Embed Size (px)
Citation preview

Molecular dynamics simulation studies forDNA sequence recognition by reactivemetabolites of anticancer compoundsKhaled M. Tumbi, Prajwal P. Nandekar, Naeem Shaikh,Siddharth S. Kesharwani and Abhay T. Sangamwar*
The discovery of novel anticancer molecules 5F-203 (NSC703786) and 5-aminoflavone (5-AMF, NSC686288) hasaddressed the issues of toxicity and reduced efficacy by targeting over expressed Cytochrome P450 1A1 (CYP1A1)in cancer cells. CYP1A1 metabolizes these compounds into their reactive metabolites, which are proven to mediatetheir anticancer effect through DNA adduct formation. However, the drug metabolite–DNA binding has not beenexplored so far. Hence, understanding the binding characteristics and molecular recognition for drug metaboliteswith DNA is of practical and fundamental interest. The present study is aimed to model binding preference shownby reactive metabolites of 5F-203 and 5-AMF with DNA in forming DNA adducts. To perform this, three differentDNA crystal structures covering sequence diversity were selected, and 12 DNA-reactive metabolite complexes weregenerated. Molecular dynamics simulations for all complexes were performed using AMBER 11 software afterdevelopment of protocol for DNA-reactive metabolite system. Furthermore, the MM-PBSA/GBSA energy calculation,per-nucleotide energy decomposition, and Molecular Electrostatic Surface Potential analysis were performed. Theresults obtained from present study clearly indicate that minor groove in DNA is preferable for binding of reactivemetabolites of anticancer compounds. The binding preferences shown by reactive metabolites were also governedby specific nucleotide sequence and distribution of electrostatic charges in major and minor groove of DNAstructure. Overall, our study provides useful insights into the initial step of mechanism of reactive metabolitebinding to the DNA and the guidelines for designing of sequence specific DNA interacting anticancer agents.Copyright © 2014 John Wiley & Sons, Ltd.Additional supporting information may be found in the online version of this article at the publisher's web site.
Keywords: DNA; molecular recognition; reactive metabolites; molecular dynamics simulations; molecular docking;MM-PBSA; per-nucleotide energy decomposition; MESP
INTRODUCTION
The specific DNA sequence plays an important role in DNAreplication, promotion, and transcription (Rosenberg and Court1979). Plethora of drug molecules targets DNA leading to theiranticancer (Hurley 2002), antibacterial (Shen et al., 1989),antiprotozoal, and antifungal activities (Ismail et al. 2004).Alkylating agents are among the first DNA-interactive anticancercompounds, which covalently bind with DNA and show theirpharmacological activity (Kohn 1996). Recent trend has shownthe design and synthesis of non-covalent, sequence selective,minor groove specific DNA binding agents such as Netropsinand Distamycin with antiviral and anticancer activity, respec-tively (Portugal and Waring 1987; Nielsen 1991; Hélène 1993).These molecules have the ability to govern the expression ofgenes responsible for progression of disease of interest (Hurley2002). Their binding is mainly governed by a set of non-covalentinteractions such as hydrogen bonding and electrostatic interac-tions with floor and walls of minor groove of DNA (Neidle 2001).Binding of the ligand to nucleotides of DNA depends on theirchemical nature and the reactive states. Apart from the well-known non-covalent binders such as HOECHST 33258,Distamycin, Anthramycin, Mytomicin C, and so on, somecovalently binding ligands also show anticancer activity.
However, their potential disadvantage is the lack of targetbinding specificity leading to severe toxicities (Izbicka andTolcher, 2004)Among the DNA binding agents, we concentrated our work on
5F-203 (NSC703786) (Trapani et al., 2003) and 5-aminoflavone(NSC686288) (Meng et al., 2005) molecules, which were reachedup to phase I clinical trial for clinical evaluation against non-smallcell lung cancer therapy (Bradshaw and Westwell 2004). Currently,molecule 5F-203 is abandoned from clinical trials (http://www.evaluategroup.com/), and the phase 1 trials of 5-aminoflavonehas finished successfully (http://www.clinicaltrials.gov). They areexhibited for having potential antitumor activity of <0.1 nM onlung cancer cell lines (Nandekar and Sangamwar 2012). The novelmechanism underlined for anticancer activity of both ligands is viageneration of reactive metabolite (nitrenium ion species) after
* Correspondence to: Abhay T. Sangamwar, Department of Pharmacoinformatics,National Institute of Pharmaceutical Education and Research (NIPER), S.A.S.Nagar, Sector 67, Mohali, Punjab PIN 160062, India.E-mail: [email protected]
K. M. Tumbi, P. P. Nandekar, N. Shaikh, S. S. Kesharwani, A. T. SangamwarDepartment of Pharmacoinformatics, National Institute of PharmaceuticalEducation and Research (NIPER) S.A.S. Nagar, Punjab, India
Research Article
Received: 18 June 2013, Revised: 19 November 2013, Accepted: 20 November 2013, Published online in Wiley Online Library
(wileyonlinelibrary.com) DOI: 10.1002/jmr.2342
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd.
138

metabolism by CYP1A1 (Figure 1) (Nandekar and Sangamwar 2012).Nitrenium ion species bindswithDNAand formsDNA-adducts leadingto cancerous cell death (O’Brien et al., 2003; Pobst and Ames 2006).In past years, molecular dynamics (MD) simulations have been
extensively used to predict the ligand-binding mode andbinding site recognition in DNA. First MD simulation on pureDNA system was traced long back to 1983 on a glance of solva-tion effects (Levitt 1983; Tidor et al., 1983; Singh et al., 1985). Onthe other hand, some computer simulations on DNA-ligandsystem were performed with focus on snug fit mechanism ofNetropsin (Zakrzewska et al., 1984; Caldwell and Kollman 1986).Moreover, with exponential increase in computational powerand well developed force fields, realm of MD simulations ofDNA increased with some distinguished studies on minor groovebinding of Netropsin, Distamysin (Martí-Renom et al., 2000),Hoechst 33258 (Bostock-Smith et al., 2001), and CGP 40215A(Nguyen et al., 2004). In the recent years, Perez et al. refinedthe traditional non-specific AMBER parm99 force field for nucleicacids and named it as parmbsc0 (Perez et al., 2007). They madechanges for correct representation of the α/γ concerted rotationin nucleic acids. Many of the previously performed studies wererepeated using this well developed and validated force field; oneof them was for Netropsin using parmbsc0 force field (Andacet al., 2011). Recently, Ascona B-DNA Consortium broke themicrosecond barriers in B-DNA simulation using parmbs0, givinga clear view on B-DNA behavior in long-run MD simulations(Beveridge et al., 2012). Rodrigo et al. have performed the MDsimulations and quantum chemical studies for prediction ofligand-binding modes of copper containing anticancer com-pounds in DNA structure (Galindo-Murillo et al., 2012). Srivastavaet al. have successfully compared various molecular dockingmethods to model minor groove binders and suggested theGOLD docking algorithm and aqueous conditions for DNA simula-tion using AMBER software (Srivastava et al., 2011). They success-fully correlated MM-PBSA/GBSA binding free energy to ΔTm(change in melting temperature of DNA with drug) and experi-mentally determined binding strength (Shaikh et al., 2004;Srivastava and Sastry 2012). Till date, several in silico studies werereported on neutral ligands as DNA binders, but the bindingpreferences shown by actual reactive metabolites of anticancercompounds have not been explored so far. This study willcertainly be helpful in designing better DNA intercalatinganticancer compounds.Herein, we have performed extensive MD simulations on three
different DNA sequences viz protein data bank ID: 226D (AT rich),1QC1 (GC rich), and 2K0V (mixed nucleotides sequence).
DNA-ligand complexes of three DNA sequences with reactivemetabolite of 5F-203 and 5-aminoflavone were selected for MDsimulation studies. The coordinates for DNA-ligand complexes inmajor and minor groove of DNA were generated using GOLD mo-lecular docking programme to ensure all possibilities of formingDNA-ligand complexes. The favorable molecular docking poseswere selected on the basis of GOLD fitness scores and hydro-gen bonding interactions. A total of 12 input DNA-ligandcomplexes were selected and MD simulations (~10 ns each)using AMBER 11 software package were performed. Further-more, the per-nucleotide energy decomposition analysis andMolec-ular Electrostatic Surface Potential (MESP) analysis were performedto observe the influence of nucleotides and charge distribution onligand binding to DNA structures. Thus, the presented computa-tional study rigorously examines the DNA binding pattern ofreactive metabolites. Finally, the paper provides important insightsfor metabolism-based drug design in anticancer drug discovery.
MATERIALS AND METHODS
Selection of ligands
The reactive metabolite of ligands 5F-203 (5F) and 5-aminoflavone(AMF) possessing DNA-binding potential was selected for molecu-lar docking studies with three different DNA structures. Thecompound 5F-203 and 5-aminoflavone are potent antitumoragents from benzothiazole and aminoflavone series, respectively,and are currently in clinical trials (Leong et al., 2003; Pobst andAmes 2006). The 5F-203 and 5-aminoflavone induce CYP1A1 by ac-tivating aryl hydrocarbon receptor pathway and subsequently getmetabolized by CYP1A1 to produce reactive metabolite that formsDNA adduct leading to cancer cell death (Figure 1). The mecha-nism of anticancer effect is mediated via generation of reactivemetabolite (Nitrenium ion species). In organic chemistry, nitreniumion is a reactive intermediate based on nitrogen with both an elec-tron lone pair and positive charge with two substitutions (R2N
+)(Gassman 1970). Nitrenium ions are isoelectronic to carbene andcan exist in either a singlet or a triplet state (Harrison and Eakers1973). The structure of nitrenium ion as reported in the literaturewas drawn and energy minimized in GAUSSIAN03 software usingHF method with 6-31G+ * basis set (Frisch et al., 2008). The partialRESP charges on all atoms of 5F (reactivemetabolite of 5F-203) andAMF (reactive metabolite of 5-Aminoflavone) were calculated andused for MD simulation studies.
Selection of DNA sequences
Structural and chemical features of DNA structures are governedby their nucleotide sequence (Steitz 1990). Minor groove of GCrich DNA structure is wider as compared with AT rich DNAsequence (Neidle 2001). Also, the minor groove of AT richsequence is more electronegative than GC rich DNA sequencestructure (Neidle 2001). Previous studies stated that DNAsequence plays a crucial role in binding of ligand to DNA strands(Nielsen 1991). Hence, it is necessary to consider maximum pos-sible chemical environments in DNA structures before selectingDNA structures for further molecular docking and MD simulationstudies. Considering the previously mentioned facts, threedifferent DNA structures were selected and retrieved fromprotein data bank as 226D (AT-rich), IQC1 (GC-rich), and 2K0V(mixed sequence) that cover the sequence space of DNAstructures (Chobe et al., 2012) (Table 1).
Figure 1. The mechanism of generation of reactive metabolites of5F-203 and 5-aminoflavone.
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
139
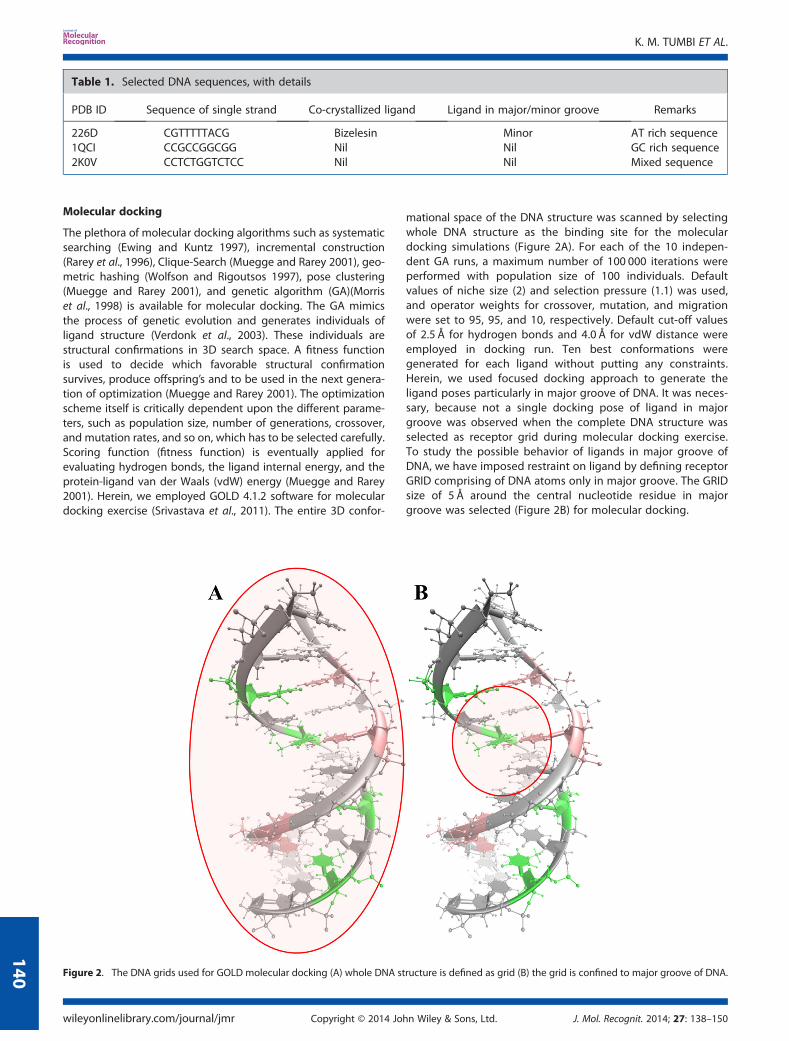
Molecular docking
The plethora of molecular docking algorithms such as systematicsearching (Ewing and Kuntz 1997), incremental construction(Rarey et al., 1996), Clique-Search (Muegge and Rarey 2001), geo-metric hashing (Wolfson and Rigoutsos 1997), pose clustering(Muegge and Rarey 2001), and genetic algorithm (GA)(Morriset al., 1998) is available for molecular docking. The GA mimicsthe process of genetic evolution and generates individuals ofligand structure (Verdonk et al., 2003). These individuals arestructural confirmations in 3D search space. A fitness functionis used to decide which favorable structural confirmationsurvives, produce offspring’s and to be used in the next genera-tion of optimization (Muegge and Rarey 2001). The optimizationscheme itself is critically dependent upon the different parame-ters, such as population size, number of generations, crossover,and mutation rates, and so on, which has to be selected carefully.Scoring function (fitness function) is eventually applied forevaluating hydrogen bonds, the ligand internal energy, and theprotein-ligand van der Waals (vdW) energy (Muegge and Rarey2001). Herein, we employed GOLD 4.1.2 software for moleculardocking exercise (Srivastava et al., 2011). The entire 3D confor-
mational space of the DNA structure was scanned by selectingwhole DNA structure as the binding site for the moleculardocking simulations (Figure 2A). For each of the 10 indepen-dent GA runs, a maximum number of 100 000 iterations wereperformed with population size of 100 individuals. Defaultvalues of niche size (2) and selection pressure (1.1) was used,and operator weights for crossover, mutation, and migrationwere set to 95, 95, and 10, respectively. Default cut-off valuesof 2.5 Å for hydrogen bonds and 4.0 Å for vdW distance wereemployed in docking run. Ten best conformations weregenerated for each ligand without putting any constraints.Herein, we used focused docking approach to generate theligand poses particularly in major groove of DNA. It was neces-sary, because not a single docking pose of ligand in majorgroove was observed when the complete DNA structure wasselected as receptor grid during molecular docking exercise.To study the possible behavior of ligands in major groove ofDNA, we have imposed restraint on ligand by defining receptorGRID comprising of DNA atoms only in major groove. The GRIDsize of 5 Å around the central nucleotide residue in majorgroove was selected (Figure 2B) for molecular docking.
Table 1. Selected DNA sequences, with details
PDB ID Sequence of single strand Co-crystallized ligand Ligand in major/minor groove Remarks
226D CGTTTTTACG Bizelesin Minor AT rich sequence1QCI CCGCCGGCGG Nil Nil GC rich sequence2K0V CCTCTGGTCTCC Nil Nil Mixed sequence
Figure 2. The DNA grids used for GOLD molecular docking (A) whole DNA structure is defined as grid (B) the grid is confined to major groove of DNA.
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
140

Molecular dynamics simulation methodology
The 12 DNA-ligand complex structures obtained from moleculardocking were used as starting structure for MD simulation studiesusing PMEMD (CUDA version) programme included in AMBER 11software package (Case et al., 2010) with GPU parallelization. TheRESP charges for reactive nitrenium ion species (5F and AMF) werecalculated using DFT method with HF/6-31G+* geometry optimiza-tion in GAUSSIAN03 software. Ligand parameters for reactive nitreniumion species were prepared in Antechembermodule of AmberTools1.5using GAFF force field (Wang et al., 2004). The parameters for DNAsystem were generated using recently developed parmbsc0 forcefield with periodic boundary conditions (Perez et al., 2008). TheDNA-ligand complex was solvated in truncated octahedral TIP3Pwater box having dimensions of 10×10×10Å (Jayaram and Jain2004). The solvated system was neutralized using Na+ counter ions(Ponomarev et al., 2004). The stepwise protocol applied for MD
simulation studies is described in Figure 3. The non-bonded interac-tion cut-off of 8Å was used for all systems as validated by Srivastavaet al. and suggested by Beck et al. (Beck et al., 2004; Srivastava et al.,2011). We followed a number of steps for system equilibration as itwas difficult to get completely equilibrated system in one or twosteps of simulations (Wang and Laughton 2007; Wang andLaughton 2010) (Figure 3).
All hydrogen-heavy atom bonds were constrained by the SHAKEmethod, and simulations were performed with a 1 fs time step andLangevin dynamics for temperature control. After heating anddensity equilibration, the completely equilibrated system wasattained after seven steps of constrained and unconstrained equil-ibration simulation (Figure 3). Once the system equilibration wasachieved, the production MD simulations of 10 ns were performedfor all DNA-ligand complexes using NVT ensemble without anypositional constrains. MM-PBSA/GBSA binding free energy
Figure 3. Flow chart showing stepwise protocol used for equilibration and molecular dynamics simulations of all DNA-ligand complexes.
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
141

calculations and per-nucleotide energy decomposition wereperformed for complete production run by extracting snapshotsat an interval of 10 frames.
RESULTS AND DISCUSSION
Molecular docking
Molecular docking of two ligands was performed using GOLD 4.1.2docking software. Molecular docking results were analyzed in Her-mes visualizer and in house DNA-Docking Analysis Tool (DDAT).The code of DDAT was written in C++ programming language.DDAT tool uses GOLD output file and gives results in terms of interatomic distance, atom ID, and nucleotide number of nucleophilic ni-trogen that are present around the ligand. DDAT provides respectiveGOLD fitness score and docking solution ID of docked pose. DDAT isuseful for user to separate the ligand docking pose with particularfeature, for example, distance of different nucleophilic centers inDNA to that of electropositive center in ligand molecule.
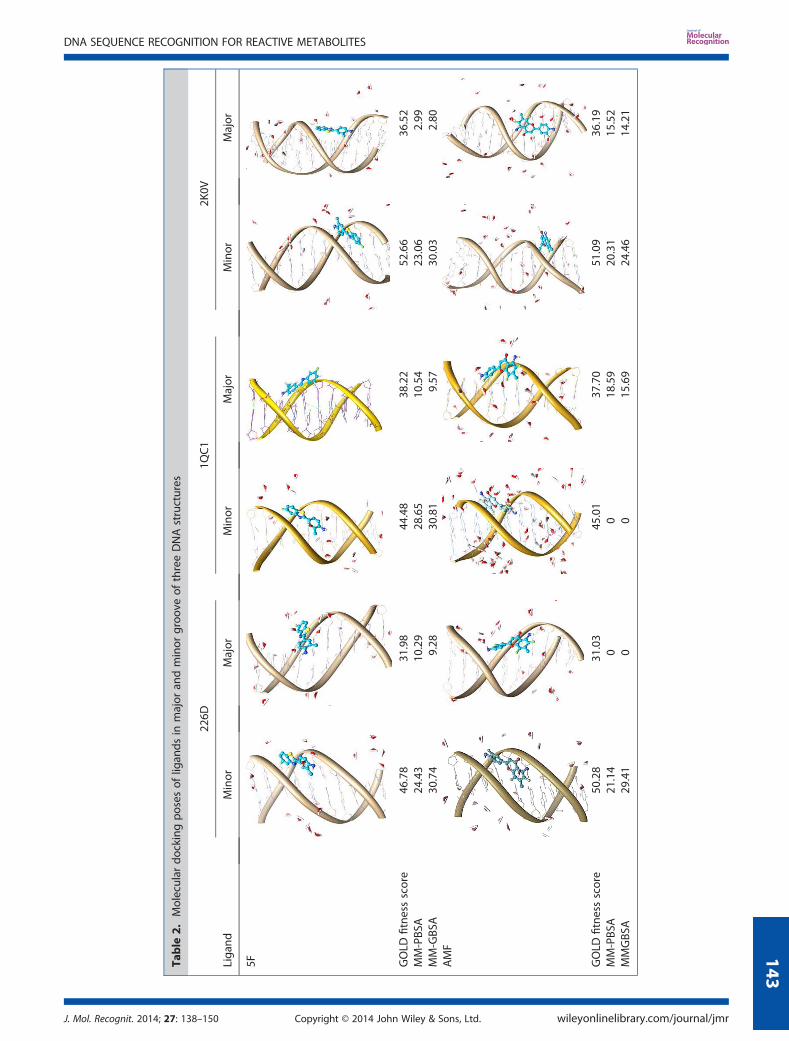
Molecular docking analysis showed that all ligands weresuccessfully docked in minor groove of DNA with low RMSDvalues to each other. The best docking pose of ligand in eachDNA structure was selected based on GOLD fitness score. TheDNA-ligand interactions were also considered to select the bestligand-binding pose. The rationale behind the discard of otherhigh-scoring poses is their low RMSD values with selected bind-ing pose. The tunnel like cavity of minor groove might be thefirst choice for planar ligands. The ligands docked in minorgroove of DNA were observed to have favorable π–stackinginteractions between aromatic ring system of ligand and thenucleotides of DNA. The molecular docking GOLD fitness scoresobtained for ligands 5F and AMF were summarized in Table 2.The GOLD fitness score for ligands 5F and AMF in minor grooveof 2K0V were 52.66 and 51.09, respectively. It clearly indicatesmore preference of ligands towards minor groove of 2K0V(mixed nucleotide sequence) structure as compared with minorgroove of 226D (46.8 and 50.28) and 1QC1 (44.48, and 45.01)sequence structures. A significant difference in GOLD fitnessscores between docking in minor groove and major groovewas observed for 5F and AMF (Table 2). For example, the scoresobtained for 5F in minor and major groove of 226D were 46.78and 31.98, respectively, showing the difference of ~15 unitswhich specifies ligand preference towards minor groove ascompared with major groove of DNA structure. Similar patternwas observed for ligands 5F and AMF in all three DNA structures.
The covalent binding of reactive species to DNA is supposed tobe biphasic event (Hurley and Needham-VanDevanter 1986).Although the reactive species binds to DNA with covalent bond,the event of covalent binding between ligand and DNA starts fromthe stable non-covalent interactions leading to the placement of li-gand in the binding cavity. It is mainly governed by electrostaticand hydrophobic interactions, or both in combination with vdWinteractions, and it determines the sequence recognition for thegiven ligand (Levitt 1984; Zakrzewska et al. 1984). Consideringthe previously mentioned facts, we have performed non-covalentmolecular docking and molecular simulations of reactive speciesof ligands in the major and minor groove of DNA.
Molecular dynamics simulations
Molecular dynamics simulations of a total of 12 DNA-ligandcomplexes in major and minor grooves of three DNA structures
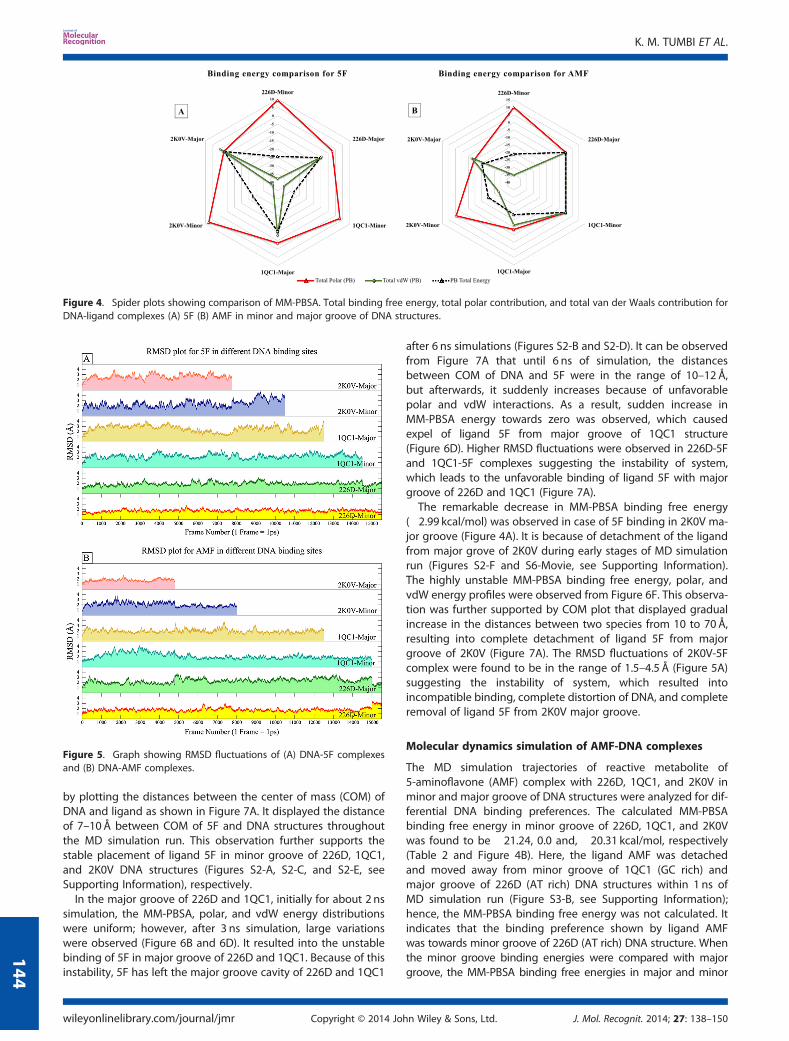
were performed in AMBER 11 software using previously de-scribed protocol (Figure 3). The MM-PBSA/GBSA total bindingfree energy, total polar contribution, and total vdW contributionfor DNA-ligand complexes are summarized in Table 2 and Fig-ure 4. In the following sections, we shall discuss about MD simu-lation trajectories analysis and the physical changes observed inthe 12 DNA-ligand complexes.
Stability of trajectories
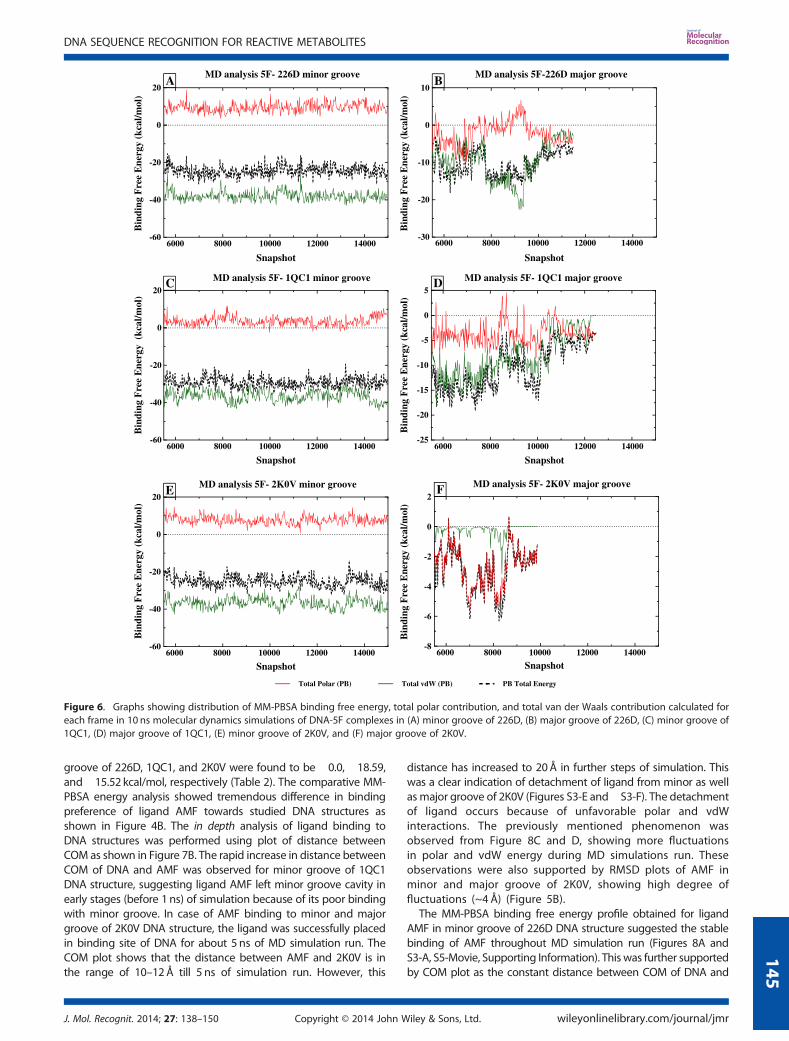
The MD simulation trajectories of DNA-ligand complexes wereanalyzed for their structural stability during 10 ns of MD simula-tion run. The total energy fluctuations in MD simulation run wereobserved to determine the energetic stability of DNA ligandcomplexes. The total energy of complex versus frame numberplots for 12 simulations are shown in Figure S1 (SupportingInformation). It was observed that the total energy of systemis oscillating around the average energy of DNA-ligand com-plex, and it was converged throughout 10 ns simulation time(Figure S1). These observations suggest the complete stability ofsystem at given conditions. Stability of trajectory can also beassessed by RMSD fluctuations of DNA-ligand complexes in MDsimulation run. RMSD fluctuations of DNA-ligand complexesversus frame numbers were plotted for all MD simulations(Figure 5A and B). RMSD graphs clearly indicate that all systemshave attained their equilibration in 5.5 ns as described in MDsimulation methodology. The observations from total energyand RMSD plots helped us to estimate whether these systemswere stabilized throughout the MD simulation run. Effective posesampling for MM-PBSA/GBSA free energy calculations wasperformed for production run of 10 ns simulations.
Molecular dynamics simulation of 5F-DNA complexes
The MD simulation trajectories of reactive metabolite of 5F-203(5F) complexes with 226D, 1QC1, and 2K0V in minor and majorgroove were analyzed for differential DNA binding preferences.The calculated MM-PBSA binding free energy in minor grooveof 226D, 1QC1, and 2K0V was found to be �24.43, �28.65, and�23.06 kcal/mol, respectively, as shown in Table 2 and Figure 4A.Results indicate that the ligand 5F has more stable binding andshow preference towards minor groove of 1QC1 (GC rich) struc-ture. When minor groove binding energies were compared withmajor groove; the MM-PBSA binding free energy was found tohave �10.29, �10.54, and �2.99 kcal/mol in major groove of226D, 1QC1, and 2K0V structures, respectively. The energiesobtained for major groove bindings were far less than energiesobtained for minor grooves, in the range of 10–15 kcal/mol. Thisimposed that ligand 5F favors the minor groove of DNA struc-tures, specifically the minor groove of GC rich sequence structure(1QC1) as compared with other structures.The MM-PBSA binding free energy, polar energy, and vdW
energy contribution were plotted against frame number to studythe binding stability of ligands throughout MD simulation run.Figure 6A, C, and E have shown that the stable binding energyprofiles were observed for ligand 5F in minor groove of 226D,1QC1, and 2K0V DNA structures. The stable polar and vdWinteraction energy profile (Figure 6A, C, and E) suggested thatthe ligand is consistently interacting with the polar as well ashydrophobic part of nucleotides present in minor groove ofDNA structures. The successful and stable placement of ligandin minor and major grooves of DNA structures was evaluated
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
142
Table
2.Molecular
dockingpo
sesof
ligan
dsin
major
andminor
groo
veof
threeDNAstructures
226D
1QC1
2K0V
Liga
ndMinor
Major
Minor
Major
Minor
Major
5F GOLD
fitnessscore
46.78
31.98
44.48
38.22
52.66
36.52
MM-PBS
A�2
4.43
�10.29
�28.65
�10.54
�23.06
�2.99
MM-GBS
A�3
0.74
�9.28
�30.81
�9.57
�30.03
�2.80
AMF
GOLD
fitnessscore
50.28
31.03
45.01
37.70
51.09
36.19
MM-PBS
A�2
1.14
00
�18.59
�20.31
�15.52
MMGBS
A�2
9.41
00
�15.69
�24.46
�14.21
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
143
by plotting the distances between the center of mass (COM) ofDNA and ligand as shown in Figure 7A. It displayed the distanceof 7–10Å between COM of 5F and DNA structures throughoutthe MD simulation run. This observation further supports thestable placement of ligand 5F in minor groove of 226D, 1QC1,and 2K0V DNA structures (Figures S2-A, S2-C, and S2-E, seeSupporting Information), respectively.
In the major groove of 226D and 1QC1, initially for about 2 nssimulation, the MM-PBSA, polar, and vdW energy distributionswere uniform; however, after 3 ns simulation, large variationswere observed (Figure 6B and 6D). It resulted into the unstablebinding of 5F in major groove of 226D and 1QC1. Because of thisinstability, 5F has left the major groove cavity of 226D and 1QC1
after 6 ns simulations (Figures S2-B and S2-D). It can be observedfrom Figure 7A that until 6 ns of simulation, the distancesbetween COM of DNA and 5F were in the range of 10–12 Å,but afterwards, it suddenly increases because of unfavorablepolar and vdW interactions. As a result, sudden increase inMM-PBSA energy towards zero was observed, which causedexpel of ligand 5F from major groove of 1QC1 structure(Figure 6D). Higher RMSD fluctuations were observed in 226D-5Fand 1QC1-5F complexes suggesting the instability of system,which leads to the unfavorable binding of ligand 5F with majorgroove of 226D and 1QC1 (Figure 7A).The remarkable decrease in MM-PBSA binding free energy
(�2.99 kcal/mol) was observed in case of 5F binding in 2K0V ma-jor groove (Figure 4A). It is because of detachment of the ligandfrom major grove of 2K0V during early stages of MD simulationrun (Figures S2-F and S6-Movie, see Supporting Information).The highly unstable MM-PBSA binding free energy, polar, andvdW energy profiles were observed from Figure 6F. This observa-tion was further supported by COM plot that displayed gradualincrease in the distances between two species from 10 to 70Å,resulting into complete detachment of ligand 5F from majorgroove of 2K0V (Figure 7A). The RMSD fluctuations of 2K0V-5Fcomplex were found to be in the range of 1.5–4.5 Å (Figure 5A)suggesting the instability of system, which resulted intoincompatible binding, complete distortion of DNA, and completeremoval of ligand 5F from 2K0V major groove.
Molecular dynamics simulation of AMF-DNA complexes
The MD simulation trajectories of reactive metabolite of5-aminoflavone (AMF) complex with 226D, 1QC1, and 2K0V inminor and major groove of DNA structures were analyzed for dif-ferential DNA binding preferences. The calculated MM-PBSAbinding free energy in minor groove of 226D, 1QC1, and 2K0Vwas found to be �21.24, 0.0 and, �20.31 kcal/mol, respectively(Table 2 and Figure 4B). Here, the ligand AMF was detachedand moved away from minor groove of 1QC1 (GC rich) andmajor groove of 226D (AT rich) DNA structures within 1 ns ofMD simulation run (Figure S3-B, see Supporting Information);hence, the MM-PBSA binding free energy was not calculated. Itindicates that the binding preference shown by ligand AMFwas towards minor groove of 226D (AT rich) DNA structure. Whenthe minor groove binding energies were compared with majorgroove, the MM-PBSA binding free energies in major and minor
Figure 4. Spider plots showing comparison of MM-PBSA. Total binding free energy, total polar contribution, and total van der Waals contribution forDNA-ligand complexes (A) 5F (B) AMF in minor and major groove of DNA structures.
Figure 5. Graph showing RMSD fluctuations of (A) DNA-5F complexesand (B) DNA-AMF complexes.
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
144
groove of 226D, 1QC1, and 2K0V were found to be �0.0, �18.59,and �15.52 kcal/mol, respectively (Table 2). The comparative MM-PBSA energy analysis showed tremendous difference in bindingpreference of ligand AMF towards studied DNA structures asshown in Figure 4B. The in depth analysis of ligand binding toDNA structures was performed using plot of distance betweenCOM as shown in Figure 7B. The rapid increase in distance betweenCOM of DNA and AMF was observed for minor groove of 1QC1DNA structure, suggesting ligand AMF left minor groove cavity inearly stages (before 1 ns) of simulation because of its poor bindingwith minor groove. In case of AMF binding to minor and majorgroove of 2K0V DNA structure, the ligand was successfully placedin binding site of DNA for about 5ns of MD simulation run. TheCOM plot shows that the distance between AMF and 2K0V is inthe range of 10–12Å till 5 ns of simulation run. However, this
distance has increased to 20Å in further steps of simulation. Thiswas a clear indication of detachment of ligand from minor as wellas major groove of 2K0V (Figures S3-E and S3-F). The detachmentof ligand occurs because of unfavorable polar and vdWinteractions. The previously mentioned phenomenon wasobserved from Figure 8C and D, showing more fluctuationsin polar and vdW energy during MD simulations run. Theseobservations were also supported by RMSD plots of AMF inminor and major groove of 2K0V, showing high degree offluctuations (~4 Å) (Figure 5B).
The MM-PBSA binding free energy profile obtained for ligandAMF in minor groove of 226D DNA structure suggested the stablebinding of AMF throughout MD simulation run (Figures 8A andS3-A, S5-Movie, Supporting Information). This was further supportedby COM plot as the constant distance between COM of DNA and
Figure 6. Graphs showing distribution of MM-PBSA binding free energy, total polar contribution, and total van der Waals contribution calculated foreach frame in 10 ns molecular dynamics simulations of DNA-5F complexes in (A) minor groove of 226D, (B) major groove of 226D, (C) minor groove of1QC1, (D) major groove of 1QC1, (E) minor groove of 2K0V, and (F) major groove of 2K0V.
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
145
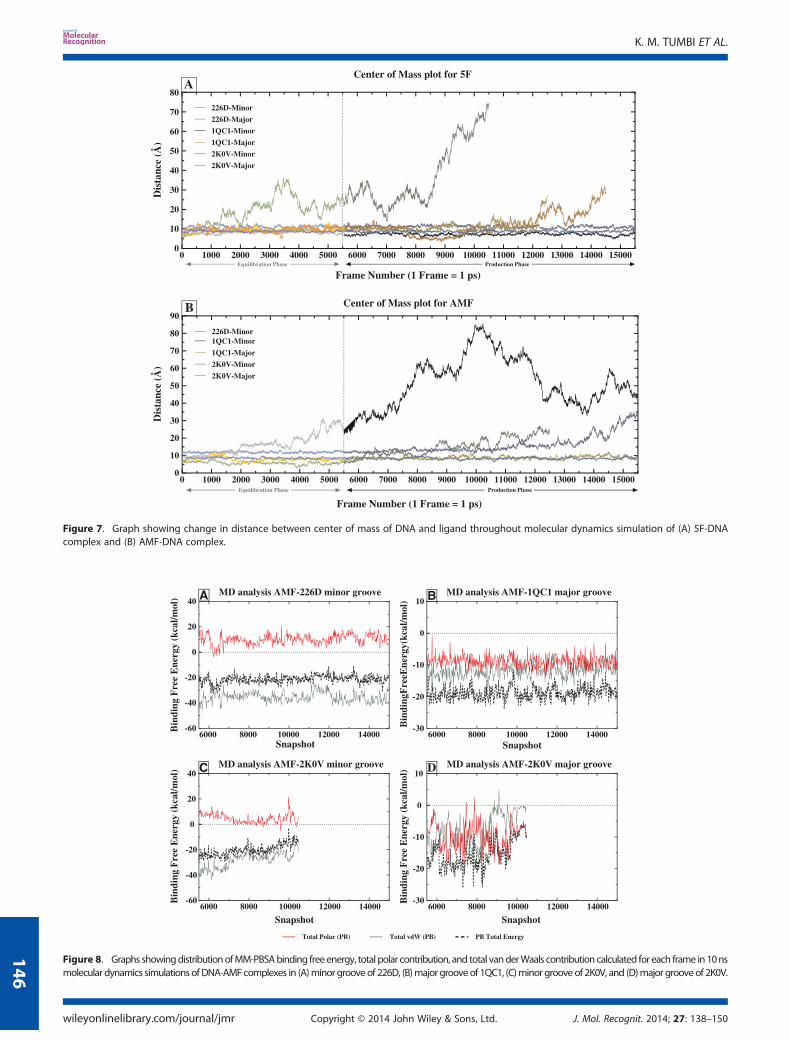
Figure 7. Graph showing change in distance between center of mass of DNA and ligand throughout molecular dynamics simulation of (A) 5F-DNAcomplex and (B) AMF-DNA complex.
Figure 8. Graphs showingdistributionofMM-PBSAbinding freeenergy, total polar contribution, and total vanderWaals contribution calculated for each frame in10nsmolecular dynamics simulations ofDNA-AMFcomplexes in (A)minor grooveof 226D, (B)major grooveof 1QC1, (C)minor grooveof 2K0V, and (D)major groove of 2K0V.
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
146
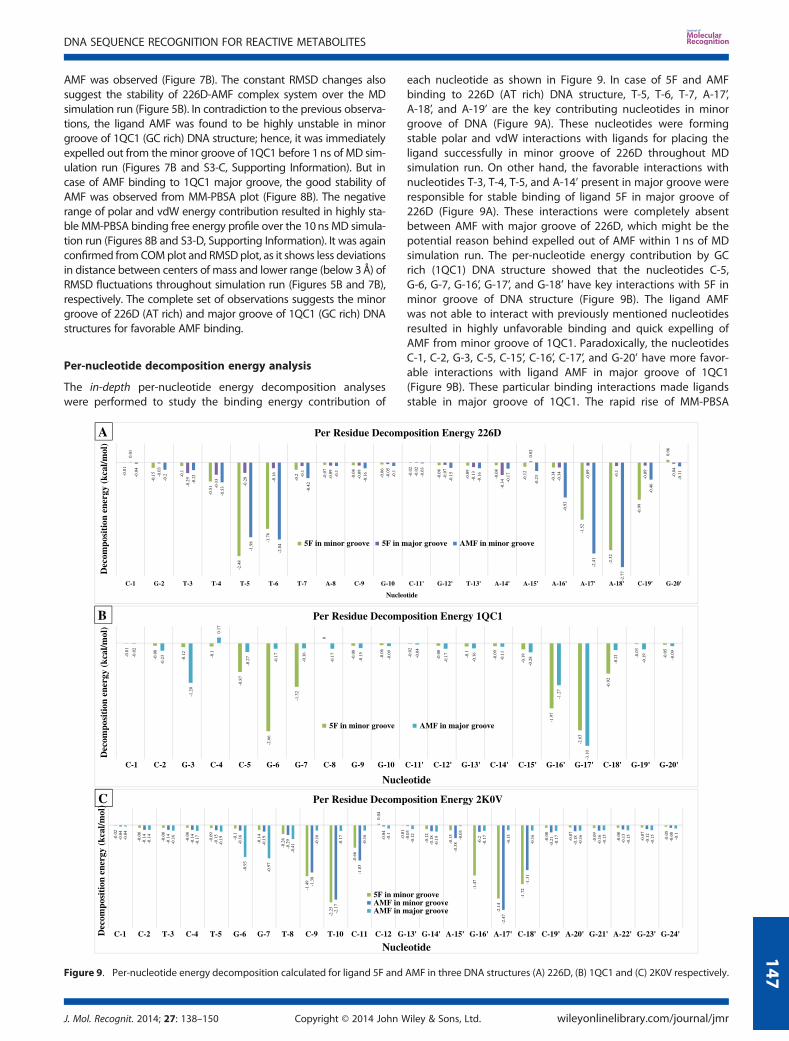
AMF was observed (Figure 7B). The constant RMSD changes alsosuggest the stability of 226D-AMF complex system over the MDsimulation run (Figure 5B). In contradiction to the previous observa-tions, the ligand AMF was found to be highly unstable in minorgroove of 1QC1 (GC rich) DNA structure; hence, it was immediatelyexpelled out from theminor groove of 1QC1 before 1ns of MD sim-ulation run (Figures 7B and S3-C, Supporting Information). But incase of AMF binding to 1QC1 major groove, the good stability ofAMF was observed from MM-PBSA plot (Figure 8B). The negativerange of polar and vdW energy contribution resulted in highly sta-ble MM-PBSA binding free energy profile over the 10ns MD simula-tion run (Figures 8B and S3-D, Supporting Information). It was againconfirmed fromCOMplot and RMSDplot, as it shows less deviationsin distance between centers of mass and lower range (below 3Å) ofRMSD fluctuations throughout simulation run (Figures 5B and 7B),respectively. The complete set of observations suggests the minorgroove of 226D (AT rich) and major groove of 1QC1 (GC rich) DNAstructures for favorable AMF binding.
Per-nucleotide decomposition energy analysis
The in-depth per-nucleotide energy decomposition analyseswere performed to study the binding energy contribution of
each nucleotide as shown in Figure 9. In case of 5F and AMFbinding to 226D (AT rich) DNA structure, T-5, T-6, T-7, A-17’,A-18’, and A-19’ are the key contributing nucleotides in minorgroove of DNA (Figure 9A). These nucleotides were formingstable polar and vdW interactions with ligands for placing theligand successfully in minor groove of 226D throughout MDsimulation run. On other hand, the favorable interactions withnucleotides T-3, T-4, T-5, and A-14’ present in major groove wereresponsible for stable binding of ligand 5F in major groove of226D (Figure 9A). These interactions were completely absentbetween AMF with major groove of 226D, which might be thepotential reason behind expelled out of AMF within 1 ns of MDsimulation run. The per-nucleotide energy contribution by GCrich (1QC1) DNA structure showed that the nucleotides C-5,G-6, G-7, G-16’, G-17’, and G-18’ have key interactions with 5F inminor groove of DNA structure (Figure 9B). The ligand AMFwas not able to interact with previously mentioned nucleotidesresulted in highly unfavorable binding and quick expelling ofAMF from minor groove of 1QC1. Paradoxically, the nucleotidesC-1, C-2, G-3, C-5, C-15’, C-16’, C-17’, and G-20’ have more favor-able interactions with ligand AMF in major groove of 1QC1(Figure 9B). These particular binding interactions made ligandsstable in major groove of 1QC1. The rapid rise of MM-PBSA
Figure 9. Per-nucleotide energy decomposition calculated for ligand 5F and AMF in three DNA structures (A) 226D, (B) 1QC1 and (C) 2K0V respectively.
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
147

energy graph was observed for AMF in major groove binding of1QC1 (Figures 8B and 9C), which was due to slightly bad interactionswith G-3, C-4, and G-17’ nucleotides (Figure 9B). These observationsprompted us to conclude that the guanine nucleotides present inminor groove of GC rich structure were responsible for goodinteractions and necessary for stable binding of ligands resulted informing DNA adducts.
Similar analysis was performed for mixed sequence (2K0V)DNA structure showing that key nucleotides were T-8, C-9,T-10, C-11, G-16’, A-17’, C-18’, and C-19’ (Figure 9C). Particularly,the ligand 5F was found to be stable only in minor groove of2K0V DNA structure (Figures 6E and 9D) and have favorable bind-ing in major groove of 2K0V (Figures 6F, 8D, and 9E; Figures S2-F,S3-E, and S4-F in Supporting Information). The per-nucleotideenergy contribution by the previously mentioned nucleotides forligand AMF was found to be less as compared with ligand 5F.These mentioned key interactions were necessary for successfulplacement of ligand in minor groove of 2K0V. In case of AMF inmajor groove of 2K0V, the nucleotides G-6 and G-7 have smallcontribution to the total binding free energy and poor interactionto hold AMF in major groove, of resulted into expelled out of AMFafter 5 ns of simulation. Previous observations helped us inconcluding the importance of the mentioned nucleotides pres-ent in major and minor groove of DNA, which were responsiblefor stable binding of ligand to the mixed nucleotide sequenceDNA structures.
Molecular electrostatic surface potential analysis
Additionally, we performed MESP analysis to explore the reasonbehind substantial binding preference of the ligands with minorgroove of DNA structure (Figure 10). The distribution of chargesin minor and major groove of DNA structures was searched bygenerating electrostatic potential maps on the Connolly sol-vent-accessible surface by using the MOLCAD program includedin SYBYL7.1 (Molecular modeling software packages, version 7.1,Tripos Associates, Inc., St. Louis). MOLCAD’s rendering techniqueallows the rapid calculation and display of property-codedsurfaces for the molecular recognition. The MESP analysis ofthree DNA structures suggested that the minor groove of 2K0V(mixed) has comparatively less electronegative surface potential(around �1300 kcal/mol) than minor groove of 226D (AT rich)and 1QC1 (GC rich) DNA structures (Figure 10).
It was also observed that the difference between electronega-tive potential at major and minor groove of 2K0V was least~250 kcal/mol as compared with 226D and 1QC1, while 226Dhas the highest difference of around ~800 kcal/mol. When the
binding preferences exhibited by studied ligands werecompared with electronegativity differences, it was observedthat the GC rich sequence structure has the highest value of elec-tronegativity of around ~9600 kcal/mol in minor groove and alsoshown to have comparatively stable binding of ligands in theminor groove of 1QC1. In case of AT rich sequence structure,the moderate difference in electronegativity was observedbetween minor and major groove of around ~700 kcal/mol.The differential stability of ligands was observed in minor andmajor groove of 2K0V. For example, ligand 5F has stable bindingprofile in minor groove, whereas ligand AMF is stable only inmajor groove. It showed the stable binding of ligands in minoras well as major groove of 2K0V DNA structure. The probablereason behind binding preferences was moderate difference inelectronegativity, which is due to different nucleotide sequencesand their orientations in minor and major groove of DNAstructures. These observations gave us an idea about theinfluence of charge distribution on ligand binding to minorand major groove of DNA structures.
CONCLUSIONS
The molecular docking, MD simulations, per-nucleotidedecomposition energy contribution, and MESP analysis wereperformed to study the binding preference of reactive metabo-lites of two anticancer compounds towards different DNAstructures. The DNA structures selected for this study weresupposed to cover the three majorly occurring fragments inDNA such as 226D (AT rich), 1QC1 (GC rich), and 2K0V (mixedsequence) structures. The results obtained from moleculardocking exercise were preliminary to explain the binding pref-erence of ligands 5F and AMF towards the minor and majorgroove of DNA structures. Hence, the study was extended toMD simulations of DNA-ligand complexes in minor and majorgroove of DNA structures. We have successfully developedand validated the protocol for reactive metabolite-DNAsimulations using AMBER 11 software as described earlier. TheMD simulation results concluded that the reactive metaboliteshave differential binding preference towards minor and majorgroove of DNA structures depending upon the nucleotideenvironment present in it. The potential anticancer moiety 5Fwas showing specific binding only in minor groove of all DNAstructures; however, the bioactive moiety AMF, which is largerin size than 5F, is stable in minor groove of AT rich and in majorgroove of GC rich nucleotides sequences. The MM-PBSAbinding energy obtained for AMF strongly suggested the
Figure 10. Molecular Electrostatic Surface Potential generated for 226D, 1QC1, and 2K0V DNA structures from MOLCAD program (Tripose SYBYL7.1)using Gasteiger-Hückel charges.
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
148

preferential binding of ligands in either major or minor grooveof sequence specific DNA structures. The crucial nucleotidespresent in the minor and major grooves were identified, whichwere responsible for successful placement of ligands, as theyhave more favorable contribution in polar and vdW interactionswith ligands. The distribution of charges in the minor and majorgroove has played a crucial role in DNA-ligand binding andshowed potential effect on binding preference of ligandstoward DNA. Finally, we concluded that DNA binding abilityof reactive metabolites is not only dependent the DNA grooves(minor and major) but also the specific nucleotide sequenceand charge environment present in the binding site ofDNA fragments.
We have successfully explained the DNA binding preferencesshown by reactive metabolites of anticancer compounds. Theidea of study of reactive metabolites binding to DNA could behelpful for molecular modeling scientists in designing of newand specific anticancer compounds in early phase of drugdiscovery. The extension of such studies for more number ofcompounds and various DNA fragments would lead to unveilthe mystery of reactive species as DNA binder.
Acknowledgements
The authors acknowledge financial support from NIPER S.A.S.Nagar and Department of Biotechnology (DBT), New Delhi.
REFERENCES
Andac CA, Miandji AM, Hornemann U, Noyanalpan N. 2011. Use of theparmbsc0 force field and trajectory analysis to study the binding ofnetropsin to the DNA fragment (5’CCAATTGG)(2) in the presence of ex-cess NaCl salt in aqueous solution. Int. J. Biol. Macromol. 48: 531–539.
Beck DAC, Armen RS, Daggett V. 2004. Cutoff size need not strongly influ-ence molecular dynamics results for solvated polypeptides. Biochem-istry 44: 609–616.
Beveridge DL, Cheatham TE, 3rd, Mezei M. 2012. The ABCs of moleculardynamics simulations on B-DNA, circa 2012. J. Biosci. 37: 379–397.
Bostock-Smith CE, Harris SA, Laughton CA, Searle MS. 2001. Induced fitDNA recognition by a minor groove binding analogue of Hoechst33258: fluctuations in DNA A tract structure investigated by NMRand molecular dynamics simulations. Nucleic Acids Res. 29: 693–702.
Bradshaw T, Westwell A. 2004. The development of the antitumourbenzothiazole prodrug, Phortress, as a clinical candidate. Curr. Med.Chem. 11: 1009–1021.
Caldwell J, Kollman P. 1986. A molecular mechanical study of netropsin-DNA interactions. Biopolymers 25: 249–266.
Case D, Darden T, Cheatham III T, Simmerling C, Wang J, Duke R, Luo R,Walker R, Zhang W, Merz K. 2010. AMBER 11. University of California,San Francisco.
Chobe SS, Dawane BS, Tumbi KM, Nandekar PP, Sangamwar AT. 2012. Anecofriendly synthesis and DNA binding interaction study of somepyrazolo [1,5-a]pyrimidines derivatives. Bioorg. Med. Chem. Lett. 22:7566–7572.
Ewing TJA, Kuntz ID. 1997. Critical evaluation of search algorithms for au-tomated molecular docking and database screening. J. Comput.Chem. 18: 1175–1189.
Frisch MJ, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Mont-gomery J, Vreven T, Kudin K, Burant J. 2008. Gaussian 03, revision C. 02.
Galindo-Murillo R, Ruiz-Azuara L, Moreno-Esparza R, Cortes-Guzman F.2012. Molecular recognition between DNA and a copper-based anti-cancer complex. Phys. Chem. Chem. Phys.: PCCP 14: 15539–15546.
Gassman PG. 1970. Nitrenium ions. Accounts. Chem. Res. 3: 26–33.Harrison JF, Eakers CW. 1973. Electronic structure of reactive intermediates.
Nitrenium ions nitrenium(+), fluoronitrenium(+), and difluoronitrenium(+). J. Am. Chem. Soc. 95: 3467–3472.
Hélène C. 1993. Sequence-selective recognition and cleavage of double-helical DNA. Curr. Opin. Biotechnol. 4: 29–36.
Hurley LH. 2002. DNA and its associated processes as targets for cancertherapy. Nat. Rev. Cancer 2: 188–200.
Hurley LH, Needham-VanDevanter DR. 1986. Covalent binding ofantitumor antibiotics in the minor groove of DNA. Mechanism ofaction of CC-1065 and the pyrrolo(1,4)benzodiazepines. Acc. Chem.Res. 19: 230–237.
Ismail MA, Brun R, Wenzler T, Tanious FA, Wilson WD, Boykin DW. 2004.Novel Dicationic Imidazo[1,2-a]pyridines and 5,6,7,8-Tetrahydro-imidazo[1,2-a]pyridines as antiprotozoal agents. J. Med. Chem.47: 3658–3664.
Izbicka E, Tolcher A. 2004. Development of novel alkylating drugs asanticancer agents. Curr. Opin. Investig. Drugs 5: 587–591.
Jayaram B, Jain T. 2004. The role of water in protein-DNA recognition.Annu. Rev. Biophys. Biomol. Struct. 33: 343–361.
Kohn KW. 1996. Beyond DNA cross-linking: history and prospects of DNA-targeted cancer treatment—Fifteenth Bruce F. Cain Memorial AwardLecture. Cancer Res. 56: 5533–5546.
Leong CO, Gaskell M, Martin EA, Heydon RT, Farmer PB, Bibby MC, Coo-per PA, Double JA, Bradshaw TD, Stevens MFG. 2003. Antitumour2-(4-aminophenyl)benzothiazoles generate DNA adducts in sensitivetumour cells in vitro and in vivo. Br. J. Cancer 88(3): 470–477.
Levitt M. 1983. Computer simulation of DNA double-helix dynamics. ColdSpring Harbor Symp. Quant. Biol. 47: 251–262.
Levitt M. 1984. Molecular dynamics of the DNA double helix. In Specificityin Biological Interactions, Chagas C, Pullman B (eds). Springer:Netherlands; 43–57.
Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Šali A. 2000.Comparative protein structure modeling of genes and genomes.Annu. Rev. Bioph. Biom. 29: 291–325.
Meng L-h, Kohlhagen G, Liao Z-y, Antony S, Sausville E, Pommier Y. 2005.DNA-protein cross-links and replication-dependent histone H2AXphosphorylation induced by aminoflavone (NSC 686288), a novel an-ticancer agent active against human breast cancer cells. Cancer Res.65: 5337–5343.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, OlsonAJ. 1998. Automated docking using a Lamarckian geneticalgorithm and an empirical binding free energy function.J. Comput. Chem. 19: 1639–1662.
Muegge I, Rarey M. 2001. Small Molecule Docking and Scoring. InReviews in Computational Chemistry, Volume 17, Lipkowitz KB, BoydDB (eds). John Wiley & Sons, Inc.: New York; 1–60.
Nandekar PP, Sangamwar AT. 2012. Cytochrome P450 1A1-mediatedanticancer drug discovery: in silico findings. Expert Opin. Drug Dis.7: 771–789.
Neidle S. 2001. DNA minor-groove recognition by small molecules. Nat.Prod. Rep. 18: 291–309.
Nguyen B, Hamelberg D, Bailly C, Colson P, Stanek J, Brun R, Neidle S,Wilson WD. 2004. Characterization of a novel DNA minor-groovecomplex. Biophys. J. 86: 1028–1041.
Nielsen PE. 1991. Sequence-selective DNA recognition by synthetic li-gands. Bioconjugate Chem. 2: 1–12.
O’Brien SE, Browne HL, Bradshaw TD, Westwell AD, Stevens MF, LaughtonCA. 2003. Antitumor benzothiazoles. Frontier molecular orbital analysispredicts bioactivation of 2-(4-aminophenyl)benzothiazoles to reactiveintermediates by cytochrome P4501A1. Org. Biomol. Chem. 1: 493–497.
Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA,Orozco M. 2007. Refinement of the AMBER force field for nucleicacids: improving the description of alpha/gamma conformers.Biophys. J. 92: 3817–3829.
Perez A, Lankas F, Luque FJ, Orozco M. 2008. Towards a molecular dynamicsconsensus view of B-DNA flexibility. Nucleic Acids Res. 36: 2379–2394.
Pobst L, Ames M. 2006. CYP1A1 activation of aminoflavone leads to DNAdamage in human tumor cell lines. Cancer Chemother. Pharmacol.57: 569–576.
Ponomarev SY, Thayer KM, Beveridge DL. 2004. Ion motions in moleculardynamics simulations on DNA. Proc. Natl. Acad. Sci. U. S. A.101: 14771–14775.
Portugal J, Waring MJ. 1987. Hydroxyl radical footprinting of thesequence-selective binding of netropsin and distamycin to DNA.FEBS Lett. 225: 195–200.
Rarey M, Kramer B, Lengauer T, Klebe G. 1996. A fast flexible dockingmethod using an incremental construction algorithm. J. Mol. Biol.261: 470–489.
DNA SEQUENCE RECOGNITION FOR REACTIVE METABOLITES
J. Mol. Recognit. 2014; 27: 138–150 Copyright © 2014 John Wiley & Sons, Ltd. wileyonlinelibrary.com/journal/jmr
149

Rosenberg M, Court D. 1979. Regulatory sequences involved in thepromotion and termination of RNA transcription. Annu. Rev. Genet.13: 319–353.
Shaikh SA, Ahmed SR, Jayaram B. 2004. A molecular thermodynamic viewof DNA–drug interactions: a case study of 25 minor-groove binders.Arch. Biochem. Biophys. 429: 81–99.
Shen LL, Mitscher LA, Sharma PN, O’Donnell T, Chu DWT, Cooper CS,Rosen T, Pernet AG. 1989. Mechanism of inhibition of DNA gyraseby quinolone antibacterials: a cooperative drug-DNA binding model.Biochemistry 28: 3886–3894.
Singh U, Weiner SJ, Kollman P. 1985. Molecular dynamics simulations of d(CGCGA) X d (TCGCG) with and without" hydrated" counterions. Proc.Natl. Acad. Sci. U. S. A. 82: 755–759.
Srivastava HK, Sastry GN. 2012. Efficient estimation of MMGBSA-based BEs forDNA and aromatic furan amidino derivatives. J. Biomol. Struct. Dyn. 5:522–537.
Srivastava HK, Chourasia M, Kumar D, Sastry GN. 2011. Comparison ofcomputational methods to model DNA minor groove binders.J. Chem. Inf. Model. 51: 558.
Steitz TA. 1990. Structural studies of protein–nucleic acid interaction: thesources of sequence-specific binding. Q. Rev. Biophys. 23: 205–280.
Tidor B, Irikura KK, Brooks BR, Karplus M. 1983. Dynamics of DNA Oligo-mers. J. Biomol. Struct. Dyn. 1: 231–252.
Trapani V, Patel V, Leong CO, Ciolino HP, Yeh GC, Hose C, Trepel JB, StevensMFG, Sausville EA, Loaiza-Perez AI. 2003. DNAdamage and cell cycle ar-rest induced by 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole (5F
203, NSC 703786) is attenuated in aryl hydrocarbon receptor deficientMCF-7 cells. Br. J. Cancer 88: 599–605.
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Im-proved protein–ligand docking using GOLD. Proteins: Struct., Funct.,Bioinf. 52: 609–623.
Wang H, Laughton CA. 2007. Molecular modelling methods for predictionof sequence-selectivity in DNA recognition. Methods (San Diego,Calif.) 42: 196.
Wang H, Laughton C. 2010. Molecular Modelling Methods to QuantitateDrug-DNA Interactions. In Drug-DNA Interaction Protocols, Fox KR(ed). Humana Press: New York; 119–131.
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. 2004. Develop-ment and testing of a general amber force field. J. Comput. Chem.25: 1157–1174.
Wolfson HJ, Rigoutsos I. 1997. Geometric hashing: An overview. Comput.Sci. & Engin., IEEE 4: 10–21.
Zakrzewska K, Lavery R, Pullman B. 1984. The solvation contribution tothe binding energy of DNA with non-intercalating antibiotics. NucleicAcids Res. 12: 6559–6574.
SUPPORTING INFORMATIONAdditional supporting information may be found in the onlineversion of this article at the publisher’s web site.
K. M. TUMBI ET AL.
wileyonlinelibrary.com/journal/jmr Copyright © 2014 John Wiley & Sons, Ltd. J. Mol. Recognit. 2014; 27: 138–150
150














![Naturally occurring triterpene Lupane exerts anticancer ... · biological functions [1]. These naturally occurring compounds fall in broad categories of primary and secondary metabolites](https://img.pdfslide.us/doc/110x75/5fd8c7dbf631af59543bd391/naturally-occurring-triterpene-lupane-exerts-anticancer-biological-functions.jpg)














