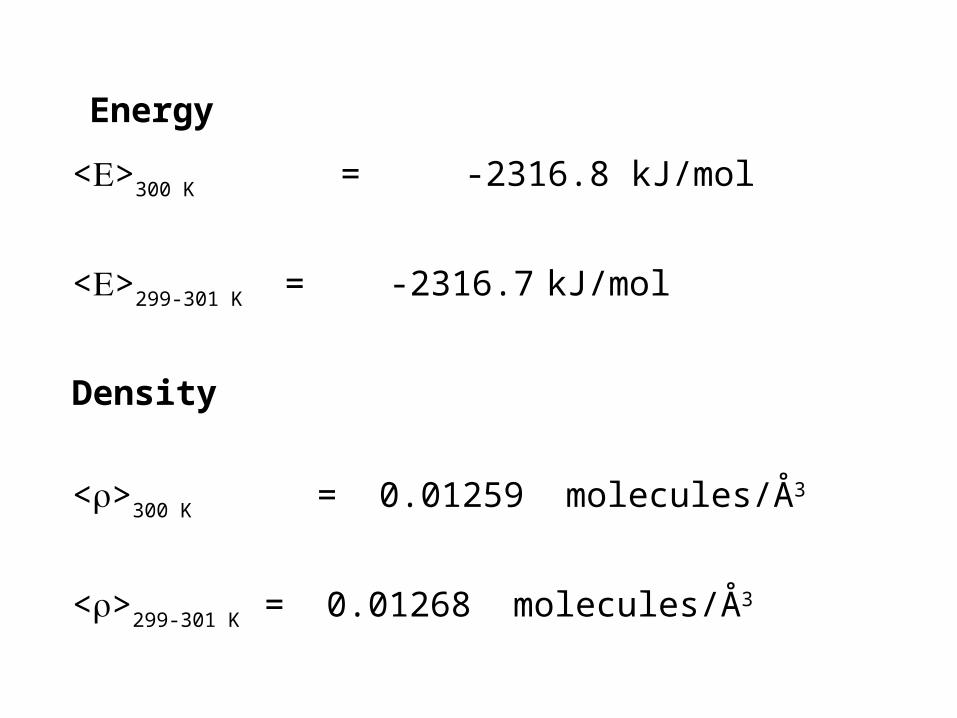
Energy
<>300 K = -2316.8 kJ/mol
<>299-301 K = -2316.7 kJ/mol
Density
<>300 K = 0.01259 molecules/Å3
<>299-301 K = 0.01268 molecules/Å3

Step 5:
We introduce and compute an order parameter.
We have 4 molecules per unit cell, and Nc =No/4 unit cells.
Order parameter:
=(1/4)i ,
4
1i
Usefulness of
i = (1/N΄) | exp(ik.rj) |.
Nc
j 1
k = 2 ( i/ax + j /ay + k/az).
for an ordered solid.
for liquid.
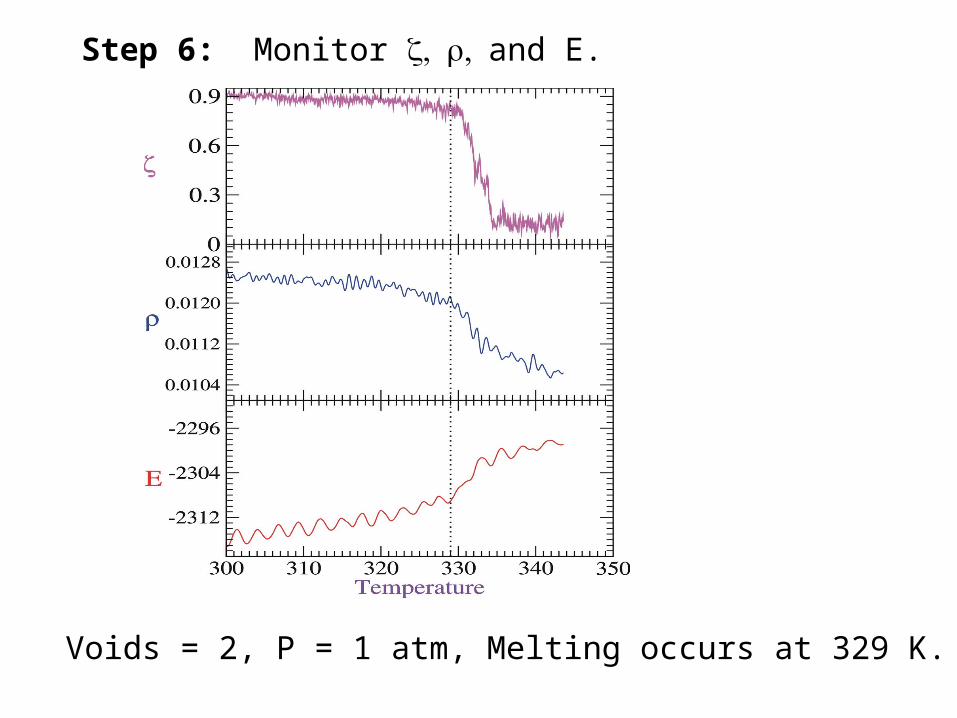
Step 6: Monitor and E.
Voids = 2, P = 1 atm, Melting occurs at 329 K.
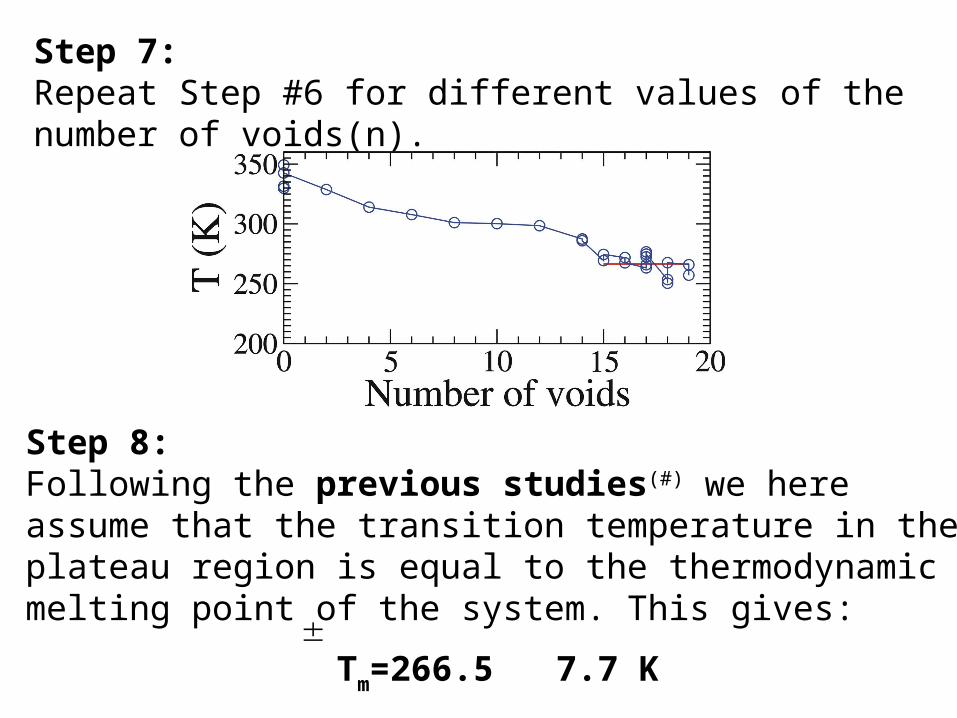
Step 7:Repeat Step #6 for different values of the number of voids(n).
Step 8: Following the previous studies(#) we here assume that the transition temperature in the plateau region is equal to the thermodynamic melting point of the system. This gives:
Tm=266.5 7.7 K

(#) Previous studies
(a) Solca, Dyson, Steinebrunner,and Kirchner, Chem. Phys.224, 253 (1997). (Melting of Ar)
Small difference (~5%) between the void nucleated melting points and those given by thermodynamics on the same potential are attributed to the hysteresis by de Koning et al.*
(b) Solca, Dyson, Steinebrunner, Kirchner and Huber, J. Chem. Phys. 108, 4107 (1998). (Melting of Ne) (c) Agrawal, Rice, and Thompson, J. Chem. Phys. 118, 9680 (2003). ( Melting of Ar) * de Koning, Antonelli, and Yip, J. Chem. Phys. 115, 11025 (2001).

(c) Agrawal, Rice, and Thompson(*)
System: Ar
(i) At 12 values of P up to 531.6 kbar.
(ii) Good agreement with the results of Zha et al.(**) computed by
the thermodynamic method using the same potential (exp-6). (iii) For the majority of data the difference is less than 3%. The
maximum difference is 11%
(iv) The results of Agrawal et al. are consistent with the Lindemann criterion for melting; in fact, in better agreement than are the thermodynamic results of Zha et al..
(*) Agrawal, Rice, and Thompson, J. Chem. Phys. 118, 9680 (2003). (**) Zha, Boehler, Young, and Ross, J. Chem. Phys. 85, 1034 (1986).

Other aspects of melting
(a) Radial distribution function
(b) Diffusion coefficient
(c) Molecular plots
(d) Movie
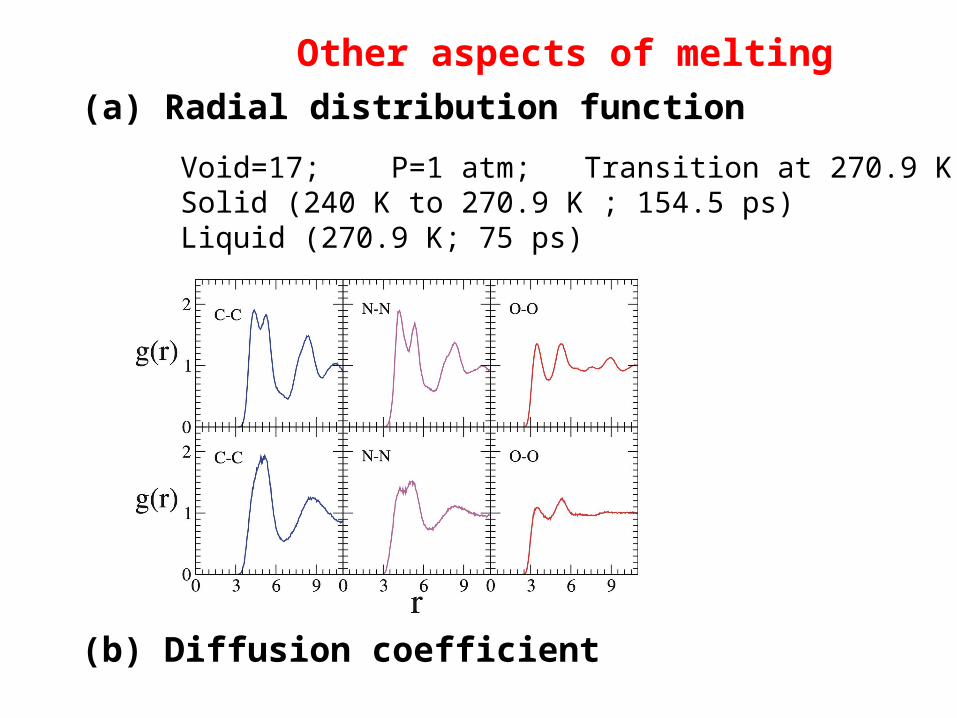
Other aspects of melting
(a) Radial distribution function
Void=17; P=1 atm; Transition at 270.9 K Solid (240 K to 270.9 K ; 154.5 ps) Liquid (270.9 K; 75 ps)
(b) Diffusion coefficient

(c) Movie
(d) Molecular plots

Method II: Simulation of coexisting phases(a)
Started with 880 molecules.
400 molecules of liquid on the top of 480 molecules of solid with periodic boundaryconditions.
(a) Morris and Song, Chem. Phys. 116, 9352 (2002).
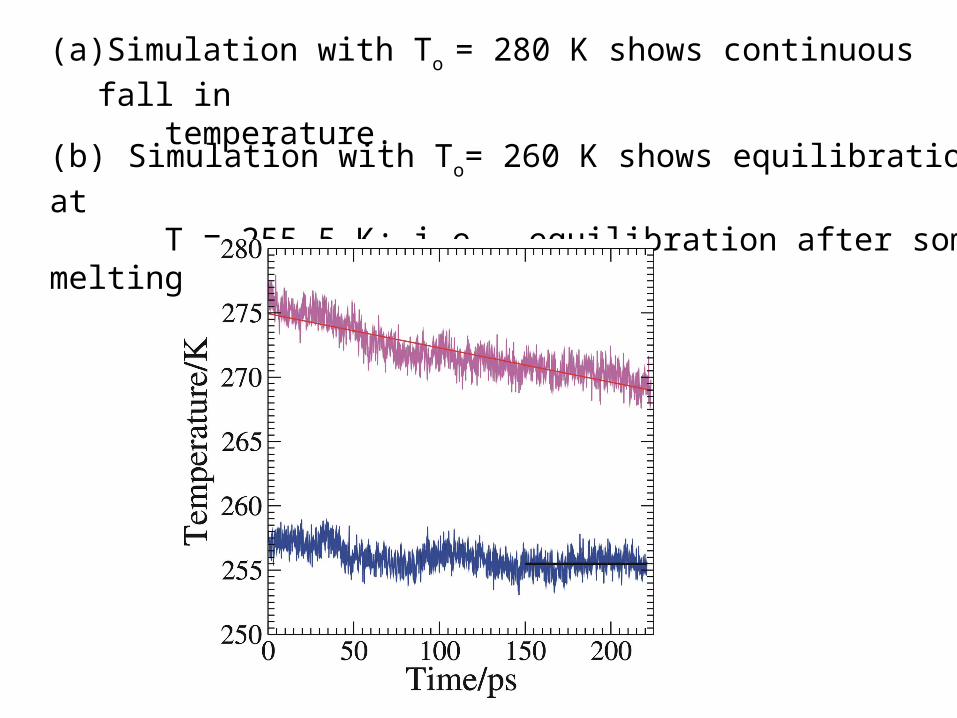
(a) Simulation with To = 280 K shows continuous fall in
temperature.
(b) Simulation with To= 260 K shows equilibration at
T = 255.5 K; i.e., equilibration after some melting.

Comparison
Melting point at P = 1 atm
Void nucleated studies: 266.5 K Coexisting liquid and solid: 255.5 K Experiment:( *) 244.73 K
Difference??
(a) Fluctuations in P and T lead to large variations ~8 K in the melting point.
(b) Hysteresis effect.
(c) Other possible causes not yet known due to the lack of theoretical basis of the void nucleated method.
(*) Jones and Giauque, J. Am. Chem. Soc. 69, 983 (1947).

Other points of interest
Limit of superheating of the solid f=Tm/Ts
Tm = True melting point or thermodynamic melting point.
Ts = Melting point of a perfect crystal without boundaries.
Here Tm = 266.5 K; Ts = 338.3 K; f = 0.79
Studies(a) show that for a large number of systems f~0.8. (a) Lutsko, Wolf, Phillpot, and Yip, Phys. Rev. B 40, 2841 (1989);Solca, Dyson, Steinebrunner, and Kirchner, Chem. Phys. 224, 253 (1997);Solca, Dyson, Steinebrunner, Kirchner, and Huber, J. Chem. Phys. 108, 4107 (1998); Agrawal, Rice, and Thompson, J. Chem. Phys. 118, 9680 (2003);Agrawal, Rice, and Thompson, J. Chem. Phys. In press;Velardez, Alavi, and Thompson J. Chem. Phys. 119, 6698 (2003);Gavezzotti, J. Mol. Struct. 485-486, 485 (1999);Lu and Li, Phys. Rev. Lett. 80, 4474 (1998);Jin, Gumbsch, Lu, and Ma, Phys. Rev. Lett. 87, 55703 (2001).
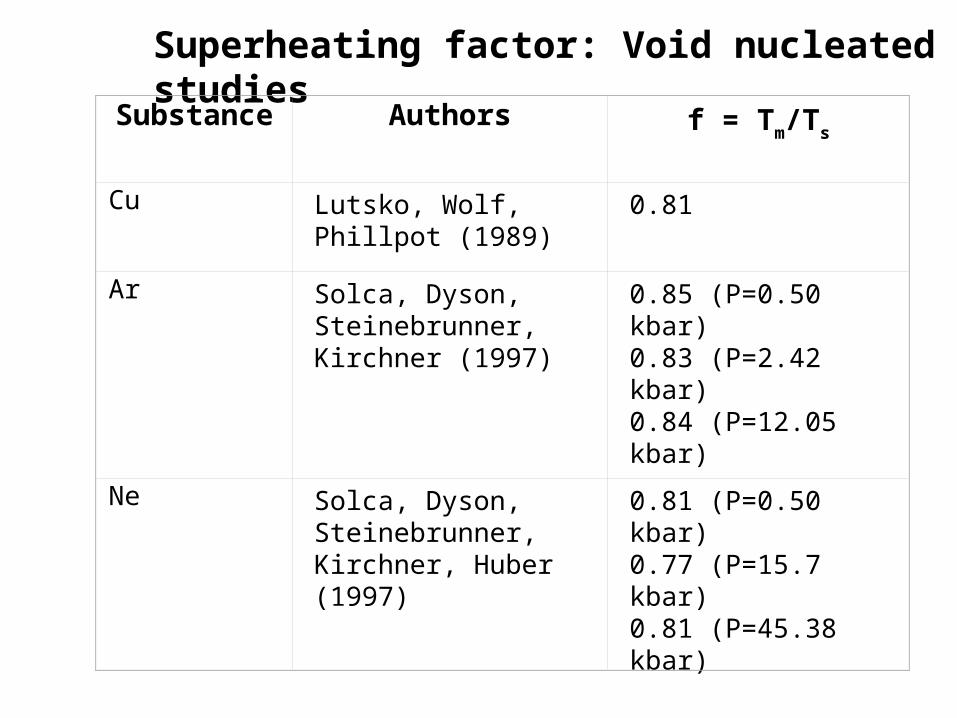
Superheating factor: Void nucleated studies
Substance Authors f = Tm/Ts
Cu Lutsko, Wolf, Phillpot (1989)
0.81
Ar Solca, Dyson, Steinebrunner, Kirchner (1997)
0.85 (P=0.50 kbar) 0.83 (P=2.42 kbar)0.84 (P=12.05 kbar)
Ne Solca, Dyson, Steinebrunner, Kirchner, Huber (1997)
0.81 (P=0.50 kbar)0.77 (P=15.7 kbar)0.81 (P=45.38 kbar)

CH3COOH Gavezzotti f0.93
Ar Agrawal, Rice, Thompson (2003 A)
0.83 to 0.87(0.094 P 531.6 kbar)
CH3NO2 Agrawal, Rice, Thompson (2003 B) (This work)
0.79
Ammoniumdinitramide
Velardez, Alavi, Thompson (2003)
0.77
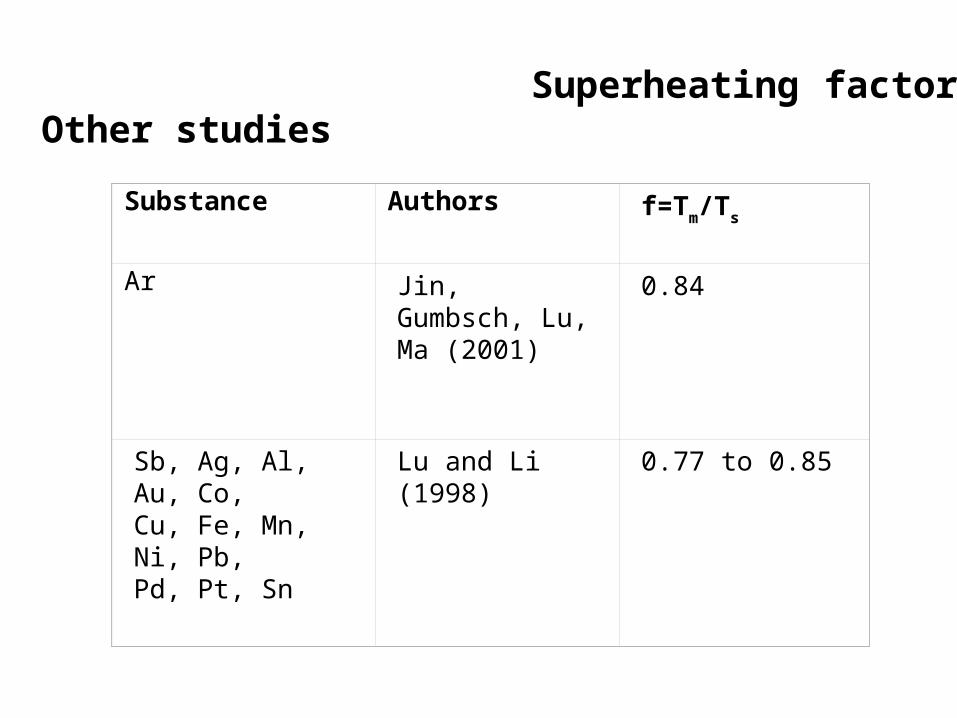
Substance Authors f=Tm/Ts
Ar Jin, Gumbsch, Lu, Ma (2001)
0.84
Sb, Ag, Al, Au, Co, Cu, Fe, Mn, Ni, Pb, Pd, Pt, Sn
Lu and Li (1998)
0.77 to 0.85
Superheating factor: Other studies

Importance of superheating factor (f=Tm/Ts)
(1) Mechanism of melting
(2) Pressure dependence studies Studies(a) show that f has little dependence on P. Here we use
Tm=0.79 × Ts
(a) Solca, Dyson, Steinebrunner, and Kirchner, Chem. Phys. 224, 253 (1997); Solca, Dyson, Steinebrunner, Kirchner, and Huber, J. Chem. Phys. 108, 4107 (1998); Agrawal, Rice, and Thompson, J. Chem. Phys. 118, 9680 (2003).

Curve: Our results fitted to the Simon-Glatzel equation: P (kbar) = aT b + c
a = 1.597 × 10-5, b = 2.322, c = -6.74
(a) Piermarini, Block, and Miller, J. Phys. Chem. 93, 457 (1989).(b) Jones and Giauque, J. Am. Chem. Soc. 69, 983 (1947).
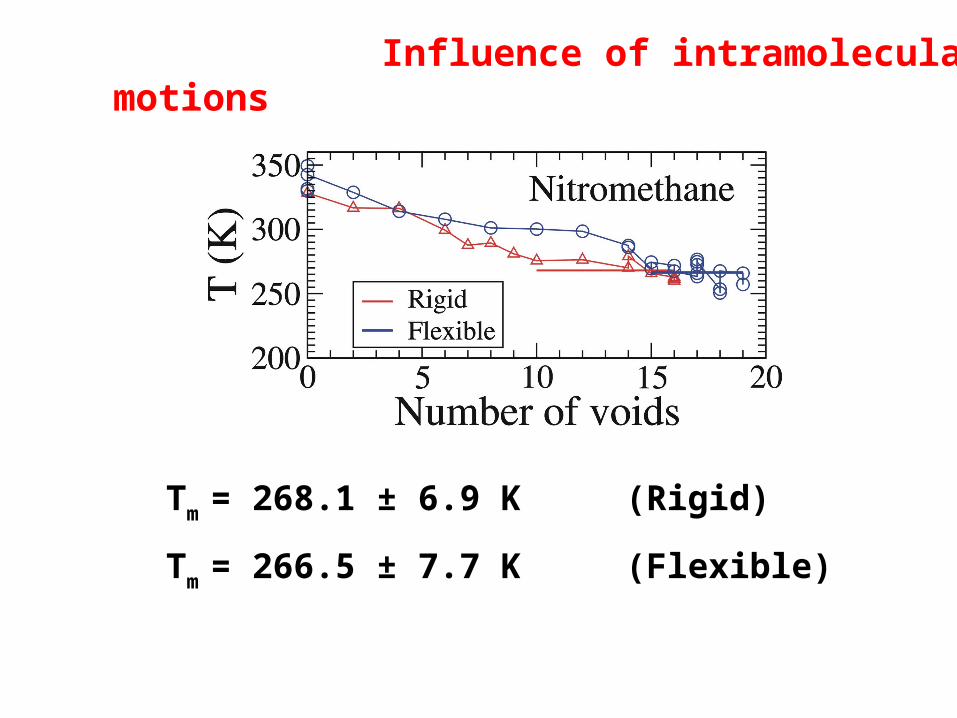
Influence of intramolecular motions
Tm = 268.1 ± 6.9 K (Rigid)
Tm = 266.5 ± 7.7 K (Flexible)

Charge dependence
(a) qi=Fqi0 (i=1,7)
F Ts (K)
1.00 338.3
0.90 303.7
0.85 287.9
0.80 278.7
(b) Used charges computed by Seminario et al.(*)
Result: Tm=230.4 K
(*) Seminario, Concha, and Politzer, J. Chem. Phys. 102, 8281(1995).

Conclusions
(1) Two convenient methods to compute Tm for molecular
solids have been investigated.
(2) Our force field for nitromethane is good. (3) Little effect of intramolecular interactions. (4) Significant role of partial charges.
(5) Gradual heating method works well.
More details:
P. M. Agrawal, B. M. Rice, and D. L. Thompson, J. Chem. Phys. in press. (preprint available at : http://theory.chem.okstate.edu/MURI/)

Future plans (1) Melting of RDX
Present stage:
(i) We are exploring the empirical intra-molecular force fields(a) with the intermolecular interactions given by Sorescu, Rice and Thompson.(b)
(ii) We observed that the results are sensitive to torsion terms as well as partial charges.
(iii) The search for the correct potential model that produces good geometry, density, melting point and enthalpy of sublimation is in progress.
(2) General Model: To extend the experience gained from the RDX studies for other systems.
(a) Ye, Tonokura, and Koshi, J. Japan Explos. Soc. 63, 104 (2002);
Cornell et al. (‘A second generation force field for the simulations of proteins, nucleic acids and organic molecules’), J. Am. Chem. Soc. 117, 5179 (1995). (b) Sorescu, Rice, and Thompson, J. Phys. Chem. B 101, 798 (1997).





























