Embed Size (px)
Citation preview

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
MOLECULAR DYNAMICS STUDIES OF THERMOPHYSICALPROPERTIES OF SUPERCRITICAL ETHYLENE
Obika C. Nwobi? Lyle N. Long! Michael M. Miccif
Department of Aerospace EngineeringThe Pennsylvania State University
University Park, PA 16802
Abstract
This work involves the determination of transport coef-ficients and equation of state of supercritical fluids bymolecular dynamics (MD) simulations on parallel com-puters using the Green-Kubo formulae and the virialequation of state, respectively. The MD program usesan effective Lennard-Jones potential, linked cell lists forefficient sorting of molecules, periodic boundary condi-tions, and a modified velocity Verlet algorithm for parti-cle displacement. Previously, simulations had been car-ried out on pure argon, nitrogen and oxygen, and thishas now been extended to ethylene, C^H^, at varioussupercritical conditions, with shear viscosity and ther-mal conductivity coefficients, and pressures computedfor most of the conditions. The results compare wellwith experimental and National Institute of Standardsand Technology (NIST) values.
Nomenclature
a = acceleration vectoraz
a = acceleration vector of a given atomd = molecular bond lengthf = intermolecular forceg = constraint forcei = unit tensorJp = stress tensorJ7 = heat tensorke = Boltzmann constantm = atomic massn = number of atoms per moleculeN = number of molecules in the system0 = time originP = pressurer = position vectorr^ = position vector of a given atom
"Research Assistant, Student Member, AIAAt Professor. Associate Fellow, AIAA°© 1998 by Nwobi, Long and Micci. Published by the
American Institute of Aeronautics and Astronautics, Inc. withpermission.
ra/3 = position vector between two atoms i, jRQ = c.m. velocity of a given moleculeRa/3 = c.m. position vector between two moleculest = time delayT — temperatureV = volumev = velocity vectorv^ = velocity vector of a given atoma,/3 = given moleculesSt = time-step sizee = Lennard-Jones energy parameter7Q = constraint between atomsA = thermal conductivityH = shear viscosity$ = intermolecular potentialp = density
IntroductionCurrent rocket motors, gas turbines, and many pro-
jected advanced combustor designs operate supercriti-cally or near critical conditions.1 Computational FluidDynamics (CFD) has been used to model droplet evapo-ration and combustion in supercritical and near criticalenvironments by several researchers including Yang etal.2 and Delplanque and Sirignano.3 Quantitative in-formation on the coefficients of mass, momentum andenergy transport, and the equation of state are very im-portant in these studies. However, in many complex sys-tems such as rarefied flows, supercritical environments,shock regions, and highly-energized plasmas, these coef-ficients are not known accurately and the experimentsto measure them are difficult and costly. As a resultthe necessary information, is lacking in the use of theCFD methods for simulating supercritical evaporationand combustion. Moreover, these techniques make use oflimiting approximations such as spherical droplet shapeand constant density which are not necessarily valid un-der these supercritical conditions. Hence, alternativemethods for modeling the evaporation of supercriticaldroplets have to be considered.

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
Molecular dynamics (MD), the technique used in thepresent study, involves the solution of the equations ofmotion of a system of molecules that interact with eachother through an intermolecular potential.4 Providedthat an accurate potential can be found for the systemof interest, MD can be used regardless of the phase andthermodynamic conditions of the substances involved.Although computationally intensive, this method re-quires no a priori assumptions regarding geometricalsymmetry, transport properties, or thermodynamic be-havior. All calculations are performed from first prin-ciples based on intermolecular potentials, and thermo-dynamic and transport properties are results instead ofassumptions. MD has been used to support research insuch fields as chemistry, biology, physics, and materialscience.5"10 It is also currently being used to model theevaporation of a submicron droplet in supercritical envi-ronments.11-12
MD simulations are computationally intensive sincethe interaction between each particle and all its neigh-bors must be taken into account. The speed of thesesimulations can be greatly increased by using parallelcomputers. The natural parallelism in MD comes fromthe fact that the force calculations and position/velocityupdates can be done simultaneously for all the molecules.
The method of parallelization currently being used isthe particle decomposition method.13 This technique isimplemented by distributing the particles evenly amongthe different processors. Each processor calculates theforces and displacements for the particles for which it isresponsible. Then the new positions and velocities fromeach processor are broadcast to all other processors withwhich the next time step begins. Using the particle de-composition method, almost perfect load balancing hasbeen achieved.11
Transport coefficients can be computed either bythe use of Green-Kubo formulae or Einstein relationsduring equilibrium molecular dynamics (EMD) simula-tions,4'14'15 or by conducting suitable non-equilibriummolecular dynamics (NEMD) simulations.16'17 TheGreen-Kubo formulae have been used successfully tocompute the transport coefficients of atomic fluids, whileonly limited success has been achieved for molecularfluids which are essential for supercritical droplet stud-ies.18"20 At various times this might be due to the inad-equate modeling of the long-time tail behavior of the rel-evant auto-correlation functions, the use of small systemsizes because of limited computer resources available, ordue to the incorrect use of potential model parameters.The NEMD emerged as a suitable simulation techniquefor computing the transport coefficients of fluids due tothe above drawbacks. Normally to compute shear vis-cosities or thermal conductivities, separate computer ex-periments have to be carried out and the codes cannotbe incorporated easily into an MD droplet evaporation
computer program to compute the relevant properties asthe droplet vaporizes. Hence, this method is not utilizedin the current study.
MD has been used to compute the equation of stateof some fluids by several researchers including Barkeret al.,21 Singer et al.,22 and Vogelsang and Hoheisel,23
for only a few state points. No comprehensive studyhas been done to calculate the pressures of ethylene andother fluids over a wide range of states.
An attempt is made herein to compute the transportproperties and pressures of supercritical fluids which arerelevant for droplet evaporation studies using the Green-Kubo formulae, and the virial equation of state, respec-tively. Previous studies had been done on argon, ni-trogen and oxygen, and work has now been extended toethylene in this paper.24-25 Successful implementation ofthe code will impact the understanding and modeling ofthe processes occurring in current and future aerospacepropulsion systems.
Molecular DynamicsMolecular dynamics (MD) has become a widely used
direct simulation technique. The advantage of this tech-nique for the solution of complex reacting flow problemsis that all attendant physical phenomena - shear viscos-ity, thermal conductivities, surface tension, evaporation,and reaction rates - come from the force potential be-tween the particles. MD does not require the use ofempirical submodels for each physical process or temper-ature dependent physical properties. It is an extremelypowerful and simple method of simulation. The accuracyof a given MD model depends mainly on the chosen in-termolecular potential, and the finite difference scheme.
Potentials
Molecular dynamics generally adopts a classical ap-proach in the modeling of matter. Typically, atoms ormolecules are represented as point masses interactingthrough forces (potentials) that depend on the separa-tion of the particles. The cornerstone of all moleculardynamics simulations is the intermolecular potential. Inorder to calculate the positions, velocities, and acceler-ations of each molecule in the system, the forces actingon each of the molecules are required. These forces arederived from the intermolecular potentials. All resultsand properties (thermodynamic, structural and trans-port) determined with MD simulations, also follow di-rectly from the potential.
An appropriate potential for MD simulations mustcapture the important features of intermolecular inter-actions: a strong repulsive force at short distances dueto orbital overlap, a negative potential well causing reg-ular structure in liquids at low temperatures, and an at-tractive tail at large distances due to dispersion forces.
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
The most commonly used potential which captures theimportant features of intermolecular interactions is theLennard-Jones (L-J) 12-6 potential.4
(1)
where <£ is the energy of interaction between the centersof two atoms i and j which are a distance r^ apart. Thetwo fundamental constants in the Lennard-Jones poten-tial are e, which accounts for energy and a, which ac-counts for size. The values of these parameters dependon the particular chemical specie that is being modeled.a represents the zero energy separation distance and hasunits of length, while 6 represents the depth of the po-tential well and has units of energy. The L-J potentialcan be used for modeling solid, liquid or gaseous states.The attraction part of the potential, -1/r6, is used tobind the system for the solid and liquid states, while therepulsive part. —1/r12 prevents collapse.
For molecular fluids additional measures have to betaken. Molecules can be modeled by one of twomeans: treating each atom in the molecule as a separateLennard-Jones site (site-site), or by defining a centralsite with corrections for angular dependency. The cen-tral site method starts with the Newton-Euler equationsof motion. Because singularities appear when written interms of Euler angles, the equations are written in termsof quaternions which are then solved in matrix form.4
The site-site method consists of the solution of the or-dinary equations of motion for each atomic site subjectto a constraint which is the fixed bond length.4'26 Thesites are located at the positions of the nuclei of the con-stituent atoms. Diatomics such as oxygen and nitrogenas well as non-diatomics such as ethylene (63 #4) canbe modeled as 2-site molecules. Because of the fact thateach carbon atom is much heavier than a hydrogen atom,each group of CH^ atoms in ethylene can be treated asa single site (composed of just the carbon atom) withthe effects of the hydrogen atoms accounted for by theuse of an effective Lennard-Jones potential. For mod-eling systems at relatively low temperatures, fixing thebond length is an excellent approximation. At such lowtemperatures, very little vibration occurs and the bondsare essentially rigid.27
The molecular interaction generally consists of pair-wise additive atom-atom interactions. For two diatomicmolecules, there are four interactions between the foursites and only interactions between atoms of differentmolecules are considered.
The intermolecular force, f , is obtained from the spa-tial gradient of the potential.
2
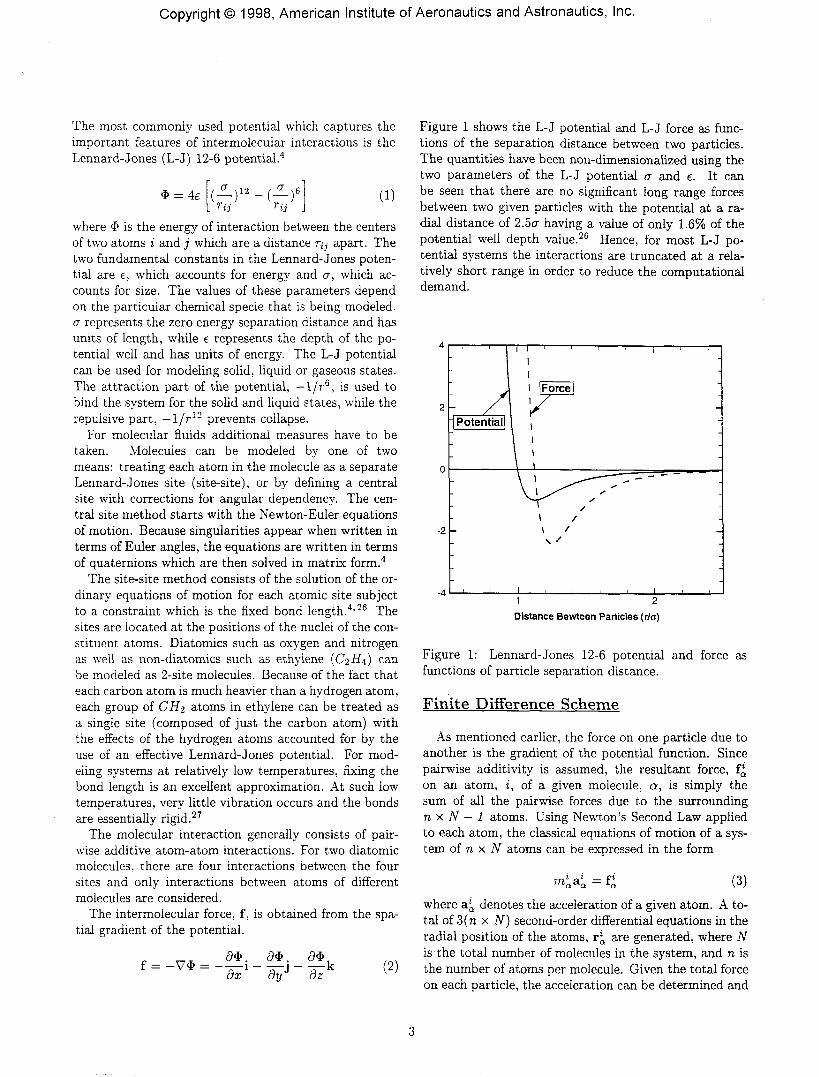
Figure 1 shows the L-J potential and L-J force as func-tions of the separation distance between two particles.The quantities have been non-dimensionalized using thetwo parameters of the L-J potential a and e. It canbe seen that there are no significant long range forcesbetween two given particles with the potential at a ra-dial distance of 2.5cr having a value of only 1.6% of thepotential well depth value.26 Hence, for most L-J po-tential systems the interactions are truncated at a rela-tively short range in order to reduce the computationaldemand.
^ . . ,f = _V$ = -— i-— J-— kdx dy dz
1 2Distance Bewteen Particles (r/c)
Figure 1: Lennard-Jones 12-6 potential and force asfunctions of particle separation distance.
Finite Difference Scheme
As mentioned earlier, the force on one particle due toanother is the gradient of the potential function. Sincepairwise additivity is assumed, the resultant force, f£on an atom, i, of a given molecule, a, is simply thesum of all the pairwise forces due to the surroundingn x N — 1 atoms. Using Newton's Second Law appliedto each atom, the classical equations of motion of a sys-tem of n x N atoms can be expressed in the form
"»X = % (3)where a^ denotes the acceleration of a given atom. A to-tal of 3(n x N) second-order differential equations in theradial position of the atoms, rl
a are generated, where Nis the total number of molecules in the system, and n isthe number of atoms per molecule. Given the total forceon each particle, the acceleration can be determined and

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
the velocity obtained. Using these velocities and acceler-ations, all the particles in the system are moved a finiteamount at each time step. An appropriate finite differ-ence scheme must be fast, require little memory, stable,accurate, and permit long time step size. The schemeshould also satisfy the known conservation laws for en-ergy and momentum, and must reproduce certain time-and space-dependent correlations to a sufficient degree ofaccuracy. Several schemes have been used in MD calcula-tions including Gears predictor-corrector and the Verletalgorithms.4
The Verlet schemes are the most widely used algo-rithms.4 There are several versions of the schemes in-cluding the Stoemer-Verlet, the leap-frog Verlet and thevelocity-Verlet. The velocity-Verlet algorithm is uti-lized in the present study and it requires the storage ofonly atomic positions, velocities and accelerations. Thescheme is a dual-step process. In the first step the po-sitions are advanced a full step, while the velocities areadvanced only a half step. After the accelerations areupdated at the current time step, the velocities are thenadvanced another half a step.
= rla(t) + 6t <(*) + l-5t2 aj,(0 (4)
5t
vj,(t + 6t) =
(5)
6t) (6)
where aj,, vla and rl
a are the acceleration, velocity,and radial position, respectively, of atom i of a givenmolecule a. £ is the simulation time and 6t is the timestep size. The velocity Verlet algorithm stores positionsand velocities at a given time (i), and the accelerationsat time (t + 6 t ) . This scheme minimizes round-off errorand the atomic velocities can be easily scaled in order tocontrol the temperature of the system.
The site-site method with constraint dynamics is usedin this study for modeling rigid diatomic molecular sys-tems. Each atom of a given molecule is treated as a sep-arate Lennard-Jones site. The technique involves writ-ing the equations of motion to include the constraintforce due to the rigid bond length. The constraints arethe fixed bond lengths which are given by the followingequation
-v —fa — r1 — rj r —ra ra| & —= 0 (7)
dij denotes the desired separation distance betweenatoms i and j of a given molecule a. r^ and r£ are theradial positions of the two atoms of a given molecules a.The equations of motion are now written as
(8)
where fa, ga, ma and ra denote the intermolecular force,constraint force, mass and radial position of atom i ofa given molecule a. The constraint forces help to keepthe desired bond lengths constant and require a few sub-iterations at each time step.
Performance Enhancements
Because of the computationally intense nature of MDsimulations, the system sizes tend to be relatively smallcompared to those of macroscopic systems. A majorobstacle to these simulations is the large fraction ofmolecules which lie on the surface boundaries. Whetheror not the simulation box is surrounded by a containingwall, molecules on the surface experience quite differentforces from molecules in the core of the system. Thesurface effects could lead to error in the properties andquantities computed during simulations and should beminimized. This can be achieved by implementing peri-odic boundary conditions.4
One great limitation of MD is the computational de-mand. Generally, each molecule experiences a pairwisecontribution from every additional molecule and its in-finite images in the system due to the use of periodicboundaries. This equates to an infinite number of com-putations which quickly exceeds the capability of thelargest supercomputers. Hence, many techniques havebeen used to increase the speed and efficiency of thecode, and these vary depending on both the system sizeand type.
Firstly, a "minimum image" convention is used to re-duce the number of force calculations, due to the in-corporation of periodic boundary conditions. This isachieved by the use of a potential cut-off radius. Sinceintermolecular forces decay rapidly beyond just a fewatomic diameters, the effect of such forces may be ne-glected for all molecules which lie outside the interactionsphere of a molecule defined by the cut-off radius. Thenumber of force calculations is reduced from infinity to| AT xn(N x n- 1).
Secondly, a cell index method is used for checkingwhich molecules need to be included in the force calcu-lation of each molecule. This is achieved by dividing thesimulation box into small cells into which the moleculesare sorted. Since interactions are only possible betweenparticles that are either in the same cell or in imme-diately adjacent cell, only molecules in the surroundingcells need to be considered for force calculations.
Thirdly, a "linked list" method is used for fast sortingof the molecules into cells.4 In the cell index method,linked lists are used to associate molecules with the cellsin which they reside at any given instant; a separate listis required for each cell.26 The "linked list" refers tothe linking of a list array with the head array during

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
the neighbor search.4 The routine is rapid and may beperformed at every time-step. This method requires onlythe additional storage of two relatively small array pairs,and its efficient use of memory is applied to track theneighbor locations. The use of above schemes with apotential cut-off reduces the force calculations to roughly250(N x n).
Property Calculations
All calculations in MD are performed from first prin-ciples based on the intermolecular potential, and vari-ous thermodynamic, structural and transport propertiescan be obtained from the simulations. The thermody-namic properties include pressure, temperature, internalenergy; structural functions include the radial distribu-tion function; while the transport properties include var-ious coefficients of diffusion, shear viscosity, and thermalconductivity.
Pressure Calculation
Pressure is related to the force per unit area actingnormal to an imaginary surface in a system. By New-ton's second law, the force can be related to the momen-tum that crosses the surface per unit time. The pressureof the system may be calculated during an MD simu-lation by means of the virial equation of state:4'28 Formolecular fluids pressure is given by the following equa-tion
1pkBT „. ...0- a=1/3=1 i=1 j=i
This mav be rewritten as
PV = NkBT - (10)a=l/3=l i=l j=l
where T. P, V and p denote the temperature, pressure,volume and density of the system. N, n are the numberof molecules in the system and the number of atomsper molecule, respectively, while kg is the Boltzmannconstant. Ra/j denotes the difference vector between thecenter of mass position vectors of two given molecules aand Q. and $ is the center-to-center potential function.
The first term on the right-hand-side of the aboveequation denotes the ideal-gas contributions. It arisesbecause of the motion of the particles, while the sec-ond term accounts for intermolecular forces, assuming apairwise additive potential. For molecular systems, allthe intramolecular contributions including those along
the bonds have to be included. At low densities the sec-ond term is negligible, but as the density increases, itscontributions becomes significant.
Correlation Functions
In computer simulations, transport coefficients can becalculated from equilibrium correlation functions, by ob-serving Einstein relations, or by conducting a suitablenon-equilibrium MD simulation.4 In the present studythe transport coefficients were computed using Green-Kubo formulae in which a correlation function is inte-grated over time.4'14'15 The autocorrelation functions(ACFs) give insight into the microscopic behavior of flu-ids and they help in the understanding of the dynamicsof liquids. An ACF can be derived from the Enskogkinetic equation and the corresponding transport coef-ficient then obtained via the linear response theory us-ing the hydrodynamic equations.15 The ACF of a realdynamic variable A measures the relation between thevalues of the variable at two different times: A(0) andA(t). It is given by the following expression:
C(t) = (A(Q)A(t)) (11)where 0 and t denote an arbitrary time (time origin)and the time difference (delay time), respectively, and(...) denotes a time average.
Green-Kubo Formulae
The shear viscosity coefficient, /j,, is a measure of theinternal fluid friction which tends to oppose any dynamicchange in the fluid motion. It gives an indication of theshear stress induced by an applied velocity. The corre-sponding correlation function contains the non-diagonalelements of the stress tensor and it is a collective prop-erty of the entire fluid.15 The Green-Kubo formula isgiven by
13VkBT (Jp(0)Jp(i)> dt (12)
where Jp is the stress tensor which depends on the veloci-ties, radial positions and potentials of the particles of thesystem. T and V denote the temperature and volume ofthe system, respectively, and kg denotes Boltzmann con-stant. Statistical precision is improved, when appropri-ate, by averaging over all the three terms (fJ-xyiVxnfJ-yz)that result from the stress tensor. In MD simulations,the shear viscosity is computed by averaging over a finitenumber of time origins.
M
(13)

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
where M is the number of time origins. For molecularfluids the stress tensor is given by
Jp = m0=1 0=1/3=1 i=l j=l
(14)where RQ denotes the center of mass velocity ofeach molecule, while RQ, denotes the difference vec-tor between the center of mass position vectors of themolecules. $ indicates the pair interaction potentialbetween the atoms of two molecules a and /3, while nand AT are the number of atoms per molecule and thetotal number of molecules in the system, respectively.The shear stress ACF is composed of a kinetic term,which measures the correlation of momentum transportcaused by atomic motions; a potential term which mea-sures the correlation of momentum transport caused byinteratomic forces, and a cross term, which measures thecoupling of atomic motions and forces. Generally, the ki-netic term dominates the ACF at low densities (gases),while the potential term dominates at higher densities(liquids). The potential and cross terms are generallycomputed within the main force subroutine in the MDcomputer code.
The thermal conductivity coefficient, A, measures thetransport of heat in a system. The correlation function isobtained from the heat current and it is also a collectiveproperty of the entire fluid.15 The Green-Kubo formulais given by
A = WkBT2 (15)
where J, is the heat current. Statistical precision is im-proved, when appropriate, by averaging over the threeterms (Ax , Ay, Xz) that result from the heat current. InMD simulations, the thermal conductivity is computedby averaging over a finite number of time origins.
M
(16)
where M is the number of time origins.For molecular fluids the heat current is given by
N
a=l
a=l t=l 0=1 j=l
where i denotes a unit tensor.The ACF is also composed of kinetic, potential, and cross(kinetic-potential) terms. The computation of thermal
conductivity takes more time than the computation ofshear viscosity because of the additional terms in the for-mula. As in the shear viscosity case, the potential andcross terms are computed within the main force subrou-tine in the MD computer code.
Molecular Dynamics SimulationsThe MD computer code is written in Fortran-77 and
it uses the message passing interface (MPI). All the runswere made on an IBM SP2 computer using 8, 16 or 32processors. The simulations were carried out in the con-ventional constant number of molecules-energy-volume(NEV) ensemble. The equations of motion were solvedusing the modified velocity Verlet algorithm with a timestep of 2xlO~15s, periodic boundary conditions, and apotential cut-off radius of 2.5cr. The simulations weredone using cubical geometries, with the length of eachside about 12 to 45fr depending on the density and thenumber of molecules in the system. These lengths aremuch larger than the critical point correlation length.20
The system sizes of the simulations carried out rangedbetween 1000 to 1200 molecules. The Lennard-Jones 12-6 (L-J) potential were used for the pair interactions andthe L-J parameters of ethylene used in the simulations,as well as the critical temperature and pressure of ethy-lene are given in Table 1.
Table 1: Relevant Parameters of Ethylene
PCMPa
TcK
(7
A KdA
5.04 282.0 3.33 137.7 2.46
Starting from an initial configuration with themolecules placed on the positions of a face-centered cubic(FCC) lattice, the first 5 x 104 time steps were used forequilibrating the system to the chosen thermodynamicstate. During the equilibration run, the velocities of themolecules are frequently rescaled in order to obtain thedesired temperature. Normally, at the end of this pe-riod, the final temperature of the system differs by lessthan 1 % of the desired value, while the total energy ofthe system would have stabilized with a fluctuation ofless than 0.2 % from the mean value. The data runswere about 2 x 106 time steps for the transport coeffi-cients and 2 x 105 for pressures. Statistical uncertaintiesin these calculations were estimated by evaluating inde-pendent averages over blocks of 1 x 105, and 1 x 104
time steps for the transport coefficients and pressures,respectively.9
The number of molecules used in the simulation, thetime step size and the length of the runs have been
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
250
5000 10000Number of Molecules
15000
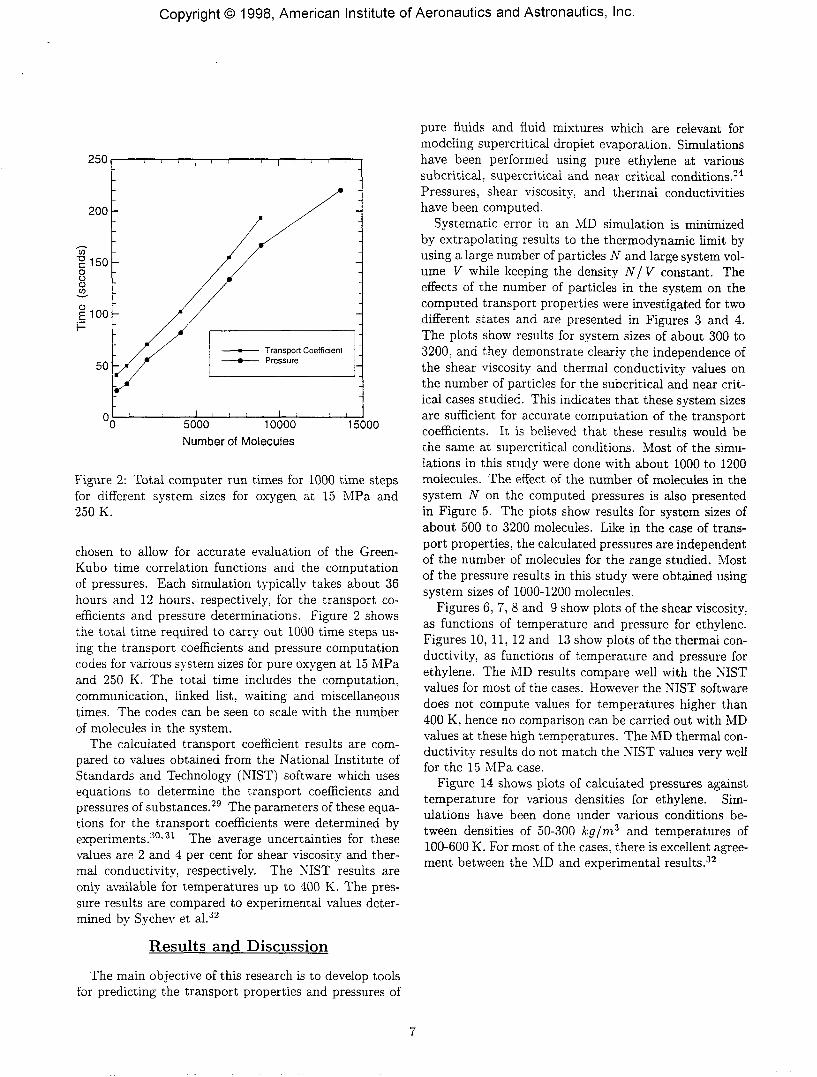
Figure 2: Total computer run times for 1000 time stepsfor different system sizes for oxygen at 15 MPa and250 K.
chosen to allow for accurate evaluation of the Green-Kubo time correlation functions and the computationof pressures. Each simulation typically takes about 36hours and 12 hours, respectively, for the transport co-efficients and pressure determinations. Figure 2 showsthe total time required to carry out 1000 time steps us-ing the transport coefficients and pressure computationcodes for various system sizes for pure oxygen at 15 MPaand 250 K. The total time includes the computation,communication, linked list, waiting and miscellaneoustimes. The codes can be seen to scale with the numberof molecules in the system.
The calculated transport coefficient results are com-pared to values obtained from the National Institute ofStandards and Technology (NIST) software which usesequations to determine the transport coefficients andpressures of substances.29 The parameters of these equa-tions for the transport coefficients were determined byexperiments.30'31 The average uncertainties for thesevalues are 2 and 4 per cent for shear viscosity and ther-mal conductivity, respectively. The NIST results areonly available for temperatures up to 400 K. The pres-sure results are compared to experimental values deter-mined by Sychev et al.32
Results and Discussion
The main objective of this research is to develop toolsfor predicting the transport properties and pressures of
pure fluids and fluid mixtures which are relevant formodeling supercritical droplet evaporation. Simulationshave been performed using pure ethylene at varioussubcritical, supercritical and near critical conditions.24
Pressures, shear viscosity, and thermal conductivitieshave been computed.
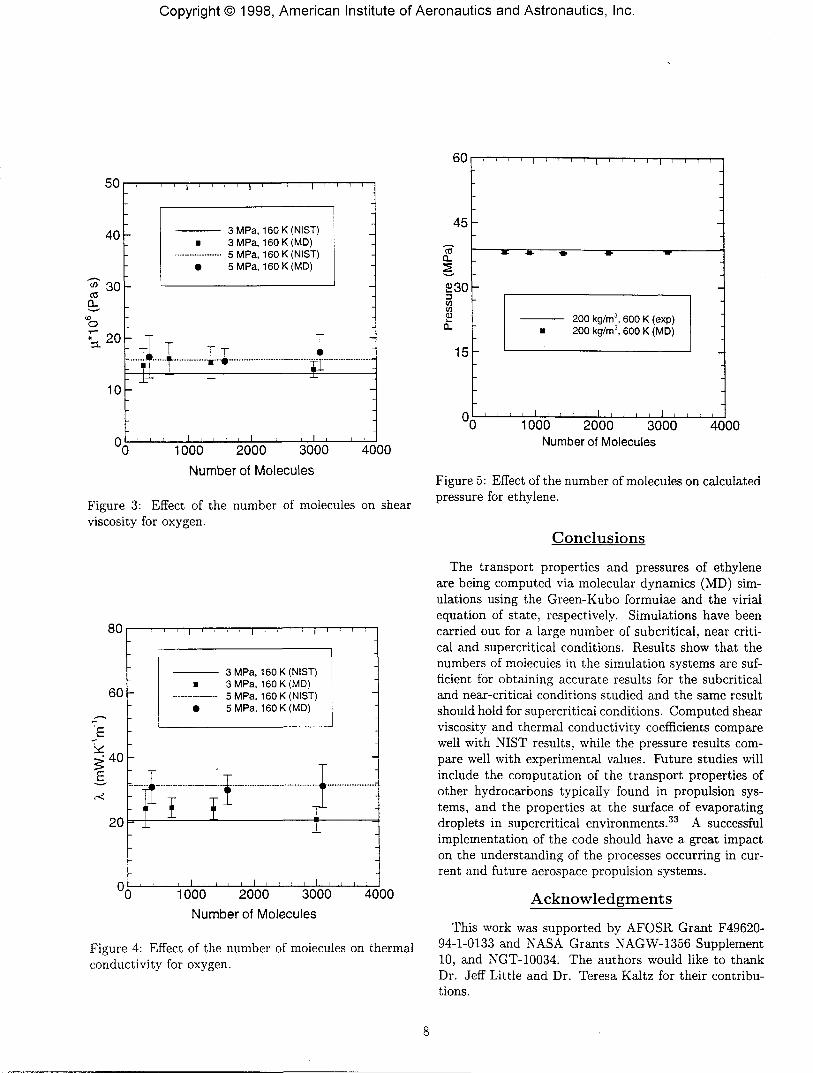
Systematic error in an MD simulation is minimizedby extrapolating results to the thermodynamic limit byusing a large number of particles N and large system vol-ume V while keeping the density N / V constant. Theeffects of the number of particles in the system on thecomputed transport properties were investigated for twodifferent states and are presented in Figures 3 and 4.The plots show results for system sizes of about 300 to3200, and they demonstrate clearly the independence ofthe shear viscosity and thermal conductivity values onthe number of particles for the subcritical and near crit-ical cases studied. This indicates that these system sizesare sufficient for accurate computation of the transportcoefficients. It is believed that these results would bethe same at supercritical conditions. Most of the simu-lations in this study were done with about 1000 to 1200molecules. The effect of the number of molecules in thesystem N on the computed pressures is also presentedin Figure 5. The plots show results for system sizes ofabout 500 to 3200 molecules. Like in the case of trans-port properties, the calculated pressures are independentof the number of molecules for the range studied. Mostof the pressure results in this study were obtained usingsystem sizes of 1000-1200 molecules.
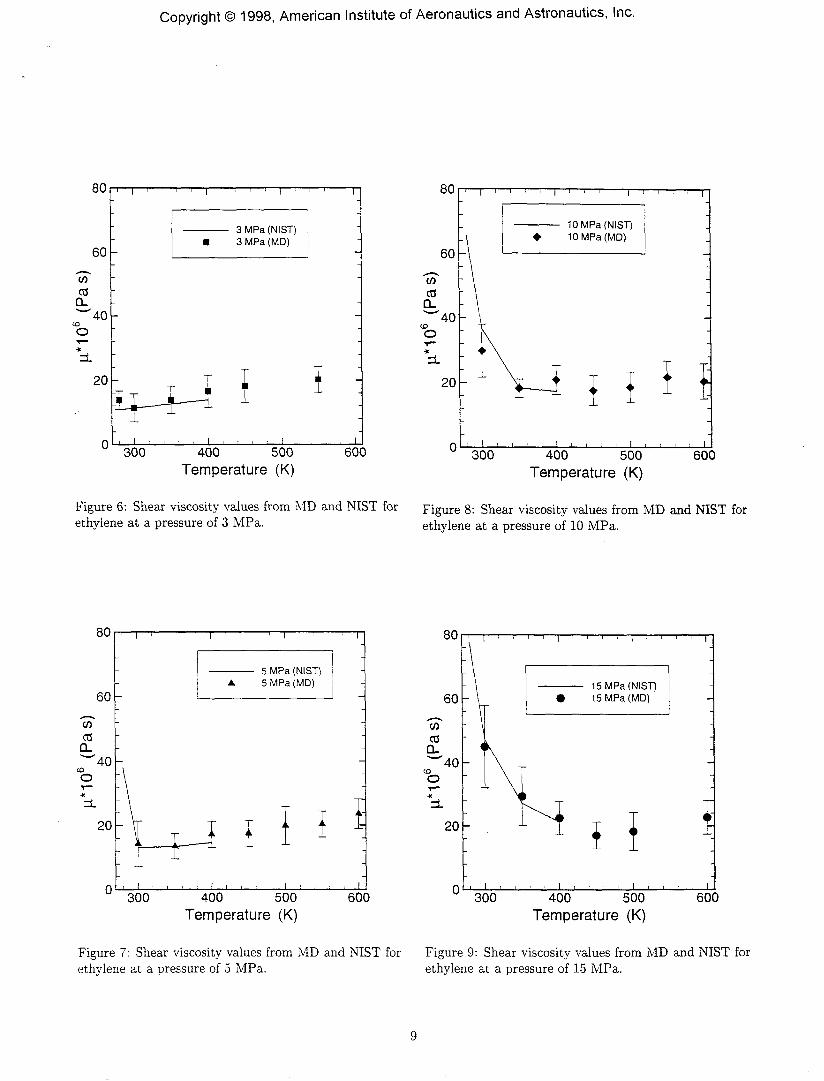
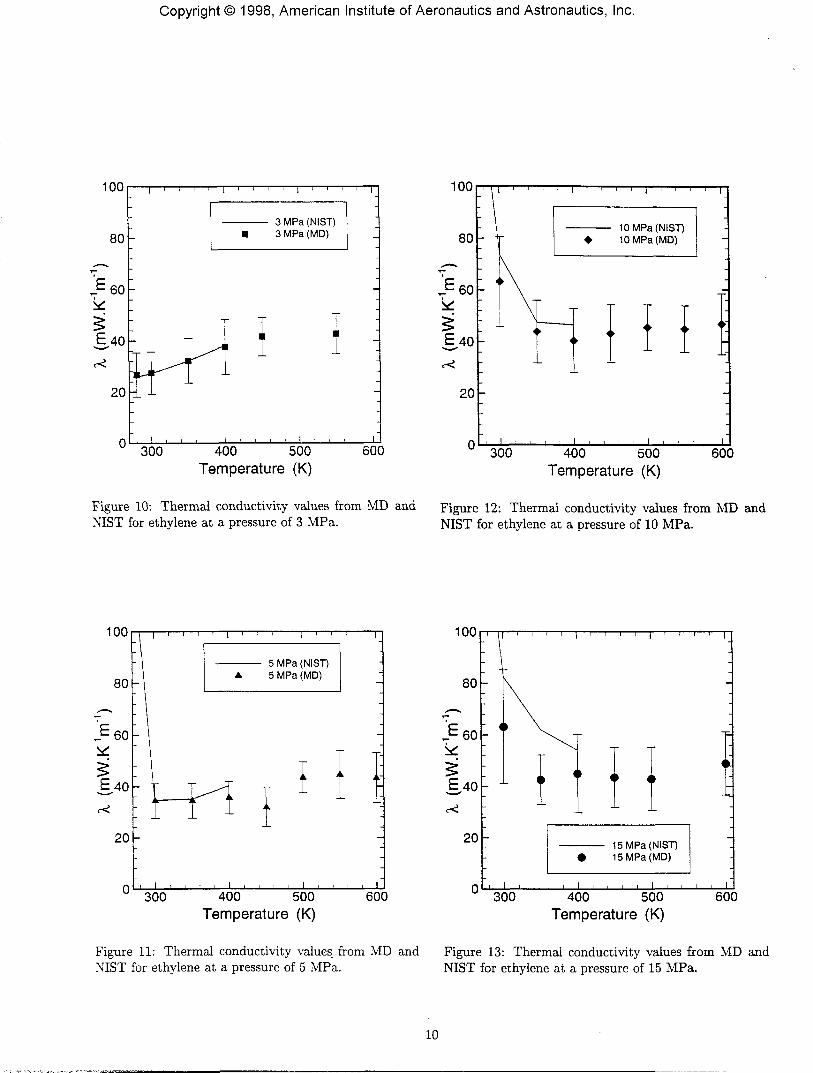
Figures 6, 7, 8 and 9 show plots of the shear viscosity,as functions of temperature and pressure for ethylene.Figures 10, 11, 12 and 13 show plots of the thermal con-ductivity, as functions of temperature and pressure forethylene. The MD results compare well with the NISTvalues for most of the cases. However the NIST softwaredoes not compute values for temperatures higher than400 K, hence no comparison can be carried out with MDvalues at these high temperatures. The MD thermal con-ductivity results do not match the NIST values very wellfor the 15 MPa case.
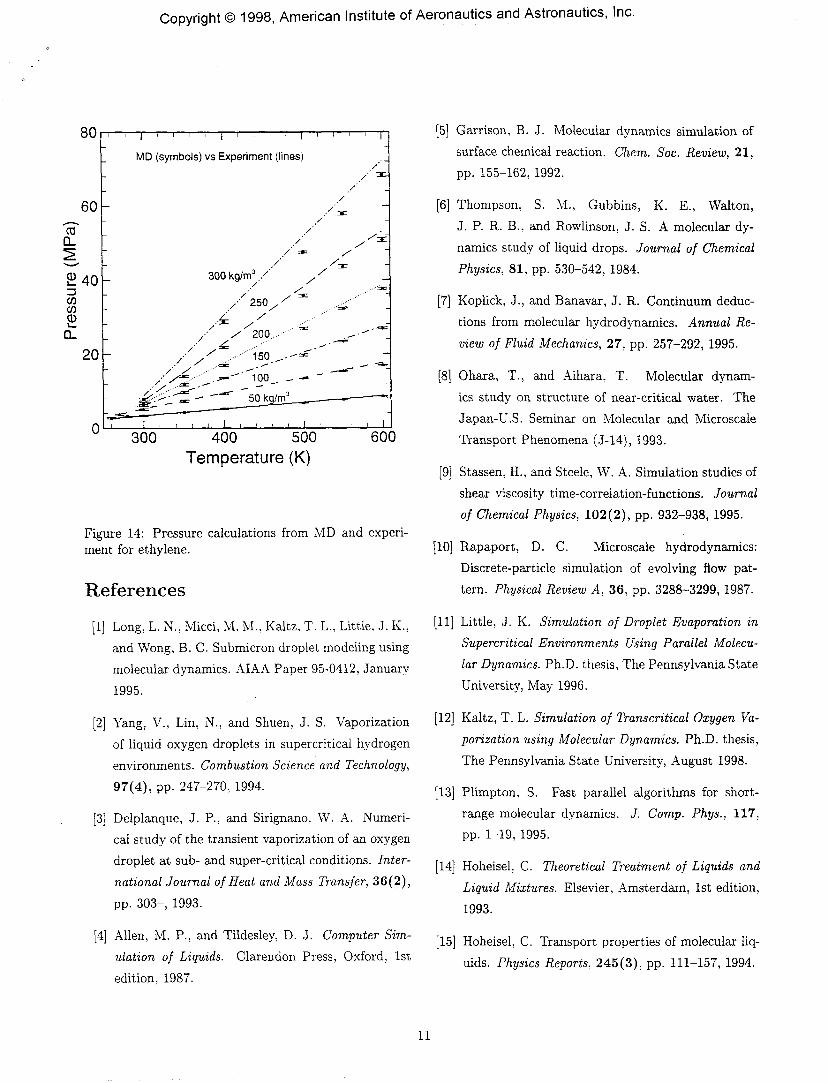
Figure 14 shows plots of calculated pressures againsttemperature for various densities for ethylene. Sim-ulations have been done under various conditions be-tween densities of 50-300 kg/m3 and temperatures of100-600 K. For most of the cases, there is excellent agree-ment between the MD and experimental results.32
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
60
50
40 -
03 30CO
o% 20
10
3MPa, 160K(NIST)3 MPa, 160K(MD)SMPa, 160 K (MIST)5MPa, 160K(MD)
1000 2000 3000Number of Molecules
4000
Figure 3: Effect of the number of molecules on shearviscosity for oxygen.
ou
60
I4"20
n
I : , I | I , , i | 1 , . , | 1 1 I ,
————— 3 MPa, 160K(NIST)• 3 MPa, 160K(MD)
....._.._.... — . 5 MPa, 160K(NIST)• 5 MPa, 160K(MD)
- " I T r: x * ~
1 . I 1 1 , , , I 1 , . ! ! 1 1 ! , ,
1000 2000 3000Number of Molecules
4000
Figure 4: Effect of the number of molecules on thermalconductivity for oxygen.
45-
COQ.
£30COCO03
15
200 kg/m3, 600 K (exp)200 kg/m3, 600 K (MD)
1000 2000 3000Number of Molecules
4000
Figure 5: Effect of the number of molecules on calculatedpressure for ethylene.
Conclusions
The transport properties and pressures of ethyleneare being computed via molecular dynamics (MD) sim-ulations using the Green-Kubo formulae and the virialequation of state, respectively. Simulations have beencarried out for a large number of subcritical, near criti-cal and supercritical conditions. Results show that thenumbers of molecules in the simulation systems are suf-ficient for obtaining accurate results for the subcriticaland near-critical conditions studied and the same resultshould hold for supercritical conditions. Computed shearviscosity and thermal conductivity coefficients comparewell with NIST results, while the pressure results com-pare well with experimental values. Future studies willinclude the computation of the transport properties ofother hydrocarbons typically found in propulsion sys-tems, and the properties at the surface of evaporatingdroplets in supercritical environments.33 A successfulimplementation of the code should have a great impacton the understanding of the processes occurring in cur-rent and future aerospace propulsion systems.
Acknowledgments
This work was supported by AFOSR Grant F49620-94-1-0133 and NASA Grants NAGW-1356 Supplement10, and NGT-10034. The authors would like to thankDr. Jeff Little and Dr. Teresa Kaltz for their contribu-tions.
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
80
60-
03Q_
<DO
20
•- 3 MPa (NIST)
3MPa(MD)
300 400 500Temperature (K)
600
Figure 6: Shear viscosity values from MD and NIST forethylene at a pressure of 3 MPa.
80
60 -
03Q.<D0
20 I I300 400 500
Temperature (K)600
Figure 8: Shear viscosity values from MD and NIST forethylene at a pressure of 10 MPa.
80r-r
60co03
Q_
COo40
20
5 MPa (NIST)5MPa(MD)
1300 400 500
Temperature (K)600
Figure 7: Shear viscosity values from MD and NIST forethylene at a pressure of 5 MPa.
80
60CO03Q.40
20
ou300 400 500
Temperature (K)600
Figure 9: Shear viscosity values from MD and NIST forethylene at a pressure of 15 MPa.
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
IUU
80
j=60
§40
20
0
i i i i i i i i i i i i i . i i
————— 3 MPa (NIST)1 • 3MPa(MD) J
-
- T " " -
~-~ ~
300 400 500 60CTemperature (K)
IUU
80
r-
J=60
§,40
20
0
1 1 | 1 ' 1 i | I I I c | I 1 1 1 |
- 1 ————— 10 MPa (NIST)- y •* 10MPa(MD)
:\ T T T f
i i i i i . i i i . , . , i300 400 500 60C
Temperature (K)
Figure 10: Thermal conductivity values from MD andNIST for ethylene at a pressure of 3 MPa.
Figure 12: Thermal conductivity values from MD andNIST for ethylene at a pressure of 10 MPa.
100
80
^60
20
A- 5 MPa (NIST)
5MPa(MD)
I300 400 500
Temperature (K)600
IUU
80
7£60
§,40
20
0
;
- <
-
. ' ' ' ' i ' i ' ' i ' ' ' ' i
•Vr T<|L
. o ° o o :
————— 15 MPa (NIST)• 15MPa(MD)
l i , i i l , i , i i i i i l300 400 500 60C
Temperature (K)
Figure 11: Thermal conductivity values from MD andNIST for ethylene at a pressure of 5 MPa.
Figure 13: Thermal conductivity values from MD andNIST for ethylene at a pressure of 15 MPa.
10
Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
80
60
COCO0)
20
MD (symbols) vs Experiment (lines)
300 kg/m3..
300 400 500Temperature (K)
600
Figure 14: Pressure calculations from MD and experi-ment for ethylene.
References
[1] Long, L. N., Micci, M. M., Kaltz, T. L., Little, J. K.,and Wong, B. C. Submicron droplet modeling usingmolecular dynamics. AIAA Paper 95-0412, January1995.
[2] Yang, V., Lin, N., and Shuen, J. S. Vaporizationof liquid oxygen droplets in supercritical hydrogenenvironments. Combustion Science and Technology,97(4), pp. 247-270, 1994.
[3] Delplanque, J. P., and Sirignano, W. A. Numeri-cal study of the transient vaporization of an oxygendroplet at sub- and super-critical conditions. Inter-national Journal of Heat and Mass Transfer, 36(2),pp. 303-, 1993.
[4] Alien, M. P., and Tildesley, D. J. Computer Sim-ulation of Liquids. Clarendon Press, Oxford, 1stedition, 1987.
[5] Garrison, B. J. Molecular dynamics simulation ofsurface chemical reaction. Chem. Soc. Review, 21,pp. 155-162, 1992.
[6] Thompson, S. M., Gubbins, K. E., Walton,J. P. R. B., and Rowlinson, J. S. A molecular dy-namics study of liquid drops. Journal of ChemicalPhysics, 81, pp. 530-542, 1984.
[7] Koplick. J., and Banavar, J. R. Continuum deduc-tions from molecular hydrodynamics. Annual Re-view of Fluid Mechanics, 27, pp. 257-292, 1995.
[8] Ohara, T., and Aihara, T. Molecular dynam-ics study on structure of near-critical water. TheJapan-U.S. Seminar on Molecular and MicroscaleTransport Phenomena (J-14), 1993.
[9] Stassen, H., and Steele, W. A. Simulation studies ofshear viscosity time-correlation-functions. Journalof Chemical Physics, 102(2), pp. 932-938, 1995.
[10] Rapaport, D. C. Microscale hydrodynamics:Discrete-particle simulation of evolving flow pat-tern. Physical Review A, 36, pp. 3288-3299, 1987.
[11] Little, J. K. Simulation of Droplet Evaporation inSupercritical Environments Using Parallel Molecu-lar Dynamics. Ph.D. thesis, The Pennsylvania StateUniversity, May 1996.
[12] Kaltz, T. L. Simulation of Transcritical Oxygen Va-porization using Molecular Dynamics. Ph.D. thesis,The Pennsylvania State University, August 1998.
[13] Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. /. Comp. Phys., 117,pp. 1-19, 1995.
[14] Hoheisel, C. Theoretical Treatment of Liquids andLiquid Mixtures. Elsevier, Amsterdam, 1st edition,1993.
[15] Hoheisel, C. Transport properties of molecular liq-uids. Physics Reports, 245(3), pp. 111-157, 1994.
11

Copyright© 1998, American Institute of Aeronautics and Astronautics, Inc.
[16] Evans, D. J., and Morriss, G. P. Non-newtonianmolecular dynamics. Computer Physics Reports, 1,pp. 297-343, 1984.
[17] Millat, J., Dymond, J. H., and Nieto de Castro,C. A. Transport Properties of Fluids: Their Corre-lation, Prediction and Estimation. Cambridge Uni-versity Press, 1st edition, 1996.
[18] Cheung, P. S. Y., and Powles, J. G. The propertiesof liquid nitrogen, part iv. a computer simulation.Molecular Physics, 30(3), pp. 921-949, 1975.
[19] Cheung, P. S. Y., and Powles, J. G. The propertiesof liquid nitrogen, part v. a computer simulationwith quadrupole interaction. Molecular Physics,32(5), pp. 1383-1405, 1976.
[20] Hoheisel, C., and Vogelsang, R. Thermal transportcoefficients for one-component and two-componentliquids from time correlation functions computedby molecular dynamics. Computer Physics Report,18(8), pp. 1-69, 1988.
[21] Barker, J. A., Fisher, R. A., and Watts, R. 0. Liq-uid argon: Monte carlo and molecular dynamics cal-culations. Molecular Physics, 21(4), pp. 657-673,1971.
[22] Singer, K., Taylor, A., and Singer, J. V. L. Thermo-dynamic and structural properties of liquids mod-elled by '2-lennard-jones centres' pair potentials.Molecular Physics, 33(6), pp. 1757-1795, 1977.
[23] Vogelsang, R., and Hoheisel, C. Comparison of var-ious potential models for simulation of the pressureof liquid and fluid nitrogen. Physics and Chemistryof Liquids, 16(3), pp. 189-203, 1987.
[24] Nwobi, 0. C., Long, L. N., and Micci. M. M. Molec-ular dynamics studies of transport properties of su-percritical fluids. AIAA Paper 97-0598, January1997.
[25] Nwobi, 0. C.. Long, L. N., and Michael, M. M.Molecular dynamics studies of properties of super-
critical fluids. Journal of Thermophysics and HeatTransfer, 12(2), pp. 1-6, 1998.
[26] Rapaport. D. C. The Art of Molecular DynamicsSimulation. Cambridge University Press, 1st edi-tion, 1995.
[27] Vincenti, W. G., and Kruger, C. H. Introduction toPhysical Gas Dynamics. Krieger Publishing Com-,pany, Malabar, Florida, 1986.
[28] Haile, J. M. Molecular Dynamics Simulation: Ele-mentary Methods. John Wiley, New York, 1st edi-tion, 1992.
[29] National Institute of Standards and Technology,Gaithersburg, MD. NIST Thermophysical Proper-ties of Pure Fluids Database: Version 3.0, 1996.
[30] Hanley, H. J. M., McCarty, R. D., and Haynes,W. M. The viscosity and thermal conductivity coef-ficients for dense gaseous and liquid argon, krypton,xenon, and oxygen. Journal of Physical ChemistryRef. Data, 3(4), pp. 979-1018, 1974.
[31] Younglove, B. A., and Hanley, H. J. M. The viscos-ity and thermal conductivity coefficients of gaseousand liquid argon. Journal of Physical ChemistryReference Data, 15(4), pp. 1323-1337, 1986.
[32] Sychev, V. V., Vasserman, A. A., Golovsky,E. A. Kozlov, A. D., Spiridonov, G. A., and Tsy-marny, V. A. Thermodynamic Properties of Ethy-lene. National Standard Reference Data Service ofthe USSR, A Series of Property Tables. HemispherePublishing Company, New York, 1987.
[33] Nwobi, 0. C. Molecular Dynamics Studies of Trans-port Properties and Equation of State of Supercrit-ical Fluids. Ph.D. thesis, The Pennsylvania StateUniversity, August 1998.
12





























